
ABSTRACT
The most common mutation in cystic fibrosis patients is a phenylalanine deletion at position 508 (ΔF508) in the CFTR (cystic fibrosis transmembrane conductance regulator) gene. This mutation impairs cell-surface trafficking of CFTR. During cellular stress, core-glycosylated CFTRΔF508 is transported to the cell surface from the endoplasmic reticulum (ER) via an unconventional route that bypasses the Golgi. However, the mechanisms for this unconventional secretory pathway of CFTR are not well delineated. Here, we report that components of the macroautophagy/autophagy and ESCRT (endosomal sorting complex required for transport) pathways are involved in unconventional secretion of CFTR. In mammalian cells, we found that autophagic pathways were modulated by conditions that also stimulate unconventional secretion, namely ER stress and an ER-to-Golgi transport blockade. Additionally, we found that knockdown of early autophagy components, ATG5 and ATG7, and treatment with pharmacological autophagy inhibitors, wortmannin and 3-methyladenine, abolished the unconventional secretion of CFTR that had been stimulated by ER stress and an ER-to-Golgi blockade. Interestingly, immunoelectron microscopy revealed that GORASP2/GRASP55, which mediates unconventional CFTR trafficking, is present in multivesicular bodies (MVB) and autophagosomal structures under ER stress conditions. A custom small-interfering RNA screen of mammalian ESCRT proteins that mediate MVB biogenesis showed that silencing of some ESCRTs, including MVB12B, inhibited unconventional CFTRΔF508 secretion. Furthermore, MVB12B overexpression partially rescued cell-surface expression and Cl− channel function of CFTRΔF508. Taken together, these results suggest that components involved in early autophagosome formation and the ESCRT/MVB pathway play a key role in the stress-induced unconventional secretion of CFTR.
Introduction
CFTR (cystic fibrosis transmembrane conductance regulator) is a chloride and bicarbonate channel in the apical membrane of epithelial cells [Citation1]. Defects in CFTR cause cystic fibrosis (CF), a life-threatening disease that affects the lungs, pancreas, and other epithelial organs. The CFTR protein undergoes a complex glycosylation process as it passes through conventional Golgi-mediated exocytosis [Citation2,Citation3]. Though more than 1,500 different mutations have been identified, the most common CFTR mutation in CF patients is a phenylalanine deletion at position 508 (ΔF508). This mutation causes protein misfolding and premature degradation by the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway, thus preventing translocation of CFTR to the plasma membrane [Citation4]. Circumvention of CFTRΔF508 arrest and premature degradation in the ER is the subject of active research, with the goal of developing a therapeutic approach to alleviating CF. CFTR is an N-glycosylated protein of which initial transfer of precursor oligosaccharide occurs in the ER (core glycosylation, also known as band B) and subsequent processing and modification of the oligosaccharide chain is carried out in the Golgi apparatus (complex glycosylation, also known as band C) [Citation5]. From a biochemical perspective, the prevailing view is that immature CFTR (band B) localizes to the ER and is rarely detected in the plasma membrane, whereas mature CFTR (band C) is predominantly present in compartments distal to the Golgi, including the apical membrane of epithelial cells [Citation5]. Therefore, CFTRΔF508 manifests as the immature band B and wild-type CFTR primarily appears as band C.
Most proteins are transported from the ER to the extracellular space or plasma membrane via the classical ER-to-Golgi pathway. However, recent studies show that some proteins reach the extracellular space or plasma membrane in a Golgi-independent manner called ‘unconventional trafficking’ [Citation6]. Unconventional translocation of the immature core-glycosylated CFTR to the plasma membrane occurs under conditions of cellular stress, particularly during ER stress or an ER-to-Golgi blockade [Citation7]. ARF1 (ADP ribosylation factor 1) is a component of the coat protein complex I (COPI) complex. Although COPI is primarily known to mediate retrograde transport from the Golgi and ER-Golgi intermediate compartment (ERGIC) membranes to the ER, ARF1 inhibition also blocks anterograde transport of vesiculotubular elements from the ER to the Golgi stacks [Citation8]. SAR1 (secretion associated Ras related GTPase 1) is an important component of the COPII complex that mediates anterograde ER-to-Golgi transport [Citation9]. STX5 (syntaxin 5) is required for the fusion of COPII transport vesicles with the acceptor Golgi membranes [Citation10]. Blockage of conventional ER-to-Golgi transport, either by ectopic expression of dominant-inhibitory forms of ARF1 and SAR1 mutants or by STX5 overexpression, stimulates unconventional trafficking of core-glycosylated wild-type and CFTRΔF508 to the plasma membrane via a GORASP/GRASP (golgi reassembly stacking protein)-mediated route [Citation7,Citation11]. Recent results indicate that SEC16A (SEC16 homolog A, endoplasmic reticulum export factor) is required for CFTR to exit the ER during unconventional secretion [Citation12]. However, the vesicular trafficking pathway that mediates this unconventional ER to cell surface transport of core-glycosylated CFTR is largely uncharacterized.
Autophagy is a conserved cellular pathway that maintains intracellular homeostasis by degrading proteins and cytosolic contents. The process requires sequestration of cytoplasmic material by a transient compartment termed the phagophore, which matures into a double-membrane autophagosome. Although autophagy primarily functions in degradative and disposal pathways, it is also involved in several biogenesis processes [Citation13,Citation14]. Recent studies have reported that proteins lacking a signal sequence, including yeast Acb1 (a homolog of Dictyostelium discoideum AcbA), IL1B (interleukin 1 beta), and IDE (insulin degrading enzyme), can be secreted via GORASP- and autophagy-dependent mechanisms [Citation14–Citation17], suggesting that the GORASP and autophagy pathways may converge. However, the functional relevance of interplay between these pathways, in unconventional trafficking and in downstream autophagy-related vesicle transport to the cell surface, remains unknown. Furthermore, their roles in unconventional trafficking of proteins containing a signal peptide, including CFTR, are not known.
In this study, we aim to elucidate the vesicular trafficking factors and pathways involved in unconventional CFTR secretion. Our results indicate that components of autophagosome formation and of the endosomal sorting complex required for transport (ESCRT) participate in the unconventional trafficking of core-glycosylated CFTR. In particular, a component of the ESCRT-I complex, MVB12B (multivesicular body subunit 12B), was important in the unconventional secretion of CFTR. We found that MVB12B depletion impaired the unconventional trafficking of CFTRΔF508 to the cell surface, and that MVB12B overexpression partially rescued surface expression and ion channel function of CFTRΔF508.
Results
ER-to-Golgi blockade induces an increase of autophagosome formation
Blockage of ER-to-Golgi transport results in an accumulation of secretory proteins in the ER lumen, which evokes ER stress and the unfolded protein response (UPR) [Citation18]. This induces unconventional secretion of CFTR in mammalian cells [Citation7]. Recent reports suggest that ER stress and UPR activation increase autophagic flux [Citation19]. To examine whether an ER-to-Golgi transport blockade induces autophagy, we monitored the conversion of MAP1LC3B/LC3 (microtubule associated protein light chain 3 beta) under ER stress (thapsigargin) and ER-to-Golgi blockade (ARF1 and SAR1 mutants) conditions in HEK293 cells (). MAP1LC3B, a mammalian homolog of yeast Atg8, is widely used to monitor autophagy, because the amount of the phosphatidylethanolamine-conjugated form of MAP1LC3B (LC3-II) is correlated with the number of autophagosomes [Citation20]. Thapsigargin induces ER stress and the UPR by depleting Ca2+ in the ER. Induction of ER stress by treatment with thapsigargin, or blockage of ER-to-Golgi transport using dominant-inhibitory forms of ARF1Q71L and SAR1T39N mutants [Citation21], both increased the LC3-II:ALDOA (aldolase, fructose-bisphosphate A) ratio in HEK293 cells, indicating an activation of autophagosome formation under these conditions (). In order to determine whether the increase in LC3-II levels is a result of either upregulation of autophagosome formation or blockage of autophagosome degradation, we monitored MAP1LC3B in the presence of the lysosomal fusion inhibitor bafilomycin A1 (Figure S1) [Citation20]. In an early period of ER-to-Golgi blockade (e.g., 24 h after transfection with ARF1Q71L and SAR1T39N), bafilomycin A1 significantly further increased LC3-II levels, indicating that autophagosome formation was increased (Figure S1A and S1B). However, in the later period of ER-to-Golgi blockade and ER stress (e.g., 48 h after transfection with ARF1Q71L and SAR1T39N, and treatment with thapsigargin for 6 h) bafilomycin A1 did not further increase LC3-II levels (Figure S1C and S1D), suggesting that these conditions may also induce the inhibition of autophgosome degradation. It has been previously reported that SAR1T39N may inhibit starvation-induced autophagy [Citation22]. Similar to the previous result, SAR1T39N partially reduced the LC3-II levels induced by starvation (Figure S2). However, apart from this partial inhibition, SAR1T39N alone strongly increased LC3-II levels (Figure S2), suggesting that the SAR1T39N-mediated ER-to-Golgi blockade independently induces autophagosome accumulation.
Figure 1. ER-to-Golgi transport blockade and ER stress induce autophagosome accumulation. (a and b) Autophagosome accumulation was measured by monitoring LC3-II conversion. HEK293 cells were transfected with ARF1Q71L- or SAR1T39N- expression plasmids (36 h), or treated with thapsigargin (1 µM, 12 h). Cells were then lysed and whole cell lysates were immunoblotted with anti-MAP1LC3B/LC3B antibodies. (a) Representative immunoblots are shown. ALDOA was used as a loading control. (b) A summary of multiple experiments (n = 4) is given. ARF1Q71L, SAR1T39N, and thapsigargin increased the amount of LC3-II relative to ALDOA. *P < 0.05, **P < 0.01, relative to the first lane. (c-g) Confocal images were taken from HEK293 cells expressing tandem mRFP-GFP fluorescent-tagged LC3 (Tf-LC3). Cells grown on glass-bottom dishes were subjected to the following conditions: (c) untreated control, (d) starvation (2 h), (e) ARF1Q71L transfection (36 h), (f) thapsigargin (1 µM, 6 h). Arrows indicating a red signal represent autolysosomes and arrowheads indicating a yellow signal represent autophagosomes. (g) A summary of multiple experiments is given (Control, n = 15; Starvation, n = 14; ARF1Q71L, n = 14; Thapsigargin, n = 16). **P < 0.01, relative to Control (yellow dots). Bar graph data are mean ± SEM.
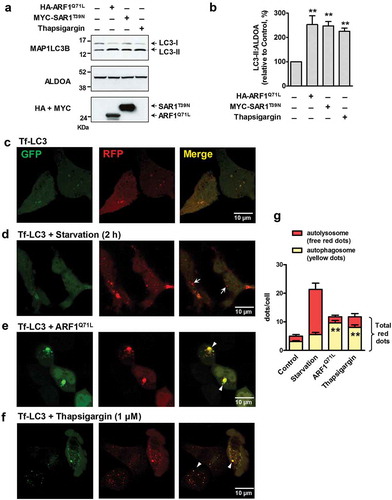
Next, we visualized autophagosome accumulation and LC3 translocation by fluorescence microscopy using a monomeric red fluorescent protein-green fluorescent protein (mRFP-GFP) tandem fluorescent-tagged LC3 (Tf-LC3) construct [Citation16]. Before fusion with lysosomes, autophagosomal Tf-LC3 produces a yellow signal in merged images, resulting from mRFP-GFP fluorescence. After fusion with lysosomes, autolysosomal Tf-LC3 produces a red signal, due to loss of the GFP signal under acidic conditions. Confocal microscopy images of control HEK293 cells showed low levels of yellow Tf-LC3 signal ()). However, starvation-induced autophagy strongly increased the number of red fluorescent puncta (), arrows), indicating autolysosome formation. Interestingly, the number of yellow fluorescent puncta increased in cells that expressed ARF1Q71L or were treated with thapsigargin, which induced unconventional trafficking of CFTRΔF508 [Citation7], (, suggesting that autophagosome formation without lysosome fusion is primarily activated during ER stress or an ER-to-Golgi blockade. A summary of the immunofluorescence results is presented in ).
Autophagosome formation is required for unconventional transport of CFTR
We then used pharmacological inhibitors to examine whether the autophagy pathway is involved in the rescue of CFTRΔF508. Wortmannin and 3-methyladenine (3-MA) are widely used autophagy inhibitors due to their inhibitory effect on class III phosphatidylinositol 3-kinase (PtdIns3K) activity [Citation23–Citation25], which is essential for membrane nucleation in autophagy [Citation26]. Both wortmannin and 3-MA treatments almost completely blocked thapsigargin-induced transport of CFTRΔF508 to the plasma membrane (). Rescued surface expression of CFTRΔF508 by GORASP1/GRASP65 or GORASP2/GRASP55 overexpression was also abolished by wortmannin or 3-MA treatment ().
Figure 2. Impairment of autophagosome formation inhibits unconventional secretion of CFTRΔF508. HEK293 cells expressing CFTRΔF508 were surface biotinylated and immunoblotted. Before surface biotinylation, wortmannin (100 nM, 12 h), 3-methyladenine (3-MA; 2 mM, 12 h), spautin-1 (5 μM, 6 h), or siRNA against PIKC3/VPS34 (100 nM 36 h) was treated to inhibit autophagosome formation by inhibiting PIK3C3. (a and b) One-day after transfection with pCMV-CFTRΔF508, cells were treated with thapsigargin (1 µM, 12 h) and surface biotinylated. Representative immunoblots (a) and a summary of 3 independent experiments (b) are shown. (c and d) HEK293 cells were transfected with plasmids expressing CFTRΔF508 and GORASP2/GRASP55 or GORASP1/GRASP65 (36 h), and surface biotinylated. Representative immunoblots (c) and a summary of 3 independent experiments (d) are shown. (e and f) HEK293 cells were transfected with plasmids expressing CFTRΔF508 and/or ARF1Q71L (36 h), and surface biotinylated. Representative immunoblots (e) and a summary of 3 independent experiments (f) are shown. (g-i) Cells were transfected with CFTRΔF508 and GORASP2 expression plasmids, and/or siRNAs against PIK3C3/VPS34 (100 nM, 36 h). Representative immunoblots (g) and a summary of 3 independent experiments (h) are shown. Individual siRNA information is given in Table S1 and the efficiency of each siRNA treatment was validated by quantitative real-time PCR (i). The fold-change in gene expression normalized to GAPDH and relative to the control sample was calculated using the 2−ΔΔCt method (n = 3). Bar graph data are mean ± SEM. **P < 0.01, relative to each control.
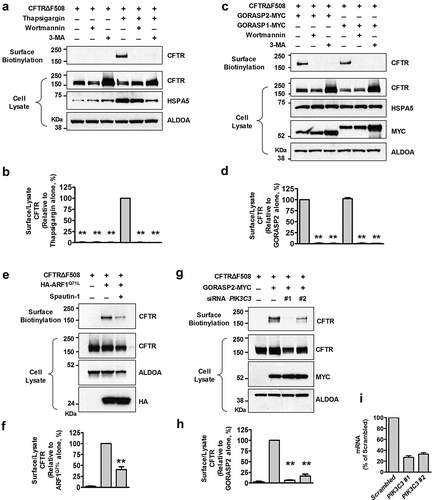
Additionally, we examined the effect of spautin-1, a more specific inhibitor of the PtdIns3K catalytic subunit (PIK3C3/VPS34) that is responsible for autophagosome formation in eukaryotic cells [Citation27]. Treatment with spautin-1 again reduced surface expression of CFTR induced by ARF1Q71L overexpression (). We then inhibited autophagy induction by knockdown of PIK3C3 using siRNAs. Depletion of PIK3C3 strongly suppressed GORASP2 overexpression-induced unconventional transport of CFTRΔF508 (,h)). These experiments indicate that autophagosome formation via the PtdIns3K signaling pathway is important for unconventional trafficking of core-glycosylated CFTR.
Autophagosome formation requires the activity of autophagy-related (ATG) proteins, which were first studied in yeast and later identified in mammals [Citation28]. Recent work indicates that ATGs are involved in unconventional secretion of some soluble cargos lacking a signal sequence [Citation14–Citation17]. We first inactivated some early autophagy regulators. ATG13 and RB1CC1/FIP200 play a key role in the initial stage of autophagy as parts of the ULK (unc-51 like autophagy activating kinase) complex [Citation29,Citation30]. Knockdown of ATG13 or RB1CC1 strongly reduced ARF1Q71L-induced surface expression of CFTRΔF508 (Figure S3).
Previously, we have suggested that some key ATGs of autophagosome formation, including ATG5 and ATG7, would be required for unconventional secretion of CFTR (Figure S6 of reference 8) [Citation7]. Therefore, in the present study, we further analyzed the involvement of these ATGs in unconventional CFTR trafficking using both biochemical and morphological approaches with wild-type CFTR as a control. ATG5 is an E3 ubiquitin ligase required for phagophore elongation [Citation31]. Depletion of ATG5 did not affect conventional surface trafficking of complex-glycosylated wild-type CFTR (; band C). However, ATG5 knockdown strongly inhibited ARF1Q71L-induced unconventional cell-surface expression of core-glycosylated wild-type and CFTRΔF508 (; band B). ATG7 is an E1-like enzyme that mediates ATG12–ATG5 conjugation and lipidation of LC3, which is essential for core autophagy in mammals [Citation32,Citation33]. Similar to the results with ATG5, ATG7 knockdown impaired trafficking of core-glycosylated wild-type and CFTRΔF508, but not complex-glycosylated wild-type CFTR (–h)). The above results were supported by immunocytochemistry (). In , co-transfection with ARF1Q71L in HEK293 cells rescued expression of CFTRΔF508 on the plasma membrane. Importantly, knockdown of ATG5 and ATG7 abolished the cell surface expression of mutant CFTR induced by ARF1Q71L (). We next examined whether general autophagy inducers such as starvation and treatment with rapamycin induce the surface expression of CFTRΔF508; however, autophagy inducers by themselves showed no effect on membrane trafficking of CFTR (). These data indicate that autophagy activation alone is not sufficient for evoking unconventional secretion of CFTR.
Figure 3. ATG5 and ATG7 are required for unconventional secretion of core-glycosylated-CFTR. HEK293 cells were transfected with the indicated plasmids or siRNAs (36 h), and then CFTR on the cell surface was biotinylated and immunoblotted. (a-d) Cells were treated with siRNAs against ATG5 (100 nM, 36 h) and cell surface expression of wild-type and CFTR∆F508 was analyzed by a surface biotinylation assay. Representative immunoblots (a and b) and a summary of 3 independent experiments (c and d) of wild-type and CFTRΔF508, respectively, are shown. (e–h) Cells were treated with siRNAs against ATG7 (100 nM, 36 h) and other experimental conditions were the same as in panels A – D. Representative immunoblots (e and f) and a summary of 3 independent experiments (g and h) of wild-type and CFTRΔF508, respectively, are shown. Immunoblots using antibodies against ATG5 and ATG7 represent knockdown efficiency of siRNA. Individual siRNA information is given in Table S1. Bar graph data are mean ± SEM. B, band B (core-glycosylated) CFTR; C, band C (complex-glycosylated) CFTR; *P < 0.05, **P < 0.01 relative to each control.
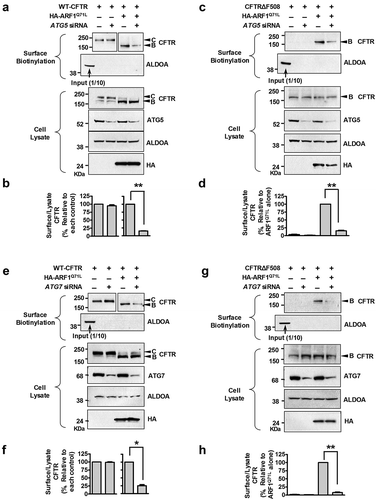
Figure 4. Immunofluorescence analysis of CFTRΔF508 in cells treated with siRNAs against ATG5 and ATG7. HEK293 cells were transfected with the indicated plasmids or siRNAs (36 h), and then immunostained with anti-CFTR (green, Alexa Flour 488) and anti-HA (red, Alexa Fluor 568) antibodies. Anti-HA immunofluorescence represents the expression levels of ARF1Q71L. Fields enclosed by boxes in CFTR images are shown at higher magnification to the left. (a) CFTRΔF508 is mostly expressed in intracellular regions. (b) Co-transfection with ARF1Q71L in HEK293 cells induced cell-surface expression of CFTRΔF508 (white arrows). (c Treatments with siRNAs against ATG5 (siATG5) and (d) treatments with siRNAs against ATG7 (siATG7) abolished the ARF1Q71L-induced cell surface expression of mutant CFTR. Four independent experiments showed similar results. Scale bar: 10 µm.
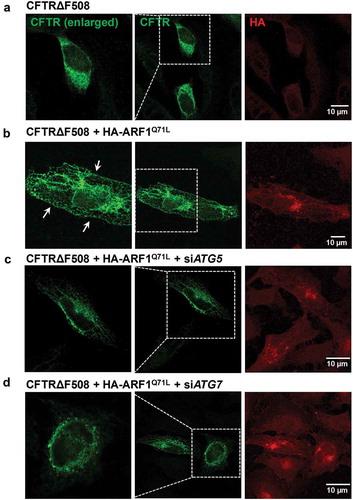
Figure 5. Autophagy inducers and an autophgosome-lysosome fusion inhibitor do not evoke unconventional trafficking of CFTRΔF508. HEK293 cells were transfected with the indicated plasmids, and then surface biotinylated and immunoblotted. (a–d) Before surface biotinylation, cells were treated with rapamycin (a and b; 300 nM, 24 h) or nutrient starvation (c and d; 2 h) was used to induce autophagy. Representative immunoblots (a and c) and a summary of 3 independent experiments (b and d), respectively, are shown. (e and f) Before surface biotinylation, bafilomycin A1 (100 nM, 1 h) treated was carried out to inhibit autophagosome-lysosome fusion. Representative immunoblots (e) and a summary of 3 independent experiments (f) are shown. Bar graph data are mean ± SEM.
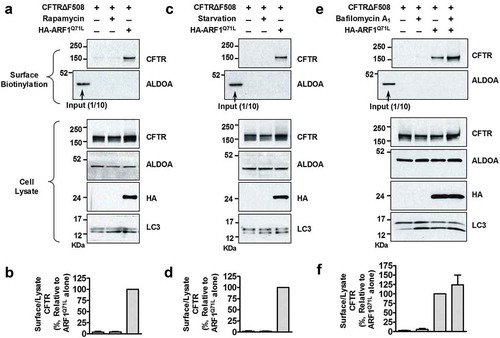
Previously, it has been shown that VAMP7 (vesicle associated membrane protein 7), a v-SNARE involved in autophagosome-lysosome fusion, is not required for the unconventional secretion of CFTR [Citation7]. Consistent with this finding, treatment with bafilomycin A1, an autophagosome-lysosome fusion inhibitor, did not affect unconventional transport of CFTR (). Moreover, knockdown of STX17 (syntaxin 17), a t-SNARE involved in autophagosome-lysosome fusion [Citation34], also did not significantly affect the rescue of CFTRΔF508 by ARF1Q71L (Figure S4A and S4B). Taken together, late autophagy components involved in autophagosome-lysosome fusion appear to be dispensable in unconventional secretion of CFTR. Interestingly, knockdown of KIF5A (kinesin family member 5A), which is a kinesin motor that associates with VAMP7 vesicles [Citation35], inhibited ARF1Q71L-induced surface expression of CFTR∆F508 (Figure S4C and S4D). However, KIF5A is not only involved in the trafficking of VAMP7 vesicles but in many other microtubule-dependent vesicular trafficking events to the cell periphery [Citation35,Citation36]. Therefore, this result suggests that KIF5A- and microtubule-dependent vesicular traffics play a role in unconventional cell surface transport of CFTR.
Unconventional trafficking of CFTR∆F508 requires components of the MVB pathway
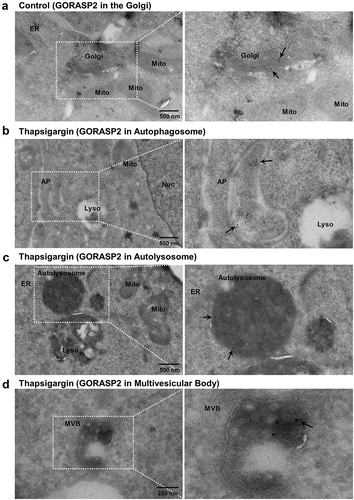
To characterize the route of unconventional CFTR secretion, we used immunogold electron microscopy (EM). We previously showed that GORASP2 physically interacts with CFTR and mediates the unconventional secretion of core-glycosylated CFTR during ER stress [Citation7,Citation11]. Because CFTR antibodies did not function in preliminary immunogold EM experiments, we labeled GORASP2 with gold and examined its intracellular localization, focusing on vesicular compartments using the EM images taken from ultrathin sections (). In control HEK293 cells, GORASP2 mostly localized to the Golgi ()). Upon thapsigargin treatment, which induces surface expression of core-glycosylated CFTR, GORASP2 was detected in crescent-shaped double-membrane structures, which are a typical shape of early autophagosomes ()). This finding supports our biochemical analyses that autophagosomes are involved in unconventional secretion of CFTR (–). However, GORASP2 did not localize exclusively to early autophagosomes, as some gold-labeled GORASP2 particles were present in autolysosomes ()). Notably, upon thapsigargin treatment, GORASP2 was also observed in membranous structures (50 – 100 nm in size) that contained many intralumenal vesicles, known as multivesicular bodies (MVBs) ()). Semi-quantification of EM images indicated that the thapsigargin treatment increased the number of autophagosomes and MVBs, as well as the proportion of GORASP2-labeling in both structures (Figure S5A). The increase in MVB structures upon thapsigargin treatment was further evidenced by immunoblot analysis of the MVB protein TSG101 (tumor susceptibility 101) (Figure S5B and S5C) [Citation37]. Consistent with the EM images, immunofluorescence analyses indicate that thapsigargin significantly increased colocalization between GORASP2 and the autophagosome marker GFP-LC3 or the MVB protein MVB12B (see below) (Figure S6).
Figure 6. Immunoelectron microscopy of GORASP2/GRASP55. Transmission electron microscopy was performed in HEK293 cells after immunogold labeling of GORASP2, as described in the Materials and Methods. Fields enclosed by boxes are shown at higher magnification to the right. (a) In control cells, GORASP2 was principally localized in the Golgi complex. Upon thapsigargin treatment (1 µM, 6 h), GORASP2 was detected in (b) crescent-shaped autophagosomes (AP), (c) autolysosomes, and (d) multivesicular bodies (MVB). Arrows indicate immunogold-labeled GORASP2. Mito, mitochondria; Nuc, nucleus; Lyso, lysosome.
Because the EM images suggested that GORASPs localized to MVBs under unconventional secretion-inducing conditions, we investigated whether the ESCRT machinery is required for unconventional trafficking of CFTRΔF508. In addition, under certain autophagy conditions, autophagosomes fuse with MVB endosomal compartments to generate hybrid organelles called amphisomes [Citation38,Citation39]. HEK293 cells were treated with siRNAs against ESCRT-related genes, and biotinylation assays were performed (). As shown in ), single knockdowns of 6 ESCRT genes (MVB12B, SNF8/VPS22, VPS25, VPS36, CHMP4B, and VPS4A) significantly reduced ARF1Q71L-induced rescue of mutant CFTR. However, knockdown of these genes did not affect cell-surface expression of complex-glycosylated wild-type CFTR (Figure S7), indicating that MVBs are not involved in the conventional Golgi-mediated route.
Figure 7. Unconventional trafficking of CFTRΔF508 requires MVB12B. (a) Results of quantification of a surface biotinylation assay using siRNAs against MVB genes. HEK293 cells were transfected with CFTRΔF508 and ARF1Q71L expression plasmids and siRNAs against the indicated MVB genes (100 nM, 36 h). Then, CFTRΔF508 on the cell surface was biotinylated, immunoblotted, and quantitatively analyzed. Individual siRNA information is given in Table S1. (b and c) HEK293 cells were transfected with the indicated plasmids and/or siRNA against MVB12B. Representative immunoblots (b) and a summary of 3 independent experiments (c) are shown. Immunoblots using antibodies against MVB12B represent the knockdown efficiency of siRNA. Depletion of MVB12B reduces the ARF1Q71L-induced surface expression of CFTRΔF508. (d and e) HEK293 cells were transfected with the indicated plasmids and coimmunoprecipitation experiments between MVB12B and CFTR were performed. Protein samples were immunoprecipitated with anti-MYC (MVB12B) and immunoblotted with anti-CFTR, anti-MYC, and anti-HA (ARF1) antibodies. MVB12B principally interacts with core-glycosylated CFTR during ARF1 inhibition. Bar graph data are mean ± SEM. B, band B (core-glycosylated) CFTR; C, band C (complex-glycosylated) CFTR; **P < 0.01, relative to each control.
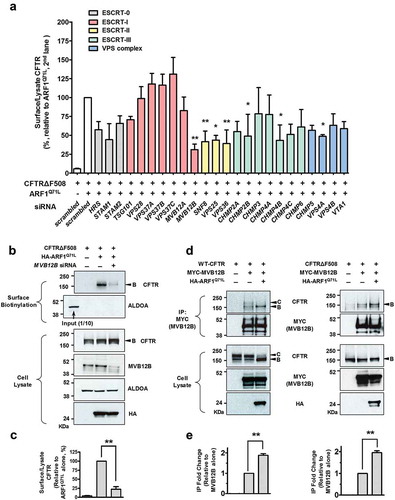
Of the 24 ESCRT genes investigated, we found that MVB12B knockdown resulted in the strongest and most significant inhibition of unconventional secretion of CFTRΔF508 (P = 0.0005). Representative results of MVB12B silencing surface biotinylation assays are shown in . MVB12B knockdown also inhibited GORASP2 overexpression-induced unconventional cell-surface expression of CFTRΔF508 (Figure S8). We then examined whether MVB12B physically interacts with CFTR (). Co-immunoprecipitation results indicated MVB12B associated with the ER form of core-glycosylated wild-type and CFTRΔF508 (band B), whereas it less likely interacted with the Golgi form of complex-glycosylated CFTR (band C). More importantly, ER-to-Golgi blockade by ARF1Q71L resulted in a stronger interaction between core-glycosylated CFTR and MVB12B, supporting the idea that MVB12B participates in the ER-to-Golgi blockade-induced unconventional secretion of the ER form of CFTR.
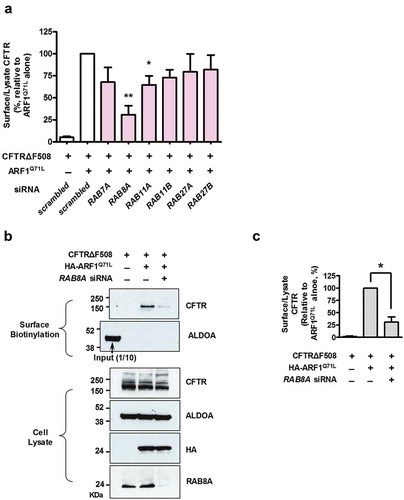
The RAB GTPases regulate many vesicular trafficking and membrane fusion events including those in the autophagy and MVB pathways. We, therefore, focused our investigation of unconventional secretion of CFTRΔF508 on RAB proteins that are reported to be involved in the autophagy and MVB pathways [Citation13,Citation16,Citation40–Citation42], using siRNA-based gene silencing (). One of these proteins, RAB8A (RAB8A, member RAS oncogene family), which plays a role in recycling endosomes [Citation43], appears to be involved in unconventional trafficking of CFTR. RAB8A knockdown markedly reduced the ARF1Q71L-induced cell-surface expression of CFTRΔF508, suggesting that cargo trafficking from MVBs to recycling endosomes may contribute to the unconventional secretion of CFTRΔF508.
Figure 8. Unconventional trafficking of CFTRΔF508 requires RAB8A. (a) Quantification of a surface biotinylation assay using siRNAs against RAB genes that are involved in the autophagy and MVB pathways. HEK293 cells were transfected with CFTRΔF508 and ARF1Q71L expression plasmids and siRNAs against the indicated RAB genes (36 h). Then, CFTRΔF508 on the cell surface was biotinylated, immunoblotted, and analyzed quantitatively. Individual siRNA information is given in Table S1. (b and c) HEK293 cells were transfected with the indicated plasmids and/or siRNA against RAB8A. Representative immunoblots (b) and a summary of 3 independent experiments (c) are shown. Immunoblots using antibodies against RAB8A represent the knockdown efficiency of siRNA. Bar graph data are shown as mean ± SEM. *P < 0.05, ***< 0.001, relative to ARF1Q71L + scrambled siRNA.
MVB12B partially rescues the cell-surface expression of CFTRΔF508
Because MVB12B is required for unconventional secretion of CFTRΔF508 ( and S8), we next examined whether upregulation of MVB12B would rescue the cell-surface expression and anion channel function of CFTRΔF508. Notably, co-expression of MVB12B in HEK293 cells resulted in surface expression of CFTRΔF508 at 33% of the level of expression induced by ARF1Q71L (). VPS28, of which knockdown did not affect the unconventional secretion of CFTRΔF508 ()), was included as a negative control. MVB12B overexpression appeared to partially increase the total MVB levels as judged by immunoblot analysis of TSG101; however, the increase was not statistically significant (Figure S5B and S5C). In cells depleted of ATG5 and ATG7, MVB12B retained significant levels of ability for inducing unconventional trafficking of CFTRΔF508 (Figure S9, compare results in ), indicating that MVB12B function is downstream of autophagy. However, depletion of ATG5 and ATG7 partially reduced the CFTR unconventional secretion (Figure S9), implying that basal levels of autophagy may enhance the MVB12B function of inducing unconventional secretion.
Figure 9. MVB12B partially rescues the cell-surface expression and chloride channel function of CFTRΔF508. (a and b) HEK293 cells were transfected with CFTRΔF508 expression plasmids and plasmids expressing the indicated proteins (mock, MVB12B, VPS28, or ARF1Q71L). Representative immunoblots (a) and a summary of 4 independent experiments (b) are shown. VPS28 was used as a negative control for an MVB gene (see ). (c-g) Whole-cell patch clamp recordings were performed in HEK293 cells transfected with the indicated plasmids. Representative traces (d–g) and a summary of multiple experiments (C) are shown (WT-CFTR, n = 7; CFTRΔF508, n = 13; CFTRΔF508 + MVB12B, n = 12; CFTRΔF508 + VPS28, n = 7; CFTRΔF508 + ARF1Q71L, n = 9). (d) Cells expressing wild-type (WT) CFTR show typical CFTR chloride currents, which are inhibited by CFTRinh-172. Cells expressing CFTRΔF508 show no discernable chloride currents. (e) Co-expression of MVB12B evokes CFTR currents in CFTRΔF508-expressing cells, (f) while cells co-expressing VPS28 do not. (g) ARF1Q71L was used as positive control for inducing CFTR currents in CFTRΔF508-expressing cells. Bar graph data are mean ± SEM. **P < 0.01, relative to CFTRΔF508 alone.
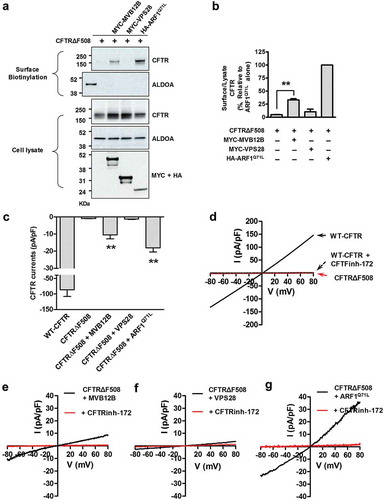
Furthermore, we confirmed that MVB12B overexpression resulted in functional rescue of CFTRΔF508 by Cl− current measurements (). ) shows the typical CFTR Cl− currents in cells expressing wild-type CFTR, which is activated by cAMP, shows a linear current-voltage relationship, and is inhibited by CFTR inhibitor-172. Cells transfected with CFTRΔF508 expression plasmids did not produce discernible CFTR currents ()). However, CFTR currents were detected in CFTRΔF508-expressing HEK293 cells that co-expressed MVB12B (), but not VPS28 ()). Currents in MVB12B co-expression cells were found to be 11% and 56% of the level of current in cells expressing wild-type CFTR ()) and CFTRΔF508 with ARF1Q71L ()), respectively. A summary of multiple experiments is provided in ).
Discussion
Most secreted proteins follow a well-characterized route from the ER to the Golgi and eventually to the plasma membrane. However, recent studies suggest that some proteins reach this final destination by alternative routes, and that an unconventional pathway can be used to transport CFTR to the plasma membrane under certain conditions [Citation7,Citation8]. There are a variety of unconventional transport routes taken by each of the known protein substrates [Citation44]. However, these unconventional secretion routes all seem to be switched on by cellular stress. In other words, unconventional secretion of most substrates is non-constitutive and triggered by stress events, including inflammation, starvation, or mechanical stress arising from cell-cell rearrangements during development [Citation44,Citation45]. Although these studies have helped refine the understanding of how cellular trafficking works, the complete mechanism underlying unconventional secretion remains undefined. This study demonstrates that several cellular trafficking components are involved in unconventional trafficking of core-glycosylated CFTR in HEK293 cells.
Notably, we found that specific ESCRT proteins are involved in the unconventional surface trafficking of core-glycosylated CFTR. In general, ESCRTs comprise a pathway of 5 distinctive complexes (ESCRT-0, -I, -II, -III, and VPS4) and are involved in the ubiquitin-dependent protein sorting pathways that deliver cargos to the vacuole or lysosome for degradation [Citation46]. In our study, MVB12B was the most relevant gene involved in unconventional CFTR secretion. Both knockdown and overexpression of this gene affected rescue of mutant CFTR ( and ). To date, MVB12B is known as a component of ESCRT-I, which functions in endocytic degradation of some membrane proteins, including EGFR (epidermal growth factor receptor) [Citation47], and also functions in human immunodeficiency virus budding [Citation48]. Human ESCRT-I is made up of TSG101, VPS28, VPS37, and MVB12, with a stoichiometry of 1:1:1:1. Additional VPS37 and MVB12 isoforms exist in mammals, although the significance of these multiple subunit isoforms is not yet known [Citation46]. As shown by the differential regulation of EGFR by MVB12A and MVB12B [Citation47], the wide variety of ESCRT-I complex combinations using 4 subunits of Vps37A-D and 3 MVB12 subunits (MVB12A, MVB12B, and UBAP1 [ubiquitin associated protein 1]) may allow the adjustment of responses to diverse cellular processes, such as cargo selection, vesicular localization (e.g., endosome vs. autophagosome), and biological functioning (e.g., degradation vs. recycling). Interestingly, although MVB12A and UBAP1, the third MVB12-like ESCRT-1 subunit, have ubiquitin-binding activity [Citation49], MVB12B does not have a ubiquitin-binding domain [Citation47]. A recent report suggests that non-ubiquitinated cargos are prone to unconventional secretion, by showing that USP19 (ubiquitin specific peptidase 19), an ER-associated deubiquitinase, promotes unconventional secretion of a misfolded GFP mutant (GFP1-10) [Citation46]. Therefore, it is plausible that non-ubiquitinated cargos are recognized by an ESCRT-I complex containing MVB12B and travel to the cell surface through an unconventional pathway during ER stress, whereas ubiquitinated cargos are recognized by an ESCRT-I complex containing MVB12A or UBAP1 and are destined for degradation.
In addition to MVB12B, gene knockdown experiments suggest that some other ESCRTs, in particular ESCRT-II components, are involved in the unconventional secretion of CFTR (). Interestingly, the ESCRT-II complex has a GLUE domain providing endosomal localization specificity by binding with PtdIns3P, which is generated in endosomes through the PtdIns3K that also plays a key role in autophagy [Citation50]. In the case of HIV-1, an ESCRT-I complex, but not ESCRT-II, is required for budding [Citation51]. Therefore, the route and molecular components of CFTR unconventional secretion appear to be different from those of HIV budding.
Autophagy and the UPR pathways usually degrade and clear unfolded proteins. Our study suggests that, in addition to degradation, autophagosomal vesicles provide an alternative route for the transport of core-glycosylated CFTR to the plasma membrane. The autophagosome is a hallmark organelle of macroautophagy, and the mechanism of autophagosome formation is relatively well-defined. Knockdown of several key factors involved in autophagosome formation, including ATG5 and ATG7, resulted in inhibition of core-glycosylated CFTR secretion ( and ), suggesting that CFTR is sequestered within autophagosomes during unconventional secretion. Interestingly, knockdown of ATG5 and ATG7 did not affect the transport of fully glycosylated wild-type CFTR to the plasma membrane (). This result implies that autophagy-related trafficking is specifically involved in the unconventional secretion route of core-glycosylated CFTR and is not part of the conventional pathway.
It appears that ER-to-Golgi blockade and ER stress also increase autophagosome accumulation by inhibiting its degradation. Treatment with bafilomycin A1 did not further increase LC3-II levels in the later period of ER-to-Golgi blockade and ER stress, indicating an inhibition of autophgosome degradation (Figure S1C and S1D), which would further contribute to the autophagosome-mediated unconventional secretion of CFTR. However, one thing we need to consider is that accumulation of autophagosomes alone by treatment with starvation, rapamycin, or bafilomycin A1 did not evoke unconventional surface transport of CFTR (). These results indicate that events other than autophagosome formation, in particular those involved in the ER exit of cargos, such as activation and redistribution of GORASP and SEC16A [Citation11,Citation12], are also required and work together with autophagosome components for unconventional transport of CFTR evoked by ER-to-Golgi blockade or ER stress.
An intriguing question that remains to be answered is the precise role of GORASP in the unconventional trafficking of CFTR and other cargos. Although GORASP was initially characterized as functioning in Golgi reassembly and stacking, later studies indicated that it is involved in unconventional trafficking of several proteins [Citation6,Citation45]. In the context of core-glycosylated CFTR trafficking, it has been shown that GORASP2 relocalizes to the ER exit sites during ER stress and ER-to-Golgi blockade [Citation12]. Therefore, it is possible that GORASP plays a role in cargo recognition during ER exit and subsequent travel to vesicles for cell-surface trafficking. Specifically, because ER exit sites are a physical and functional core of autophagosome biogenesis [Citation52], GORASP may help tether CFTRΔF508 and shuttle it to autophagosomal structures at the ER exit site. In fact, EM images in our study revealed that GORASP2 localized to autophagosomes during ER stress ()). Another possibility is that GORASP2 itself might affect autophagic flux. Several reports have suggested this possibility, after the discoveries that GORASPs and autophagosome components are involved in unconventional secretion of yeast Acb1 [Citation15] and macrophage IL1B [Citation13]. However, our previous studies did not identify any toxicities caused by GORASP overexpression in mammalian cells and mice [Citation7,Citation11]. These results suggest that GORASP may not simply evoke a global increase in autophagic flux, and that further research is necessary to better understand the interplay between GORASP and autophagic activation.
Interestingly, our EM analyses showed that GORASP2 was also present in autolysosomes, albeit less frequently than those observed in autophagosomes ()). Therefore, it appears that ER-to-Golgi blockade or ER stress signals do not completely block autophagosome-lysosome fusion, and a certain level of autophagic flux is maintained; as shown in , a certain level of Tf-LC3 red fluorescent puncta representing autolysosomes remained present under these conditions, suggesting that the association between GORASP and autophagy machinery may also contribute to relieve the protein burden in the ER lumen via a degradation pathway in addition to unconventional secretion. Another interesting observation in the EM image is that GORASP2 appears to localize on the inner vesicle membrane of the autophagosome and MVB upon ER stress (). According to the classical concept of the vesicle fusion process, cargos on the outer membrane rather than those of the inner membrane would be suitable for localizing on the plasma membrane. Further research is needed to understand how the cargos on the inner vesicle membrane can reach the cell surface. A possibility is the reuptake of extracellular vesicles after shedding, as has been shown in a variety of endocytic pathways [Citation53].
Because several RAB proteins play roles in autophagy and MVB pathways, we examined the role of these RABs in unconventional secretion of CFTRΔF508 (). Notably, siRNA silencing of RAB8A resulted in the strongest reduction of unconventional secretion of CFTRΔF508 (). RAB8A regulates post-Golgi vesicle traffic from the trans-Golgi network to the cell surface, and deletion of RAB8A in intestinal epithelial cells induces missorting of apical proteins into lysosomes [Citation13,Citation43]. In addition, RAB8A is required for autophagy-dependent secretion of cytosolic proteins, including IL1B and IDE (insulin degrading enzyme) [Citation13,Citation16]. Therefore, it appears that RAB8A regulates vesicle traffic to the common recycling endosomes from MVBs, as well as from the trans-Golgi network. It also seems likely that the transporting vesicles with membrane cargos, such as CFTR, and with soluble protein cargos, such as IL1B/IL-1β and IDE, share this RAB8A-mediated recycling route to the cell surface. RAB11 is necessary for autophagosome-MVB fusion [Citation41], and RAB27 is involved in MVB docking at the plasma membrane [Citation42]. However, single knockdown of these RABs had only a partial or no effect on the unconventional trafficking of CFTRΔF508 ()). Therefore, it is possible that the multiple isoforms of these RABs are redundant in mammalian cells, or that RABs involved in autophagosome-MVB fusion may be more important to MVBs directed to the degradation pathway than those directed to the secretion pathway.
A model for unconventional secretion of core-glycosylated CFTR based on our findings is provided in Figure S10. Under ER stress or ER-to-Golgi blockade conditions, the core-glycosylated CFTR leaves the ER and is subsequently recruited to the autophagic vacuoles and MVBs. Although some (presumably poly-ubiquitinated) CFTRs are routed to degradation pathways, other (presumably non-ubiquitinated or deubiquitinated) CFTRs travel to recycling vesicles via MVB12B- and RAB8A-mediated mechanisms. These cargos eventually travel to their final destination on the cell surface. Though further research is needed to validate this model, our findings provide valuable insight into the big picture of unconventional secretion of CFTR, and may also inform the understanding of unconventional secretion of other proteins, including soluble cytosolic cargos.
Materials and methods
Plasmids, cell culture, chemicals, and siRNA
pCIneo-HA-STX5 and pCIneo-MYC- SAR1 (gifts from B. L. Tang at National University of Singapore, Singapore), pcDNA3-HA- ARF1Q71L (a gift from S. Wakatsuki at the Kyoto University, Japan), pCMV-CFTR (pCMVNot6.2) (a gift from J. Rommens at the Hospital for Sick Children, Canada), tandem fluorescent-tagged LC3 (Tf-LC3) [Citation16], pCMV-MYC-MVB12B (Origene, RC222537), pCMV-MYC-VPS28 (Origene, RC200025), and pCMV-GORASP2-MYC (GRASP55-MYC) [Citation11] plasmids were used for transfection experiments. HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium-HG (Gibco, 11995–065) supplemented with 10% fetal bovine serum, penicillin (50 IU/mL), and streptomycin (50 μg/mL). For nutrient starvation, cells were starved for 2 h in EBSS (Gibco, 14155) before harvest. For visualization of lysosomes, 50 nM LysoTracker Red (Invitrogen, L7528) was added for 30 min in the culture medium before fixation. Plasmids were transiently transfected into HEK293 cells using Lipofectamine Plus Reagent (Invitrogen, 10963–013). Rapamycin (Sigma Aldrich, R8781), bafilomycin A1 (Sigma Aldrich, B1793), spautin-1 (Sigma Aldrich, SML0440), thapsigargin (Sigma Aldrich, T9033), wortmannin (Cayman, 10010591), and 3-methyladenine (Sigma Aldrich, M9281) were purchased from commercial sources. The control scrambled siRNAs and siRNAs against each gene were purchased from Dharmacon (Lafayette, CO, USA) and Bioneer (Daejeon, Korea). The information for siRNAs used in this study is listed in Table S1. siRNAs were transiently transfected into HEK293 cells using DharmaFECT1 (Dharmacon, T-2001).
Immunoblotting, immunoprecipitation and immunofluorescence staining
For immunoprecipitation, cell lysates were mixed with the anti-HA-conjugated (Sigma Aldrich, E6654) or anti-MYC-conjugated (Sigma Aldrich, E6779) beads and incubated overnight at 4°C in a lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% (v:v) Nonidet P-40 (USB Corporation, 19628), and complete protease inhibitor mixture (Roche Applied Science, 04693116001). Immune complexes were washed 4 times with lysis buffer prior to electrophoresis. For immunoblotting, cells were lysed with a lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% [v:v] Nonidet P-40, 0.25% [v:v] sodium deoxycholate [Fisher Scientific, 5285], and a complete protease inhibitor mixture). For biochemical analysis of LC3 and TSG101, cells were lysed with RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% [v:v] Nonidet P-40, 0.5% [v:v] sodium deoxycholate, 2 mM MgCl2, 2 mM EDTA, 1% [v:v] sodium dodecyl sulfate [Sigma Aldrich, L3771], and a complete protease inhibitor mixture). After centrifugation, protein samples were suspended in SDS buffer and separated by SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred to a nitrocellulose membrane and blotted with appropriate primary and secondary antibodies. Protein bands were detected by enhanced chemiluminescence (GE Healthcare, Amersham®, RPN2106).
For immunocytochemistry, HEK293 cells were cultured on coverslips and immunostained 2 days after transfection. Cells grown on coverslips were fixed and permeabilized by incubation in cold acetone for 10 min at −20°C. Nonspecific binding sites were blocked by incubation for 1 h at room temperature with a blocking medium composed of 0.1 ml of phosphate-buffered saline (PBS; Bioneer, C9024) containing 5% horse serum (Thermo Scientific, 26050070), 1% bovine serum albumin (BSA; Sigma Aldrich, A3059), and 0.1% gelatin (Sigma Aldrich, G1890). After blocking, cells were stained by incubation with the indicated primary antibodies and then treated with fluorophore-tagged secondary antibodies. Anti-CFTR M3A7 (Millipore, 05–583), anti-GORASP2 (Abcam, ab74579), anti-MAP1LC3B (Cell Signaling Technology, 2775), anti-ATG5 (Cell Signaling Technology, 2630), anti-ATG7 (Abcam, ab52472), anti-MVB12B (Novus, NBP2-13979), anti-CANX/calnexin (Abcam, ab22595), anti-HSPA5 (Cell Signaling Technology, 3177), anti-HA (Cell Signaling Technology, 2367), anti-MYC (Cell Signaling Technology, 2276), anti-GAPDH (Santa Cruz Biotechnology, sc-20358), anti-ALDOA (Santa Cruz Biotechnology, sc-12059) and anti-TSG101 (GeneTex, GTX70255) antibodies were purchased from commercial sources. Alexa Fluor 488 goat anti-mouse (Invitrogen, A11001), Alexa Fluor 488 goat anti-rabbit (Invitrogen, A11008), Alexa Fluor 568 goat anti-mouse (Invitrogen, A11004) and Alexa Fluor 568 goat anti-rabbit (Invitrogen, A11036) secondary antibodies were from Invitrogen. Fluorescence images were obtained with a Zeiss LSM 780 confocal microscope (Carl Zeiss, Berlin, Germany).
Surface biotinylation assay
After transfection with the indicated plasmids, HEK293 cells were washed 3 times with PBS. The plasma membrane proteins were then biotinylated by gently shaking for 30 min cells in a borate buffer containing sulfo-NHS-SS-biotin (Thermo, 21331). After biotinylation, cells were washed extensively with quenching buffer (1% BSA solution) to remove excess biotin and then washed twice with PBS. The cells were then lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% [v:v] Nonidet P-40, 0.25% [v:v] sodium deoxycholate, and complete protease inhibitor mixture) and incubated overnight at 4°C with avidin solution (UltraLink Immobilized NeutrAvidin Beads 10%; Thermo, 53150). Avidin-bound complexes were pelleted (15,700 x g, 20 min) and washed 3 times with lysis buffer. Biotinylated proteins were eluted in a 2X SDS sample buffer for 40 min at 37°C, resolved by SDS-PAGE, electrotransferred, and immunoblotted with the indicated antibodies.
Measurement of Cl− channel activities
Whole-cell recordings of Cl− current were performed on CFTR-expressing HEK293 cells. Glass coverslips containing cells transfected with plasmids for expressing GFP and the indicated proteins were transferred to a chamber on the stage of an inverted microscope (IX71, Olympus, Tokyo, Japan). Whole-cell currents were measured using an Axon 200B amplifier at a holding potential of −60 mV. The bath solution contained: 146 mM HCl (Sigma Aldrich, 320331), 146 mM N-methyl D-glutamine (NMDG; Sigma Aldrich, M2004), 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.4. The pipette solution contained: 148 mM HCl, 148 mM NMDG, 5 mM EGTA, 1 mM MgCl2, 3 mM Mg-ATP (Sigma Aldrich, A9287), 10 mM HEPES, pH 7.2. Pipettes were pulled from borosilicate glass with a resistance of 3–5 MΩ after fire polishing. Seal resistances were between 3 and 10 GΩ. After establishing the whole-cell configuration, CFTR was activated by addition of forskolin (10 µM; Tocris, 1099) and IBMX (Calbiochem, 410957). Whole-cell currents were elicited by applying hyperpolarizing and depolarizing voltage pulses from a holding potential of 0 mV to potentials between −100 and + 100 mV in a ramp pulse. Recordings were made at room temperature using an Axopatch-200B (Axon Instruments, Union City, CA, USA). Currents were digitized with a Digidata 1440A converter (Axon Instruments) and filtered at 5 kHz. Command potential and data acquisition were controlled by pClamp 10.2 software (Axon instruments). Data analysis was performed with Clampfit 10.2.
Immunoelectron microscopy
Immunogold labeling of ultrathin frozen sections was performed as previously described [Citation54]. HEK 293 cells grown in Petri dishes were immersion-fixed in situ with a mixture of 2% formaldehyde (freshly prepared from paraformaldehyde) and 0.1% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2 for 30 min at 37°C. After rinsing with buffer, cells were mechanically removed, sedimented by centrifugation, enclosed in liquefied 15% gelatin (Dr. Oetker, Gold extra Gelatine Sheets), infiltrated stepwise with 0.6 M, 1.2 M and 2.3 M sucrose (Sigma Aldrich, M6385), and frozen in liquid nitrogen. Ultrathin frozen sections (100 nm) were cut with cryo diamond knifes (Diatome) at −110°C using a Leica ultramicrotome equipped with a cryochamber. Before immunolabeling, sections attached to nickel grids were incubated with 2% gelatin in PBS at 40°C for 15 min, rinsed with PBS, and conditioned with PBS containing 1% BSA and 0.01% Tween 20 (Thermo Scientific, 28320).
For immunogold labeling, sections were incubated for 2 h at ambient temperature with rabbit anti-GORASP2 (40 μg/ml; Abcam, ab118728) diluted in PBS containing 1% BSA and 0.01% Tween 20. After rinsing with buffer, sections were incubated for 1 h with 12-nm Colloidal Gold AffiniPure Donkey Anti-Rabbit IgG (H + L) (EM Grade; Jackson ImmunoRes Labs., 711–205-152) diluted to an absorbance of 0.1 at 520 nm in PBS containing 1% BSA and 0.01% Tween 20. After rinsing with PBS and a final rinse in distilled water, sections were post-fixed with 1% glutaraldehyde for 5 min and embedded in 2% methylcellulose (USB corporation, 21938) containing 0.3% uranyl acetate. Immunogold-labeled sections were analyzed using a HitachiH-7650 electron microscope at 80 kV, and images were acquired with a CCD XR611-M digital camera (Advanced Microscopy Techniques, Woburn, MA, USA).
Data analysis and statistics
For western blot quantification, densitometric analyses were performed using Multi Gauge V3.0 software. Results represent the means ± SEM for the indicated number of experiments. Statistical analysis was performed using the Student’s t tests or with analysis of variance followed by Tukey’s multiple comparison test using GraphPad Prism7 software.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ALDOA | = | aldolase, fructose-bisphosphate A |
| ARF1 | = | ADP ribosylation factor 1 |
| ATG | = | autophagy-related |
| CF | = | cystic fibrosis |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| ΔF508 | = | deletion of phenylalanine at 508 |
| DMEM | = | Dulbecco’s modified Eagle medium |
| EBSS | = | Earle’s balanced salt solution |
| EM | = | electron microscopy |
| ER | = | endoplasmic reticulum |
| ESCRT | = | endosomal sorting complex required for transport |
| FBS | = | fetal bovine serum |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| GORASP1/GRASP65 | = | Golgi reassembly stacking protein 1 |
| GORASP2/GRASP55 | = | Golgi reassembly stacking protein 2 |
| HSPA5 | = | heat shock protein family A (Hsp70) member 5 |
| KIF5A | = | kinesin family member 5A |
| MAP1LC3 | = | microtubule associated protein 1 light chain 3 |
| MVB | = | multivesicular body |
| MVB12B | = | multivesicular body subunit 12B |
| PBS | = | phosphate-buffered saline |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| PIK3C3 | = | phosphatidylinositol 3-kinase catalytic subunit type 3 |
| RAB | = | member RAS oncogene family |
| RB1CC1/FIP200 | = | RB1 inducible coiled-coil 1 |
| SAR1 | = | secretion associated Ras related GTPase 1 |
| SEC16A | = | SEC16 homolog A, endoplasmic reticulum export factor |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| STX5 | = | syntaxin 5 |
| STX17 | = | syntaxin 17 |
| Tf-LC3 | = | tandem fluorescent-tagged LC3 |
| TG | = | thapsigargin |
| UPR | = | unfolded protein response |
| TSG101 | = | tumor susceptibility 101 |
| UBAP1 | = | ubiquitin associated protein 1 |
| VAMP7 | = | vesicle associated membrane protein 7 |
| Wort | = | wortmannin |
| WT | = | wild-type |
Supplemental Material
Download Zip (35.1 MB)Acknowledgments
We thank Dong Soo Chang for editorial assistance and the Yonsei-Carl Zeiss Advanced Imaging Center for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material can be accessed here.
Additional information
Funding
Related Research Data
References
- Park HW, Nam JH, Kim JY, et al. Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology. 2010 Aug;139(2):620–631. PMID:20398666.
- Gee HY, Tang BL, Kim KH, et al. Syntaxin 16 binds to cystic fibrosis transmembrane conductance regulator and regulates its membrane trafficking in epithelial cells. J Biol Chem. 2010 Nov 12;285(46):35519–35527. PMID:20826815.
- Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999 Jan;79(1 Suppl):S3–s22. PMID:9922374.
- Amaral MD. CFTR and chaperones: processing and degradation. J Mol Neurosci. 2004;23(1–2):41–48. PMID:15126691.
- Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003 Jul;285(1):C1–C18. PMID:12777252.
- Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009 Feb;10(2):148–155. PMID:19122676.
- Gee HY, Noh SH, Tang BL, et al. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011 Sep 2;146(5):746–760. PMID:21884936.
- Yoo JS, Moyer BD, Bannykh S, et al. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J Biol Chem. 2002 Mar 29;277(13):11401–11409. PMID:11799116.
- Russell C, Stagg SM. New insights into the structural mechanisms of the COPII coat. Traffic. 2010 Mar;11(3):303–310. PMID:20070605.
- Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005 Jul 10;1744(3):493–517. PMID:16038056.
- Kim J, Noh SH, Piao H, et al. Monomerization and ER Relocalization of GRASP is a requisite for unconventional secretion of CFTR. Traffic. 2016 Jul;17(7):733–753. PMID:27062250.
- Piao H, Kim J, Noh SH, et al. Sec16A is critical for both conventional and unconventional secretion of CFTR. Sci Rep. 2017 Jan 09;7:39887. PMID:28067262.
- Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 2011 Nov 30;30(23):4701–4711. PMID:22068051.
- Bruns C, McCaffery JM, Curwin AJ, et al. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol. 2011 Dec 12;195(6):979–992. PMID:22144692.
- Manjithaya R, Anjard C, Loomis WF, et al. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010 Feb 22;188(4):537–546. PMID:20156962.
- Son SM, Cha MY, Choi H, et al. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy. 2016 Mar 10;12:784–800. PMID:26963025.
- Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015 Nov 02;4. doi:10.7554/eLife.11205. PMID:26523392.
- Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999 Jul;79(3):683–701. PMID:10390516.
- Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. PMID:16822172.
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007 Nov-Dec;3(6):542–545. PMID:17611390.
- Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994 Jan 14;269(2):1437–1448. PMID:8288610.
- Zoppino FC, Militello RD, Slavin I, et al. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010 Sep;11(9):1246–1261. PMID:20545908.
- Blommaart EF, Krause U, Schellens JP, et al. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997 Jan 15;243(1–2):240–246. PMID:9030745.
- Vlahos CJ, Matter WF, Hui KY, et al. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994 Feb 18;269(7):5241–5248. PMID:8106507.
- Powis G, Bonjouklian R, Berggren MM, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994 May 1;54(9):2419–2423. PMID:8162590.
- Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001 Feb 5;152(3):519–530. PMID:11157979.
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PMID:21801009.
- Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009 May 01;284(18):12297–12305. PMID:19258318.
- Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009 Apr;20(7):1992–2003. PMID:19225151.
- Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007 Dec 28;282(52):37298–37302. PMID:17986448.
- Tanida I, Mizushima N, Kiyooka M, et al. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999 May;10(5):1367–1379. PMID:10233150.
- Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998 Sep 24;395(6700):395–398. PMID:9759731.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 07;151(6):1256–1269. PMID:23217709.
- Burgo A, Proux-Gillardeaux V, Sotirakis E, et al. A molecular network for the transport of the TI-VAMP/VAMP7 vesicles from cell center to periphery. Dev Cell. 2012 Jul 17;23(1):166–180. PMID:22705394.
- Nakajima K, Yin X, Takei Y, et al. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron. 2012 Dec 6;76(5):945–961. PMID:23217743.
- Kowal J, Tkach M, Théry C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014 Aug 1;29: 116–125.
- Fader CM, Colombo MI. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy. 2006 Apr-Jun;2(2):122–125. PMID:16874060.
- Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008 Jul;10(7):776–787. PMID:18552835.
- Fader CM, Sanchez D, Furlan M, et al. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008 Feb;9(2):230–250. PMID:17999726.
- Khandelwal P, Ruiz WG, Balestreire-Hawryluk E. Rab11a-dependent exocytosis of discoidal/fusiform vesicles in bladder umbrella cells. Proc Natl Acad Sci USA. 2008 Oct 14;105(41):15773–15778. PMID:18843107.
- Ostrowski M, Carmo NB, Krumeich S, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010 Jan;12(1):19–30; sup pp 11–13. PMID:19966785.
- Nakajo A, Yoshimura S, Togawa H, et al. EHBP1L1 coordinates Rab8 and Bin1 to regulate apical-directed transport in polarized epithelial cells. J Cell Biol. 2016 Feb 01;212(3):297–306. PMID:26833786.
- Rabouille C. Pathways of unconventional protein secretion. Trends Cell Biol. 2017 Mar;27(3):230–240. PMID:27989656.
- Giuliani F, Grieve A, Rabouille C. Unconventional secretion: a stress on GRASP. Curr Opin Cell Biol. 2011 Aug;23(4):498–504. PMID:21571519.
- Lee JG, Takahama S, Zhang G, et al. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016 Jul;18(7):765–776. PMID:27295555.
- Tsunematsu T, Yamauchi E, Shibata H, et al. Distinct functions of human MVB12A and MVB12B in the ESCRT-I dependent on their posttranslational modifications. Biochem Biophys Res Commun. 2010 Aug 20;399(2):232–237. PMID:20654576.
- Morita E, Sandrin V, Alam SL, et al. Identification of human MVB12 proteins as ESCRT-I subunits that function in HIV budding. Cell Host Microbe. 2007 Jul 12;2(1):41–53. PMID:18005716.
- Agromayor M, Soler N, Caballe A, et al. The UBAP1 subunit of ESCRT-I interacts with ubiquitin via a SOUBA domain. Structure. 2012 Mar 07;20(3):414–428. PMID:22405001.
- Babst M, Katzmann DJ, Snyder WB, et al. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell. 2002 Aug;3(2):283–289. PMID:12194858.
- Langelier C, von Schwedler UK, Fisher RD, et al. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J Virol. 2006 Oct;80(19):9465–9480. PMID:16973552.
- Graef M, Friedman JR, Graham C, et al. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell. 2013 Sep;24(18):2918–2931. PMID:23904270.
- Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracellular Vesicles. 2014 Aug;3(1):24641. PMID:PMC4122821.
- Roth J, Bendayan M, Orci L. Ultrastructural localization of intracellular antigens by the use of protein A-gold complex. J Histochem Cytochem. 1978 Dec;26(12):1074–1081. PMID:366014.