
ABSTRACT
The interruption of endoplasmic reticulum (ER)-mitochondrial Ca2+ communication induces a bioenergetic crisis characterized by an increase of MTOR-independent AMPK-dependent macroautophagic/autophagic flux, which is not sufficient to reestablish the metabolic and energetic homeostasis in cancer cells. Here, we propose that upon ER-mitochondrial Ca2+ transfer inhibition, AMPK present at the mitochondria-associated membranes (MAMs) activate localized autophagy via BECN1 (beclin 1). This local response could prevent the proper interorganelle communication that would allow the autophagy-derived metabolites to reach the necessary anabolic pathways to maintain mitochondrial function and cellular homeostasis.
Abbreviations: 3MA: 3-methyladenine; ADP: adenosine diphosphate; AMP: adenosine monophosphate; ATG13: autophagy related 13; ATG14: autophagy related 14; ATP: adenosine triphosphate; BECN1: beclin 1; Ca2+: calcium; DNA: deoxyribonucleic acid; ER: endoplasmic reticulum; GEF: guanine nucleotide exchange factor; ITPR: inositol 1,4,5-trisphosphate receptor; MAMs: mitochondria-associated membranes; MCU: mitochondrial calcium uniporter; MTOR: mechanistic target of rapamycin kinase; MTORC1: MTOR complex 1; OCR: oxygen consumption rate; PtdIns3K: class III phosphatidylinositol 3-kinase; RB1CC1/FIP200: RB1 inducible coiled-coil 1; RPTOR: regulatory associated protein of MTOR complex 1; RYRs: ryanodine receptors; STK11/LKB1: serine/threonine kinase 11; TCA: tricarboxylic acid; TSC2: TSC complex subunit 2; ULK1: unc-51 like autophagy activating kinase 1; V-ATPase: vacuolar-type H+-ATPase; VDAC: voltage dependent anion channel; XeB: xestospongin B
The juxtaposition between the endoplasmic reticulum (ER) and mitochondria is the most characterized interorganelle interaction in cells [Citation1]. This interaction occurs through specialized microdomains termed mitochondria-associated membrane (MAMs), and, today, many aspects of its structural and functional features are known [Citation2]. Calcium (Ca2+) released from the ER, through the ITPRs (inositol 1,4,5-trisphosphate receptors) and RYRs (ryanodine receptors) is the principal ion messenger connecting the ER and mitochondria. Ca2+ crosses the outer mitochondrial membrane through VDAC (voltage dependent anion channel) and, in a second step, enters the inner mitochondrial membrane through the MCU (mitochondrial calcium uniporter) complex [Citation3]. Ca2+ in the mitochondrial matrix regulates the activity of 3 rate-limiting dehydrogenase enzymes of the tricarboxylic acid (TCA) cycle [Citation4], the ATP synthase [Citation5] and the malate-aspartate shuttle [Citation6]. In 2010, our group demonstrated that interruption of Ca2+ flux between the ER and mitochondria compromises cell bioenergetics in several cell lines as a consequence of limited function of the Ca2+-sensitive pyruvate dehydrogenases, generating a metabolic stress. This phenomenon consequently induces a pro-survival autophagy, even in nutrient-rich conditions [Citation7]. Interestingly, this is an MTOR (mechanistic target of rapamycin kinase)-independent AMP-activated protein kinase (AMPK)-dependent autophagy.
ER-mitochondrial Ca2+ communication, as well as proteins that participate in this ‘conversation’, have many roles in the biology of cancer cells [Citation8]. Our group showed that cancer cells are ‘addicted’ to the Ca2+ flux between ER and mitochondria [Citation9] and are more susceptible to death once ER-mitochondrial Ca2+ communication is disturbed [Citation10]. Besides supporting ATP production, the transfer of Ca2+ into the mitochondrial matrix sustains the generation of TCA intermediates and the role of mitochondria as ‘stations of building blocks’ that uphold the synthesis of nucleotides (and likely lipids and proteins) essential for the high growth-rate demands of cancer cells [Citation10] (Figure 1(a)). Evidently, the MTOR-independent autophagy produced after the interruption of ER-mitochondrial Ca2+ communication is not enough to overcome the metabolic stress produced in cancer cells [Citation10], because the coupling between the autolysosome and mitochondria is missing/inadequate or because the generation of metabolites by autophagy is insufficient (). MTOR-dependent autophagy has traditionally been viewed as a catabolic process; however, recently it is increasingly perceived as part of the anabolic machinery, generating building blocks that exit the autolysosome to rapidly synthesize nucleotides, fatty acids, membranes and proteins [Citation8,Citation9] (). In addition, it has been described that insufficient and/or disrupted MTOR-dependent autophagy generates mitochondrial dysfunction [Citation11,Citation12]. However, the increase of autophagic flux in response to the interruption of Ca2+ flux between the ER and mitochondria is not sufficient in metabolic terms, forsaking the cells to die before they reach cell division, through a phenomenon called ‘mitotic catastrophe’ [Citation13]. The induction of this mechanism is principally associated with DNA damage [Citation13], and has been linked to mitochondrial dysfunction [Citation14] and the limitation of available nucleotides for DNA synthesis [Citation13].
Figure 1. MTOR-dependent and independent autophagy and its effect on mitochondrial function. (a) In normal physiological conditions, the entry of metabolites (i.e., glucose and amino acids) to the mitochondria plus the Ca2+ transfer from the ER mediated by ITPR sustain the activity of the TCA cycle and therefore the generation of ATP. In this scenario, the AMP:ATP ratio is maintained at a low level preventing the activation of AMPK and the activation of autophagy mediated by both MTORC1 and BECN1. (b) In the absence of metabolites (induced by e.g. starvation), the activity of the TCA cycle is reduced, as is the ATP level, causing an increase in the AMP:ATP ratio and activation of AMPK. The V-ATPase at the surface of the lysosome facilitates docking of the complex AXIN1-AMPK, which inhibits the guanine nucleotide exchange factor (GEF) activity of the RAGULATOR toward RRAG family GTPases causing the dissociation from the lysosome and inactivation of MTORC1. MTORC1 inactivation unleashes MTOR-dependent autophagy, which provides metabolites to the mitochondria allowing cancer cell survival. Note that these events occur in the presence of Ca2+ transfer to mitochondria. (c) The inhibition of Ca2+ transfer to the mitochondria, even in the presence of nutrients, triggers a reduction in the activity of the TCA cycle and ATP levels, causing an increase in the AMP:ATP ratio that is rapidly sensed by a mitochondrial pool of AMPK. Active mitochondrial AMPK phosphorylates BECN1, which recruits PtdIns3K and ATG14 to activate MTOR-independent autophagy, which is insufficient to provide the necessary metabolites to mitochondria, generating a loss of homeostasis and leading to cancer cell death.
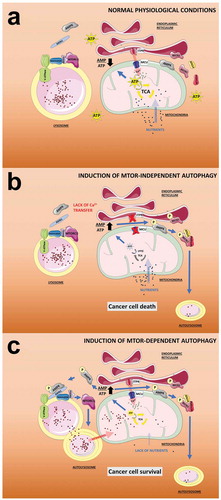
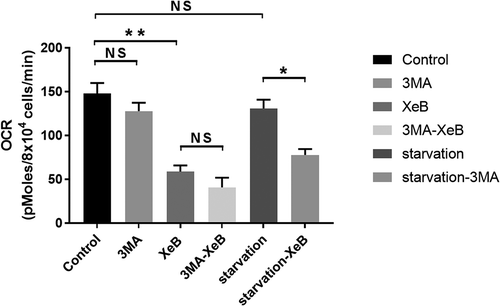
We think that the MTOR-independent autophagy-derived metabolites generated during the interruption of Ca2+ flux between the ER and mitochondria cannot reach or be redirected to the necessary anabolic process to maintain cellular homeostasis, as occurs with the metabolites generated by MTOR-dependent autophagy shown to maintain the TCA cycle in starved RAS-driven lung cancer cells [Citation15] (). When ER-mitochondrial Ca2+ communication is inhibited with the ITPR inhibitor xestospongin B (XeB), mitochondrial function, as determined by oxygen consumption rate (OCR) (), remains almost unchanged when autophagy is inhibited by 3-methyladenine (3MA), whereas the OCR in starved cells decreases significantly in the presence of 3MA, confirming our assumption.
Figure 2. Effect of MTOR-dependent and -independent autophagy in mitochondrial respiration. Basal oxygen consumption rates (OCR) in HeLa cells exposed to the ITPR inhibitor XeB (5 µM, 6 h) or starvation (6 h) in the presence or absence of 3MA (10 mM). n = 3, mean ± SEM. **p < 0.01, *p < 0.05, NS: not significant (Student’s t test).
MTOR, the master regulator of cell growth in eukaryotic cells, is a serine/threonine protein kinase that forms the catalytic subunit of 2 distinct protein complexes, with complex 1 being the most characterized (MTORC1) [Citation16]. In response to amino acid availability MTORC1 is recruited to the lysosomal membrane, the major site of MTORC1 activation [Citation17], from where it promotes cellular anabolic processes including protein, pyrimidine, and lipid biosynthesis [Citation18]. The shortage of amino acids causes the inactivation of MTORC1 and its translocation to the cytosol [Citation19], allowing the activation of the kinase ULK1 which in turn phosphorylates ATG13 and RB1CC1/FIP200 initiating the MTOR-dependent autophagy program [Citation20,Citation21] sustaining mitochondrial function [Citation22].
Upon inhibition of ER-mitochondrial Ca2+ communication, AMPK, a highly conserved sensor of intracellular adenosine nucleotide levels and therefore the main sensor of the energy status of the cell, is activated as the ATP production decreases resulting in a relative increase in AMP or ADP [Citation4]. Once activated, AMPK increases cellular catabolic activities to generate more ATP meanwhile reducing energy consumption by switching off anabolic pathways [Citation23]. As part of the energy saving mechanism, AMPK inhibits MTORC1 by activating TSC2 or by direct phosphorylation of RPTOR (regulatory associated protein of MTOR complex 1) and subsequent inhibition of MTORC1 [Citation22]. However, why is MTORC1 not inhibited by AMPK after ITPR inhibition?
A mislocalization of AMPK could be the reason that MTORC1 is not inhibited after ITPR inhibition. During a widespread energy stress such as glucose starvation or the exogenous addition of AMP, the affinity of myristoylated AMPK for the scaffold protein AXIN1 increases, simultaneously binding to STK11/LKB1. This leads to the formation of an AXIN1-STK11-AMPK complex with the subsequent phosphorylation and activation of AMPK on the late endosome/lysosome, where MTORC1 is located [Citation24]. In this scenario, the likelihood that MTORC1-activity is inhibited by AMPK and that MTOR-dependent autophagy is triggered, is very high. However, under interruption of the ER-mitochondrial Ca2+ communication, glucose consumption increases rather than decreases. This may be one of the key signaling events missing for the localization of AMPK on the lysosome/endosome, the MTOR-dependent autophagy, and subsequent connection to the autolysosome ().
Given that AMPK can be located in different organelles, such as mitochondria, ER and lysosomes/endosomes, where it forms ‘clusters’ that can be differentially activated [Citation25], we propose that a specific pool of AMPK could be located between the ER and mitochondria, specifically in MAM microdomains. Here, AMPK could sense fast changes in the AMP:ATP ratio induced by mitochondrial malfunction, become activated and phosphorylate BECN1 [Citation26], initiating autophagy in an AMPK-dependent and MTORC1-independent fashion (), and most likely preventing a mitochondrial-autolysosome coupling. BECN1 has been described as part of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex responsible for the vesicle nucleation that gives rise to autophagosomes [Citation27]. In line with this idea, the presence of a PtdIns3K inhibitor significantly reduces the increased autophagy generated by blocking ER-mitochondrial Ca2+ communication, suggesting that PtdIns3K and possibly the activation of a BECN1-PtdIns3K complex via AMPK are involved in the MTOR-independent autophagosome formation [Citation10]. The idea that AMPK is located in the MAMs is consistent with the fact that MAMs are important platforms for autophagosome formation [Citation28]. Further experiments are necessary to prove this mechanism.
Deciphering the MTOR-independent autophagy signaling generated by the bioenergetic stress induced by inhibition of ER-mitochondrial Ca2+ transfer will help unveil the complexity of intra-organelle crosstalk, especially under stress conditions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Rowland AA, Voeltz GK. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625.
- Giacomello M, Pellegrini L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ. 2016;23:1417–1427.
- De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem. 2016;85:161–192.
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316.
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973.
- Del Arco A, Contreras L, Pardo B, et al. Calcium regulation of mitochondrial carriers. Biochim Biophys Acta. 2016;1863:2413–2421.
- Cárdenas C, Miller RA, Smith I, et al. Essential regulation of cell bioenergetics by constitutive InsP(3) receptor Ca(2+) transfer to mitochondria. Cell. 2010;142:270–283.
- Bustos G, Cruz P, Lovy A, et al. Endoplasmic reticulum-mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer, a novel potential target. Front Oncol [Internet]. 2017. Available from: http://journal.frontiersin.org/article/10.3389/fonc.2017.00199/abstract
- Ivanova H, Kerkhofs M, La Rovere RM, et al. Endoplasmic reticulum–mitochondrial Ca2+ fluxes underlying cancer cell survival [Internet]. Front Oncol. 2017;7:70. Available from: http://journal.frontiersin.org/article/10.3389/fonc.2017.00070
- Cárdenas C, Müller M, McNeal A, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2017;14:2313–2324.
- Gunst J, Derese I, Aertgeerts A, et al. Insufficient autophagy contributes to mitochondrial dysfunction, organ failure, and adverse outcome in an animal model of critical illness. Crit Care Med. 2013;41:182–194.
- Wu JJ, Quijano C, Chen E, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY). 2009;1:425–437.
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19:107–120.
- Wang H, Sharma L, Lu J, et al. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget. 2015;6:15857–15870.
- Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30:1704–1717.
- Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713–1719.
- Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–162.
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322.
- Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406.
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991.
- Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003.
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472.
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262.
- Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7.
- Miyamoto T, Rho E, Sample V, et al. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep. 2015;11:657–670.
- Zhang D, Wang W, Sun X, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12:1447–1459.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393.