
ABSTRACT
Macroautophagy/autophagy (bulk autophagy) is a catabolic process that nonselectively degrades cytoplasmic proteins and organelles. In contrast to bulk autophagy, selective types of autophagy target specific cellular components as cargos, whereas their specific receptor proteins play central roles in cargo selection. In the yeast Saccharomyces cerevisiae, receptor proteins for the cytoplasm-to-vacuole targeting pathway, mitophagy, and pexophagy are phosphoregulated by kinases. This phosphorylation facilitates interaction with the scaffold/adaptor protein Atg11, subsequently recruiting core autophagy proteins to initiate autophagosome formation. However, the molecular mechanism inhibiting this phosphorylation to prevent unrequired selective autophagy remains unknown. Our recent study revealed that the protein phosphatase 2A-like protein phosphatase Ppg1 and its associated Far complex cooperatively inhibit mitophagy by counteracting casein kinase 2-mediated phosphorylation of the mitophagy receptor Atg32. Herein, we summarize our findings regarding Ppg1 and pose unanswered questions.
Mitochondrial synthesis of adenosine triphosphate (ATP) via oxidative phosphorylation produces most of the ATP required for various cellular activities. Additionally, this process generates reactive oxygen species as byproducts, which can damage the mitochondria. Therefore, damaged mitochondria must be eliminated to maintain mitochondrial homeostasis. Mitochondrial autophagy (mitophagy) contributes to maintaining mitochondrial quality and quantity via selective degradation of mitochondria. Recent studies revealed the molecular mechanisms of mitophagy in yeast by identifying the mitophagy receptor Atg32. Upon mitophagy induction, Atg32 is phosphorylated on Ser114 and Ser119 by casein kinase 2 (CK2), which is an essential trigger for Atg32-Atg11 interaction. Subsequently, the autophagy core machinery is recruited to initiate autophagosome formation that delivers mitochondria to the vacuole for degradation. Although CK2 is a constitutively active and abundant kinase, Atg32 is phosphorylated only under mitophagy-inducing conditions (i.e., a shift from respiratory medium to nitrogen starvation or continuous culture in a respiratory medium), suggesting that Atg32 phosphorylation is negatively regulated by unknown mechanisms to prevent unrequired mitophagy.
Initially, we screened the potential negative regulators of Atg32 phosphorylation using a comprehensive set of protein phosphatases from yeast mutants and their associated proteins. The screening was performed based on mitophagy-induction-independent puncta formation of N-terminal green fluorescent protein (GFP)-tagged Atg32 on mitochondria and Atg32 phosphorylation, which are sensitive indicators of the initial stage of mitophagy. The screening identified the PP2A-like protein phosphatase Ppg1, whose deletion affects the phosphorylation status of Atg32, unlike that of the cytoplasm-to-vacuole targeting pathway or the pexophagy receptors Atg19 and Atg36. Deletion of the PPG1 gene results in a constitutive Atg32-Atg11 interaction and accelerated mitophagy. Importantly, under growing conditions, mitophagy is not observed at a detectable level in ppg1Δ cells, but artificial activation of the autophagy core machinery through exogenous expression of a non-phosphorylatable Atg13 induces mitophagy in ppg1Δ cells, indicating that activation of the autophagy core machinery is also required for the progression of mitophagy following Atg32 phosphorylation.
PP2A (Pph21 and Pph22) and PP2A-like (Pph3 and Sit4) protein phosphatases interact with multiple associated proteins, such as Tpd3 (scaffold subunit) and Cdc55/Rts1 (regulatory subunits). However, these proteins are dispensable for Ppg1 function to dephosphorylate Atg32, suggesting that Ppg1 is associated with specific proteins. Using a mass spectrometry-based proteomics approach, we attempted to identify Ppg1-associated proteins; one of the identified proteins was Far8, a component of the Far complex. The Far complex comprises Far3, Far7, Far8, Far9, Far10, and Far11 and is involved in pheromone-induced cell cycle arrest, the TORC2 signaling pathway, and human CASP10/caspase-10-induced toxicity in yeast. Similar to ppg1Δ cells, yeast cells lacking any of the Far complex components (except Far10) show the same phenotypes (Atg32 phosphorylation, punctate formation of GFP-Atg32, and accelerated mitophagy), suggesting that Ppg1 and the Far complex cooperatively dephosphorylate Atg32 to inhibit mitophagy.
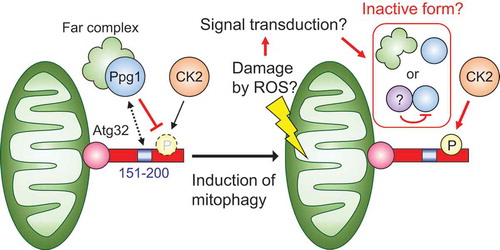
We were unable to detect a direct interaction between Atg32 and Ppg1 probably because of the transient nature of their interaction. Instead, we attempted to determine the region of Atg32 required for its dephosphorylation through Ppg1. Analysis of partial deletion mutants of Atg32 revealed that the Atg32 mutant lacking the amino acid (aa) 151–200 region shows constitutive interaction with Atg11, punctate formation without mitophagy induction, and accelerated mitophagy. Deletion of the aa 151–200 region of Atg32 induces a similar effect as that induced by the absence of Ppg1, suggesting that Ppg1 dephosphorylates Atg32 via this region.
Identification of Ppg1 as a negative regulator of Atg32 phosphorylation added to the information regarding the regulatory mechanism of mitophagy in yeast. We speculate that the phosphatase activity of the Ppg1-Far complex toward Atg32 is inactivated by unidentified mechanisms upon mitophagy induction, consequently leading to Atg32 phosphorylation by CK2 (). This hypothesis raises further questions. Which signal regulates the Ppg1-Far complex? Is this signal derived from damaged mitochondria and through signal transduction systems? Does the signal activate the core machineries of mitophagy and autophagy in a coordinated or independent manner? Further studies investigating the Ppg1-Far complex can address these questions and provide insights into the mechanisms of recognition of mitochondrial malfunction and removal through mitophagy.
Figure 1. Model for the regulatory mechanism of Atg32 phosphorylation. Under normal conditions, Ppg1 with the Far complex dephosphorylates Atg32 via its amino acid 151–200 region to prevent unrequired mitophagy. Upon mitophagy induction, the activity of Ppg1 is suppressed through unidentified mechanisms (such as dissociation of Ppg1 and the Far complex and binding of an inhibitory protein to Ppg1), leading to Atg32 phosphorylation by CK2.
Primarily, there are 2 types of mitophagy reported in mammals: (1) mitochondrial serine/threonine kinase PINK1 and E3 ligase PRKN/PARK2/Parkin-mediated mitophagy and (2) receptor-mediated mitophagy, such as BNIP3L/Nix, BNIP3, FKBP8, FUNDC1, and BCL2L13. Although mitophagy is a conserved phenomenon among eukaryotes, there is no homology identified between Atg32 and factors of mammalian mitophagy. Only yeast Far complex proteins have a mammalian homologue termed STRiatin-Interacting Phosphatase And Kinase (STRIPAK) complex. Investigation of the role of this complex on mitophagy may advance our understanding of this process in mammals.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.