
ABSTRACT
Mutations in C9orf72 leading to hexanucleotide expansions are the most common genetic causes for amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). A phenotype resembling ALS and FTD is seen in transgenic mice overexpressing the hexanucleotide expansions, but is absent in C9orf72-deficient mice. Thus, the exact function of C9orf72 in neurons and how loss of C9orf72 may contribute to neuronal dysfunction remains to be clearly defined. Here, we showed that primary hippocampal neurons cultured from c9orf72 knockout mice have reduced dendritic arborization and spine density. Quantitative proteomic analysis identified C9orf72 as a component of the macroautophagy/autophagy initiation complex composed of ULK1-RB1CC1-ATG13-ATG101. The association was mediated through the direct interaction with ATG13 via the isoform-specific carboxyl-terminal DENN and dDENN domain of C9orf72. Furthermore, c9orf72 knockout neurons showed reduced LC3-II puncta accompanied by reduced ULK1 levels, suggesting that loss of C9orf72 impairs basal autophagy. Conversely, wild-type neurons treated with a ULK1 kinase inhibitor showed a dose-dependent reduction of dendritic arborization and spine density. Furthermore, expression of the long isoform of human C9orf72 that interacts with the ULK1 complex, but not the short isoform, rescues autophagy and the dendritic arborization phenotypes of c9orf72 knockout neurons. Taken together, our data suggests that C9orf72 has a cell-autonomous role in neuronal and dendritic morphogenesis through promotion of ULK1-mediated autophagy.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are adult-onset neurodegenerative diseases, predominantly affecting the motor and cognitive systems, respectively, and are recognized as one disease spectrum with each manifesting a distinct set of symptoms. Besides the clinical overlap, recent advances further establish that ALS and FTD share common genetic causes, molecular signatures and pathological hallmarks [Citation1–Citation3]. Among these genetic causes, repeat hexanucleotide (GGGGCC) expansion within the C9orf72 (chromosome 9 open reading frame 72) gene is the most common genetic cause for ALS and FTD [Citation4–Citation6], accounting for up to ~40% of familial ALS, ~7% of sporadic ALS, ~20% of familial FTD and ~80% of familial ALS-FTD [Citation1,Citation7]. Multiple non-mutually-exclusive pathogenic mechanisms, including gain-of-function toxicity due to the expanded repeats and partial loss of C9orf72 function due to the silencing of the mutant allele, have been proposed [Citation2,Citation8,Citation9]. A number of laboratories have addressed the physiological role of C9orf72 and whether loss of C9orf72 may contribute to ALS and FTD pathogenesis. Specifically, acute knockdown of C9orf72 in the central nervous system (CNS) of mice using antisense oligonucleotides (ASOs) does not affect general motor activity [Citation10]. Furthermore, CNS-deletion of C9orf72 [Citation11] as well as full-body C9orf72 knockout in mice does not cause motor neuron degeneration but results in progressive splenomegaly and lymphadenopathy leading to systemic immune dysfunctions [Citation12–Citation16]. Together, the evidence suggests that the loss of C9orf72 function is not sufficient to cause motor neuron disease. Nevertheless, c9orf72 knockout mice show an age-dependent reduction in social interaction, indicative of a FTD-like phenotype [Citation12]. Thus, the functions of C9orf72 in the CNS remain to be defined.
Alternative splicing of exon 5 in the human C9orf72 gene results in 2 protein isoforms. The short isoform of C9orf72 has a terminal lysine at position 222, whereas the remaining 221 amino acids (aa) are identical to the 481-aa long isoform [Citation4,Citation17]. Initial bioinformatics and structural studies predicted that the long isoform of C9orf72 belongs to a family of DENN (differentially expressed in neoplastic versus normal cells)-domain containing GDP/GTP exchange factors (GEFs) for RAB GTPases [Citation18,Citation19], which regulates intracellular membrane trafficking [Citation20]. In contrast, the short isoform, lacking part of the core DENN and dDENN (downstream DENN) domains, associates with the nuclear envelope [Citation17]. Thus, the two C9orf72 isoforms appear to function differently. Indeed, recent works have found that the long C9orf72 isoform interacts with RAB1, RAB5, RAB7, RAB29/RAB7L1, RAB8A, RAB11, and RAB39B to regulate membrane trafficking and autophagy functions [Citation21–Citation25]. Long C9orf72’s role in autophagy has been further supported by its association with ULK1/Atg1 (unc-51 like autophagy activating kinase 1) [Citation22–Citation24,Citation26,Citation27] and/or through MTOR-dependent TFEB (transcription factor EB) signaling [Citation28]. Conversely, Sivadasan and colleagues found that the long C9orf72 isoform interacts with CFL1 (cofilin 1) and modulates the small GTPases ARF6 and RAC1. This proposed interaction links C9orf72 to actin dynamics and axon outgrowth in cultured motor neurons [Citation29]. Therefore, the long C9orf72 isoform may regulate both membrane trafficking and cytoskeleton organization, whereas the functions of the short C9orf72 isoform remain poorly defined.
Autophagy is an intricate and finely regulated biodegradation process that typically degrades long-lived proteins, membrane proteins, and organelles via the lysosome [Citation30,Citation31], and can be further classified into 3 primary types: macroautophagy, microautophagy and chaperone-mediated autophagy [Citation32]. In macroautophagy, damaged organelles and proteins for degradation are enclosed by a double-membrane compartment (termed the phagophore); the phagophore expands and matures to form an autophagosome, which subsequently fuses with the lysosome to allow degradation of internal material within the autolysosome. One of the canonical signaling pathways for the activation of macroautophagy (hereafter referred as ‘autophagy’) is through ULK1, which forms a complex with RB1CC1/FIP200 (RB1 inducible coiled-coil 1), ATG13 and ATG101 [Citation33,Citation34]. ULK1 is in a feedback loop with 2 key master regulators, MTOR (mechanistic target of rapamycin kinase) and (AMP-activated protein kinase (AMPK), which controls biosynthetic and catabolic metabolism, respectively [Citation35,Citation36]. These 3 kinases, MTOR, AMPK and ULK1, form an intricate signaling loop to instruct how cells switch between anabolic and catabolic metabolism in response to the environmental status [Citation37]. ULK1 initiates the autophagy program by phosphorylating the downstream class III phosphatidylinositol 3-kinase (PtdIns3K) complex composed of BECN1/Beclin-1, ATG14, PIK3R4, NRBF2 and PIK3C3/VPS34 for phagophore formation and the subsequent autophagy cascade. Thus, the ULK1-RB1CC1-ATG13-ATG101 complex, by integrating various cellular and environmental signals, is the key component in autophagy initiation [Citation34].
Recent reports have clearly demonstrated a role of the long C9orf72 isoform in autophagy [Citation22–Citation24,Citation26–Citation28], although the exact mechanisms, such as how multiple putative RABs interact with C9orf72 to regulate autophagy, remain to be clarified. In addition, in these published studies, siRNAs were used to downregulate C9orf72 in assessing the functional impact on autophagy. Thus, how complete loss of C9orf72 may contribute to neuronal dysfunction and whether there are any functional differences between the 2 C9orf72 isoforms have not yet been addressed, which are the main focus of this study. Here, primary hippocampal neurons cultured from c9orf72 knockout mice showed reduced dendritic arborization and spine density, indicating a cell-autonomous effect of C9orf72 in neurons. In search for potential mechanisms, quantitative proteomic and biochemical analysis identified C9orf72 and SMCR8 as components of the autophagy initiation complex. The associations were mediated through direct interactions via the isoform-specific carboxyl-terminal DENN and dDENN domains of C9orf72 with ATG13. Consistent with the biochemical data, c9orf72 knockout neurons showed apparent defective autophagy accompanied by reduced ULK1 levels. Conversely, when wild-type neurons were treated with a ULK1 inhibitor, they showed a dose-dependent reduction of dendritic arborization and spine density similar to c9orf72 knockout neurons. Critically, expression of the long C9orf72 isoform that interacts with the ULK1 autophagy initiation complex, but not the short C9orf72 isoform, partially rescued the autophagy and dendritic arborization phenotype, demonstrating functional differences between these 2 C9orf72 isoforms. In summary, our data suggest a cell-autonomous physiological role of C9orf72 in neurons, where loss of C9orf72 affects neuronal morphology and spine density through defective ULK1-mediated autophagy, highlighting the concept that autophagy may contribute to neuronal morphogenesis as well as synaptic structures and functions.
Results
Reduced dendritic arborization and spine density in c9orf72 knockout neurons
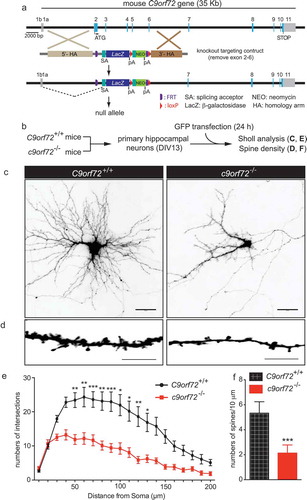
To address whether complete loss of C9orf72 contributes to neuronal dysfunction in a cell-autonomous manner, primary hippocampal neurons from wild-type (C9orf72+/+) and c9orf72 knockout (c9orf72−/-) mice [Citation12] were isolated and cultured in vitro. The neurons from C9orf72+/+ and c9orf72−/- mice were transfected with GFP at 13 days in vitro (DIV13) and their dendritic arborization and spine density was subsequently quantified ()). Sholl analysis, which measures the numbers of dendritic branch points as a function of the distance from the neuronal cell body, revealed reduced numbers of intersections of neurites in c9orf72−/- hippocampal neurons, indicating less complex dendritic arborization as compared to C9orf72+/+ neurons (). Furthermore, the spine density of c9orf72−/- hippocampal neurons was significantly decreased by ~60% when compared with C9orf72+/+ control neurons (). Thus, diminished dendritic spine numbers and dendritic arborization in the complete absence of C9orf72 in cultured hippocampal neurons suggests that neuronal C9orf72 is required for normal dendritic development and formation or maintenance of dendritic spines.
Figure 1. Reduced dendritic arborization and spine density in c9orf72 knockout hippocampal neurons. (a) Schematic of c9orf72 knockout mice showed that exons 2–6 were deleted and resulted in a null allele. (b) Primary hippocampal neurons were cultured from wild-type (C9orf72+/+) and c9orf72 knockout (c9orf72−/-) mice and transfected with a plasmid encoding GFP to visualize the neuronal and dendritic morphology. (c) Images of GFP-transfected hippocampal neurons from control and c9orf72 knockout mice. The images are presented in gray scale and inverted color. Scale bar: 50 µm. (d) High-magnification renderings of dendritic segments. The dendritic images were taken with 0.3-µm step z-section and then stacked as a maximum projection. Scale bar: 10 µm. (e) Sholl analysis of dendritic arborization. (f) Reduced spine density in the c9orf72 knockout neurons. (at least 3 independent experiments, N > 4 neurons per genotype per experiment, 17 total neurons were quantified for E and F, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
C9orf72 associates with the autophagy initiation complex via the isoform-specific DENN-dDENN domain
To understand how C9orf72 may contribute to the observed neuronal phenotype, we sought to identify the C9orf72 interacting proteins using our previous approach that combines affinity purification and quantitative mass spectrometry [Citation38,Citation39]. To this end, we first generated an inducible stable cell line expressing a single copy of the long C9orf72 isoform tagged with a localization and affinity purification (LAP)-tag, which is comprised of GFP and the 6xHis+FLAG-epitopes separated by a PreScission cleavage site ()). Immunoblot of C9orf72 revealed that the LAP-tagged C9orf72 was expressed at a level comparable to endogenous C9orf72 (). A stable isotope labeling by amino acids in cell culture (SILAC) approach coupled with affinity purification was used to identify specific interactors, where eluates from PreScission cleavage after GFP immunoprecipitation were subjected to mass spectrometry (). Using the criteria that (1) all the proteins must have been identified with more than 2 unique peptides, and (2) all peptide signals (heavy:light isotope labeling) for each protein must have at least 8-fold enrichment in the C9orf72 sample compared with the control (purified from the parental HeLa cell line), 5 proteins were identified: SMCR8 (Smith-Magenis syndrome chromosome region, candidate 8), C9orf72, IARS (isoleucyl-tRNA synthetase), WDR41 (WD repeat domain 41) and RB1CC1 (). Among these, RB1CC1 is a scaffold protein that associates with ULK1, and ATG13 (autophagy related 13) to form the autophagy initiation complex [Citation34]. Consistent with recent reports [Citation22–Citation24,Citation26,Citation27], the putative C9orf72-associating proteins, including RB1CC1, ULK1, ATG13, SMCR8 and WDR41, were confirmed by immunoblotting in C9orf72 immunoprecipitation samples (). Furthermore, reciprocal immunoprecipitations using HA-tagged RB1CC1, ATG13 and ATG101 [Citation40] validated the interaction between RB1CC1, ATG13, ATG101 and C9orf72 ( and S1).
Figure 2. C9orf72 associates with the autophagy initiation complex. (a) Schematic of the LAP-tagged long C9orf72 isoform. (b) Immunoblotting of C9orf72 in an inducible stable cell line expressing a single copy of the long C9orf72 isoform. Lysates from MEFs generated from wild-type and c9orf72 knockout (c9 KO) mice were used to identify the correct immune-reactive band as endogenous C9orf72, as multiple bands were recognized by commercially available C9orf72 antibody. The LAP-tagged C9orf72 long isoform expresses at a comparable level to the endogenous protein. (c) Schematic of SILAC-affinity purification to identify C9orf72-associating proteins. Cell lines stably expressing the LAP-tagged C9orf72 were grown in isotopically ‘heavy’ medium containing 13C6,15N4-arginine and 13C6,15N2-lysine, while the parental line (i.e., no transgene) was grown in ‘light’ medium containing normal arginine and lysine. Elution after the PreScission cleavage step was used for mass spectrometry analysis. (d) Graphic representation of the C9orf72 interactome. Y-axis displays the average ratio of peptides identified in the heavy vs light labeling, while the X-axis represents individual proteins; a protein was considered to be C9orf72-associated if it was at least 8-fold above background signal. The protein ID, median SILAC ratio, and numbers of unique peptides identified are listed. (e) Confirmation of mass spectrometry results by IP-western blots, including RB1CC1, ULK1, ATG13, SMCR8 and WDR41. (f) Reciprocal immunoprecipitation using HA-tagged ATG13 and ATG101. Cells were transfected with plasmids as indicated with pcDNA6 as a control. HA-tagged ATG13 and ATG101 pulled down the LAP-tagged long C9orf72 isoform. (g) Schematic representation of in situ proximity-ligation assay. (h) Representative results for in situ proximity-ligation assay. Phalloidin staining, which labels F-actin, was used to outline the cells. Scale bar: 20 µm. (i) Quantification of in situ PLA results (N = 3, cell numbers >20 per experiment, ***, p < 0.001). C9, C9orf72.
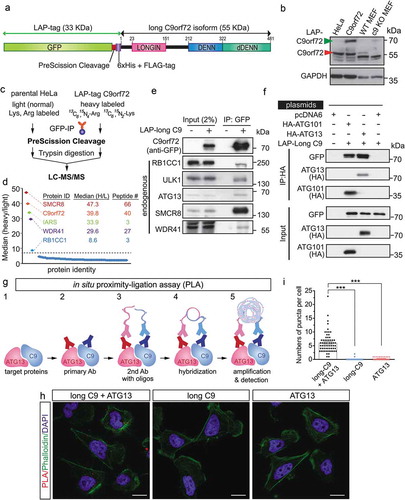
To further confirm the in vivo association of C9orf72 and the autophagy initiation complex, an in situ proximity-ligation assay (PLA) [Citation41] was employed. In this assay, cells were fixed with cell membranes gently lysed, and subsequently incubated with primary antibodies against C9orf72 (in this case, we used a FLAG antibody recognizing LAP-tagged C9orf72) and endogenous ATG13. The secondary antibodies conjugated with different oligonucleotides (1 for each of the 2 different primary antibodies) were used. When the distance between the 2 different oligonucleotides is less than 40–50 nm, they can be hybridized and act as primers for rolling-circle amplification. Subsequently, fluorescent-labeled oligonucleotides were hybridized with the amplification products and the signals observed with a fluorescence microscope (). PLA signals were observed in the cytosol when a combination of anti-FLAG (C9orf72) and anti-ATG13 antibodies were used, but not in 2 negative control conditions where only 1 primary antibody was used (). Thus, the biochemical and imaging data indicate that C9orf72 associates with the autophagy initiation complex in vivo.
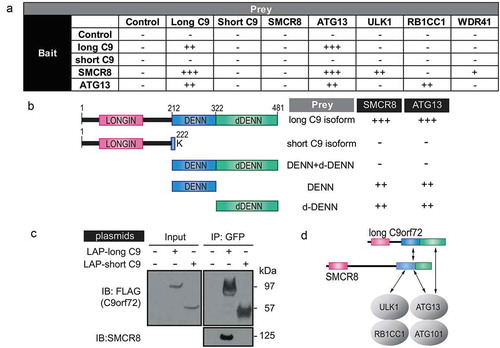
To delineate how C9orf72 and SMCR8 associate with the autophagy initiation complex, yeast two-hybrid assays were performed to determine the binary interactions between different components of the autophagy initiation complex. In addition, the short isoform of C9orf72 that contains the first 221 amino acids with a terminal lysine residue at 222 aa was used to determine the specificity of the interaction ()). The long, but not the short, isoform of C9orf72 interacted with itself, SMCR8 and ATG13, whereas SMCR8 interacted with the long C9orf72 isoform, ATG13 and ULK1. The interaction between ATG13 and the long C9orf72 isoform was confirmed using ATG13 as bait. Because the long, but not the short, C9orf72 isoform interacted with SMCR8 and ATG13, we reasoned that the C-terminal DENN-dDENN domain of C9orf72 may be required for the direct interactions. To test this, the C-terminal DENN and dDENN domain of C9orf72 were further divided into DENN, dDENN, and DENN-dDENN fragments in the yeast two-hybrid assay. The individual DENN or dDENN domain alone, but not the DENN-dDENN fragment, interacted with SMCR8 and ATG13 (). To further corroborate the isoform-specific interaction between C9orf72 and SMCR8, we performed GFP immunoprecipitation with transient transfection of the long and short C9orf72 isoforms, respectively. The long, but not the short, C9orf72 isoform immunoprecipitated SMCR8 (). Taken together, our data indicated that the isoform-specific DENN-dDENN domain of C9orf72 are involved in direct interactions with the autophagy initiation complex via ATG13 and SMCR8, the latter of which also directly interacted with ATG13 and ULK1 ().
Figure 3. C9orf72 associates with the autophagy initiation complex via isoform-specific DENN-dDENN domains. (a) Yeast two-hybrid (Y2H) mapping of binary interaction pairs. + is used to represent the strength of the X-gal staining and, thus, the strength of the interaction. – means that no cells survived in the 4-drop out conditions. (b) Y2H mapping of C9orf72 domains with SMCR8 and ATG13. Domain representation of long and short C9orf72 isoform, DENN-dDENN, DENN and dDENN domains and their association with SMCR8 and ATG13. (c) GFP immunoprecipitations were performed using the transiently expressed long or short C9orf72 isoform in cells and probed with SMCR8. Long and short C9orf72 isoforms were probed with FLAG antibody. The long, but not the short, C9orf72 isoform co-immunoprecipitated with SMCR8. (d) Proposed interactions among long C9orf72, SMCR8, and the autophagy initiation complex. The carboxyl terminus of the DENN-dDENN domains of C9orf72 is critical for their direct interaction with ATG13 and SMCR8, the latter of which also directly interacts with ULK1 and ATG13. C9, C9orf72.
Defective ULK1-mediated autophagy in c9orf72 knockout neurons underlies the reduced dendritic arborization phenotype
Because of the association between C9orf72 and members of the autophagy initiation complex, we next examined autophagy using a MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)-based assay in primary neurons from wild type (C9orf72+/+) and c9orf72 knockout (c9orf72−/-) mice. During autophagy, LC3 undergoes a post-translational modification, in which the extreme 5 carboxyl-terminal amino acids are first cleaved, and then the newly exposed C-terminal glycine is conjugated to a lipid (phosphatidylethanolamine, PE) group. The terms, LC3-I and LC3-II, are used to distinguish the unconjugated and lipid-modified form of LC3, respectively. The accumulated LC3-II levels and puncta serve as indicators of autophagy.
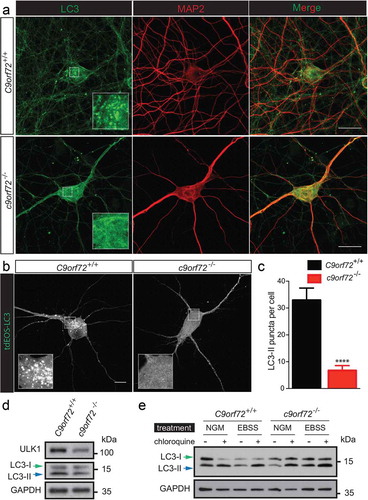
To correlate the changes in dendritic morphology and spine density observed in the c9orf72 knockout neurons with autophagy, LC3-II puncta and levels were examined using primary hippocampal neurons cultured from wild-type and c9orf72 knockout mice. Immunostaining of endogenous LC3 revealed a reduction of LC3-II puncta ()). However, due to the elaborated neuronal processes, it is difficult to reliably quantify the LC3-II puncta for any given neuron. To circumvent this problem, C9orf72+/+ and c9orf72−/- hippocampal neurons were transfected with a photo-convertible fluorescent protein (tdEOS)-tagged LC3, and LC3-II puncta were quantified. c9orf72−/- hippocampal neurons showed an ~70% reduction of LC3-II puncta (). Consistent with the image data, reduced LC3-II accumulation was observed in c9orf72−/- cortical neurons, indicative of altered autophagy (). Furthermore, the reduced LC3-II levels correlated with a reduced ULK1 expression level in primary cortical neurons (). To address whether autophagy flux is also affected in the c9orf72−/- neurons, autophagy was induced by nutrient deprivation in the presence and absence of chloroquine, a lysosome activity inhibitor that neutralizes lysosomal pH. Increased LC3-II accumulation was also observed upon starvation and in the presence of chloroquine, suggesting that starvation-induced autophagy and autophagy flux were normal (). Taken together, our data suggest that reduced basal autophagy in c9orf72 knockout neurons may act via a ULK1-dependent mechanism.
Figure 4. Reduced autophagy in c9orf72 knockout neurons accompanied by reduced ULK1 levels. (a) Immunofluorescence images of endogenous LC3 and MAP2/MAP2A on primary hippocampal neurons cultured from wild-type and c9orf72 knockout mice. Scale bar: 20 μm. (b) Fluorescent images of wild-type and c9orf72 knockout hippocampal neurons transfected with photo-convertible GFP (tdEOS)-tagged LC3. Insets show a scaled-up image of the boxed region. Wild-type neurons showed visible LC3-II puncta. Scale bar: 50 µm. (c) Quantification of LC3-II puncta in wild type and C9orf72 knockout hippocampal neurons (at least 3 independent experiments, N > 4 neurons per genotype per experiment, ***, p < 0.001, ****, p < 0.0001). (d) Immunoblots of ULK1, LC3 and GAPDH on lysates of wild-type (C9orf72+/+) and c9orf72 knockout (c9orf72−/-) cortical neurons. Green and blue arrows indicate LC3-I and LC3-II, respectively. Levels of ULK1 and LC3-II were reduced in c9orf72 knockout neurons. (e) Immunoblots of LC3 and GAPDH on lysates of wild type (C9orf72+/+) and c9orf72 knockout (c9orf72−/-) cortical neurons treated with normal growth medium (NGM) or Earle’s balance salt solution with or without chloroquine (CQ). LC3-II accumulated in the presence of chloroquine, indicating the autophagy flux is normal in the c9orf72 knockout neurons.
ULK1 initiates the autophagy program by phosphorylating the downstream class III PtdIns3K complex composed of BECN1, ATG14, NBRF2, PIK3R4 and PIK3C3. Thus, if reduced ULK1-mediated autophagy accounts for the dendritic arborization and spine density phenotype observed in c9orf72 knockout neurons, one would expect that reducing ULK1 kinase activity in wild-type neurons could give rise to a phenotype similar to that observed in the c9orf72 knockout neurons. To test this, a selective ULK1 competitive inhibitor, SBI-0206965 [Citation42], was used to treat primary hippocampal neuron cultures from wild-type mice ()). Although high doses (10 and 50 μM) of SBI-0206965 were effective in inhibiting direct ULK1 substrates using in vitro biochemical assays [Citation42], they were very toxic to neurons as no neurons survived the treatment (data not shown). In contrast, low dose (1 and 2 μM) of SBI-0206965 treatment produced a dose-dependent reduction in dendritic arborization and spine density in wild-type hippocampal neurons ().
Figure 5. Inhibition of ULK1 kinase activity reduces dendritic arborization and spine density in wild-type hippocampal neurons. (a) Schematic of ULK1 kinase inhibitor (SBI-0206965) treatment in wild-type hippocampal neurons. Wild-type neurons were treated with 1, 2, 5, 10, and 50 μM of SBI-0206965 at DIV7 with a fresh dose every 3 days and transfected with a plasmid encoding GFP at DIV13 to visualize the neuronal morphology. Sholl analysis and spine density was quantified. Ten and 50 µM of SBI-0206965 treatment was toxic to neurons as no neurons survived, and thus, was excluded from the analysis. (b) Images of GFP-transfected hippocampal neurons from control mice treated with DMSO (control), or 1 and 2 µM of SBI-0206965. The images are presented in gray scale and inverted color. Scale bar: 50 μm. (c) High-magnification renderings of dendritic segments. The dendritic images were taken with 0.3-µm step z-section and then stacked as a maximum projection. Scale bar: 5 µm. (d,e) Dose-dependent reduction of dendritic arborization (d) and spine density (e) in the wild-type neurons treated with SBI-0206965 (at least 3 independent experiments were performed, total numbers of neurons scored are indicated, *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001). (f,g) Immunoblots of BECN1, phospho-Ser93 BECN1, MTOR, phospho-Ser2448 MTOR, ULK1 and GAPDH in HeLa cells (f) and primary cortical neurons (g) treated with varying concentrations (1, 2, 5, 10, 50 µM) SBI-0206965.
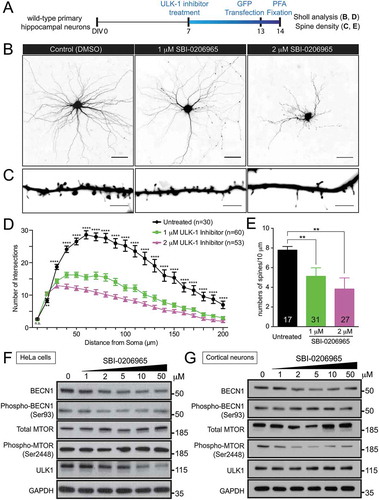
To confirm the specificity and effectiveness of the ULK1 kinase inhibitor in inhibiting autophagy, the phosphorylation status and protein level of BECN1, a direct ULK1 substrate [Citation42] and a crucial molecule in phagophore formation, was examined. At all the doses (1, 2, 5, 10, and 50 μM) examined, ULK1 inhibitor SBI-0206965 treatment was effective in reducing phospho-Ser93 BECN1 levels, corresponding to a putative ULK1 phosphorylation site in HeLa cells. Furthermore, a consistent reduction of BECN1 protein levels was also observed (). The reduction of BECN1 protein level upon ULK1 inhibition is consistent with published work [Citation42]. Although there was no clear reduction of phospho-Ser93 BECN1 and ULK1, the reduction of BECN1 were observed in primary neurons treated with varying concentrations (1, 2, 5, 10, and 50 μM) of ULK1 inhibitor (), suggesting that inhibiting ULK1 kinase activity, at least in part, reduces autophagy via a BECN1-mediated pathway.
To further test whether autophagy inhibition contributes to the dendritic arborization phenotype, an autophagy inhibitor, spautin-1, which triggers BECN1 degradation, was used [Citation43]. As spautin-1 has been shown to be effective in inhibiting starvation-induced autophagy [Citation43], we first determined the conditions in which spautin-1 inhibits autophagy using LC3-II accumulation as a readout in HeLa cells. A combination of either nutrition deprivation (by Earle’s balanced salt solution treatment) or MTOR inhibition (by rapamycin) with spautin-1 were more effective than spautin-1 alone in reducing LC3-II accumulation (Figure S2(a)). However, primary hippocampal neurons were very susceptible to the combination of short-term starvation of culture medium without B27 supplement with spautin-1 treatment, as very few neurons survived this treatment (data not shown), whereas the combination of rapamycin and spautin-1 was tolerated. Under these conditions, rapamycin treatment did not affect dendritic arborization, whereas combined rapamycin and spautin-1 treatment led to reduced dendritic arborization (Figure S2(b,c)). Taken together, our data suggest that defective ULK1-BECN1-mediated autophagy may, at least in part, underlie the reduced dendritic arborization phenotype observed in c9orf72 knockout neurons.
The long, but not the short, isoform of C9orf72 rescues the dendritic arborization and autophagy phenotype
To further determine whether C9orf72-mediated autophagy regulation is required for neuronal morphology as described above, and whether the 2 putative C9orf72 isoforms perform separate functions, c9orf72−/- hippocampal neurons were transfected with either the long or short isoform of C9orf72; only the former associates with the autophagy initiation complex (). The rescue constructs contain a membrane-bound form of mCherry (mCherryCAAX) to visualize the neuronal morphology ()). The long, but not the short, isoform of C9orf72 partially rescued the dendritic arborization phenotype, but not the spine density of c9orf72−/- hippocampal neurons (). Importantly, expression of the long, but not the short, C9orf72 isoform restored the accumulation of LC3-II puncta (). Because reduced ULK1 levels were observed in c9orf72 knockout neurons (), we also attempted to rescue the dendritic arborization phenotypes of c9orf72 knockout neurons by overexpressing ULK1. Unfortunately, transient high-level expression of ULK1 in c9orf72 knockout neurons appeared to be toxic to neurons, with surviving neurons exhibiting dramatically reduced dendritic arborization phenotypes (Figure S3).
Figure 6. The long, but not the short, isoform of C9orf72, partially rescues the autophagy and the dendritic arborization phenotype. (a) Images of transfected hippocampal neurons from control and c9orf72 knockout mice. Nuclear-localized GFP and membrane-bound mCherry linked by P2A peptide (GFPNLS-P2A-mCherryCAAX, pSYC97), or long or short C9orf72 isoform replacing GFPNLS were examined for their effects on dendritic arborization and autophagy in c9orf72 knockout neurons. Genotypes of neurons are listed in the top panel. The second panels indicate the rescue constructs used: GFPNLS (GFPNLS-P2A-mCherryCAAX, long C9orf72 (long C9ORF72-P2A-mCherryCAAX), and short C9orf72 (short C9ORF72-P2A-mCherryCAAX). The images are presented in gray scale and inverted color. Scale bar: 20 µm. (b,c) Sholl analysis of dendritic arborization (b) and spine density (c) of the rescue experiments (at least 4 independent experiments, N > 3 neurons per genotype per experiment, n.s.: not significant, *, p < 0.05; **, p < 0.01; ***, p < 0.001). (d) Fluorescent images of c9orf72 knockout hippocampal neurons transfected with photo-convertible GFP (tdEOS)-tagged LC3 without or with long C9orf72 (long C9ORF72-P2A-mCherryCAAX) or with short C9orf72 (short C9ORF72-P2A-mCherryCAAX) rescue constructs. Insets show a scaled-up image of the boxed region. Scale bar: 10 µm. (e) Quantification of LC3-II puncta in c9orf72 knockout hippocampal neurons with or without the long C9orf72 rescue construct (at least 3 independent experiments N > 3 neurons per genotype per experiment, ***, p < 0.001). (f) LMNB1-immunofluorescent images of hippocampal neurons from control and c9orf72 knockout mice. Nuclear envelopes were apparently normal in both wild-type and c9orf72 knockout hippocampal neurons. Scale bar: 10 µm. (g) Relative mRNA expression levels of C9orf72, Ulk1, Rb1cc1, Atg13, and Atg101 in primary cortical neurons cultured from wild-type and c9orf72 knockout mice. At least 3 independent cultures were used (n.s.: not significant; ***, p < 0.001). (h) Immunoblots of RB1CC1, ATG13, SQSTM1, TARDBP and GAPDH on protein lysates of primary cortical neurons cultured from wild-type and c9orf72 knockout mice.
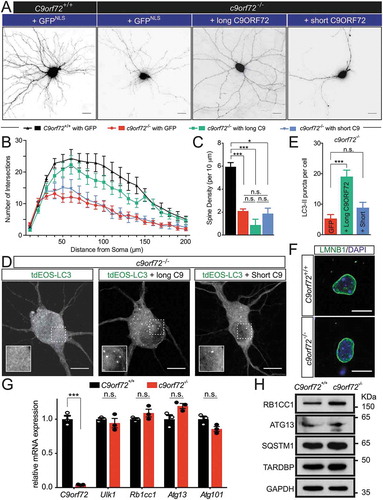
So far, the function of the short C9orf72 isoform is not known, but this isoform associates with the nuclear envelope [Citation17], which implies a potential nuclear function. Immunofluorescent staining of LMNB1 (lamin B1) was performed in primary neurons from wild-type and c9orf72 knockout neurons. No obvious deformations in nuclear envelope were observed (). In addition, a reduced ULK1 protein level () raised the possibility that ULK1 protein reduction may be due to the unknown nuclear C9orf72 function affecting the Ulk1 transcript. To this end, qRT-PCR using RNA extracted from wild-type and c9orf72 knockout neurons was performed. Comparable Ulk1 mRNA levels were observed with or without C9orf72 (). Furthermore, mRNA levels for Rb1cc1, Atg13 and Atg101 remained unchanged (). Consistent with the mRNA expression, RB1CC1 and ATG13 protein levels were also comparable in wild-type and c9orf72 knockout neurons (). Furthermore, the protein levels for SQSTM1/p62 and TARDBP/TDP-43 were also similar. Therefore, the data indicate that C9orf72 may regulate ULK1 protein reduction via a post-transcriptional mechanism. Taken together, restored dendritic morphology and autophagy functions with the C9orf72 isoform that interacts with the autophagy initiation complex suggests that (i) C9orf72-mediated autophagy is required for normal dendritic arborization, and (ii) the 2 C9orf72 isoforms may function differently.
Discussion
In this study, we examined the physiological role of C9orf72 in primary neurons in culture. Our results showed that C9orf72 plays a cell-autonomous role in neuronal and dendritic morphogenesis potentially via an autophagy-mediated mechanism. Biochemically, C9orf72 together with SMCR8 are components of the autophagy initiation complex composed of ULK1, RB1CC1, ATG13, and ATG101, which is consistent with recent published work [Citation22–Citation24,Citation26,Citation27]. We further showed that the isoform-specific DENN-dDENN domain of C9orf72 is required for the direct interaction among C9orf72, SMCR8 and ATG13. Furthermore, the observed defects in autophagy and the reduction in ULK1 levels in the c9orf72 knockout neurons further supports the notion that C9orf72 regulates autophagy. The reduction of dendritic arborization and spine density can be recapitulated by pharmacological inhibition of ULK1 kinase activity in wild-type neurons. Functionally, expression of the long C9orf72 isoform that interacts with the autophagy initiation complex partially rescues the dendritic arborization and autophagy phenotypes, unlike the short isoform, which does not interact with components of the autophagy initiation complex, suggesting the long and short C9orf72 isoforms may have different functions. Although there were previous descriptions for C9orf72’s function in autophagy using knockdown and cell lines [Citation22–Citation24,Citation26–Citation28], our current work highlights the following points: (i) there is a cell-autonomous role of C9orf72 in neurons based on the use of primary neurons cultured from c9orf72-null mice, (ii) there is a link between ULK1-mediated autophagy with neuronal morphogenesis, (iii) there are functional differences between the 2 C9orf72 isoforms.
Our biochemical findings are consistent with recent work showing that C9orf72 and SMCR8 are components of the ULK1 autophagy initiation complex () [Citation22–Citation24,Citation26,Citation27]. Using a yeast two-hybrid assay, we showed that both C9orf72 and SMCR8 interact directly with the components of the ULK1 autophagy initiation complex: C9orf72 interacting with ATG13; and SMCR8 with ATG13 and ULK1. In addition, C9orf72 also interacts directly with SMCR8. Due to the auto-activation of RB1CC1 in the yeast two-hybrid assay (not shown), we were not able to map the interaction between RB1CC1 and C9orf72 or SMCR8. Nevertheless, using a co-immunoprecipitation assay with various RB1CC1 fragments, the amino-terminal 450 amino acids of RB1CC1 appeared to be critical for the association with C9orf72 (Figure S1).
Both C9orf72 and SMCR8 belong to a family of proteins containing the DENN (differentially expressed in neoplastic vs normal cells) domain family proteins, which typically are comprised of the upstream DENN (uDENN, also called longin domain), core DENN, and the downstream DENN (dDENN) as tripartite DENN modules [Citation18,Citation44,Citation45]. The DENN and dDENN domains of C9orf72 appeared to be critical for the direct interaction between C9orf72 with SMCR8 and ATG13 (). Our biochemical data are consistent with the co-immunoprecipitations with DENN-deleted C9orf72 and SMCR8 constructs [Citation24,Citation46] and in vitro affinity isolation assays using recombinant proteins of C9orf72 with RB1CC1, ATG13 and ULK1 [Citation23]. However, it should be noted that in contrast to our data as well as 2 other published studies [Citation24,Citation46], Webster and colleagues showed that both long and short C9orf72 isoforms interact with the ULK1 complex, albeit the short isoform interacts with an apparent lower affinity [Citation23]. Our interpretation that only the long isoform interacts with the autophagy initiation complex is further bolstered by the yeast two-hybrid data, which is an experimental paradigm not utilized by the other 3 studies, whereas the differences in the co-immunoprecipitation results may be simply methodological in origin, although it is interesting to note that even the Webster study confirms a preferential interaction of ULK1 with the long C9orf72 isoform [Citation23].
It is worth mentioning that either the DENN or dDENN domain of C9orf72 could interact directly with SMCR8 and ATG13. However, expression of the DENN and dDENN portion of C9orf72 does not appear to interact with SMCR8 or ATG13. This could be explained by at least 2 possible scenarios. It is conceivable that there are intramolecular interactions between DENN and dDENN, and the interaction may prevent intermolecular DENN and dDENN interactions. The other possibility may be that the orientation of the interaction prevented the activation of downstream transcription in the yeast two-hybrid assay. Using various SMCR8 fragments expressed in cells for co-immunoprecipitation, Jung and colleagues mapped the interactions of SMCR8 with C9orf72 and ATG13 to the N-terminal domain of SMCR8 [Citation27]. Thus, the exact importance of the DENN and dDENN domain of C9orf72 and SMCR8 will require further structure-function studies.
Functionally, reduced LC3-II levels and puncta in c9orf72 knockout neurons () suggests defects in autophagy. Our results are consistent with previous studies using shRNA to knock down C9orf72 in primary neurons and organotypic slice cultures [Citation22,Citation23]. More strikingly, the steady-state ULK1 level was reduced () with comparable mRNA levels () in the c9orf72 knockout neurons, suggesting that C9orf72 regulates ULK1 expression via a post-transcriptional mechanism. The reduction of the level of phospho-serine 757 of ULK1, which is a measurement of ULK1 inhibition, has been observed under C9orf72 knockdown conditions, although ULK1 levels were not investigated in one study using mouse embryonic fibroblasts (MEFs) [Citation24], whereas ULK1 levels were not affected in another study using HEK293 cells [Citation23]. The discrepancy between our results and those of Webster and colleagues may be due to the cell type used and the knockdown efficiency, as in our case, the C9orf72 gene was completely knocked out. Curiously, SMCR8 appears to have an antagonistic effect with C9orf72, as Smcr8 deletion showed enhanced ULK1 serine 757 phosphorylation [Citation24]. SMCR8 has been recently shown to be able to regulate transcription of several autophagy genes [Citation27]. Furthermore, Nazio and colleagues showed a delicate regulatory mechanism for Ulk1 transcription and translation to ensure autophagy oscillation and prevent constitutively active autophagy-induced cell death [Citation47]. The exact mechanism by which C9orf72 regulates the ULK1 level requires further investigation.
Although the ULK1 function in autophagy is well-established [Citation33,Citation34], ULK1 is also involved in membrane transport, including ER-to-Golgi trafficking [Citation48]. ULK1 is the mammalian homolog of the nematode C. elegans’ unc-51 (uncoordinated-51) gene, mutations of which gave rise to various axon abnormalities [Citation49]. The axonal phenotypes are not observed for constitutive knockout of other autophagy genes, such as Atg5 [Citation50], Atg7 [Citation51], and Atg3 [Citation52], suggesting that UNC-51/Atg1/ULK1 has autophagy-independent functions. Indeed, subsequent work demonstrated that unc-51/Atg1/ULK1 regulates axonal outgrowth via at least 2 different mechanisms: unc-51/Atg1/ULK1 interacts with SYNGAP1 and SDCBP/Syntenin to regulate RAB5-mediated endocytic pathways [Citation53], and with UNC-76/FEZ1, an adaptor protein for the kinesin heavy chain, to regulate axonal transport [Citation54]. Similar to the nematode work, Atg1/UNC-51/ULK1 regulates axonal and dendritic development via kinesin-mediated vesicle transport in Drosophila [Citation55]. Because the ULK1 level is reduced in the c9orf72 knockout neurons, it is formally possible that the reduced dendritic arborization and spine density may also be due to the above-mentioned autophagy-independent functions of ULK1. Nevertheless, previous work indeed supports the notion that autophagy is involved in neuronal morphogenesis and spine maintenance. In particular, autophagy inhibition using 3-methyladenine reduces the viability and branching complexity of rat dorsal root ganglia (DRG) neurons [Citation56], and ATG7 appears to regulate axon extension in rat primary cortical neurons [Citation57]. Furthermore, MTOR-dependent autophagy is required for synapse pruning [Citation58]. It is also worth noting that the optimal level and activity of ULK1 is critical for neuronal health, as either high dose of ULK1 inhibitor () or transient ULK1 overexpression (Figure S3) appear to be toxic to neurons. The latter is consistent with the notion that constitutively active autophagy induces cell death [Citation47]. In this study, 2 independent autophagy inhibitors, SBI-0206965 and spautin-1, that target ULK1 and BECN1, respectively, were used to determine whether autophagy inhibition could give rise to the phenotypes similar to those observed in c9orf72 knockout neurons. In both cases, autophagy inhibition and reduced arborization phenotypes were observed. Because (i) reduced ULK1 levels were accompanied by reduced LC3-II puncta in c9orf72 knockout neurons (), (ii) inhibition of ULK1 kinase activity in wild-type neurons reduces dendritic arborization and spine density similar to c9orf72 knockout neurons (), and (iii) the autophagy defects and dendritic arborization phenotype could be rescued by expressing the C9orf72 isoform that associates with the ULK1 complex (), our data suggest that C9orf72 may regulate neuronal morphogenesis and spine density via ULK1-dependent autophagy.
It is well-recognized that disruption of protein homeostasis (known as proteostasis), including autophagy dysfunction, contributes to neurodegenerative diseases [Citation59,Citation60]. Moreover, recent genetic studies on ALS and FTD identified several causal genes involved in proteostasis regulation, such as VCP [Citation61], OPTN [Citation62], UBQLN2 [Citation63], SQSTM1 [Citation64], TBK1 [Citation65,Citation66], and CHMP2B [Citation67,Citation68]. We and others have shown that C9orf72 plays a role in autophagy regulation [Citation22–Citation24,Citation26–Citation28]. Our study further highlights a cell-autonomous role of C9orf72 in neuronal morphogenesis and dendritic function via autophagy. Intriguingly, knockdown of C9orf72 in zebrafish and cultured motor neurons showed an axonal growth and branching phenotype [Citation29,Citation69], further supporting the role of C9orf72 in neuron morphogenesis. Taken together, although the loss of C9orf72 function may not be sufficient to trigger motor neuron disease, C9orf72 may be required for optimal neuronal functions such as synaptic structures and functions via cell-intrinsic autophagy mechanisms.
Materials and methods
c9orf72 knockout mice
c9orf72 knockout mice were generated as described previously [Citation12] and maintained in a C57BL/6 background. All studies were carried out under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the National University of Singapore, and were in compliance with Association for Assessment of Laboratory Animal Care (AAALAC) guidelines for animal use.
Constructs
The Flp-In LAP-tag construct based on pcDNA5/FRT/TO was described previously [Citation38,Citation39]. N-terminal FLAG peptide (DYKDDDDK)-tagged long and short human C9orf72 isoform was amplified by PCR from the cDNA clone (Dharmacon, MGC human sequence-verified cDNA, clone ID: 5298741, MHS6278-202809072) and inserted into pcDNA5/TO/FRT/LAP using BamHI and XhoI sites. FLAG-tagged long and short C9orf72 isoforms and HA-tagged ULK1 were cloned into pSYC-97 (Addgene, 31565; deposited by Seok-Yong Choi) to generate FLAG-long C9ORF72-P2A-mCherryCAAX, FLAG-short C9ORF72-P2A-mCherryCAAX and HA-ULK1-P2A-mCherryCAAX. Full-length human ATG13 and ATG101 were amplified from cDNA (Addgene 22875 and 22877, respectively; deposited by Noboru Mizushima) and inserted into the pRK5-HA-ULK1 vector (Addgene, 31963, deposited by Do-Hyung Kim) with SalI and NotI sites. Long isoform, short isoform, DENN-alpha, DENN and the alpha domain of C9orf72, SMCR8 (MGC sequence-verified cDNA clone:164968, PlasmID Repository at Harvard Medical School, HsCD00347993), ATG13, ULK1, RB1CC1/FIP200 (MGC sequence-verified cDNA clone MGC:9519, PlasmID Repository at Harvard Medical School, HsCD00327488), and WDR41 (Dharmacon, MGC human sequence-verified cDNA, clone ID: 3447269, MHS6278-202755585) were amplified by PCR and cloned into yeast two-hybrid vectors, pGADT7-AD (Takara, 630442) and pGBKT7 (Takara, 630443). LC3 was amplified by PCR and subcloned into tdEos-Tubulin-6 (Addgene, 57682; deposited by Michael Davidson) to generate ptdEOS-LC3. All constructs were confirmed by sequencing the entire open reading frame.
Primary hippocampal neuronal culture, treatment, transfection, image acquisition, image and morphological analysis
Primary hippocampal neurons were cultured as described previously [Citation70]. In brief, hippocampi were harvested from the brains of pups at postnatal day 0 or 1 (P0 or P1) from non-transgenic animals or homozygous c9orf72 knockout mice, and dissociated using 0.125% trypsin. The hippocampal cultures were seeded at a density of 300 cells/mm2 on poly-L-lysine-coated coverslips and maintained in Neurobasal medium (Thermo Fisher Scientific, 21103049) supplemented with 2% B27 supplement (Thermo Fisher Scientific, 17504044), 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070063), GlutaMax (Thermo Fisher Scientific, 35050061), sodium pyruvate (Thermo Fisher Scientific, 11360070) and 50 mM HEPES, pH 7.4 (GE Healthcare HyClone, SH30237.01). Cells were grown in 5% CO2 at 37ºC. The neurons were transfected with plasmids encoding EGFP, GFPNLS-P2A-mCherryCAAX, or C9ORF72-P2A-mCherryCAAX to visualize dendritic spines using calcium phosphate precipitation or Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) at DIV13 as indicated in the figure legends. SBI-0206965 (Sigma-Aldrich, SML1540), rapamycin (Sigma-Aldrich, 37094) and spautin-1 (Sigma-Aldrich, SML0440) were diluted in DMSO for the final concentrations as indicated in the figure legends. DMSO alone was used for the control. Neurons treated with DMSO or mock treatment were indistinguishable and pooled for quantification purpose. The neurons were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, 15713), 4% sucrose (VWR, VWRC0335) in 1x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and analyzed within 24 h after the transfection. A Zeiss LSM700 inverted confocal microscope and a 63×/1.15 N.A. oil immersion objective was used for visualization and image acquisition. Images were captured using an AxioCam MRm monochromatic CCD camera (Zeiss) run by Zeiss Zen software. EGFP- or mCherryCAAX-labeled neurons were chosen randomly for imaging from neuronal cultures from at least 3 coverslips. For all dendritic spine analyses, the region of the apical dendrites after the first branch point was selected (secondary dendrite). Dendritic spine density was scored from 3 randomly chosen areas per neuron. Z-sections were taken at 0.3-μm intervals and were stacked with maximum projection. Data were expressed as means ± SEM from independent replicates. Sholl analysis was done with the Fiji plug-in for ImageJ. The image (8-bit) threshold was adjusted prior to analysis. The starting and end point for the analyzed area around the cell were manually chosen. For quantification of LC3-II puncta, the image (8-bit) threshold was adjusted to remove the background of diffuse LC3-I. Numbers of LC3-II puncta within the cell body were manually counted. For in vitro studies in primary hippocampal neurons, all experiments were repeated 3 to 5 times. The spine density was compared by one-way ANOVA with Tukey’s post-hoc test when compared between genotypes and different treatments. The unpaired t-test was used to compare the 2 treatments within the same genotype of neurons. All data were represented as mean ± SEM. All statistical analyses were carried out using Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Generation of stable cell lines, MEFs, cell culture maintenance, and transfection
The generation of an inducible single-copy transgene using Flp-In TRex-HeLa cells was as described previously [Citation38,Citation39]. In brief, pcDNA5/FRT/TO-LAP-long C9orf72 was cotransfected with pOG44 (Thermo Fisher Scientific, V600520), a Flp recombinase expression plasmid, and selected with 250 μg/ml hygromycin (Thermo Fisher Scientific, 10687010). All cell lines used in this study were maintained in DMEM (GE Healthcare HyClone, SH30243.01) with 10% FBS (GE Healthcare HyClone, SV30160.03) and 1% Pen-Strep (Thermo Fisher Scientific, 15070063) at 37°C and 5% atmospheric CO2. Tetracycline (Sigma-Aldrich, T7660) was added (100 μg/ml) for 18 h to induce the expression of C9orf72. All plasmid transfections in cell lines were performed using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) or TurboFect (Thermo Fisher Scientific, R0531) as per the manufacturer’s instruction. E13 embryos from wild-type and c9orf72 knockout mice were used to generate MEFs. Passage 2 MEFs were cotransfected with SV40LT (a kind gift from Dr. Boon Seng Wong, National University of Singapore) and pcDNA6 (Thermo Fisher Scientific, V22120) plasmids using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) for 24 h. Stable cells were selected for using blasticidin (Thermo Fisher Scientific, R21001) for 10 days with media changes every 3 days. Confluent cells were split at 1:10 dilution 5 times to select for fast-growing immortalized cells.
Affinity purification and quantitative mass spectrometry
The procedure for tandem affinity purification coupled with quantitative mass spectrometry was performed as described previously [Citation38,Citation39]. In brief, cells were grown in SILAC DMEM (Thermo Fisher Scientific, 88364) supplemented with 10% dialyzed FBS (Thermo Fisher Scientific, 30067334) and penicillin-streptomycin (Thermo Fisher Scientific, 15070063) with 0.4 mM of L-arginine and 0.8 mM of L-lysine. Normal (‘light’) L-arginine (69.2 μg/ml; Sigma-Aldrich, A8094) and L-lysine (116.6 μg/ml; Sigma-Aldrich, L5501) were added to the ‘light’ growth medium, and ‘heavy’ L-Arg-13C6,15N4.HCl (87.8 μg/ml; Sigma-Aldrich, 608033), and L-Lys-13C6,15N2.HCl (152 μg/ml; Sigma-Aldrich, 608041) were added for the ‘heavy’ growth medium. Cells were passaged in SILAC media for at least 5 ~ 6 doublings to ensure complete incorporation of isotopic amino acids. Cells were harvested and cell extracts were prepared as described previously [Citation38,Citation39]. Cells were lysed in lysis buffer (50 mM HEPES, pH 7.5, 150 mM KCl, 1 mM MgCl2, 1 mM EGTA, 1% NP-40 (Sigma-Aldrich, 74385), 10% glycerol, 1 mM dithiothreitol, 50 μM latrunculin B (Enzo Life Sciences, BML-T110-0001), 50 μM cytochalasin D (Enzo Life Sciences, BML-T109-0001) supplemented with complete protease inhibitors and PhosSTOP (Roche, 04693124001 and 04906845001, respectively] and sonicated to obtain total cell extracts. Clarified lysate or high-speed supernatants (HSS) were prepared by spinning the total cell extract at 100,000 xg for 20 min at 4ºC. The protein concentrations of HSS were measured using the BCA assay (Thermo Fisher Scientific, 23227) and normalized for both ‘light’ and ‘heavy’ samples. 20 mg of total proteins were used for tandem affinity purification. GFP immunoprecipitation were carried out with 1/20 volume (of HSS) of GFP-binder beads (in-house [Citation38,Citation71]) at 4ºC for 2 h. PreScission protease (GE healthcare, 27084301) was added to liberate C9orf72 and its associated proteins from the GFP-binder beads. Eluted fractions from the PreScission protease step were used for LC-MS/MS analysis as described previously [Citation38,Citation39].
Immunoprecipitation and immunoblotting
Tetracycline-induced Flp-In C9orf72 HeLa cells or transiently transfected cells were grown in 10-cm dishes to 80–90% cell density. The cells were lysed in ice-cold modified lysis buffer containing 50 mM Tris HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100 (Sigma-Aldrich, T9284) and supplemented with protease inhibitors (Roche, 04693124001). Cells were lysed for 20 min and centrifuged for 10 min at 15,000 xg. The protein concentration of the supernatant was determined using a BCA reagent kit (Thermo Fisher Scientific, 23227). A total of 1 g of protein was pre-cleared using Protein A/G PLUS-agarose (Santa Cruz Biotechnology, sc-2003) for 30 min at 4°C. The pre-cleared supernatant was added to either 10 μL of GFP-Binder [Citation71] or 10 μL of HA-magnetic beads (Thermo Fisher Scientific, 88836) and rotated at 4°C for at least 2 h. The beads were washed in 1x PBS with 0.1% Triton X-100. The proteins were eluted using 1X sample loading buffer (62.5 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue, 0.1 M dithiothreitol). A total of 30 μL of protein sample was analyzed using western blotting.
Total cell lysates were prepared in ice-cold radio immunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate (Sigma-Aldrich, 30970), 0.1% SDS (Bio-Rad, 1610302), 50 mM Tris, pH8.0 supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific, A32961). Cells were lysed for 20 min and centrifuged for 10 min at 15,000 xg. The protein concentration of the soluble fractions was determined using BCA reagent (Thermo Fisher Scientific, 23227). A total of 20–70 μg of protein was resolved using 10–12% denaturing polyacrylamide gel electrophoresis (Bio-Rad, CA, USA) and transferred to PVDF membrane (Bio-Rad, 1620177). Proteins on PVDF membranes were blocked using 5% low-fat milk in TBS (20 mM Tris, pH 7.4, 150 mM NaCl) containing 0.1% Tween-20 (Sigma-Aldrich, P7949) and incubated with primary antibody in SuperBLOCK (Thermo Fisher Scientific, 37535) in TBS overnight at 4°C. The primary antibodies used in this study were: LC3 (Proteintech, 14600-1-AP; 1:1000), RB1CC1/FIP200 (Cell Signaling Technology, 12436S; 1:1000; and Proteintech, 17250-1-AP; 1:1000), ULK1 (Cell Signaling Technology, 8054; 1:1000), ATG13 (Abnova, PAB19256; 1:1000), GAPDH (Proteintech, 60004-1-Ig; 1:5000), SMCR8 (Sigma-Aldrich, HPA021557; 1:1000), ATG101 (Abcam, ab105387; 1:1000), BECN1 (Cell Signaling Technology, Clone D40C5, 3495P; 1:1000), phospho-Ser93 BECN1 (Cell Signaling Technology, 14717; 1:1000), MTOR (Cell Signaling Technology, clone 7C10, 2983; 1:1000), phospho-Ser2448 MTOR (Cell Signaling Technology, clone D9C2, 5536; 1:1000), TARDBP/TDP-43 (in-house, clone FL4 [Citation38]), SQSTM1/p62 (Bethyl Laboratory, A302-857A; 1:1000) and HA-tag (Bethyl Laboratory, A190-238A; 1:5000). The membranes were washed in TBST (20 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween-20). Anti-rabbit or anti-mouse horseradish peroxidase (HRP) conjugated secondary antibodies (Thermo Fisher Scientific, 31460 and 31430, respectively; 1:10000) in 5% milk in TBST were incubated with the membranes for 1 h at room temperature. The membranes were incubated with WestPico or WestFemto chemiluminescent substrate (Thermo Fisher Scientific, 34080 and 34095, respectively) and exposed to x-ray films (FujiFilm, 4741019289). The films were developed using a Konica Minolta film developer. Protein band intensity was quantified using LI-COR Image Studio Lite.
In situ proximity-ligation assay
In situ proximity ligation assays were done following the manufacturer’s protocol (Sigma-Aldrich, SUO92101). Anti-FLAG (clone M2, Sigma-Aldrich, F1804), which detected LAP-tag C9orf72, was paired with anti-ATG13 (Proteintech, 18258-1-AP) for the primary antibodies. Anti-FLAG or anti-ATG13 only, served as negative controls.
Yeast two-hybrid assay
Yeast two-hybrid assay was performed following the manufacturer’s protocol (Takara, 630489). In brief, the prey (pGADT7-AD-based plasmids) and the bait (pGBDT7-based plasmids) were transformed into Y187 and AH109 yeast strains, respectively. The colonies from prey and bait plates were mated and selected using double-dropout (-Leu/-Trp) plates. The strength of interactions was assayed by serial dilution on quadruple-dropout (-Ade/–His/–/Leu/-Trp) with and without X-α-Gal (Takara, 630407).
RNA extraction and quantitative RT-PCR
RNA was isolated using TRIZOL (Thermo Fisher Scientific, 15596018) as per the manufacturer’s instructions. First strand cDNA synthesis and quantitative PCR were performed using a Thermo Maxima cDNA synthesis kit (Thermo Fisher Scientific, K1641) and Maxima SYBR Green Master mix (Thermo Fisher Scientific, K0223), respectively. The forward and reverse primer sequences were based on pre-validated primers from PrimerBank (https://pga.mgh.harvard.edu/primerbank/) and listed below: mouse C9orf72, 5ʹ- TTC CAT TTG AAG CCT GGC CT-3ʹ, 5ʹ-TCC CCT TCT GCG TAT CAT CC-3ʹ; mouse Ulk1, 5ʹ-AAG TTC GAG TTC TCT CGC AAG-3ʹ and 5ʹ-CGA TGT TTT CGT GCT TTA GTT CC-3ʹ; mouse Rb1cc1, 5ʹ-GAC ACT GAG CTA ACT GTG CAA-3ʹ, 5ʹ-GCG CTG TAA GTA CAC ACT CTT C-3ʹ; mouse Atg13, 5ʹ-CCA GGC TCG ACT TGG AGA AAA-3ʹ, 5ʹ-AGA TTT CCA CAC ACA TAG ATC GC-3ʹ, mouse Atg101, 5ʹ-CAC GGG CAA GTT TCA CTA CA-3ʹ, 5ʹ-CCA TCA CCA CCT GAG TTC CT-3ʹ.
Statistical analysis
All data are expressed as mean ± S.E.M. Statistical analysis was performed using Student’s two-tailed t-test or One-Way ANOVA with post-hoc analysis and considered statistically significant when p < 0.05.
Abbreviations
| ALS | = | amyotrophic lateral sclerosis; |
| C9orf72 | = | chromosome 9 open reading frame 72; |
| DENN | = | differentially expressed in neoplastic versus normal cells; |
| FTD | = | frontotemporal dementia; |
| GEFs | = | GDP/GTP exchange factors; |
| LAP | = | localization and affinity purification; |
| MEFs | = | mouse embryonic fibroblasts; |
| PLA | = | proximity-ligation assay; |
| RB1CC1 | = | RB1 inducible coiled-coil 1; |
| SMCR8 | = | Smith-Magenis syndrome chromosome region, candidate 8; |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
Supplemental Material
Download Zip (26.3 MB)Acknowledgments
We thank Drs. Yi-Ping Hsueh, Yijuang Chern and Karen Chiang for their comments on the early stage of the manuscript. We thank Noboru Mizushima (University of Tokyo, Japan) for the gift of ATG13 and ATG101 plasmids, Vladimir Gelfand (Northwestern University, USA) for the gift of ptdEOS-tubulin plasmid, Do-Hyung Kim (University of Minnesota, USA) for the gift of ULK1 plasmid, Seok-Yong Choi (Chonnam National University Medical School, Korea) for the gift of pSYC-97 plasmid. We thank Dr. Wei Guo for her assistance in cloning of the yeast two-hybrid constructs, and Peter Low and Dr. Greg Tucker-Kellogg for the technical assistance on the proximity-ligation assay. We also thank all of the Ling laboratory members for support, discussion, and suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Pottier C, Ravenscroft TA, Sanchez-Contreras M, et al. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138(Suppl 1):32–53.
- Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438.
- Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256.
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268.
- Gijselinck I, Van Langenhove T, Zee JVD, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65.
- Cruts M, Gijselinck I, Van Langenhove T, et al. Current insights into the C9orf72repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36:450–459.
- Moens TG, Partridge L, Isaacs AM. Genetic models of C9orf72: what is toxic? Curr Opin Genet Dev. 2017;44:92–101.
- Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem. 2016;138(Suppl 1):145–162.
- Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Nat Acad Sci. 2013;110:E4530–9.
- Koppers M, Blokhuis AM, Westeneng H-J, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78:426–438.
- Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550.
- O’Rourke JG, Bogdanik L, Yáñez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329.
- Sudria-Lopez E, Koppers M, de Wit M, et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016;132(1):145–147.
- Atanasio A, Decman V, White D, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204.
- Burberry A, Suzuki N, Wang J-Y, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra93.
- Xiao S, MacNair L, McGoldrick P, et al. Isoform specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568–583.
- Zhang D, Iyer LM, He F, et al. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet. 2012;3:283.
- Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29:499–503.
- Marat AL, Dokainish H, McPherson PS. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 2011;286:13791–13800.
- Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–3595.
- Sellier C, Campanari M-L, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35:1276–1297.
- Webster CP, Smith EF, Bauer CS, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. Embo J. 2016;35:1656–1676.
- Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2:e1601167.
- Aoki Y, Manzano R, Lee Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140:887–897.
- Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51.
- Jung J, Nayak A, Schaeffer V, et al. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife. 2017;6:2.
- Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12:e1006443–26.
- Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19:1610–1618.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741.
- Shen H-M, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;39:61–71.
- Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361.
- Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol. 2016;39:61–68.
- Wong P-M, Puente C, Ganley IG, et al. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9:124–137.
- Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Investig. 2015;125:25–32.
- Hardie DG. AMPK—sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952.
- Alers S, Löffler AS, Wesselborg S, et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11.
- Ling S-C, Albuquerque CP, Js H, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Nat Acad Sci. 2010;107:13318–13323.
- Sun S, Ling S-C, Qiu J, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun. 2015;6:6171.
- Hosokawa N, Sasaki T, Iemura S-I, et al. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5:973–979.
- Söderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000.
- Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–297.
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234.
- Levivier E, Goud B, Souchet M, et al. uDENN, DENN, and dDENN: indissociable domains in Rab and MAP kinases signaling pathways. Biochem Biophys Res Commun. 2001;287:688–695.
- Yoshimura S-I, Gerondopoulos A, Linford A, et al. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. 2010;191:367–381.
- Xiao S, MacNair L, McLean J, et al. C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res. 2016;1647:43–49.
- Nazio F, Carinci M, Valacca C, et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol. 2016;215:841–856.
- Joo JH, Wang B, Frankel E, et al. The noncanonical role of ULK/ATG1 in ER-to-golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62:491–506.
- Ogura K, Wicky C, Magnenat L, et al. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994;8:2389–2400.
- Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434.
- Sou Y-S, Waguri S, Iwata J-I, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–4775.
- Tomoda T, Kim JH, Zhan C, et al. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558.
- Toda H, Mochizuki H, Flores R, et al. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–3307.
- Mochizuki H, Toda H, Ando M, et al. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the drosophila brain. PLoS ONE. 2011;6:e19632–15.
- Clarke J-P, Mearow K. Autophagy inhibition in endogenous and nutrient-deprived conditions reduces dorsal root ganglia neuron survival and neurite growth in vitro. J Neurosci Res. 2016;94:653–670.
- Ban BK, Jun MH, Ryu HH, et al. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33:3907–3919.
- Tang G, Gudsnuk K, Kuo S-H, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–1143.
- Balch WE, Morimoto RI, Dillin A, et al. Adapting proteostasis for disease intervention. Science. 2012;319:1–5.
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034.
- Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864.
- Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226.
- Deng H-X, Chen W, Hong S-T, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215.
- Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446.
- Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441.
- Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636.
- Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808.
- Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67:1074–1077.
- Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74:180–187.
- Tyan S-H, Shih AY-J, Walsh JJ, et al. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci. 2012;51:43–52.
- Trinkle-Mulcahy L, Boulon S, Lam YW, et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol. 2008;183:223–239.