
ABSTRACT
Macroautophagy/autophagy is a physiological mechanism that is essential for the maintenance of cellular homeostasis and stress adaptation. Defective autophagy is associated with many human diseases, including cancer and neurodegenerative disorders. The emerging implication of epigenetic events in the control of the autophagic process opens new avenues of investigation and offers the opportunity to develop novel therapeutic strategies in diseases associated with dysfunctional autophagy-lysosomal pathways. Accumulating evidence reveals that several methyltransferases and demethylases are essential regulators of autophagy, and recent studies have led to the identification of the lysine demethylase KDM1A/LSD1 as a promising drug target. KDM1A/LSD1 modulates autophagy at multiple levels, participating in the transcriptional control of several downstream effectors. This review summarizes our current understanding of the role of KDM1A/LSD1 in the autophagy regulatory network.
Introduction
Accessibility of promoter elements to the transcriptional machinery is regulated by post-translational modifications of histone tails, that modify chromatin structure and nucleosome organization. Histone methylation, acetylation, ubiquitination, phosphorylation, sumoylation, and ADP ribosylation are reversible modifications to endogenous and external stimuli that accommodate transient cellular transcriptional responses [Citation1]. These modifications are finely and dynamically regulated during cellular processes. The specificity of the epigenetic pattern is endorsed by the interactions between histone-modifying enzymes and transcription factors, which strategically recruit specific epigenetic modifiers to drive the deposition of histone marks at discrete genetic loci. Such an intricate regulatory network is essential for physiological responsive activation and/or repression of genes involved in cell proliferation, embryonic differentiation and homeostatic processes, including macroautophagy (hereafter called autophagy).
Autophagy is an evolutionarily conserved, lysosomal-degradative process used for degradation of damaged organelles and protein aggregates and for recycling of cellular components [Citation2]. Autophagy represents a major mechanism for maintaining cellular homeostasis and acts constitutively at low basal levels, but could be also induced by several stimuli such as oxidative stress, hypoxia, DNA damage and nutrient starvation [Citation3]. Given its role in the maintenance of cellular homeostasis, stress response and cell-fate decision, it is not surprising that autophagy needs to be tightly controlled.
Regulation of the autophagic process is mostly dependent on the activity of energy and nutrient sensors, such as MTORC1 (mechanistic target of rapamycin kinase complex 1) and 5ʹ AMP-activated protein kinase (AMPK) [Citation4], that not only control activation but also regulate the long-term outcome of autophagy, leading to the specific cytosol-nucleus shuttling of different transcription factors involved in the control of lysosome- and autophagy-specific genes, including the master regulator TFEB (transcription factor EB) [Citation5]. Over the past several years, a wide number of transcription factors [Citation6], including the FOXO (forkhead box O) family members [Citation7], TP53 [Citation8], HIF1A [Citation9,Citation10], E2F1 [Citation11] and SOX2 [Citation12], have been linked to the control of autophagy in response to various stresses. These transcription factors likely work in cooperation with epigenetic modifiers, but our understanding of the integration of epigenetic and transcriptional control of autophagy is only at the beginning [Citation13].
Genome-wide investigations reveal that the autophagic process encompasses an intricate epigenetic program [Citation14]. Autophagy induction is associated with H4K16 deacetylation and concomitant downregulation of KAT8/MOF lysine acetyltransferase. Importantly, H4K16 acetylation status seems to participate in the autophagic survival-or-death decision [Citation15], because inhibition of H4K16ac downregulation upon autophagy, by overexpression of KAT8/MOF or inhibition of its counterpart, SIRT1 (sirtuin 1) deacetylase, increases autophagic flux and promotes cell death. In parallel with the deacetylation of H4K16, a global reduction of H3K4me3 is observed in both mammalian and yeast cells upon autophagy induction [Citation15], in accordance with the established molecular connection between these histone marks [Citation16], but responsible regulators have not yet been uncovered. Glucose starvation increases genome-wide H3R17 dimethylation, linked to transcriptional activation, through the stabilization of the H3R17 methyltransferase CARM1 [Citation17]. H2Bub1 (H2B mono-ubiquitination) is downregulated during starvation; reduction in H2Bub1 results in autophagy activation, and the de-ubiquitinase USP44 is likely the cause of the starvation-induced decrease in H2Bub1 [Citation18]. In yeast, treatment with rapamycin, an established TOR/MTOR inhibitor, is accompanied by the reduction of H3K56ac residues [Citation19].
Indeed, changes in protein levels of key histone modifiers, such as KAT8/MOF [Citation15] and CARM1 [Citation17], occur during autophagy, leading to global chromatin modulation; conversely, histone modifications have been reported to be actively involved in long-term modulation of autophagy, driving a regulatory feedback loop which contributes to the control of the autophagic flux, metabolic direction and cell-fate decision, through the transcriptional regulation of autophagy-related (ATG) genes [Citation13].
Here we provide a comprehensive coverage on the relative contribution of histone methyltransferases and demethylases in autophagy regulation, and summarize our current understanding of the emerging role of the lysine-specific demethylase KDM1A/LSD1 in the autophagy regulatory network. Histone methyltransferases and demethylases linked to the regulation of autophagy, with their mode of action, are listed in .
Table 1. Histone methyltransferases (HMTs) and lysine demethylases (KDMs) currently known to regulate autophagy.
Regulating autophagy by histone methyltransferases
Enzymatic histone methylation occurs at specific lysine or arginine residues on histones H3 and H4 and is catalyzed by lysine methyltransferases and arginine methyltransferases, with S-adenosyl-L-methionine as cofactor and methyl donor. Histone methylation can involve the transfer of 1, 2 or 3 methyl groups, resulting in mono-, di-, or tri-methylated lysine, respectively, and in mono- or di-methylated arginine, leading to silencing or activation of target gene promoters [Citation20]. Histone methyltransferases function as regulators of essential biological processes, such as cell cycle, genome stability and lineage development, and have emerged as an integral part of the autophagic process [Citation21].
CARM1 (coactivator associated arginine methyltransferase 1), which catalyzes histone H3R17 dimethylation, has a genome-wide role in regulating the autophagic process and acts as TFEB transcriptional co-activator of autophagy and lysosomal genes upon glucose starvation [Citation17]. Importantly, the CARM1 level is tightly regulated by nutrient availability. Under glucose-rich conditions, nuclear CARM1 is degraded by the SKP2- containing Skp, Cullin, F-box containing E3 ubiquitin ligase. Glucose starvation results in AMPK accumulation in the nucleus. Once in the nucleus, AMPK phosphorylates FOXO3 and positively regulates its transcriptional activity; FOXO3, in turn, transcriptionally represses SKP2, leading to CARM1 protein stabilization and subsequent increases in histone H3R17 dimethylation and autophagy induction () [Citation17].
Figure 1. Methyltransferases linked to regulation of autophagy. (a) Starvation results in AMPK-dependent CARM1 protein stabilization and subsequent increases in histone H3R17 dimethylation and autophagy induction. (b) H3K27 trimethylation by EZH2 represses the expression of several MTOR pathway negative regulators, resulting in MTOR activation and the consequent inhibition of autophagy. (c) Under nutrient-rich conditions H3K9 dimethylation by EHMT2 suppress expression of genes essential for the autophagic process, maintaining autophagy at a low basal level. Induction of autophagy by starvation results in EHMT2 displacement, decreasing the H3K9me2 repressive mark and enhancing ATG gene expression. Dashed arrows indicate repression of gene expression.
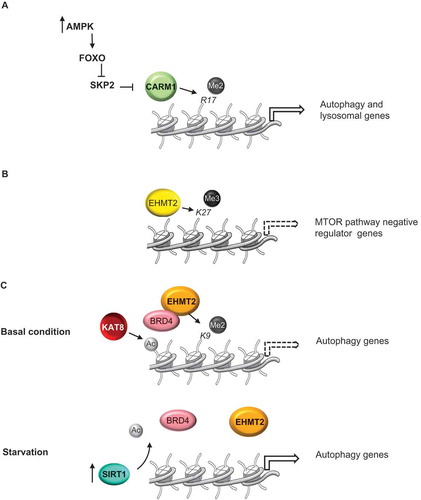
While CARM1-dependent H3R17 dimethylation is a transcriptional activation mark for autophagy, H3K9 dimethylation by EHMT2/G9a and H3K27 trimethylation by EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) have been reported to suppress expression of genes essential for the autophagic process, playing an important role in maintaining autophagy at low basal level under nutrient-rich conditions.
EZH2 is a histone H3 lysine 27 methyltransferase that acts as a transcriptional repressor. EZH2 is recruited, via MTA2 (metastasis associated 1 family member 2), to the promoter region of several MTOR pathway negative regulators, including TSC2, DEPTOR, RHOA, and GPI [Citation22]. Thus, transcriptional repression by EZH2 results in MTOR activation and, consequently, inhibition of autophagy ().
EHMT2 methyltransferase associates with the promoter regions of LCB3, TP53INP2/DOR, and WIPI1 genes, involved in the autophagic process, and represses their expression [Citation23]. Significantly, EHMT2 is displaced upon the induction of autophagy by starvation, resulting in decreased H3K9me2 repressive mark level and enhanced ATG gene expression. Accordingly, pharmacological inhibition or knockdown of EHMT2 promotes formation of autophagosomes resulting in the occurrence of autophagy [Citation23].
Recently, it has been reported that the epigenetic reader BRD4 (bromodomain containing protein 4), interacting with EHMT2, negatively regulates autophagy and lysosome gene expression [Citation24]. Under normal conditions, BRD4 binds to the promoter regions of ATG genes via histone H4K16 acetylation by KAT8/MOF (). Then, BRD4 recruits EHMT2, which dimethylates H3K9 and represses autophagy gene transcription. During starvation, SIRT1, activated via an AMPK-cascade, deacetylates H4K16, leading to BRD4 displacement from ATG gene promoters and consequent denied recruitment of EHMT2, thereby activating autophagy [Citation24].
Regulating autophagy by lysine demethylases
KDMs (lysine demethylases) cluster in the KDM1 subfamily, containing the lysine-specific demethylase enzymes, and the KDM2-KDM7 subfamilies, consisting of the Jumonji C domain-containing enzymes [Citation20]. The KDM1 subfamily members, KDM1A/LSD1 and KDM1B/LSD2, are flavin adenine dinucleotide (FAD)-dependent amine oxidases which rely on a lone electron pair within the lysine for catalysis and thereby can catalyze demethylation reactions on only mono- and di-methylated lysines. In contrast, the Jumonji C domain-containing KDMs directly oxidize methyl groups and can demethylate mono-, di-, or tri-methylated lysines [Citation25]. Lysine demethylases are involved in mammalian embryonic development and cell reprogramming playing an important role in the regulation of cellular processes essential to cell plasticity and homeostasis, including autophagy [Citation26].
The H3K27me3 demethylase KDM6A/UTX antagonizes polycomb-group protein-mediated silencing, in particular EZH2, by removing the repressive methylation mark from H3K27me3 and establishing an active chromatin state [Citation27,Citation28]. It has been reported that the Drosophila KDM6A ortholog, Utx/dUTX, coordinates temporal regulation of key apoptosis and autophagy genes, during steroid hormone-mediated programmed cell death of the salivary glands [Citation29]. Developmental cell death in Drosophila could occur in an autophagy-dependent manner, and Utx represents a molecular link between survival versus death during metamorphosis, modulating expression of both apoptosis and autophagy-related genes [Citation29].
The H3K36 demethylase Rph1/KDM4A is a transcriptional negative regulator of several ATG genes and a repressor of autophagy induction under nutrient-replete conditions, although its role in autophagy is independent of histone demethylase activity [Citation30]. Overexpression of Rph1/KDM4A strongly suppresses autophagy induction after starvation and such an effect is highly conserved from yeast to mammals. Consistently, KDM4A depletion has been reported to promote autophagy in human cell lines [Citation30,Citation31].
As reported by Bernard and colleagues [Citation30], the DNA binding domain of Rph1 is necessary for its function in autophagy, suggesting that Rph1 negatively regulates the expression of ATG genes by preventing the recruitment of the transcriptional activators at their promoter regions. Importantly, phosphorylation of Rph1 is a prerequisite to the induction of ATG gene transcription and autophagy upon nutrient limitation, and blocking Rph1 phosphorylation strongly impairs autophagy induction. Rim15, a protein kinase that integrates signals from different nutrient sensors, mediates Rph1 phosphorylation, leading to its partial autophagy-independent protein degradation and inhibition of its activity [Citation30].
Multiple lines of evidence point to KDM1A as an essential regulator of autophagy [Citation32–Citation34]. KDM1A can demethylate mono- and di-, but not tri-methylated, lysines 4 and 9, of histone H3 [Citation35–Citation37], acting as co-repressor or co-activator in a target-specific manner [Citation38,Citation39]. KDM1A target specificity is regulated by its association with different partners. For example, when it interacts with transcriptional co-repressors, such as RCOR/CoREST (REST corepressor), HDAC1/2, CTBP and NuRD (nucleosome remodeling and deacetylase) complexes, KDM1A demethylates H3K4me1/2, thus repressing gene transcription [Citation40–Citation42]. In contrast, when KDM1A interacts with the AR (androgen receptor) and ESR (estrogen receptor) nuclear hormone receptors, its enzymatic specificity switches to H3K9me1/2, then promoting transcription [Citation43,Citation44].
KDM1A is essential for mammalian embryonic development [Citation45] and plays a pivotal role in many important aspects of cell biology, such as cell proliferation [Citation46], chromosome segregation [Citation47], epithelial–mesenchymal transition [Citation48], spermatogenesis [Citation49], adipogenesis [Citation50], stem cell pluripotency [Citation51] and differentiation [Citation52], and its dysregulation is often associated with both solid tumors and hematological malignancies [Citation51].
As described in detail below, a number of studies identified KDM1A as a critical negative regulator of genes essential for the autophagic process. Accordingly, inactivation of KDM1A, by depletion or pharmacological inhibition, induces autophagy in a range of different human cancer cell lines [Citation32,Citation53–Citation57].
At a mechanistic level, KDM1A acts at several places in the modulation of autophagy, regulating expression of key players involved in signaling, phagophore elongation and autophagosome formation. These studies, collectively, indicate that KDM1A orchestrates an intricate network via transcriptional control of several downstream effectors, with different and non-overlapping functions, converging in autophagy modulation.
KDM1A/LSD1 regulates autophagy in neuroblastoma through the SESN2-MTORC1 pathway
Nutrient starvation and stress signals induce autophagy through inhibition of MTORC1, which controls the balance between cellular catabolic processes and cellular energetic status [Citation58]. Recently, we found that KDM1A depletion triggers autophagy by blocking the MTORC1 cascade in neuroblastoma cells [Citation32]. We identified SESN2 (sestrin 2) as a KDM1A-repressed target gene involved in MTORC1 pathway control. SESN2 is a member of the SESN1/PA26-related protein family, involved in cellular response to different stress conditions [Citation59]. SESN2 inhibits MTORC1 activity through the regulation of the GATOR complex, suppressing the RRAG-dependent recruitment of MTORC1 to the lysosomal membrane [Citation60,Citation61].
Mechanistically, we have shown that KDM1A binds to the promoter region of the SESN2 gene and represses its expression [Citation32]. Genetic depletion or pharmacological inhibition of KDM1A is accompanied by a significant increase in histone H3 acetylation and decrease in H3 lysine 27 trimethylation, consistent with the induction of SESN2 gene expression (), although the mechanisms through which KDM1A depletion induces such changes remain to be elucidated. Knockdown also increases the H3K4me2 level at the transcriptional start site of SESN2, whereas the H3K9Me2 signature appears not to be affected, according to its repressive function.
Figure 2. KDM1A coordinates the expression of several autophagy-related genes in neuroblastoma. KDM1A is recruited to the promoter region of SESN2 (by an unknown factor) and CLU (by MYCN) genes and represses their expression. KDM1A depletion triggers a structural remodeling in the chromatin landscape, leading to the accumulation of activating histone marks, transcriptional activation of CLU and SESN2 expression, and enhanced autophagy. Dashed arrows indicate repression of gene expression.
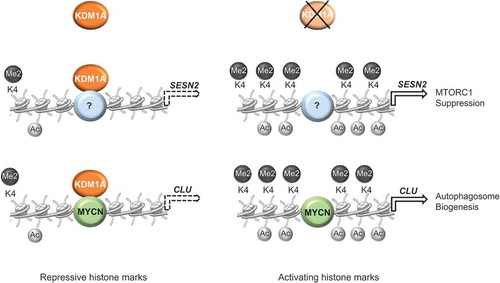
Thus, KDM1A depletion triggers a structural remodeling of the chromatin at the promoter region of the SESN2 gene, leading to transcriptional activation of SESN2 expression, which in turn inhibits MTORC1 activity, leading to enhanced autophagy. Consistently, SESN2 enhanced expression promotes autophagy in neuroblastoma cells, and SESN2 silencing attenuates MTORC1 inhibition and partially suppresses the autophagy induction obtained by KDM1A inhibition [Citation32].
SESN2 transcription is under the control of TP53, which activates its expression in response to DNA damage and oxidative stress [Citation62]. Nevertheless, following KDM1A inhibition, activation of SESN2 expression occurs in SK-N-BE(2) neuroblastoma cells, in which TP53 carries a missense mutation at codon 135, converting cysteine to phenylalanine, and the mutated TP53 fails to activate CDKN1A/p21 gene expression [Citation63]. Thus, KDM1A could regulate SESN2 transcription in both TP53-dependent and TP53-independent manners. Because KDM1A does not bind to DNA alone [Citation39], but requires association with DNA-binding interaction partners, it remains to be determined as to the presence of a DNA-binding factor capable of recruiting KDM1A at the SESN2 promoter.
Furthermore, in neuroblastoma cells, KDM1A, together with MYCN, inhibits transcription of the molecular chaperone CLU (clusterin) [Citation64], which is involved in the protein quality-control system and degradation of misfolded proteins [Citation65]. CLU is an integral part of the autophagic process because it increases autophagosome biogenesis through the stabilization of the MAP1LC3/LC3-Atg3 heterodimer [Citation66]. It is likely that CLU participates in the activation of autophagy that occurs after KDM1A inhibition in neuroblastoma cells.
Together, these findings reveal that KDM1A, by remodeling the chromatin landscape, coordinates the expression of several genes involved in the autophagic process ().
KDM1A/LSD1 mediates hepatic post-prandial autophagy
In liver, autophagy is mainly under the control of the fed-state sensing nuclear receptor NR1H4/FXR (nuclear receptor subfamily 1 group H member 4) and the fasting transcriptional activator CREBBP/CREB (CREB binding protein), which negatively or positively modulate ATG genes expression, respectively [Citation67]. In the fasting state, the CREB-CRTC2 complex, together with the histone acetyltransferase EP300/p300, activates transcription of autophagy-related genes. In the early fed-state, NR1H4 directly interacts with CREB and leads to the dissociation of EP300 and CRTC2, resulting in a reduction of activating histone marks H3K9/14ac and gene repression () [Citation67].
Figure 3. KDM1A mediates hepatic post-prandial epigenetic repression of autophagy. In the fasting state, the CREBBP-CRTC2 complex, together with the histone acetyltransferase EP300, activates transcription of autophagy-related genes. In the early fed-state, NR1H4/FXR directly interacts with CREBBP and leads to the dissociation of EP300 and CRTC2, resulting in a reduction of activating histone marks and gene repression. In the late fed-state, NR0B2/SHP recruits KDM1A to a subset of CREBBP-bound autophagy genes. The presence of the KDM1A-NR0B2/SHP-CREBBP complex triggers an epigenetic repression state by accumulation of repressive histone marks, involving additional unknown epigenetic modifiers, sustaining postprandial repression of autophagy. Dashed arrows indicate repression of gene expression.
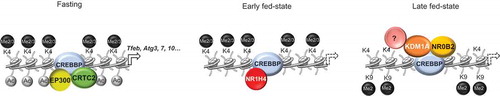
Very recently, it has been demonstrated that KDM1A participates in the post-prandial epigenetic repression of hepatic autophagy [Citation34]. In this work, the authors showed that, in response to FGF15/FGF19 signaling in the late fed-state, the nuclear receptor NR0B2/SHP (nuclear receptor subfamily 0 group B member 2) binds and recruits KDM1A to a subset of CREBBP-bound autophagy genes, including Tfeb, Atg3, Atg7, and Atg10. FGF19 treatment or feeding of mice increases occupancy of KDM1A at NR0B2/SHP-target autophagy genes. The presence of the KDM1A-NR0B2/SHP-CREBBP complex decreases the occupancy by POLR2 and initiates an epigenetic cascade by accumulation of H3K9me2 repressive marks and removal of the activating histone marks H3K4me2, H3K4me3, and H3K9/14ac, suggesting the existence of multiple molecular interaction between KDM1A and other epigenetic enzymes responsible for sustaining postprandial repression of autophagy (). Consistently, ectopic expression of KDM1A decreases autophagy, whereas downregulation of KDM1A increases basal and post-prandial autophagy in both hepatic cells and mouse liver [Citation34]. Thus, KDM1A participates in temporal modulation of hepatic autophagy after feeding, and plays a key role in maintaining cellular homeostasis, mediating the establishment of a stable epigenetic repression of several autophagy-related genes.
Noteworthy, Byun and collaborators [Citation34] showed that KDM1A is recruited at the TFEB promoter and regulates its expression. TFEB is a master regulator of autophagy and lysosomal biogenesis [Citation68,Citation69] and controls expression of the coordinated lysosomal expression and regulation gene network [Citation70]. Modulation of TFEB expression, and consequently of the TFEB-dependent transcriptional program, represents an important molecular driver of the autophagic process. Thus, KDM1A seems to be involved in autophagy participating in different and critical layers of regulation; on the one hand, participating in the CREBBP transcriptional program, and on the other hand, modulating TFEB expression. Further studies will determine whether these KDM1A functions operate also in other cell types and tissues.
Non-epigenetic regulation of autophagy by KDM1A/LSD1
Beyond its direct activity on chromatin structure, KDM1A modulates global or specific gene expression patterns through demethylation of lysine residues at several non-histone proteins, such as TP53 [Citation71], HIF1A [Citation72], DNMT1 [Citation73], E2F1 [Citation74] and PPP1R12A/MYPT1 [Citation75], modulating their activity or stability. It has been demonstrated, by Periz and coworkers, that KDM1A and UBE4B (ubiquitination factor E4B) synergistically regulate the cellular protein quality control system, and that knockdown of both UBE4B and KDM1A activates proteasomal degradation and autophagy [Citation33]. Such an effect appears to be highly conserved across species, because inactivation of KDM1A and UBE4B orthologs, ufd-2 and spr-5, suppresses neurotoxicity associated with protein aggregates in Caenorhabditis elegans [Citation33].
It was shown that TP53 acts downstream of UBE4B and KDM1A to activate degradation machineries and to improve the clearance of misfolded proteins. Mechanistically, TP53 stability and activity are negatively regulated at the post-translational level by both UBE4B and KDM1A. Whereas UBE4B promotes TP53 ubiquitination and degradation [Citation76,Citation77], KDM1A specifically demethylates the TP53 dimethylated K370 residue and represses its activity, preventing the interaction with its coactivator TP53BP1 [Citation71]. Thus, loss of UBE4B and KDM1A leads to TP53 stabilization and the activation of the TP53-mediated transcriptional program, which, in turn induces autophagy. According to its subcellular localization, TP53 is involved in the control of autophagy through 2 opposite mechanisms. Cytoplasmic TP53 is a potent inhibitor of autophagy [Citation78], conversely, nuclear TP53 binds the promoter region of several autophagy-related genes, including ATG7 and ULK1, and activates their expression [Citation79]. Moreover, TP53 activity establishes the major link between autophagy and DNA damage through the transcriptional activation of several genotoxic stress responsive factors, such as AEN/ISG20L1 [Citation80], SESN family genes [Citation62] and DRAM1 (DNA damage regulated autophagy modulator 1) [Citation81]. Thus, this study highlights an active involvement of KDM1A in the autophagic pathway via direct modulation of a TP53-mediated transcriptional program.
Very recently, it has been shown that KDM1A tethers and destabilizes the autophagy receptor and substrate SQSTM1/p62, a ubiquitin-binding scaffold protein that interacts with ubiquitinated proteins and enables their selective degradation in the lysosome [Citation57]. KDM1A is required for SQSTM1/p62 ubiquitination in uterine and ovarian cancer cells; albeit the mechanisms by which KDM1A drives SQSTM1/p62 degradation remain unclear, the authors found that KDM1A knockdown decreases ubiquitin-conjugated SQSTM1/p62 levels with consequent stabilization and accumulation of SQSTM1/p62. Interestingly, SQSTM1/p62 ubiquitination seems to not rely on direct demethylation by KDM1A, but requires its catalytic activity, because treatment with enzymatic inhibitors also increases SQSTM1/p62 protein stability [Citation57]. The impact of SQSTM1/p62 expression on autophagy is apparently cell-type dependent: whereas in HeLa cells SQSTM1/p62 is not required for autophagosome formation under basal conditions and starvation [Citation82], in cardiomyocytes, deletion of the corresponding gene results in impaired LC3-II and autophagosomes formation and leads to misfolded protein stress, affecting cell survival [Citation83]. More in-depth studies will clarify the significance of this newly identified KDM1A function and its effect on autophagy.
Regulation of KDM1A/LSD1 during autophagy
KDM1A seems to participate in the establishment of a stable epigenetic repression state of several ATG promoters. However, how exactly KDM1A recruitment to these promoters is regulated, by specific upstream autophagic stimuli, remains to be defined. Because KDM1A does not directly bind to DNA [Citation39], its occupancy at the chromatin strictly depends on the interaction with specific transcriptional partners. As described above, nutrient state affects occupancy of KDM1A at CREBBP-bound autophagy genes via NR0B2/SHP-dependent recruitment, and a similar indirect mechanism of regulation could hypothetically concern other KDM1A interactors related to autophagy.
Furthermore, KDM1A is a FAD-dependent amine oxidase and FAD levels regulate KDM1A activity under certain cellular conditions [Citation72,Citation84]. Lowering FAD content abrogates KDM1A activity and causes reduction of KDM1A protein without affecting mRNA level, suggesting that binding to FAD might stabilize KDM1A [Citation84]. We found that inhibition of KDM1A by tranylcypromine, a mechanism-based irreversible inhibitor that forms a covalent adduct with FAD in the active-site cavity of the enzyme, reduces KDM1A recruitment at the SESN2 promoter, suggesting that inhibition of FAD activity might debilitate deposition of KDM1A at target promoters [Citation32]. Intriguingly, pharmacological inhibition of MTOR activity has been found to decrease FAD levels [Citation85]. It is likely that intracellular FAD levels might affect KDM1A-mediated regulation of autophagy-related gene expression.
Finally, several studies reported that KDM1A is subjected to different post-translational modifications, which may alter its activity or stability [Citation86,Citation87]. It has been shown that acetylation of KDM1A by KAT8/MOF impairs KDM1A ability to associate with nucleosomes, resulting in enhanced expression of epithelial genes and suppressing epithelial-to-mesenchymal transition [Citation88]. Similarly, a regulatory feedback loop might occur during autophagy, linking the downregulation of KAT8/MOF and the KDM1A-mediated repression of ATG genes, to protect cells against overstimulation of autophagic flux.
Conclusions and future perspectives
As a major cytoprotective mechanism in response to environmental adversity, autophagy is crucial for cellular homeostasis and organismal survival under physiological conditions. Dysfunction and dysregulation of this process contribute to the pathogenesis in various human diseases, such as infections, neurodegenerative, cardiovascular and immune disorders, diabetes, and cancer [Citation89–Citation91]. Targeting autophagy is an attractive strategy for the treatment of many complex diseases. Chemical inducers of autophagy were successfully employed against hepatic fibrosis [Citation92] and infections [Citation93,Citation94] and have also been shown to exert a beneficial effect on the clearance of mutant SNCA/α-synuclein and protect cells against apoptosis in Alzheimer disease models [Citation95].
Over the past several years, a nuclear network has emerged in which a plethora of transcription and epigenetic factors have a role in the control of autophagy-related gene expression [Citation13,Citation14]. As dysfunctions of the autophagy process are implicated in several diseases, it would be compelling to understand how these transcriptional programs are dysregulated in pathological contexts. Alterations in either epigenetic or transcriptional regulation might coordinately drive aberrant autophagy gene expression, contributing to disease states, while their restoration or modification could be beneficial to improve autophagy-based clinical treatments.
Nuclear events represent suitable drug targets and provide ways to both positively and negatively control autophagy. Whereas transcription factors are generally considered inappropriate targets for drug design, and their inhibition is often irreversible and ineffective, epigenetic enzymes are suitable and promising drug targets for a wide variety of diseases, including cancer, neurodegenerative disorders and rheumatic diseases [Citation96].
Epigenetic targeting is emerging as an efficient way to regulate autophagy-related gene expression, and several small molecule inhibitors of histone modifications have been shown to be able to modulate autophagy so far (). For example, S-adenosyl-L-methionine-competitive EZH2 inhibitors are potent autophagy inducers [Citation22,Citation97]; histone deacetylase inhibitors modulate autophagy on several levels [Citation98–Citation101]; sirtinol, a class III histone deacetylase inhibitor, induces autophagic cell death by the downregulation of SIRT1/2 expression in breast cancer cells [Citation102]; ellagic acid treatment inhibits CARM1-induced autophagy occurrence [Citation17]; EHMT2 inhibition by small molecules, such as BIX-01294, induces autophagy in a wide number of cancer cells [Citation103,Citation104]. These studies strongly suggest that pharmacological modulation of epigenetic patterns could be a promising therapeutic strategy for diseases associated with defects in autophagy-lysosomal pathways through the long-term regulation of autophagy-related gene expression.
Table 2. Epigenetic-based drugs reported to affect the autophagy pathway.
As highlighted in this review, recent publications have suggested that KDM1A takes an important part in such a regulatory network, attending different molecular mechanisms that affect the autophagy gene programs [Citation32–Citation34]. Although observed in a limited number of cell types, these findings reveal that KDM1A participates in transcriptional repression of genes involved in the autophagic process, identifying KDM1A as a key modulator of autophagy under physiological conditions. It is tempting to investigate whether, in pathological conditions, aberrant expression of KDM1A could contribute to its unscheduled recruitment to these promoter elements, with subsequent establishment of vicious epigenetic patterns.
Given its enzymatic activity and its association with many human malignancies, including cancer [Citation51] and neurological disorders [Citation105–Citation108], there has been great interest in developing valuable and specific pharmacological inhibitors of KDM1A. Several studies show that these small molecules mimic KDM1A knockdown in autophagy activation in various model systems () [Citation32,Citation53–Citation57]. Among them, tranylcypromine, a MAO (monoamine oxidase) inhibitor previously used in the treatment of depression, and its derivatives induce autophagy in a wide range of human cancer cell lines [Citation32,Citation53,Citation56,Citation57]; JL1037, a selective and reversible KDM1A inhibitor, increases autophagosome and autolysosome formation in acute myeloid leukemia cell lines as well as in primary cells from acute myeloid leukemia patients [Citation54]; treatment with NCL-1, a specific KDM1A inhibitor, induce apoptosis and autophagy in prostate cancer cells [Citation55].
Collectively, these results strongly argue in favor of future evaluation of KDM1A inhibitors for therapeutic intervention in human disorders associated with defective autophagy-lysosomal pathways. Further studies will improve our understanding of the role of KDM1A-mediated epigenetic regulation of autophagy and its therapeutic potential in human diseases.
Abbreviations
| AMPK | = | 5ʹ AMP-activated protein kinase; |
| ATG | = | autophagy related; |
| BRD4 | = | bromodomain containing protein 4; |
| CARM1 | = | coactivator associated arginine methyltransferase 1; |
| CREBBP/CREB | = | CREB binding protein; |
| DRAM1 | = | DNA damage regulated autophagy modulator 1; |
| EZH2 | = | enhancer of zeste 2 polycomb repressive complex 2 subunit; |
| HDAC | = | histone deacetylase; |
| FAD | = | flavin adenine dinucleotide; |
| FOXO | = | forkhead box O; |
| H2Bub1 | = | H2B mono-ubiquitination; |
| KDM1A/LSD1 | = | lysine demethylase 1A; |
| MAO | = | monoamine oxidase; |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3; |
| MTA2 | = | metastasis associated 1 family member 2; |
| MTORC1 | = | mechanistic target of rapamycin kinase complex 1; |
| NR1H4/FXR | = | nuclear receptor subfamily 1 group H member 4; |
| NuRD | = | nucleosome remodeling and deacetylase complex; |
| RCOR/COREST | = | REST corepressor; |
| TFEB | = | transcription factor EB; |
| UBE4B | = | ubiquitination factor E4B; |
| ULK | = | unc-51 like autophagy activating kinase |
Acknowledgments
We thank Luigi Lania for helpful discussions, constructive criticisms and critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 2016;32:42–56.
- Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26:624–635.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141.
- Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914.
- Füllgrabe J, Ghislat G, Cho D-H, et al. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci. 2016;129:3059–3066.
- Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–169.
- White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6:a026120.
- Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903.
- Yang Z, Zhao T, Zou Y, et al. Hypoxia induces autophagic cell death through hypoxia-inducible factor 1α in microglia. PLoS One. 2014;9:e96509.
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4864.
- Cho -Y-Y, Kim DJ, Lee HS, et al. Autophagy and cellular senescence mediated by Sox2 suppress malignancy of cancer cells. PLoS One. 2013;8:e57172.
- Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Biol. 2014;15:65–74.
- Füllgrabe J, Heldring N, Hermanson O, et al. Cracking the survival code: autophagy-related histone modifications. Autophagy. 2014;10:556–561.
- Füllgrabe J, Lynch-Day MA, Heldring N, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468–471.
- Katoh H, Qin ZS, Liu R, et al. FOXP3 Orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell. 2011;44:770–784.
- Shin H-JR, Kim H, Oh S, et al. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534:553–557.
- Chen S, Jing Y, Kang X, et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res. 2017;45:1144–1158.
- Chen H, Fan M, Pfeffer LM, et al. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012;40:6534–6546.
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507.
- Wei F-Z, Cao Z, Wang X, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. 2015;11:2309–2322.
- Artal-Martinez de Narvajas A, Gomez TS, Zhang J-S, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol. 2013;33::3983–3993.
- Sakamaki J, Wilkinson S, Hahn M, et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell. 2017;66:517–532.e9.
- Walport LJ, Hopkinson RJ, Schofield CJ. Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol. 2012;16:525–534.
- Labbé RM, Holowatyj A, Yang Z-Q. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2013;6:1–15.
- Hong S, Cho Y-W, Yu L-R, et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci. 2007;104:18439–18444.
- Lee MG, Villa R, Trojer P, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450.
- Denton D, Aung-Htut MT, Lorensuhewa N, et al. UTX coordinates steroid hormone-mediated autophagy and cell death. Nat Commun. 2013;4:2916.
- Bernard A, Jin M, González-Rodríguez P, et al. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr Biol. 2015;25:546–555.
- Wang B, Fan X, Ma C, et al. Downregulation of KDM4A suppresses the survival of glioma cells by promoting autophagy. J Mol Neurosci. 2016;60:137–144.
- Ambrosio S, Saccà CD, Amente S, et al. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene. 2017;36:6701–6711.
- Periz G, Lu J, Zhang T, et al. Regulation of protein quality control by UBE4B and LSD1 through p53-mediated transcription. PLoS Biol. 2015;13:e1002114.
- Byun S, Kim Y, Zhang Y, et al. A postprandial FGF19-SHP-LSD1 regulatory axis-mediates epigenetic repression of hepatic-autophagy. Embo J. 2017;36:1755–1769.
- Forneris F, Binda C, Battaglioli E, et al. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci. 2008;33:181–189.
- Forneris F, Binda C, Vanoni MA, et al. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–2207.
- Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953.
- Shi Y-J, Matson C, Lan F, et al. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864.
- Maiques-Diaz A, Somervaille TC. LSD1: biologic roles and therapeutic targeting. Epigenomics. 2016;8:1103–1116.
- Lee MG, Wynder C, Cooch N, et al. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435.
- Shi Y, Sawada J, Sui G, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738.
- Wang Y, Zhang H, Chen Y, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–672.
- Perillo B, Ombra MN, Bertoni A, et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206.
- Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439.
- Foster CT, Dovey OM, Lezina L, et al. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol. 2010;30:4851–4863.
- Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007;282:15471–15475.
- Lv S, Bu W, Jiao H, et al. LSD1 is required for chromosome segregation during mitosis. Eur J Cell Biol. 2010;89:557–563.
- Ambrosio S, Saccà CD, Majello B. Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim Biophys Acta. 2017;1860:905–910.
- Godmann M, Auger V, Ferraroni-Aguiar V, et al. Dynamic regulation of histone H3 methylation at lysine 4 in mammalian spermatogenesis1. Biol Reprod. 2007;77:754–764.
- Musri MM, Carmona MC, Hanzu FA, et al. Histone demethylase LSD1 regulates adipogenesis. J Biol Chem. 2010;285:30034–30041.
- Amente S, Lania L, Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta. 2013;1829:981–986.
- Adamo A, Sesé B, Boue S, et al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol. 2011;13:652–659.
- Feng S, Jin Y, Cui M, et al. Lysine-specific demethylase 1 (LSD1) inhibitor S2101 induces autophagy via the AKT/mTOR pathway in SKOV3 ovarian cancer cells. Med Sci Monitor. 2016;22:4742–4748.
- Liu S, Lu W, Li S, et al. Identification of JL1037 as a novel, specific, reversible lysine-specific demethylase 1 inhibitor that induce apoptosis and autophagy of AML cells. Oncotarget. 2017;8:31901–31914.
- Etani T, Suzuki T, Naiki T, et al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses prostate cancer without adverse effect. Oncotarget. 2015;6:2865–2878.
- Wang Z, Long Q-Y, Chen L, et al. Inhibition of H3K4 demethylation induces autophagy in cancer cell lines. Biochim Biophys Acta. 2017;1864:2428–2437.
- Chao A, Lin C-Y, Chao A-N, et al. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget. 2017;8:74434–74450.
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976.
- Pasha M, Eid AH, Eid AA, et al. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid Med Cell Longev. 2017;2017:3296294.
- Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–48.
- Parmigiani A, Nourbakhsh A, Ding B, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9:1281–1291.
- Maiuri MC, Malik SA, Morselli E, et al. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8:1571–1576.
- Goldschneider D, Horvilleur E, Plassa L-F, et al. Expression of C-terminal deleted p53 isoforms in neuroblastoma. Nucleic Acids Res. 2006;34:5603–5612.
- Amente S, Milazzo G, Sorrentino MC, et al. Lysine-specific demethylase (LSD1/KDM1A) and MYCN cooperatively repress tumor suppressor genes in neuroblastoma. Oncotarget. 2015;6:14572–14583.
- Wyatt AR, Yerbury JJ, Berghofer P, et al. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell Mol Life Sci. 2011;68:3919–3931.
- Zhang F, Kumano M, Beraldi E, et al. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014;5:5775.
- Seok S, Fu T, Choi S-E, et al. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature. 2014;516:108–111.
- Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866.
- Huang J, Sengupta R, Espejo AB, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108.
- Yang S-J, Park YS, Cho JH, et al. Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1α protein stability. Embo J. 2017;36:1011–1028.
- Wang J, Hevi S, Kurash JK, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129.
- Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39:152–160.
- Cho H-S, Suzuki T, Dohmae N, et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011;71:655–660.
- Wu H, Pomeroy SL, Ferreira M, et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat Med. 2011;17:347–355.
- Du C, Wu H, Leng RP. UBE4B targets phosphorylated p53 at serines 15 and 392 for degradation. Oncotarget. 2016;7:2823–2836.
- Morselli E, Shen S, Ruckenstuhl C, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10:2763–2769.
- Kenzelmann Broz D, Spano Mello S, Bieging KT, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031.
- Eby KG, Rosenbluth JM, Mays DJ, et al. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer. 2010;9:95.
- Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134.
- Shvets E, Fass E, Scherz-Shouval R, et al. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121:2685–2695.
- Zheng Q, Su H, Ranek MJ, et al. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308.
- Hino S, Sakamoto A, Nagaoka K, et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. 2012;3:758.
- Soliman GA, Steenson SM, Etekpo AH. Effects of metformin and a mammalian target of rapamycin (mTOR) ATPCompetitive inhibitor on targeted metabolomics in pancreatic cancer cell line. J Postgenomics Drug Biomarker Dev. 2016;6:183.
- Han X, Gui B, Xiong C, et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development. Mol Cell. 2014;55:482–494.
- Peng B, Shi R, Jiang W, et al. Phosphorylation of LSD1 by PLK1 promotes its chromatin release during mitosis. Cell Biosci. 2017;7:15.
- Luo H, Shenoy AK, Li X, et al. MOF acetylates the histone demethylase LSD1 to suppress epithelial-to-mesenchymal transition. Cell Reports. 2016;15:2665–2678.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335.
- White E. The role for autophagy in cancer. J Clin Investig. 2015;125:42–46.
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034.
- Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant 1-Antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232.
- Liao W-C, Huang M-Z, Wang ML, et al. Statin decreases helicobacter pylori burden in macrophages by promoting autophagy. Front Cell Infect Microbiol. 2016;6:203.
- Parihar SP, Guler R, Khutlang R, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209:754–763.
- Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338.
- Moosavi A, Motevalizadeh Ardekani A. Role of epigenetics in biology and human diseases. Iran Biomed J. 2016;20:246–258.
- Liu T-P, Lo H-L, Wei L-S, et al. S-Adenosyl-L-methionine-competitive inhibitors of the histone methyltransferase EZH2 induce autophagy and enhance drug sensitivity in cancer cells. Anticancer Drugs. 2015;26:139–147.
- Zhang J, Ng S, Wang J, et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11:629–642.
- Gilardini Montani MS, Granato M, Santoni C, et al. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol (Dordr). 2017;40:167–180.
- Ji -M-M, Wang L, Zhan Q, et al. Induction of autophagy by valproic acid enhanced lymphoma cell chemosensitivity through HDAC-independent and IP3-mediated PRKAA activation. Autophagy. 2015;11:2160–2171.
- Gammoh N, Lam D, Puente C, et al. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci. 2012;109:6561–6565.
- Wang J, Kim TH, Ahn MY, et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:1101–1109.
- Fan J-D, Lei P-J, Zheng J-Y, et al. The selective activation of p53 target genes regulated by SMYD2 in BIX-01294 induced autophagy-related cell death. PLoS One. 2015;10:e0116782.
- Kim Y, Kim Y-S, Kim DE, et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy. 2013;9:2126–2139.
- Ambrosio S, Majello B. Targeting histone demethylase LSD1/KDM1a in neurodegenerative diseases. J Exp Neurosci. 2018;12:117906951876574.
- Christopher MA, Kyle SM, Katz DJ. Neuroepigenetic mechanisms in disease. Epigenetics Chromatin. 2017;10:47.
- Christopher MA, Myrick DA, Barwick BG, et al. LSD1 protects against hippocampal and cortical neurodegeneration. Nat Commun. 2017;8:805.
- Fiszbein A, Kornblihtt AR. Histone methylation, alternative splicing and neuronal differentiation. Neurogenesis. 2016;3:e1204844.