
ABSTRACT
Macroautophagy/autophagy is critical for normal appressorium formation and pathogenicity of the rice blast fungus Magnaporthe oryzae, but the molecular base of autophagy linked to pathogenicity remains elusive in this or other pathogenic fungi. We found that MoHat1, a histone acetyltransferase (HAT) homolog, had a role in the regulation of autophagy through the acetylation of autophagy related proteins MoAtg3 and MoAtg9. We also found that MoHat1 was subject to regulation by the protein kinase MoGsk1 that modulated the translocation of MoHat1 from the nucleus to the cytoplasm with the assistance of MoSsb1, a protein chaperone. The alternation of intracellular location affected MoHat1 in the modification of cytosolic autophagy proteins that maintained normal autophagy. Furthermore, we provided evidence linking acetylation of MoAtg3 and MoAtg9 by MoHat1 to functional appressorium development and pathogenicity. Together with the first report of MoAtg9 being subject to acetylation regulation by MoHat1, our studies depicted how MoHat1 regulated autophagy in conjunction with MoGsk1 and how normal autophagy was linked to appressorium formation and function and pathogenicity of M. oryzae.
Abbreviations: A/Ala: alanine; AP: autophagosome; Atg genes/proteins: autophagy-related genes/proteins; BiFC: bimolecular fluorescence complementation; co-IP: co-immunoprecipitation; DAPI: 4ʹ, 6-diamidino-2-phenylindole; D/Asp: aspartic acid; GFP: green fluorescent protein; GSK3: glycogen synthase kinase 3; HAT: histone acetyltransferase; Hsp70: heat-shock protein 70; IH: invasive hyphae; K/Lys: lysine; MMS: methyl methanesulfonate; Mo: Magnaporthe oryzae; PAS: phagophore assembly site; PE: phosphatidylethanolamine; PtdIns3K: phosphatidylinositol 3-kinase; R/Arg: arginine; S/Ser: serine; T/Thr: threonine; TOR: target of rapamycin; WT: wild type; YFP: yellow fluorescent protein
Introduction
Autophagy is a common and evolutionarily conserved process where proteins, membranes and organelles are degraded and recycled to maintain energy homeostasis in eukaryotic cells. This process is essential for cellular responses to environmental stress, cellular remodeling during development and differentiation, resistance to pathogens, and the extension of lifespan [Citation1–Citation3]. The initiation of autophagy begins with the de novo formation of cup-shaped membranes in the cytoplasm that gradually expand to become double-membrane vesicles, the autophagosomes (APs). In the budding yeast Saccharomyces cerevisiae, this conserved process is precisely regulated by a serious of autophagy-related (Atg) proteins [Citation4,Citation5], including Atg8 that serves as the most critical and useful marker to follow autophagy induction and related vesicular compartmentation [Citation6–Citation9]. The Atg8 conjugation system is also one of the core regulatory systems essential for AP formation [Citation7]. Newly synthesized Atg8 is a precursor protein with a carboxyl-terminal sequence that is later cleaved by Atg4 exposing the reactive Gly residue [Citation10,Citation11]. Consequently, the formation of the Atg8–phosphatidylethanolamine (PE) conjugate is regulated by Atg7 and Atg3, which are the E1 and E2 enzymes, respectively, participating in the ubiquitination reaction [Citation7]. Although our cognition of the molecular mechanism of autophagy has substantially increased through discovering and initial characterizing the Atg proteins, the complicated mechanism involved in the regulation of autophagy remains unclear. Studies have shown that transcriptional, post-transcriptional and post-transcriptional regulation all are used to modulate autophagy under different types of environmental stress [Citation12–Citation14] and several ATG proteins are acetylated [Citation15–Citation18].
Chromatin composed of nucleosomes condenses to form chromosomes during cell division [Citation19]. The core of the nucleosome is an octamer of 4 histone proteins H2A, H2B, H3 and H4. These histones not only are important to the structure of DNA but also can adjust the cellular processes through histone N-terminal modification, such as acetylation, phosphorylation, methylation, and ubiquitination [Citation20]. Histone acetyltransferases (HATs) function on the acetylation of histones on specific lysine residues and play a crucial role in transcriptional activation, gene silencing, cell cycle regulation, and DNA replication [Citation21–Citation27]. In addition to histone acetylation, recent studies have shown that the HAT Esa1/TIP60 participates in autophagy regulation through direct acetylation of certain autophagy proteins [Citation15,Citation28].
The rice blast fungal pathogen Magnaporthe oryzae forms the appressorium that employs enormous turgor pressure to rupture the outer cuticle of rice leaf for infection [Citation29,Citation30]. Appressorium maturation needs the mass transfer of lipid bodies to the developing appressorium that is accompanied with autophagic cell death in the conidium and rapid lipolysis at the onset of appressorial turgor generation [Citation31–Citation35]. A recent study uncovered a glucose-ABL1-TOR signaling regulation pattern that modulates autophagy during appressorium formation via cell cycle tuning in M. oryzae [Citation36], indicating that the autophagy process contributes to the appressorial development. Meanwhile, various Atg genes were identified in M. oryzae that showed critical roles in autophagy, functional appressorium formation and successful infection, since 16 of ATG genes deletion mutants were unable to cause the rice blast [Citation37]. In this study, we have found that the histone acetyltransferase MoHat1 had a role in the acetylation of autophagy proteins important for pathogenicity of M. oryzae. MoHat1 acetylated not only MoAtg3 to promote the formation of the Atg8–PE conjugate but also MoAtg9 increasing its binding ability to vesicles. In addition, MoHat1 could shuttle between the nucleus and the cytoplasm in the presence of MoSsb1 (a Hsp70 protein) upon nutrient deprivation, and the MoHat1-MoSsb1 interaction was influenced by phosphorylation levels of MoHat1. Taken together, our study revealed the conserved nature of how MoHat1 acetylates MoAtg3 and MoAtg9 in order to regulate infection related autophagy and also links Atg-mediated autophagy to appressorium development and pathogenicity of M. oryzae.
Results
Identification of MoHat1 as a HAT component
Previous studies have shown that acetylation is essential for autophagy regulation in budding yeast Saccharomyces cerevisiae and normal autophagy is critical for the formation of appressorium and pathogenicity in M. oryzae [Citation34,Citation37–Citation40]. To understand the underlying mechanisms, we studied whether M. oryzae HATs also play a role in autophagy and how such a role linked to pathogenicity. In our previous transcriptome profiling by RNA-Seq, we found that the expression of the HAT complex was upregulated during the infection process. We then carried out qRT-PCR validations to further confirm these findings. Among the six HAT homologs in M. oryzae, we were more curious about genes whose expression pattern fitting two characteristics: an increasing tendency during the early infection stage (0 ~ 8 hpi) and significantly higher expressions during infection. Based on these criteria, we found that MoHAT1 and MoGCN5 were most likely to be associated with appressoria formation and pathogenicity (Figure S1). However, the expression of MoESA1 and MoSAS2/3 was consistently low, and the expression of MoELP3 and MoRTT109 had no obvious upward trends during early infection stages (0 ~ 8 hpi) despite the high expression during infection process (Figure S1). Since MoGcn5 has been previously characterized [Citation18], we then decided to focus on MoHat1 in autophagy.
MoHAT1 (MGG_06375) encodes a 468-amino acid polypeptide containing a conserved N-terminal HAT domain. Phylogenetic analysis revealed that Hat1 proteins were highly conserved among several fungi, with MoHat1 being most homologous to the ‘take-all’ fungus Gaeumannomyces graminis (Figure S2A).
MoHat1 is important for appressorial penetration and pathogenicity
In S. cerevisiae, histone H3 and ScHat1 contribute to DNA double-strand break repair and the ∆Schat1 mutant is sensitive to DNA-damaging agent methyl methanesulfonate (MMS) [Citation41]. To test for any functional conservation between MoHat1 and ScHat1, we expressed the MoHAT1 gene in the ΔSchat1 mutant strain using the yeast expression vector pYES2 and found that MoHat1 could partially rescue the sensitivity defect of Schat1 to MMS (Figure S2B-S2C).
To characterize functions of MoHat1 in M. oryzae, we generated a ∆Mohat1 deletion mutant by replacing the coding region with the hygromycin-resistance cassette (HPH) and also obtained the complemented mutant strain (Figure S3A-S3B). To examine the role of MoHat1 in virulence, conidial suspensions (5 × 104 spores/ml) were sprayed onto 2-week-old rice seedlings (cv. CO-39). Despite the presence of very few typical lesions, the ∆Mohat1 mutant caused fewer and smaller lesions mostly on rice leaves 7 days after inoculation, in contrast to numerous typical lesions caused by Guy11 and complemented strains (). The lesions were then quantified by a ‘lesion-type’ scoring assay which divided the lesions into 1–5 types according to their severity (type 0, no lesion; type 1, pinhead-sized dark brown specks without visible centers; type 2, small brown lesions of 1 mm in diameter; type3, 2–3 mm gray spots with brown margins; type 4, elliptical gray spots > 3–4 mm; type 5, large eyespot lesions that coalesced infecting 50% or more of the leaf area). Results showed that the ∆Mohat1 mutant produced mostly lesion type 1–3 and no lesion type 4–5 was produced (). Consistent with this result, quantitative statistics analysis of the diseased lesion area (%) [Citation42] also showed that ∆Mohat1 mutant caused significantly less disease area (6.3%) than that of the wild-type Guy11 (41.6%) and the complemented strains (39.7%) (). In addition, a relative fungal growth assay [Citation43] confirmed that the content of fungal DNA in 1.5 g diseased rice leaves was seriously decreased infected by ∆Mohat1 mutant (). Similar results were obtained on the detached barley cultivar Four-arris leaves dropped with different concentrations of conidial suspensions (104, 103, 102 spores/ml), which found that the ∆Mohat1 mutant lesions were smaller than the wild-type (WT) (). Moreover, conidial suspensions (5 × 104 spores/ml) were also injected into 3-week-old rice sheath that showed fewer and smaller lesions by the ∆Mohat1 mutant than the control ().
Figure 1. MoHat1 is important for pathogenicity in M. oryzae. (a) Rice spraying assays. Four milliliters of conidial suspension (5 × 104 spores/ml) of each strain were sprayed on two-week old rice seedlings (Oryza sativa cv. CO39). Diseased leaves were photographed after 7 days incubation. (b) Quantification of lesion types (per 1.5 cm2) on susceptible rice spayed with conidia of wild-type Guy11, ∆Mohat1, and ∆Mohat1/MoHAT1 complemented strains. Disease lesions were quantified by a ‘lesion-type’ scoring assay which divided the lesions into 1–5 types according to their severity (type 0, no lesion; type 1, pinhead-sized dark brown specks without visible centers; type 2, small brown lesions that are approximately 1 mm in diameter; type3, 2- to 3-mm gray spots with brown margins; type 4, elliptical gray spots longer than approximately 3–4 mm; type 5, large eyespot lesions that coalesced infecting 50% or more of the leaf area). Error bars represent SD and asterisks represent significant differences (P < 0.01). (c) Diseased leaf area analysis. Data are presented as a bar chart showing percentage of lesion areas analyzed by ImageJ. Error bars represent SD and asterisks represent significant differences (P < 0.01). (d) Severity of blast disease was evaluated by quantifying M. oryzae genomic 28S ribosomal gene (rDNA) relative to rice genomic RUBQ1 DNA (7 days post-inoculation). Mean values of three determinations with standard deviations are shown. The asterisks indicate a significant difference from the wild-type Guy11 (P < 0.01). (e) Conidial suspension (2 × 105 spores/ml) of each strain was injected into rice sheath of 3-week-old seedlings and 30 healthy rice sheaths were used for each strain. The arrow points out the injection site. (f) Detached barley cultivar Four-arris leaves were dropped with different concentrations of conidial suspensions (104, 103, 102 spores/ml) and diseased leaves were photographed 5 days after inoculation. Experiments were performed more than 3 times. (g) Close observation for penetration assay with rice sheath. Excised rice sheath from 4-week-old rice seedlings was inoculated with conidial suspension. Images show invasive growth in rice sheath epidermal cells at 24 and 36 hpi. CO, conidium; Ap, appressorium; IH, invasive hyphae. Scale bar: 10 μm. (h) The penetration rate was counted for more than 100 appressoria in (g) and the experiment was repeated 3 times. Error bars represent SD and asterisk represents significant difference (P < 0.01). (i) Detailed observation and statistics for infectious growth in rice sheath cell at 24 and 36 hpi. Appressorium penetration sites (n = 100) were observed and invasive hyphae (IH) were rated from type 1 to 4 (type1, no hyphal penetration with only appressoria formation; type2, IH with 1 or 2 short branches; type3, IH with at least 3 branches but the IH are short and extending within a plant cell; type 4, IH that has numerous branches and fully occupies the plant cell or even extended to an adjacent plant cell.). The arrow points out the appressorium (Ap) and the invasive hyphae (IH); asterisks indicate IH extended to surrounding cells. Error bars represent SD from three independent experiments. Scale bar: 10 μm.
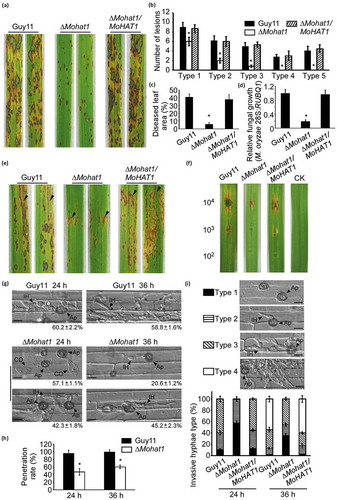
To further examine the virulence attenuation in the ∆Mohat1 mutant, we performed an excised leaf sheath assay to test the appressorial penetration rates and infectious hyphae extension ability within the host cell. After incubation with spore suspension (2 × 105 spores/ml) for 24 hours, only a few appressoria from the ∆Mohat1 mutant (42.9%) penetrated the rice tissue, compared to 95% by the WT strain (). In fact, the penetration rate of ∆Mohat1 mutant remained at a relative low level (65.8%) in comparison to that of the WT (99.8%) even when the incubation was extended to 36 h (). We then divided the invasive hyphae (IH) into 4 types (type1, no penetration with only appressoria; type 2, only with a penetration peg or a single invasive hypha with no branch; type 3, IH extended but was limited in one plant cell; type 4, IH with numerous branches and extended to surrounding cells) for further elaborate observations. We found that the WT strain had type 2 (30.5%) and type 3 (60.2%) IH 24 h post-inoculation (hpi), whereas more than half of the ∆Mohat1 mutant (57.5%) had type 1 IH with no penetration (). At 48 hpi, the IH of the ∆Mohat1 mutant was still limited within one plant cell for type 2 and type 3 in contrast to freely spreading to the surrounding cells by the control strains (). The result of onion epidermis infection assay was mostly consistent, showing that 68% of the infectious hypha produced by the ∆Mohat1 mutant were type 1 and type 2, which did not infect successfully or only generate a single IH, while 70% type 3 and type 4 infectious hypha were produced by the WT strain (Figure S4A-S4B). These results above conclusively demonstrated that MoHat1 was important in pathogenicity of M. oryzae and that virulence attenuation was due to the defect of the ∆Mohat1 mutant in appressorial penetration and infectious hyphal growth.
MoHat1 is required for appressorium turgor pressure
Appressorium requires enormous internal turgor pressure to penetrate the rice leaf cuticle, and this pressure is generated by the accumulation of glycerol [Citation32,Citation44,Citation45]. Considering that the germinating and appressorial formation were normal in the ∆Mohat1 mutant (Table S1), we measured the internal concentration of solute and the turgor pressure within appressoria by the incipient cytorrhysis (cell collapse) assay using 1–4 molar of glycerol [Citation45]. At 2 M glycerol, the appressorium collapse rate was around 60% in the ∆Mohat1 mutant compared with 18% in the WT (Figure S5). As the external glycerol concentration increased, the proportion of collapsed appressoria kept higher in the mutant than in Guy11 (WT) (Figure S5), indicating that the turgor pressure in appressoria of ∆Mohat1 mutant was reduced seriously. These results suggested that the defects in appressorial turgor pressure of the ∆Mohat1 mutant contributed to the attenuation in pathogenicity.
In M. oryzae, the appressorial maturation and appressorium-mediated host penetration largely depend on the effective transfer of glycogen and triacylglycerol [Citation31–Citation35]. Therefore, we examined the cellular distribution of glycogen and lipid bodies during appressorium development. Upon staining with I2 and KI, abundant glycogen was seen in the conidia, germ tubes, and appressoria. We also found that the mobilization of glycogen was delayed with glycogen depletion in conidia until 10 h in the ∆Mohat1 mutant, compared to 6–8 h in the WT strain (Figure S6B and S6D). We then investigated the distribution of lipid bodies by Nile red staining and found that the intracellular lipid degradation in the ∆Mohat1 mutant was significantly delayed. In Guy11, the lipid bodies were completely degraded after germinating 10 h in > 90% appressoria. However, the degradation progress was retarded in the ∆Mohat1 mutant with large number of lipid remains after 24 h in more than 50% of mature appressoria (Figure S6A and S6C).
MoHat1 is involved in autophagy and conidial nuclear degradation
Autophagy in eukaryote cells is an evolutionarily conserved process where cytosol and organelle components can be degraded and recycled [Citation2,Citation3,Citation46]. Previous studies show that the autophagic cell death is important for the elaboration of appressoria [Citation34,Citation37,Citation47]. Because MoHAT1 was upregulated during the early infection stage and the deletion of MoHAT1 caused an aberrant development of appressoria, we hypothesized that this might be related to a conserved role of MoHat1 in autophagy. To monitor the autophagy level, we transformed GFP-MoAtg8, a widely used marker for nonspecific autophagy, into the ∆Mohat1 and WT strains. During germination, GFP-MoAtg8-labeled APs were mostly accumulated in the conidia and steadily decreased accompanied by appressorium formation in the WT strain (). In contrast, the number of APs in the ∆Mohat1 mutant was significantly decreased in conidia and appressoria stages in all 3 independent transformants ( and Figure S7).
Figure 2. MoHat1 is involved in autophagy. (a) Cellular location of APs during infection-related appressorium development. Conidia were from both wild-type Guy11 and ∆Mohat1 mutant expressing GFP-MoATG8 gene fusion, inoculated onto hydrophobic interface and observed by epifluorescence microscopy at different times (scale bar: 10 μm. (b) Bar chart showing mean AP numbers present in conidia, germ tube and appressorium at 0, 2 and 4 h after germination. Three ∆Mohat1 transformants expressing GFP-MoATG8 gene fusion (∆Mohat1 #1, #2, #3) were used for statistical analysis. Error bars represent SD and asterisks represent significant differences (P < 0.01). (c) The Guy11 (WT) and ∆Mohat1 mutant strains transformed with GFP-MoAtg8 were cultured in MM-N (nitrogen starvation minimal medium) for 2 h, and the autophagy intensity was observed by Axio Observer A1 Zeiss inverted microscope. The arrow points to the vacuole. Scale bar: 5 μm. (d) Autophagy intensity was assessed by means of translocation of GFP-MoAtg8 into vacuoles (n = 100). Bars with asterisks represent significant differences (Duncan’s new multiple range method p < 0.01). (e and f) Immunoblotting was performed with anti-GFP and anti-Actin antibodies. The extent of autophagy was estimated by calculating the amount of free GFP compared with the total amount of intact GFP-Atg8 and free GFP (the numbers underneath the blot). (g) Spores of Guy11 H1-RFP and ∆Mohat1 H1-RFP strains were inoculated at a concentration of 5 × 104 spores/ml onto artificial hydrophobic surfaces and imaged by Axio Observer A1 Zeiss inverted microscope at 24 hpi. Asterisks indicate the nucleus. Scale bar: 10 μm. (h) Percentage of spores containing different number of nuclei in both Guy11 H1-RFP and ∆Mohat1 H1-RFP strains. Mean values were calculated from three independent replicates by counting the nuclei from 100 spores per strain for each replicate. Error bars are standard deviation.
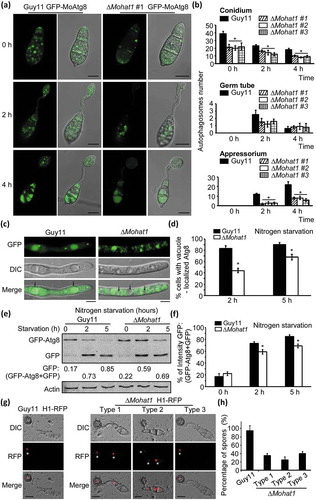
When autophagy is induced by nutrient deprivation (under MM-N treatment), autophagic bodies, accompanied with GFP-MoAtg8, are delivered into the vacuole for degradation. Upon MM-N treatment, the vacuolar GFP signal was found to be 82% (2 h) and 89% (5 h) in Guy11, and 46% (2 h) and 71% (5 h) in the ∆Mohat1 mutant ( and Figure S8). The GFP signal was also detected in the cytoplasm but not the vacuole in ∆Mohat1 (). In addition, the autophagy level was also quantitatively measured by immunoblot. The extent of autophagy was estimated by calculating free GFP compared with the total of the intact GFP-Atg8 and free GFP together. The autophagy level in the ∆Mohat1 mutant was remarkably reduced since the proportion of free GFP was lower than that in Guy11 following nutrition starvation of 2 h and 5 h (). These results indicated that MoHat1 had a role in autophagy during appressorial development and starvation-induced conditions.
To form a functional M. oryzae appressorium, the apical nucleus of each germinating conidium undergoes one round of mitosis in the germ tube and one daughter nucleus then migrates into the incipient appressorium while the other returns to the conidium undergoing degeneration through autophagic cell death [Citation34,Citation36]. Considering that the ∆Mohat1 mutant had a defect in autophagy, we were curious about if the relevant nucleus degradation process has also been interfered. We therefore generated histone H1 fused to a red fluorescent protein (H1-RFP) [Citation48] construct and introduced it into the ∆Mohat1 mutant and WT strains. Conidia of the positive transformants were dropped onto the artificial hydrophobic coverslips and observed by epifluorescence microscopy after germinating for 24 hours. Results showed that > 95% of germinating Guy11 H1-RFP spores carried a single appressorial nucleus (), indicating that the autophagy and conidial nuclear degradation process was complete. However, the degradation process was significantly impaired in the ∆Mohat1 H1-RFP strain since more than half of the mutant spores still remained two or three nuclei (type 1 and type 2) at 24 hpi ().
MoHat1 interacts with MoAtg3 and MoAtg9
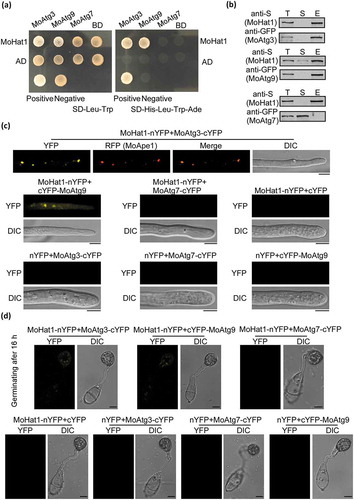
To further understand how MoHat1 participates in autophagy regulation, yeast-two hybrid was used to screen protein encoded by 22 Atg genes in M. oryzae, including 16 that are important for pathogenicity [Citation37]. We found that MoHat1 interacted with MoAtg3, a specific E2 enzyme for Atg8–PE formation that is acetylated in S. cerevisiae [Citation7,Citation11,Citation15,Citation49]. In addition, MoHat1 also interacted with MoAtg9 (). The interactions with both MoAtg3 and MoAtg9 were verified by bimolecular fluorescence complementation (BiFC) and co-immunoprecipitation (co-IP) assays ( and Figure S9A-S9D). MoAtg7 that is acetylated by another acetyltransferase MoGcn5 was used as a negative control ( and Figure S9A-S9D). In BiFC assays, we found that MoHat1 interacted with MoAtg3 mostly at the phagophore assembly site (PAS), where most Atg proteins are located under starvation conditions, and MoHat1 interacted with MoAtg9 in both the cytoplasm and the PAS (). Moreover, interactions between MoHat1-MoAtg3 and MoHat1-MoAtg9 were also found during appressorium formation ( and Figure S9A-S9D).
Figure 3. MoHat1 interacts with MoAtg3 and MoAtg9. (a) Yeast two-hybrid analysis for the interaction between MoHat1 and Atg proteins. The pGADT7 and pGBKT7 fused with specific genes were co-introduced into the yeast AH109 strain, and transformants were plated on SD-Leu-Trp as control and on selective SD-Leu-Trp-His-Ade for 5 days. The pair of plasmids pGBKT7-Lam and pGADT7-T, empty pGADT7 and pGBKT7 were used as the negative controls. MoAtg7 was used also as a negative control that had no interaction with MoHat1. (b) Co-IP assays. Western blots of total proteins, suspensions and proteins eluted from anti-S agarose from transformants co-transformed MoAtg3-GFP and MoHat1-S, MoAtg9-GFP and MoHat1-S, and also MoAtg7-GFP and MoHat1-S were detected with anti-S or anti-GFP antibodies. T, total; S, suspensions; E, elution. (c and d) BiFC assay for the patterns of MoHat1-MoAtg3, MoHat1-MoAtg9 and MoHat1-MoAtg7 in vivo. Both aerial hypha tips and 16 h appressoria were examined by DIC and fluorescence microscopy. Strains expressing MoHat1-nYFP and MoAtg7-cYFP, MoHat1-nYFP and empty cYFP, MoAtg3-cYFP and empty nYFP, MoAtg9-cYFP and empty nYFP, MoAtg7-cYFP and empty nYFP were used as negative controls. MoApe1-RFP was used as a phagophore assembly site (PAS) marker. Scale bar: 10 μm.
Mohat1 acetylates MoAtg3 in vivo and in vitro
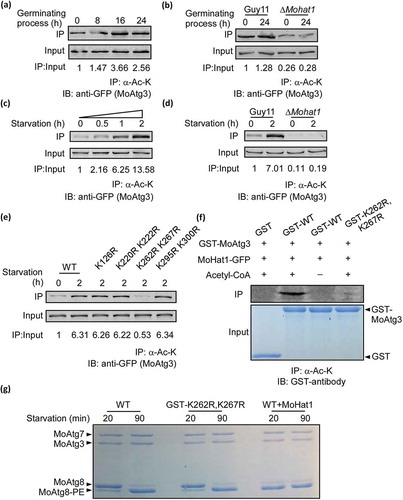
To examine whether MoHat1 acetylates MoAtg3, we generated a MoATG3-GFP construct and introduced it into both Guy11 and the ∆Mohat1 mutant strains. During germination, MoAtg3 acetylation was detected in Guy11 and the acetylation intensity was enhanced as the germinating process progressed and peaked at around 16 hours after germinating (). However, this was not found in the ∆Mohat1 mutant (). MoAtg3 acetylation was also detected when Guy11 was grown in liquid CM medium (nutrient-rich condition) and the acetylation gradually increased upon starvation induction (). This suggested that MoAtg3 was acetylated in vivo and time-dependent. However, the acetylation level of MoAtg3 in the ∆Mohat1 mutant was reduced significantly following cultured in MM-N for 0 or 2 h (). To further verify whether MoHat1 plays a role in the acetylation of MoAtg3, the in vitro acetylation assay was performed using purified GST, GST-MoAtg3, and MoHat1-GFP fusion proteins (immunoprecipitation from the ∆Mohat1/MoHAT1 complemented strain) that showed MoHat1 could acetylate MoAtg3 in vitro ().
Figure 4. Acetylation of MoAtg3 by MoHat1. (a and c) Acetylation of MoAtg3 during both appressorium development and nutrient starvation treatment. Acetylation of MoAtg3-GFP in hyphae cells treated with MM-N medium for 0, 0.5, 1 and 2 h, and the germinating conidia were harvested after germination for 0, 8, 16 and 24 h. Detected by immunoprecipitation (IP) with the antibody to acetylated-lysine and immunoblotting (IB) with the antibody to GFP. The acetylation intensity was monitored by calculating the amount of acetylated MoAtg3 compared with the total MoAtg3 protein (IP/Input). (b and d) Acetylation of MoAtg3 in ∆Mohat1 mutant. Wild-type Guy11 and ∆Mohat1 mutant strains expressing MoAtg3-GFP were starved for 2 h or germinating for 24 h before protein extraction. Proteins were purified and immunoprecipitated with the antibody to acetylated-lysine followed by immunoblot with the antibody to GFP. (e) Acetylation of MoAtg3 mutations. ∆Moatg3/MoATG3 (WT), ∆Moatg3/MoATG3K126R (K126R), ∆Moatg3/MoATG3K220,222R (K220R K222R), ∆Moatg3/MoATG3K262,267R (K262R K267R), ∆Moatg3/MoATG3K295,300R (K295R K300R) were starved for 2 h and proteins were immunoprecipitated with antibody to acetylated-lysine followed by immunoblotting with antibody to GFP. (f) In vitro acetylation of the purified GST, GST-MoAtg3 WT (GST-WT), GST-MoAtg3K262,267R (GST-K262R,K267R) proteins was measured by means of immunoprecipitation with antibody to acetylated-lysine followed by immunoblotting with GST antibody. (g) Lipidation of MoAtg8. Purified MoAtg3 and GST-MoAtg3K262,267R (GST-K262R,K267R) mutant and also MoAtg3 incubated with MoHat1 (WT+MoHat1), MoAtg7, MoAtg8, and liposomes were incubated for in vitro MoAtg8 lipidation and analyzed by SDS-PAGE with urea.
We further tested whether the acetylation of MoAtg3 maintains a high level during continuously starvation and found that MoAtg3 acetylation was gradually increased accompanied by a strengthened interaction between MoHat1 and MoAtg3. It peaked at 2 to 5 hours following starvation induction (Figure S10A-S10D). This finding indicated that M. oryzae needed certain acetylation levels even under continuous nutrient deprivation.
K262-K267 is essential for the acetylation of MoAtg3
To determine the acetylation sites of MoAtg3, we purified the MoAtg3-GFP fusion protein from Guy11. For unknown reasons, mass spectrometry could not cover the entire MoAtg3 protein after several attempts. For the regions that were covered, we found that MoAtg3 has an N-terminal acetylation on its first M (M: Met) (Figure S11A) and excluded 11 of K (K: Lys; R: Arg) sites for acetylation (Figure S11A-S11B). Since there are 18 K residues in MoAtg3, we divided the rest K sites into 4 groups and generated 4 mutation vectors (K126R, K220R K222R, K262R K267R, K295R K300R). Further in vivo and in vitro acetylation analysis showed that the acetylation level of MoAtg3 was weakened only in the K262R K267R mutation strain ().
Acetylation of MoAtg3 is important for autophagy and pathogenicity
In S. cerevisiae, Atg3 is an E2-like enzyme that functions together with the E1-like Atg7 and E3-like Atg12–Atg5 complex covalently conjugate phosphatidylethanolamine (PE) to Atg8 (Atg8–PE formation system) during autophagy [Citation49–Citation52]. Studies have shown that Atg8 lipidation can be reconstituted in vitro with recombinant Atg7 and Atg3 proteins [Citation49]. In order to eliminate the possibility that K-R mutation might affect autophagy by directly disturbing the MoAtg3 enzymatic activity rather than reducing MoAtg3 acetylation, we measured the enzymatic activity recombinant variants of MoAtg3 through an in vitro Atg8 lipidation. Results showed that mutation of K262R K267R did not affect Atg8 lipidation (), indicating that mutation on K262-K267 did not disturb the enzymatic activity of MoAtg3.
To verify if these acetylation sites play a role in autophagy, we detected the degradation of GFP-MoAtg8 in the K262R K267R mutant strain. We found that GFP-MoAtg8 could be delivered into the vacuole for degradation normally in Guy11 through MM-N induction; however, the localization and degradation of GFP-MoAtg8 were disturbed in the K262R K267R mutant strain (). Meanwhile, western blot analysis also showed that the autophagy level in the K262R K267R mutant strain was reduced significantly (). Fluorescence observation also found that GFP-MoAtg8-marked APs in the K262R K267R mutant decreased remarkably during germination ( and Figure S12). These results indicated that the acetylation of MoAtg3 was essential for autophagy during both appressorium development and nutrient starvation.
Figure 5. MoAtg3 acetylation affects autophagy and pathogenicity in M. oryzae. (a and b) Autophagy in Guy11, ∆Moatg3 and ∆Moatg3/MoATG3K262,267R (K262R K267R) mutant strains treated in MM-N (nitrogen starvation minimal medium) for 2 or 5 h were analyzed by western blot analysis for (a) GFP-MoAtg8 cleavage and (b) translocation of GFP-MoAtg8 into vacuoles (n = 100). Bars with asterisks represent significant differences (Duncan’s new multiple range method p < 0.01). (c) Cellular location of APs during infection-related appressorium development in Guy11, ∆Moatg3 and ∆Moatg3/MoATG3K262,267R (K262R K267R) mutant strains. (d) Bar charts showing mean APs numbers present in conidia, germ tube and appressorium at 0, 2 and 4 h after germination. Two transformants of ∆Moatg3 (∆Moatg3 #3 and ∆Moatg3 #13) and ∆Moatg3/MoATG3K262,267R (K262R K267R) (K262R K267R #11 and K262R K267R #16) mutant strains expressing GFP-MoATG8 construct were used for statistical analysis, respectively. Asterisks represent significant differences (p < 0.01). (e and f) Rice spraying assays and detailed observation with statistical analysis for infectious growth in rice sheath cells at 24 hpi. Four milliliters of conidial suspension (5 × 104 spores/ml and 1 × 105 spores/ml) of each strain were used for spraying and injection, respectively. Appressorium penetration sites (n = 100) were observed and invasive hyphae were rated from type 1 to 4. Ap, appressorium; IH, invasive hyphae. Error bars represent SD from 3 independent experiments. (g) Effect of MoAtg3 acetylation on MoAtg3-MoAtg8 interaction. Association of MoAtg3 and MoAtg8 in the wild-type (Guy11) strain, ∆Moatg3 and K262R K267R mutant strains expressing GFP-MoAtg8 and MoAtg3-S was incubated with GFP beads and analyzed by western blot analysis to detect the amount of coimmunoprecipitated MoAtg3 using the S tag antibody. Quantitation of the co-immunoprecipitated MoAtg3 intensity was analyzed by the ODYSSEY infrared imaging system (application software Version 2.1). T, total; S, suspensions; E, elution.
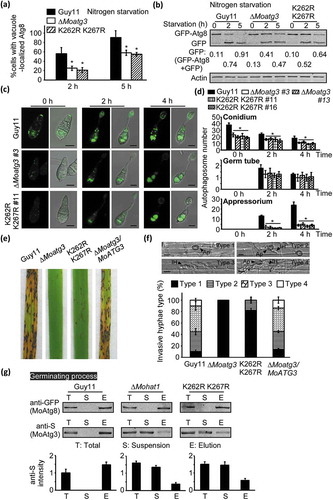
We were then curious about whether this acetylation-induced defect in autophagy is linked to the function of appressorium and pathogenicity of M. oryzae. Conidial suspensions of the K262R K267R mutant were dropped on the artificial surface for germination. Nile red, and I2 and KI staining showed that the mutation of K262R K267R significantly delayed the transfer of glycogen and intracellular lipid degradation (Figure S13A-S13B). Consistent with these results, conidial suspensions were sprayed on to rice seedlings or injected to the rice sheath and showed that the K262R K267R mutant strain had a significantly reduced infectious hyphae growth and attenuated pathogenicity ().
Acetylation of MoAtg3 affects the MoAtg3-MoAtg8 interaction
Yeast Atg8 conjugation to PE requires the interaction between Atg3 and Atg8 [Citation53]. In the ∆Mohat1 mutant, the interaction between MoAtg3 and MoAtg8 was weaker than that in the WT Guy11 ( and Figure S14). Consistent with these results, we found that K262R K267R mutation of MoAtg3 also weaken the interaction between MoAtg3 and MoAtg8 under both nutrient starvation and germinating ( and Figure S14). This suggested that MoHat1-mediated MoAtg3 acetylation influenced autophagy through controlling MoAtg3-MoAtg8 interaction. However, MoHat1 might also acetylate additional proteins to affect autophagy.
MoHat1 also acetylates MoAtg9
Atg9 is the only integral membrane protein that is essential for autophagy [Citation54,Citation55]. In yeast, unlike most Atg proteins that are localized to the PAS, Atg9 has multiple punctate locations that are detectable at the PAS along with the peripheral sites called tubulovesicular clusters, including Golgi apparatus and mitochondria, and Atg9 normally cycles between these organelles and PAS and help with the increase of AP numbers [Citation56–Citation58].
Given that MoHat1 interacted with MoAtg9 and the fact that MoHat1 is a member of HAT complexes, we wondered if MoHat1 directly acetylates MoAtg9, and thus introduced a GFP-MoATG9 construct into Guy11 and the ∆Mohat1 mutant strain, respectively. In the Guy11 strain, MoAtg9 acetylation was detected in both nutrient-rich (CM) and starvation conditions, and acetylation was increased upon nutrient starvation (). However, the acetylation of MoAtg9 was completely deprived in the ∆Mohat1 mutant (). Through bioinformatics analysis, we found seven potential acetylation sites and generated a seven-site mutation construct (7K-R) and 6 site-mutation mutants (K85R, K428R K438R, K492R, K621R, K693R, and K859R) for transformation studies (). Surprisingly, we found that the acetylation of MoAtg9 was nearly disturbed in both 7K-R and K621R mutation strains ( and Figure S15). Simultaneously, in vitro acetylation assays also proved that MoAtg9 could be acetylated by MoHat1 and that the K621 site was essential for its acetylation ().
Figure 6. MoHat1 acetylates MoAtg9 to regulate autophagy and pathogenicity. (a) Acetylation of GFP-MoAtg9 protein in Guy11 and ∆Mohat1 mutant. GFP-MoAtg9 in Guy11, ∆Mohat1 mutant and ∆Moatg9/MoATG9K621R (K621R) strains were starved for 0 or 2 h. Proteins were immunoprecipitated with the antibody to acetylated-lysine followed by immunoblotting with the antibody to GFP. (b) Prediction of acetylation sites of MoAtg9. Bioinformatics forecast through a prediction website PAIL (Prediction of Acetylation on Internal Lysines) (http://bdmpail.biocuckoo.org/prediction.php) found 7 potential acetylation sites. (c) In vitro acetylation of MoAtg9. Purified GST, GST-MoAtg9 WT (GST-WT), GST-MoAtg9K621R (GST-K621R) proteins were measured by means of immunoprecipitation with antibody to acetylated-lysine followed by immunoblotting with GST antibody. (d) Diagrammatic sketch of MoAtg9 function in autophagy regulation. The Golgi apparatus is one of the origins for the autophagosomal membrane and Atg9-containing membrane from the Golgi apparatus is important for increasing the AP number. (e) The binding ability of MoAtg9 to membrane structures. Under CM or MM-N conditions, vesicle protein of Guy11 (WT), ∆Mohat1, ∆Moatg9/MoATG9K621R (K621R) and the complemented strain ∆Moatg9/MoATG9 expressing MoSft2-RFP were extracted using ultracentrifugation and detected with antibodies to GFP and RFP. MoSft2 was used as a Golgi apparatus marker. (f and g) Acetylation of MoAtg9 is important for autophagy. Autophagy levels in Guy11, ∆Moatg9 and ∆Moatg9/MoATG9K621R (K621R) mutant strains under starvation were analyzed by means of western blot for (F) GFP-MoAtg8 cleavage and (g) translocation of GFP-MoAtg8 into vacuoles (n = 100). Bars with asterisks represent significant differences (Duncan’s new multiple range method p < 0.01). (h) Rice spraying assays. Four milliliters of conidial suspension (5 × 104 spores/ml) of each strain was used for spraying and photographed 7 days after inoculation. (i) Total DNA was extracted from per 1.5 g disease leaves from (h) and test by qRT-PCR with M. oryzae 28S rDNA and rice genomic RUBQ1 primers. Asterisks indicate statistically significant differences (Duncan’s new multiple range test, p < 0.01). (j) Detailed statistical analysis for infectious growth in rice sheath cells at 24 hpi. Appressorium penetration sites (n = 100) were observed and invasive hyphae were rated from type 1 to 4. Error bars represent SD from 3 independent experiments.
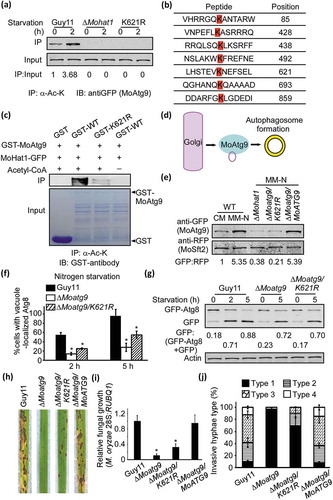
Acetylation of MoAtg9 regulates its binding to vesicles
Recent studies in yeast show that the Golgi apparatus is one of the origins for the autophagosomal membrane and Atg9-containing membrane from Golgi apparatus has a role in promoting the formation of autophagosome [Citation58,Citation59]. Considering MoAtg9 could be acetylated and MoAtg9 plays an essential role in increasing AP, we then wondered whether this acetylation by MoHat1 could affect the binding ability of MoAtg9 with the membrane structure that might interfere with the formation of APs. We used MoSft2, a transport protein homolog, as a Golgi marker to evaluate the binding ability of MoAtg9 to the membranes (). MoSft2-RFP and GFP-MoAtg9 were co-expressed in Guy11, the ∆Mohat1 mutant, and the K621R mutation strains, respectively. Through extraction of vesicle proteins and western blotting analysis, we found that the binding ability of MoAtg9 to vesicles was enhanced significantly during nutrient starvation induction in Guy11, but not in the ∆Mohat1 and K621R mutation strains ().
Acetylation of MoAtg9 is important for autophagy and pathogenicity
To further verify if the acetylation of MoAtg9 plays a role in autophagy, we detected the degradation of GFP-MoAtg8 in both the ∆Mohat1 and K621R mutant strains. During conidia and germination stages, numbers of GFP-MoAtg8-labeled APs were largely decreased in both the ∆Moatg9 and K621R mutant strains, compared with Guy11 (Figure S16A-16B), indicating the acetylation of MoAtg9 was essential for autophagy. Consistent with the result during germination, the localization and degradation of GFP-MoAtg8 were also interfered in the K621R mutation strains under starvation conditions (). Furthermore, the immunoblot assay confirmed that the autophagy levels in the ∆Moatg9 and K621R mutant strains were significantly reduced ().
We then tested whether this MoAtg9-acetylation-induced block of autophagy is also related to the development of appressorium and pathogenicity of M. oryzae. Again, Nile red, and I2 and KI staining revealed that the mutation of K621R in MoAtg9 slowed down the transfer of glycogen and intracellular lipid degradation that then interfered with turgor generation in the appressorium (Figure S13A-S13B and Table S1). Furthermore, conidial suspensions were sprayed on rice seedlings or injected to the rice sheath that further confirmed the defect in pathogenicity of the MoAtg9 K621R mutation strain ().
Cellular locations of MoHat1 in nutrient rich and starvation conditions
As a histone acetyltransferase that might display more conventional functions in the nucleus, we were also curious how MoHat1 acetylates Atg proteins in the cytoplasm. To investigate the subcellular localization of MoHat1 in M. oryzae, a MoHAT1-GFP construct was introduced into the ∆Mohat1 mutant. The resulting transformants that successfully expressed MoHat1-GFP were observed under a fluorescence microscope. Strong GFP signal was observed in the nucleus of the hyphae in nutrient-rich CM condition (). To confirm this localization, a DAPI (4ʹ, 6-diamidino-2-phenylindole) staining assay was performed and showed that the GFP signal was colocalized with DAPI (). However, under MM-N starvation induction, some of the GFP signals were translocated from the nucleus to the cytoplasm (78.56 ± 4.32%) (). Further quantitative analysis was carried out through western blotting assays. Equal amounts of total, nucleus and cytoplasm proteins were separated by SDS-PAGE and detected by GFP antibody. Results showed that the MoHat1-GFP protein could only be detected in the nucleus under nutrient-rich condition, whereas it appeared to arise in both nucleus and cytoplasm upon starvation (MM-N) (). Similar results were found during the germinating process of M. oryzae (Figure S17A-S17B), indicating that MoHat1 had an essential role in acetylating Atg proteins, including MoAtg3 and MoAtg9, in the cytoplasm during both nutrient starvation and germination.
Figure 7. Localization of MoHat1 during different nutrient conditions. (a) Confocal fluorescence microscope (Zeiss LSM710, 63x oil) observation showing that MoHat1-GFP completely localized to the nucleus (stained with DAPI [4ʹ, 6-diamidino-2-phenylindole]) in nutrient-rich conditions and some of them translocate to the cytoplasm during nutrient starvation treatment (MM-N medium) for 3 h. The asterisks stand for the nucleus and the white arrows indicate the cytoplasm. Scale bar: 5 μm. (b and c) Linescan graph consistent with the nucleus localization under nutrient-rich conditions and both nucleus and cytoplasm during nutrient starvation treatment. (d) Equal weight of hyphae powder with (MM-N) or without MM-N (CM) treatment were divided into 3 parts for extraction of total, nucleus and cytoplasm proteins, respectively. Nuclear and cytoplasmic protein were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit, according to the instructions of the manufacturer. Equal amounts of total, nucleus and cytoplasm proteins were separated by SDS-PAGE and detected by the anti-GFP antibody. The relative intensities of western blot bands were quantified with the ODYSSEY infrared imaging system (application software Version 2.1). Bars denote standard errors from 3 independent experiments.
![Figure 7. Localization of MoHat1 during different nutrient conditions. (a) Confocal fluorescence microscope (Zeiss LSM710, 63x oil) observation showing that MoHat1-GFP completely localized to the nucleus (stained with DAPI [4ʹ, 6-diamidino-2-phenylindole]) in nutrient-rich conditions and some of them translocate to the cytoplasm during nutrient starvation treatment (MM-N medium) for 3 h. The asterisks stand for the nucleus and the white arrows indicate the cytoplasm. Scale bar: 5 μm. (b and c) Linescan graph consistent with the nucleus localization under nutrient-rich conditions and both nucleus and cytoplasm during nutrient starvation treatment. (d) Equal weight of hyphae powder with (MM-N) or without MM-N (CM) treatment were divided into 3 parts for extraction of total, nucleus and cytoplasm proteins, respectively. Nuclear and cytoplasmic protein were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit, according to the instructions of the manufacturer. Equal amounts of total, nucleus and cytoplasm proteins were separated by SDS-PAGE and detected by the anti-GFP antibody. The relative intensities of western blot bands were quantified with the ODYSSEY infrared imaging system (application software Version 2.1). Bars denote standard errors from 3 independent experiments.](/cms/asset/2eabcd85-df6c-463f-a647-daf7e80aa202/kaup_a_1580104_f0007_oc.jpg)
MoHat1 translocates from the nucleus to the cytoplasm with the assistance of MoSsb1 upon starvation
To verify how MoHat1 translocates from the nucleus to the cytoplasm under nutrient starvation condition, we screened its interacting proteins using co-IP assays under MM-N treatment and identified MoSsb1, a member of Hsp70 heat shock protein family (Table S2). We then validated this interaction by co-IP assay. MoHAT1-S and MoSSB1-GFP fusion constructs were co-introduced into the protoplasts of Guy11 and positive transformants were selected. Total proteins were extracted from both hyphae under MM-N induction and germinating conidia. MoHat1 was detected in proteins eluted from anti-GFP beads using the anti-S antibody, suggesting that MoHat1 indeed interacted with MoSsb1 (). This interaction was also confirmed by a yeast two-hybrid assay (). However, the interaction could not be detected in conidia and nutrient-rich (in liquid CM cultures) conditions ().
Figure 8. MoHat1 translocates from the nucleus to the cytoplasm chaperoned by MoSsb1. (a and b) Co-IP assays of MoHat1-S and MoSsb1-GFP. western blot analysis of total proteins, suspensions and proteins eluted from anti-GFP agarose in transformants co-transformed MoHat1-S and MoSsb1-GFP were detected with anti-S or anti-GFP antibodies. Proteins were extracted from germinating conidia and hyphae under starvation treatment, respectively. (c) Yeast two-hybrid analysis for the MoHat1-MoSsb1 interaction. pGADT7 and pGBKT7 fused with specific genes were co-introduced into the yeast AH109 strain, and transformants were plated on SD-Leu-Trp as control and on selective SD-Leu-Trp-His-Ade for 5 days. The pair of plasmids pGBKT7-Lam and pGADT7-T was used as negative control. (d) Confocal fluorescence microscopy (Zeiss LSM710, 63x oil) observation showing that MoHat1-GFP is completely localized to the nucleus in the ∆Mossb1 mutant with or without nutrient starvation (MM-N). Scale bar: 5 μm. (e and f) Linescan graph consistent with the nucleus localization of MoHat1 in the ∆Mossb1 mutant under nutrient-rich (CM) or starvation (MM-N). (g) Western blot analysis of MoHat1-GFP distribution in the ∆Mossb1 mutant during nutrient-rich or starvation conditions. Nuclear and cytoplasmic protein were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit, according to the instructions of the manufacturer. ‘WT’ indicates the wild-type Guy11 strain expressing MoHat1-GFP, ‘∆’ indicates the ∆Mossb1 mutant expressing MoHat1-GFP.
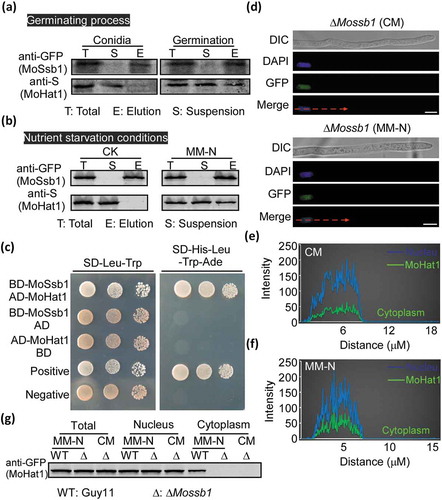
Studies have showed that as a ribosome associated chaperone protein, heat-shock protein Ssb1 assist protein cycling between the cytoplasm and the nucleus in budding yeast S. cerevisiae [Citation60–Citation63]. Recently in M. oryzae, we discovered that MoSsb1 could help with the translocation of protein phosphatase MoYvh1 to the nucleus in response to oxidative stress [Citation64]. Given that MoHat1 interacted conditionally with MoSsb1, we wondered if the translocation was also due to the role of MoSsb1. Thus, the MoHAT1-GFP construct was introduced into the ∆Mossb1 mutant and the GFP signal was detected only in the nucleus even under starvation in the resulting transformants (), in compared to in both cytoplasm and nucleus of Guy11. These results suggested that MoSsb1 aided in MoHat1 translocation from the nucleus to the cytoplasm during germination and starvation through a direct interaction.
Phosphorylation of MoHat1 affects its subcellular localization through interfering the interaction between MoHat1 and MoSsb1
Considering that MoHat1 translocated from the nucleus to the cytoplasm upon nutrient deprivation and the germinating process with the assistance of MoSsb1, we then investigated whether any protein modifications of MoHat1 would lead to the change in its protein interaction. Through protein sequence analysis, we identified 9 possible phosphorylation sites in MoHat1 (Figure S18). Mn2+-Phos-tag SDS-PAGE was thus performed to detect the phosphorylation level of MoHat1. Phosphorylated proteins in Mn2+-Phos-tag SDS-PAGE are visualized as slower migrating bands compared with the corresponding unphosphorylated proteins [Citation65]. We extracted the MoHat1-GFP protein from the nucleus and the cytoplasm of the ∆Mohat1/MoHAT1-GFP strain in the presence of a phosphatase inhibitor. Proteins were then separated in Mn2+-Phos-tag SDS PAGE and western blotting analyzed with the anti-GFP antibody. The nuclear band of MoHat1-GFP cultivated in both nutrient-rich and starvation conditions treated with the inhibitor migrated slower than that in the cytoplasm (), indicating that MoHat1 was phosphorylated in the nucleus and relatively dephosphorylated in the cytoplasm.
Figure 9. Phosphorylation of MoHat1 affects its subcellular localization. (a) Phosphorylation analysis of MoHat1. Nuclear and cytoplasmic proteins were extracted separately in the presence of phosphatase inhibitors and detected by Mn2+-Phos-tag SDS-PAGE and normal SDS-PAGE, respectively, and followed by immunoblotting with anti-GFP antibody. S8A, constitutively unphosphorylated mutation; S8D, phosphomimetic mutation. (b) Phosphorylated peptides of MoHat1 identified by prediction and seriatim sites mutation. MoHat1 (S8A) and MoHat1 (S8D) indicate constitutively unphosphorylated and phosphomimetic mutation strain, respectively. (c) Localization of MoHat1 and its mutant strains (S8A and S8D) during nutrient-rich conditions. Scale bars: 5 μm. (d) Quantification of the percentage of GFP fluorescence signal localized in the nucleus and cytoplasm in ∆Mohat1/MoHAT1 (MoHat1 WT), ∆Mohat1/MoHAT1S8A (MoHat1S8A) and ∆Mohat1/MoHAT1S8D (MoHat1S8D) strains. Asterisks represent significant differences (Duncan’s new multiple range test, p < 0.01). (e) Western blot analysis of MoHat1 WT, S8A, and S8D mutant strains. Total, nuclear and cytoplasmic proteins were extracted separately using the Nuclear and Cytoplasmic Protein Extraction Kit and detected with the GFP antibody. The western blots were quantified with the ODYSSEY infrared imaging system (application software Version 2.1). Bars denote standard errors from 3 independent experiments. (f) Co-IP analysis for the MoHat1-MoSsb1 interaction in MoHat1 WT, S8A, and S8D mutant strains. Western blots of total proteins, suspensions and proteins eluted from anti-GFP agarose from transformants co-expressing MoSsb1-GFP and MoHat1-S, MoSsb1-GFP and MoHat1S8A-S, and MoSsb1-GFP and MoHat1S8D-S were detected with anti-S or anti-GFP antibodies, respectively. T, total; S, suspensions; E, elution.
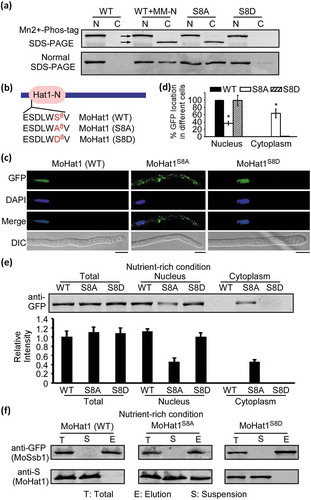
Additionally, we screened the 9 hypothetical phosphorylation sites for possible site-specific mutations, which might affect the localization and phosphorylation levels of MoHat1 (Figure S18). Interestingly, constitutively unphosphorylated mutations on these Ser sites (S to A) showed that Ser-8 (S8A) affected its localization dispersion in the cytoplasm even under nutrient-rich condition (). We then introduced a phosphomimetic MoHat1S8D-GFP (S8D) construct into the ∆Mohat1 mutant and found that a similar nuclear localization pattern as MoHat1-GFP WT (). Further Mn2+-Phos-tag SDS-PAGE assay showed that constitutively unphosphorylated MoHat1S8A strain presented a similar distribution as the WT MoHat1 under starvation (). However, the phosphomimetic MoHat1S8D strain remained similar to the WT MoHat1 in nutrient-rich condition, which was only presented in the nucleus for a relative phosphorylated state (). Furthermore, we found the de-phosphorylation of MoHat1 (either under nutrient starvation conditions or as a constitutively unphosphorylated mutation of Ser-8) promoted the interaction between MoHat1 and MoSsb1 and resulted in the translocation of MoHat1 from the nucleus to the cytoplasm in order to acetylate MoAtg3 and MoAtg9 ( and Figure S19A-S19B).
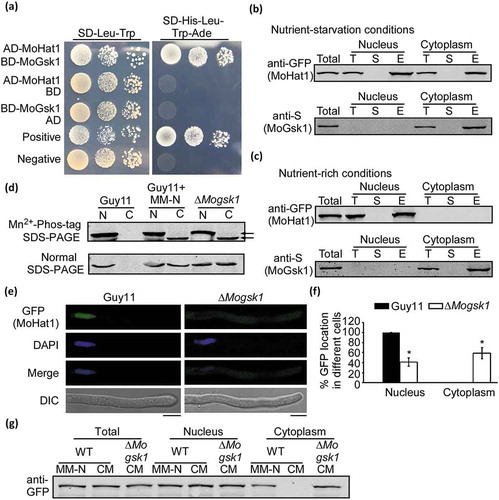
Phosphorylation of MoHat1 by MoGsk1 is important for autophagy and pathogenicity
The Ser-8 residue located in the phosphorylation motif S/TXXXS/T (S/T represent for serine/threonine, and X can be any amino acid) that can be recognized by GSK3 (glycogen synthase kinase 3) in mammals. A GSK3 homolog, MoGsk1, was previously found to be essential for fungal development, conidiation, and pathogenicity in M. oryzae [Citation66]. Incidentally, we also discovered this MoGsk1 as a MoHat1-interacting protein through the protein affinity purification approach (Table S2). To verify the interaction between MoHat1 and MoGsk1, we carried out yeast two-hybrid and in vivo co-IP assays and found that there was indeed an interaction between the two proteins in the cytoplasm (). Meanwhile, the localization of MoHat1 in the ∆Mogsk1 mutant showed partially dispersion into the cytoplasm without nutrient starvation and this portion of MoHat1 exhibited analogous unphosphorylation to the MoHat1S8A mutation strain (). These results suggested that MoHat1 was subject to phosphorylation by MoGsk1.
Figure 10. Phosphorylation of MoHat1 by MoGsk1 is important for its nuclear localization. (a) Yeast two-hybrid analysis for the interaction of MoHat1 and MoGsk1. Plasmids pGBKT7-Lam and pGADT7-T were used as a negative control. (b and c) Co-IP analysis for the interaction of MoHat1-MoGsk1. Nucleus and cytoplasm proteins were extracted separately using the Nuclear and Cytoplasmic Protein Extraction Kit under nutrient-rich and starvation conditions and incubated with anti-GFP or anti-S agarose and then eluted for western blot detecting using anti-S or anti-GFP antibodies. (d) Nucleus and cytoplasm proteins were extracted separately and the phosphorylation level of MoHat1 in the ∆Mogsk1 mutant was analyzed by Mn2+-Phos-tag SDS-PAGE and normal SDS-PAGE, respectively. (e) Localization of MoHat1 in the ∆Mogsk1 mutant was observed by confocal fluorescence microscope (Zeiss LSM710, 63x oil). Scale bars: 5 μm. (f) Quantification of GFP fluorescence localized in the nucleus and the cytoplasm in wild-type Guy11 and the ∆Mogsk1 mutant. Asterisks represent significant differences (Duncan’s new multiple range test, p < 0.01). (g) Western blot analysis of MoHat1 in Guy11 (WT) and ∆Mogsk1 mutant strains. Total, nuclear and cytoplasmic proteins were extracted separately and detected with the GFP antibody. ‘CM’ indicates the nutrient-rich conditions and ‘MM-N’ indicates nutrient starvation conditions.
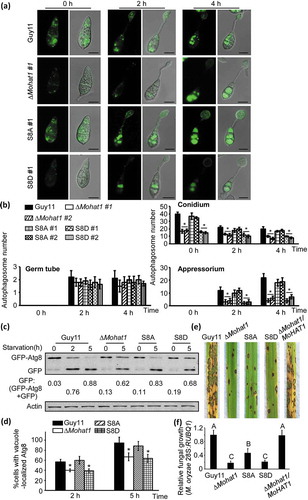
To further explore whether this phosphorylation is related to autophagy and pathogenicity, we observed the numbers of GFP-MoAtg8-labeled APs during appressorium development. As shown during the germinating process, APs were decreased significantly in the phosphomimetic MoHat1S8D strain, but were similar to the WT in the constitutively unphosphorylated MoHat1S8A strain ( and Figure S20). Similarly, the localization transformation and degradation of GFP-MoAtg8 during nutrient starvation (MM-N) conditions were blocked in the MoHat1S8D strain, whereas partially restored in the MoHat1S8A strain (). Again, Nile red, and I2 and KI staining found that the MoHat1S8D strain had a similar defect in functional appressorium development, but the MoHat1S8A strain exhibits more of a phenotype in appressorium function to the wild-type Guy11 (Figure S13A-S13B and Table S1). Further pathogenicity analysis found that the MoHat1S8D strain was significantly attenuated in pathogenicity (), indicating that the dephosphorylation of MoHat1 at Ser-8 was essential for functional appressorium development and pathogenicity. As for the MoHat1S8A strain, the pathogenicity defect was not fully restored despite it had almost normal autophagy levels (), suggesting that MoHat1 might also have an important role in the nucleus in addition to regulating Atg proteins in the cytoplasm.
Figure 11. Phosphorylation of MoHat1 is important for autophagy and pathogenicity in M. oryzae. (a) Cellular location of APs during infection-related appressorium development in Guy11, ∆Mohat1 and ∆Mohat1/MoHAT1S8A (S8A) and ∆Mohat1/MoHAT1S8D (S8D) mutant strains. Scale bar: 10 μm. (b) Bar chart showing mean APs numbers present in conidia, germ tube and appressorium at 0, 2 and 4 hours after germination. Two transformants of each kind of mutant strain (∆Mohat1 #1 and ∆Mohat1 #2, S8A #1 and S8A #2, S8D #1 and S8D #2) were used for statistical analysis. Asterisks represent significant differences (p < 0.01). (c and d) Autophagy levels in different strains treated in MM-N (nitrogen starvation minimal medium) for 2 or 5 h were analyzed by western blot analysis for (c) GFP-MoAtg8 cleavage and (d) translocation of GFP-MoAtg8 into vacuoles (n = 100). Bars with asterisks represent significant differences (Duncan’s new multiple range method p < 0.01). (e) Rice spraying assays. Four milliliters of conidial suspension (5 × 104 spores/ml) of each strain were used for spraying and examination was made 7 days after inoculation. (f) Total DNA was extracted from 1.5 g disease leaves each from (e) and tested by qRT-PCR with M. oryzae 28S rDNA and rice genomic RUBQ1 primers. Different letters indicate statistically significant differences (Duncan’s new multiple range test, p < 0.01).
Discussion
The chromatin structure is known to be a barrier to DNA repair and HATs are co-activators essential for transcriptional activation through modifying histones and remodeling nucleosomes to facilitate repair [Citation67,Citation68]. In this study, we found a component of the HAT complex, MoHat1, was involved in functional appressorium formation and pathogenicity of M. oryzae through acetylation of Atg proteins. Our studies were concordant with several recent studies that characterized HAT protein functions in saprophytic and pathogenic fungi. The taxol-producing fungus Pestalotiopsis microspora Hat1 regulates secondary metabolism, conidiation, and cell wall integrity [Citation69]. In Candida albicans, the Hat1 homolog is involved in stress resistance and virulence [Citation70]. Another component of the HAT complex, the Gcn5 protein, has also been found to play a role in phototrophy and starvation induced autophagy of M. oryzae [Citation18]. Gcn5 homologs are essential for morphological transition and virulence in Ustilago maydis and C. albicans [Citation71,Citation72]. However, reports on the functions of the HAT complex and associated regulatory mechanisms remain limited, and additional functions of Hat1 proteins in regulating acetylation besides histone modification have also not been verified. Our studies not only identified the acetylation function of MoHat1 on MoAtg3 and MoAtg9 during autophagy, but also provided an insight into the mechanism of how MoHat1 translocates to the cytoplasm during conidial germination and nutrient starvation to regulate appressorium development and pathogenicity of M. oryzae.
As a component of the HAT complex, MoHat1 was localized in the nucleus in nutrient-rich conditions and the conidia stage where much research was centered on the nuclear functions of its homologs, including histone-modification for co-regulating gene expression [Citation68,Citation73–Citation75]. However, under germination and starvation conditions, we noticed that MoHat1 translocated from the nucleus to the cytoplasm. We further found that the translocation of MoHat1 was assisted by MoSsb1 as a chaperone protein. Consistent with this result, our lab has recently found that MoSsb1 could help with the translocation of protein phosphatase MoYvh1 to the nucleus in response to oxidative stress [Citation64]. Taken with these two findings above, it seemed that the chaperone protein MoSsb1 in M. oryzae might have a role in assisting protein nucleo-cytoplasmic shuttling under various conditions/stresses.
We also found that the phosphorylation of MoHat1 by glycogen synthase kinase MoGsk1 under nutrient-rich conditions affected its binding to MoSsb1 leading to its nuclear localization, whereas under nutrient starvation conditions or appressorial development, some of MoHat1 was dephosphorylated and interacted with MoSsb1 in order to be translocated into the cytoplasm to function on acetylating autophagy proteins (MoAtg3 and MoAtg9). This kind of protein-protein interaction regulated by phosphorylation has been reported in several studies [Citation76–Citation78]. For example, S. cerevisiae Atg13, a regulatory subunit of Atg1-complex in autophagy regulation, is phosphorylated in a TORC1-dependent manner under nutrient-rich conditions and immediately dephosphorylated in response to starvation and then associated with Atg1 leading to the up-regulation of the kinase activity of Atg1 [Citation77,Citation78]. Moreover, we found the Ser-8 phosphorylation site of MoHat1 by the protein kinase MoGsk1 was essential for its nuclear localization. Although the constitutively unphosphorylated MoHat1S8A strain could completely restore the acetylation and autophagy defect during both conidial germination and nutrient starvation conditions, the pathogenicity defect could not be fully suppressed, indicating that, in addition to functions of acetylation on Atg proteins in the cytoplasm, MoHat1 might also have an essential role in the nucleus, such as modifying histones for transcriptional activation. Consistent with our findings, the hyperacetylation of histone is associated with transcriptional downregulation of several autophagy-essential Atg genes in Drosophila melanogaster that might restrict autophagic activities, in addition to direct modification on the cytosolic autophagy proteins [Citation18,Citation79].
Autophagy is a fundamental function of eukaryotic cells and is well conserved from yeasts to mammals. The most remarkable feature of autophagy is the synthesis of double membrane-bound autophagosome that sequester organelles or proteins to be degraded. Core autophagy related genes have been grouped into several regulating subgroups, including Atg1 kinase and its regulators [Citation78], Atg8 conjugation system [Citation7], the Atg12 conjugation system [Citation80], the autophagy-specific phosphatidylinositol 3-kinase (PtdIns3K) complex [Citation81], and a subgroup of functionally unknown proteins [Citation55,Citation82–Citation84]. Previous studies found that Esa1 (yeasts)/TIP60 (mammals), a member of the HAT complex, regulates autophagy by directly acetylating the autophagy protein Atg3 or ULK1 (Atg1 homolog in mammals) [Citation15,Citation28]. However, MoEsa1, a yeast Esa1 homolog of M. oryzae, has been found not to be involved in regulating autophagy [Citation18]. Interestingly, we found MoHat1 participated in the regulation of autophagy during both nutrient starvation and conidial germination of M. oryzae that might take over the role of MoEsa1 in acetylation. Furthermore, besides of a E2-enzyme in the ubiquitination reaction, which is essential for conjugation of Atg8, acetylated Atg3 is shown to be a tethering factor that brings Atg8 and the PE-containing membrane together to facilitate lipidation [Citation85]. MoHat1 directly acetylated MoAtg3 and had a role in the MoAtg3-MoAtg8 interaction in vivo since the interaction was significantly reduced in the ∆Mohat1 mutant. Meanwhile, it was found that exogenous MoAtg3 incubated with purified endogenous MoHat1 could prominently promote the conjugation of MoAtg8 compared with exogenous MoAtg3 alone (), indicating a similar promotion of acetylated MoAtg3 on MoAtg8 lipidation. Thus, MoHat1-mediated MoAtg3 acetylation seemed to affect autophagy through promoting MoAtg8 conjugation in M. oryzae.
In addition to acetylating MoAtg3, MoHat1 could also acetylate another core Atg protein, MoAtg9, which is the only transmembrane protein identified in yeast core macroautophagy machinery required for formation of APs [Citation54,Citation55]. It has been found that Atg9 not only localizes to the PAS and cytosolic pool like most Atg proteins, but also to the peripheral sites [Citation56]. Atg9 seems to move between these peripheral sites and the PAS, and the cycling of Atg9 is essential for AP formation [Citation56,Citation86–Citation88]. Here, we found that MoHat1 translocated from the nucleus to the cytoplasm and directly acetylated MoAtg9 under nutrient starvation in the cytoplasm. Although several reports recently focused on the modification of Atg9 especially for phosphorylation to affect its movement to the PAS and the rate of AP formation [Citation89,Citation90], acetylation of Atg9 has not been reported previously. Moreover, we found that the acetylation of MoAtg9 affects its binding ability to vesicles structures that increase the formation of APs and finally affect autophagy, indicating that, besides phosphorylation, MoAtg9 acetylation is also essential for its vesicle-binding ability and APs formation. Accordingly, it appeared that MoHat1 acetylated MoAtg3 and MoAtg9 to participate in two different ways to regulate AP formation, respectively (). Meanwhile, we noticed a fact that ∆Moatg3 and ∆Moatg9 deletion mutants were more severely reduced in infection and pathogenicity than the ∆Mohat1 mutant. Similar to our findings, the histone acetyltransferase MoGcn5 regulates phototrophy and starvation induced autophagy of M. oryzae through acetylating MoAtg7, and the defect of its deletion mutant in pathogenicity also seems not as serious as that of the MoATG7 knockout strain [Citation18,Citation37]. This difference might be due to that the subjects of HAT might be various (e.g. histone proteins, autophagy proteins) [Citation18,Citation79] and modifications on histone proteins might result in several genes expression alteration including positive or negative regulatory factors that showed opposite functions on pathogenicity. Among these, MoHat1-dependent acetylation on Atg proteins only had a partial effect. Accordingly, we are attempting to further investigate the complicated MoHat1-associated regulating network for searching potential additional regulatory factors in pathogenicity.
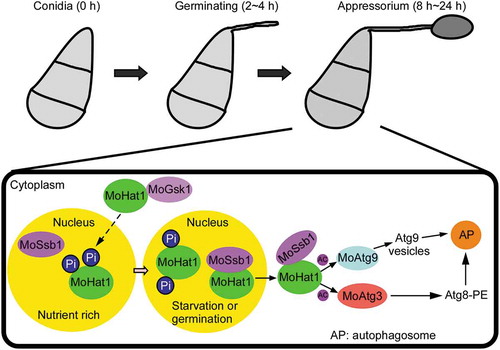
Figure 12. Model of MoHat1 acetylates Atg proteins to regulate functional appressorium formation and pathogenicity. Germination or starvation conditions induce acetylation-related autophagy, accompanied with the acetylation of MoAtg3 and MoAtg9, to form functional appressorium in M. oryzae. The yellow region indicates the nucleus and the other area in the black frame represent the cytoplasm. Under nutrient-rich conditions, MoHat1 keeps in a high phosphorylation level that affects its binding to MoSsb1 leading to its nuclear localization. During nutrient starvation conditions or appressorial development (germination), some MoHat1 alters the phosphorylation level itself in order to enhance its interaction with the chaperone protein MoSsb1, which is then translocated into the cytoplasm to function on acetylating MoAtg3 and MoAtg9. Acetylation of MoAtg9 enhances its binding ability with vesicles and acetylation on MoAtg3 promotes MoAtg8 conjugation to PE that regulates the autophagy process collectively. AP, autophagosome; Ac, acetylation.
Furthermore, our results showed that conidial germination of M. oryzae mimicked nutrient starvation, and the germinating process was accompanied by a high level of autophagy because the acetylation of MoAtg3 and MoAtg9 was significantly upregulated. This is consistent with the findings that autophagy is essential for M. oryzae to elaborate its specialized infection structure appressorium [Citation37]. Meanwhile, disturbing the acetylation of MoAtg3 and MoAtg9 affected autophagy both under germination and nutrient starvation conditions, and also led to delayed transfer of glycogen and lipid degradation resulting in abnormal appressorium development and attenuated pathogenicity. This indicated that M. oryzae needed moderate acetylation for its Atg proteins to maintain normal appressorium development and pathogenicity.
In summary, we proposed that conidial germination or starvation could trigger acetylation-related autophagy that was accompanied with the acetylation of MoAtg3 and MoAtg9 to form functional appressorium. During these conditions, histone acetyltransferase MoHat1 altered the phosphorylation level in order to enhance its interaction with MoSsb1 that then translocated into the cytoplasm to acetylate MoAtg3 and MoAtg9. We demonstrated that the acetylation of MoAtg3 and MoAtg9 were both critical for appressorium development and pathogenicity of M. oryzae. This is the first report of MoAtg9 being subject to acetylation regulation. Considering the critical role that MoHat1 played in sustaining the autophagy process and pathogenicity, further studies will focus on MoHat1-interacting proteins in order to enrich the regulatory network of MoHat1 in M. oryzae.
Materials and methods
Strains and culture conditions
The M. oryzae Guy11 strain was used as wild type (WT) in this study. All strains were cultured on complete medium (CM) for 3–15 days in the dark at 28°C [Citation91]. For vegetative growth, small agar blocks were cut from the edge of 7-day-old cultures and placed onto fresh media, followed by incubation in the dark at 28°C. The radial growth was measured after incubation for 7 days. Mycelia were harvested from liquid CM medium and used for DNA, RNA as well as protein extractions. For conidia production, strains were maintained on straw decoction and corn (SDC: 100 g of straw, 40 g of corn powder, 15 g of agar in 1 L of distilled water) agar media at 28°C for 7 days in the dark followed by 3 days of continuous illumination under fluorescent light [Citation92].
Appressorium formation, appressorium turgor and glycogen/lipid staining
For appressorium formation, conidia harvested from 10-day-old SDC cultures were filtered through two-layers of Miracloth and washed with double-distilled water (ddH2O) for three times. Droplets (30 μl) of conidial suspension (5 × 104 spores/ml) were placed on microscope cover glass (Fisher Scientific, 16938) under humid conditions at 28°C in darkness and the samples were microscopically observed at intervals [Citation93,Citation94]. The percentages of conidia germinating and conidia forming appressoria were determined by microscopic examination of at least 100 conidia for more than three times. The appressorium turgor was measured using an incipient cytorrhysis (cell collapse) assay with a 1.0 to 4.0 M glycerol solution [Citation95]. After germinating for 24 h, the water surrounding the conidia was removed carefully and then replaced with an equal volume (30 μl) of glycerol in concentrations ranging from 1.0 to 4.0 M. The number of appressoria that had collapsed after 3 min was recorded. The experiments were repeated three times, and more than 100 appressoria were observed for each replicate.
The glycogen metabolism in the germinating conidia and appressoria of strains were visualized by staining these tissues with glycogen staining solution containing 60 mg/ml KI and 10 mg/ml I2 [Citation32]. Once the samples become yellowish-brown, the glycogen deposits can be visualized in bright field optics with Zeiss Axio Observer A1 inverted microscope. For the lipid droplet observation, germinating conidia and appressoria of strains were visualized by staining these tissues with a Nile red solution consisting of 50 mM Tris/maleate buffer (pH 7.5) and 2.5 mg/ml Nile red Oxazone (Sigma-Aldrich, N3013) [Citation31,Citation32]. After 3 min incubation in darkness, the lipid droplets in the conidia and appressoria began to fluoresce and were observed under Zeiss Axio Observer A1 inverted microscope.
Virulence assay
Conidia were harvested from 10-day-old SDC agar cultures, filtered through two layers of Miracloth (EMD Millipore Corporation, 475855-1R) and resuspended to a concentration of 5 × 104 spores/ml in a 0.2% (w:v) gelatin solution. Two-week-old seedlings of rice (Oryza sativa cv. CO39) and 7-day-old seedlings of barley (Hordeum vulgare cv. Four-arris) were used for pathogenicity assays. For spray inoculation, 5 ml of conidial suspension of each treatment were sprayed onto rice with a sprayer. Inoculated plants were kept in a growth chamber at 28°C with 90% humidity and in the dark for the first 24 h, followed by a 12 h-12 h light-dark cycle. The disease severity was assessed at 7 day after inoculation. The disease lesions were quantified by a ‘lesion-type’ scoring assay which divided the lesions into 1–5 types according to their severity (type 0, no lesion; type 1, pinhead-sized dark brown specks without visible centers; type 2, small brown lesions that are approximately 1 mm in diameter; type3, 2–3 mm gray spots with brown margins; type 4, elliptical gray spots longer approximately 3–4 mm; type 5, large eyespot lesions that coalesced infecting 50% or more of the leaf area). For the injection assay, same concentrations of conidial suspension (5 × 104 spores/ml) were injected into 3-week-old rice sheath and incubation in conditions as the same as spray inoculation assay [Citation96]. For the spot test, a 30-μl droplet (concentrations of conidial suspensions: 104, 103, 102 spores/ml) was placed onto the upper side of the detached barley leaves maintained on 4% (w:v) water agar plates. The leaves were observed after 5 days of incubation at 25°C. For ‘relative fungal growth’ assay, total DNA was extracted from 1.5 g disease leaves and tested by qRT-PCR (ChamQTM SYBR® qPCR Master Mix (Vazyme Biotech Company, Q311-02/03)) with M. oryzae 28S ribosomal gene (rDNA) and RUBQ1 primers [Citation97]. Each experiment was repeated for more than 3 times and the experimental conditions were controlled to be consistent (e.g., temperature, humidity, illumination and the age of the plants).
Rice sheath and onion epidermis penetration assays
For the infection assay with rice tissues, conidia were resuspended to a concentration of 2 × 105 spores/ml in a 0.2% (w:v) gelatin solution. Rice cultivar CO-39 (3- to 4-weeks old) was inoculated with 100 μl of conidial suspension on the inner leaf sheath cuticle cells and incubation under humid conditions at 28°C. The leaf sheaths were observed under Zeiss Axio Observer A1 inverted microscope at 24 and 36 hpi. The penetration assay were further quantitatively analyzed by observing more than 100 appressoria for each strain and classifying the invasive hyphae (IH) into 4 types: type1, no hyphal penetration with only appressoria formation; type2, IH with 1 or 2 short branch; type3, IH with at least 3 branches but the IH are short and extending within a plant cell; type 4, IH that has numerous branches and fully occupies the plant cell or even extended to an adjacent plant cell.
The onion penetration was performed as previously described [Citation98]. Briefly, onion epidermal strips were isolated and 40 μl of conidial suspension was inoculated onto the adaxial surface. After 48 hpi of incubation, onion epidermal strips and infection hyphae were observed with Zeiss Axio Observer A1 inverted microscope.
Target gene deletion and complementation
The MoHAT1 gene deletion mutant was generated using the standard one-step gene replacement strategy. First, 2 fragments with 1.0 kb of sequences flanking the targeted gene were PCR amplified with primer pairs, then, the resulting PCR products of MoHAT1 was digested with restriction endonucleases and ligated with a hygromycin-resistance cassette (HPH) released from pCX62. Finally, the recombinant insert was sequenced. The 3.4-kb fragment, which includes the flanking sequences and the HPH cassette, was amplified and introduced into Guy11 protoplasts. Putative mutants were screened by PCR and further confirmed by Southern blotting analysis (Figure S3). The complement fragment, which contains the entire MoHAT1 gene coding region and its native promoter region, was amplified by PCR with primers using 2× Planta Master Mix (Vazyme Biotech Company, P511-02) and inserted into pYF11 (bleomycin resistance; Thermo Fisher Scientific, R25001) to complement the mutant strain.
Quantitative RT-PCR analysis
For qRT-PCR, total RNA was reverse transcribed into first-strand cDNA using the oligo (dT) primer and HiScript II Q RT SuperMix for qPCR (Vazyme Biotech Company, R233-01). qRT-PCR reactions were performed following previously established procedures [Citation97]. qRT-PCR was performed with 3 independent pools of tissues in 3 sets of experimental replicates.
Bimolecular fluorescence complementation (BiFC) assay
The MoAtg3-cYFP, MoAtg9-cYFP and MoAtg7-cYFP fusion constructs were generated by cloning the MoATG3, MoATG9 and MoATG7 fragments into pHZ68, respectively [Citation99]. Similarly, MoHat1-nYFP fusion constructs were generated by cloning the MoHAT1 fragment into pHZ65. Construct pairs of MoHat1-nYFP and MoAtg3-cYFP, MoHat1-nYFP and MoAtg9-cYFP, MoHat1-nYFP and MoAtg7-cYFP were co-introduced into protoplasts of wild-type Guy11, respectively. Transformants resistant to both hygromycin (Solarbio® Life Sciences, H8080) and zeocin (Thermo Fisher Scientific, R25001) were isolated and confirmed by PCR analyses. Because the empty vector negative controls in the BiFC assay are not as stable as fused constructs [Citation100], we tested the expression levels of each BiFC construct in every transformant (including negative controls) (Figure S9A-S9D). YFP signals were examined with a Zeiss LSM710, 63x oil.
Epifluorescence microscopy
M. oryzae cells (hyphae, conidia or appressorium) expressing fluorescent protein-fused chimera were incubated under appropriate conditions. The constructs including GFP-MoAtg8, MoHat1-GFP, MoHat1S8A-GFP, MoHat1S8D-GFP, and other phosphorylation mutations were transformed into ∆Mohat1 mutant or the wild-type Guy11 strain. Epifluorescence microscopy was performed using microscope of Zeiss LSM710, 63x oil. MoApe1-RFP was introduced into the BiFC transformants to mark the phagophore assembly site (PAS) where most Atg proteins are located on under starvation conditions. To visualize the nucleus, mycelia were stained with 1 μg/mL DAPI (Sigma-Aldrich, D9542) at room temperature in darkness for 5 min, followed by washing with 1× PBS (diluted from 10× PBS [Beyotime Biotechnology, ST476]) 3 times.
Protein extraction and in vivo acetylation analysis
Strains were cultured in ample liquid nutrient-rich conditions (CM media) for 36 h and then moved into nutrition starvation conditions (MM-N media: 0.52 g KCl, 0.152 g MgSO4·7H2O, 1.52 g KH2PO4, 0.01 g vitamin B1, 1 mL trace elements [2.2 g ZnSO4·7H2O, 1.1 g H3BO3, 0.5 g MnCl2·4H2O, 0.5 g FeSO4·7H2O, 0.17 g CoCl2·6H2O, 0.16 g CuSO4·5H2O, 0.15 g Na2MnO4·2H2O, 5 g Na4EDTA in 1 L of distilled water], 10 g D-glucose in 1 L of distilled water) for different hours before mycelia of each strain were harvested. For the germinating process protein, we collected germinating conidia on inductive surface at different time points (0, 8, 16, 24 h) and frozen with liquid nitrogen prepared for protein extraction. For total protein extraction, mycelia or germinating conidia were ground into a fine powder in liquid nitrogen and resuspended in 1 mL lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40 [Sigma-Aldrich, I3021]) with 2 mM PMSF (Beyotime Biotechnology, ST506-2), proteinase inhibitor cocktail (Sigma-Aldrich, 11836170001) and deacetylation inhibitors (50 mM nicotinamide, 50 mM sodium butyrate, 5 mM trichostatin A [Sigma-Aldrich, T1952]). Especially for GFP-MoAtg9 protein extraction, RIPA lysis buffer II (Sangon Biotech, C510006) was used because MoAtg9 protein could bind to membrane structures which made it difficult to extract. The lysates were placed on the ice for 30 min and shaken once every 10 min for protein lysing. Cell debris was removed by centrifugation at 13, 000 g for 10 min at 4°C. The supernatant lysates were then incubated with 5 μL anti-acetylated-lysine antibody (Cell Signaling Technology, 9441S) for 2 h and followed with PureproteomeTM protein G magnetic beads (MEMD Millipore Corporation, LSKMAGG02) for an additional 1 h. Beads were washed extensively 5 times in 1× PBS (diluted from 10 × PBS [Beyotime Biotechnology, ST476]). Samples were analyzed by 12% SDS-PAGE followed by western blotting with anti-GFP (mouse, 1:5000; Abmart, 293967), secondary antibody was anti-mouse (1:10,000; LI-COR Biosciences, C70301-02) followed by detection using the ODYSSEY infrared imaging system (application software Version 2.1). For mass spectrometry analyzing the acetylation sites, we thank the Shanghai Applied Protein Technology Co., Ltd., for technological assistance.
To prepare the cytoplasmic fraction, the mycelia powder was lysed using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, P0027) according to the manufacturer’s instructions. The lysates were centrifuged at 13, 000 g for 10 min at 4°C. The clear supernatants were collected as the cytoplasmic fraction (be sure not to touch the precipitate). For the precipitate, we removed the remnants of the supernatant completely and added nuclear protein extraction reagents with 2 mM PMSF. The lysates were again placed on the ice for 30 min and shaken once every 10 min for protein lysing. Cell debris was removed by centrifugation at 13, 000 g for 10 min at 4°C. The clear supernatants were collected as the nuclear fraction.
Co-immunoprecipitation (co-IP)
To confirm the interactions of MoHat1-MoAtg3, MoHat1-Atg9, MoAtg3-MoAtg8, MoHat1-MoSsb1, and MoHat1-MoGsk1 in vivo, the MoHat1-S, MoAtg3-GFP, MoAtg9-GFP, MoAtg8-GFP, MoAtg3-S, MoSsb1-GFP, MoHat1S8A-S, MoHat1S8D-S, MoHat1-GFP, and MoGsk1-S fusion constructs were prepared. Then different pairs of specific constructs were co-transformed into protoplasts of the WT strain. Total proteins were isolated from different positive transformants and incubated with anti-GFP agarose (Chromo Tek, gta-20) or anti-S agarose (Abcam, ab19369) at 4°C for 2 to 12 h with gently shaking. Proteins bound to the beads were eluted after a serious of washing steps by 1× PBS (diluted from 10× PBS [Beyotime Biotechnology, ST476]). Elution buffer (200 mM glycine, pH 2.5) and neutralization buffer (1 M Tris base, pH 10.4) were used for the elution process. Total, suspension, and also eluted protein were then analyzed by western blot using anti-GFP (mouse, 1:5000; Abmart, 293967) or anti-S (rabbit, 1:5000; Abcam, ab183674).
Mohat1-complex purification and in vitro acetylation assays
Purification of the MoHat1-complex was carried out with GFP-Trap (Chromo Tek, gta-20) and eluted with elution buffer (200 mM glycine, pH 2.5) and neutralization buffer (1 M Tris base, pH 10.4). In vitro HAT reactions were performed for 1 h at 30°C in a 400-μL reaction mixture containing ~8 μg of GST-MoAtg3 protein (WT and K262R K267R) or GST-MoAtg9 protein (WT and K621R), 1 mM acetyl-CoA (Sigma-Aldrich, A2056), 5 mM nicotinamide, 1 mM PMSF and 1 mM DTT in HAT buffer (50 mM Tris-HCl, pH 7.5, 5% glycerol [Sigma-Aldrich, G5516], 0.1 mM EDTA, 50 mM KCl), and 2 μg of immune-purified MoHat1-complex [Citation101]. The reaction mixture was immune-precipitated with anti-acetylated-lysine antibody (Cell Signaling Technology, 9441S) at 4°C for 3 h, and followed by incubation with PureproteomeTM protein G magnetic beads (MEMD Millipore Corporation, LSKMAGG02) for an additional 1 h. Beads were washed extensively 5 times in 1× PBS. Samples were analyzed by 12% SDS-PAGE followed by western blotting with anti-GFP antibody.
In vitro MoAtg8–PE conjugation assay
E. coli strain BL21 (DE3) expressing GST-MoATG7, GST-MoATG3, and GST-MoATG8 constructs were collected and treated with lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Triton X-100 [Sigma-Aldrich, T8787]). To confirm expression of the GST fusion proteins, bacterial lysates were separated by SDS-PAGE gel followed by Coomassie Brilliant Blue staining. The purified MoAtg7 (1 μM), MoAtg3 (1 μM: WT or K262R K267R) and MoAtg8 (10 μM) were mixed with liposomes (350 μM lipids: Liposome Kit [Sigma-Aldrich, L4395-1VL]) in the presence of 1 mM DTT and 1 mM ATP (Sigma-Aldrich, FLAAS-1VL) in Tris-HCl buffer and incubated at 30°C for 20 min and 90 min, respectively [Citation85]. The reaction was finally stopped by 5× SDS-PAGE sample buffer and boiled at 95°C for 10 min. Samples were then separated on 0.15 g/ml urea (Sigma-Aldrich, U5378) SDS-PAGE and proteins were identified by Coomassie Brilliant Blue staining.
Phosphorylation analysis through Phos-tag gel
The MoHat1-GFP, MoHat1S8A-GFP, MoHat1S8D-GFP fusion constructs were introduced into ∆Mohat1 mutant, respectively. The proteins extracted from mycelium (un-treatment or under nutrient starvation treatment) were resolved on 8% SDS-polyacrylamide gels prepared with 50 µM acrylamide-dependent Phos-tag ligand and 100 µM MnCl2 as described [Citation65,Citation94,Citation102]. Gel electrophoresis was run at a constant voltage of 80 V for 3–6 h. Before transferring, gels were equilibrated in transfer buffer with 5 mM EDTA for 20 min 2 times and followed with transfer buffer without EDTA for another 20 min. Protein transfer from the Mn2+-phos-tagTM acrylamide (NARD institute Limited company, 18D-01) gel to the PVDF membrane was performed for ~36 h (depend on different proteins) at 80 V at 4°C, and then the membrane was analyzed by western blotting using the anti-GFP antibody.
Accession number
The genes from this article can be found in the GenBank database under the following accession numbers: MoHat1 (MGG_06375), MoAtg3 (MGG_02959), MoAtg9 (MGG_09559), MoAtg8 (MGG_01062), MoSft2 (MGG_05708), MoApe1 (MGG_07536), MoSsb1 (MGG_11513), MoGsk1 (MGG_12122).
Supplemental Material
Download Zip (23.1 MB)Acknowledgments
This research was supported by the key program of Natural Science Foundation of China (Grant No: 31530063, ZZ), National Science Foundation for Distinguished Young Scholars of China (Grant No.31325022 to ZZ), the China National Funds for Innovative Research Groups (Grant No.31721004) and Innovation Team Program for Jiangsu Universities (2017). The Wang lab research was supported by US NIH grants AI121451 and AI121460. We thank Dr. Zonghua Wang of Fujian Agriculture and Forestry University for providing the ∆Mogsk1 mutant, Dr. Xiaohong Liu of Zhejiang University for providing plasmid GFP-MoAtg8 and the ∆Moatg9 mutant, Dr. Yizhen Deng of South China Agricultural University for helpful suggestions on optimizing in vivo acetylation assay and offering MoAtg7-RFP, pGADT7-MoATG7 constructs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18.
- Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–21.
- Klionsky DJ. The molecular machinery of autophagy and its role in physiology and disease. Semin Cell Dev Biol. 2010;21:663.
- Klionsky DJ, Cregg JM, Dunn WA Jr., et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545.
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937.
- Klionsky DJ. For the last time, it is GFP-Atg8, not Atg8-GFP (and the same goes for LC3). Autophagy. 2011;7:1093–1094.
- Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492.
- Kabeya Y, Mizushima N, Uero T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728.
- Kirisako T, Baba M, Ishihara N, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–446.
- Kirisako T, Ichimura Y, Okada H, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276.
- Kim J, Huang WP, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64.
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720.
- Feng YC, Yao ZY, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25:354–363.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45.
- Yi C, Ma M, Ran L, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336:474–477.
- Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284:6322–6328.
- Zhao Y, Yang J, Liao WJ, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675.
- Zhang S, Liang M, Naqvi NI, et al. Phototrophy and starvation-based induction of autophagy upon removal of Gcn5-catalyzed acetylation of Atg7 in Magnaporthe oryzae. Autophagy. 2017;13:1318–1330.
- Schnitzler GR. Isolation of histones and nucleosome cores from mammalian cells. Curr Protoc Mol Biol. 2001;50:1–12.
- Verreault A, Kaufman PD, Kobayashi R, et al. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr Biol. 1998;8:96–108.
- Zerler B, Roberts RJ, Mathews MB, et al. Different functional domains of the adenovirus E1A gene are involved in regulation of host cell cycle products. Mol Cell Biol. 1987;7:821–829.
- Reifsnyder C, Lowell J, Clarke A, et al. Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nat Genet. 1996;14:42–49.
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606.
- Otero G, Fellows J, Li Y, et al. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell. 1999;3:109–118.
- Brown CE, Lechner T, Howe L, et al. The many HATs of transcription coactivators. Trends Biochem Sci. 2000;25:15–19.
- Choy JS, Kron SJ. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol Cell Biol. 2002;22:8215–8225.
- Grienenberger A, Miotto B, Sagnier T, et al. The MYST domain acetyltransferase Chameau functions in epigenetic mechanisms of transcriptional repression. Curr Biol. 2002;12:762–766.
- Lin SY, Li TY, Liu Q, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336:477–481.
- Talbot NJ. On the trail of a cereal killer: exploring the biology of Magnaporthe grisea. Annu Rev Microbiol. 2003;57:177–202.
- Zhang H, Zheng X, Zhang Z. The Magnaporthe grisea species complex and plant pathogenesis. Mol Plant Pathol. 2016;17:796–804.
- Wang ZY, Soanes DM, Kershaw MJ, et al. Functional analysis of lipid metabolism in Magnaporthe grisea reveals a requirement for peroxisomal fatty acid beta-oxidation during appressorium-mediated plant infection. Mol Plant Microbe Interact. 2007;20:475–491.
- Thines E, Weber RW, Talbot NJ. MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell. 2000;12:1703–1718.
- Davis DJ, Burlak C, Money NP. Biochemical and biomechanical aspects of appressorial development on Magnaporthe grisea. Dev Plant Pathol. 2000;15:248–256.
- Veneault-Fourrey C, Barooah M, Egan M, et al. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science. 2006;312:580–583.
- Foster AJ, Ryder LS, Kershaw MJ, et al. The role of glycerol in the pathogenic lifestyle of the rice blast fungus Magnaporthe oryzae. Environ Microbiol. 2017;19:1008–1016.
- Marroquin-Guzman M, Sun G, Wilson RA. Glucose-ABL1-TOR signaling modulates cell cycle tuning to control terminal appressorial cell differentiation. PLoS Genet. 2017;13:e1006557.
- Kershaw MJ, Talbot NJ. Genome-wide functional analysis reveals that infection-associated fungal autophagy is necessary for rice blast disease. Proc Natl Acad Sci USA. 2009;106:15967–15972.
- Deng Y, Qu Z, Naqvi NI. Role of macroautophagy in nutrient homeostasis during fungal development and pathogenesis. Cells. 2012;1:449–463.
- Liu XH, Chen SM, Gao HM, et al. The small GTPase MoYpt7 is required for membrane fusion in autophagy and pathogenicity of Magnaporthe oryzae. Environ Microbiol. 2015;17:4495–4510.
- Liu XH, Xu F, Snyder JH, et al. Autophagy in plant pathogenic fungi. Semin Cell Dev Biol. 2016;57:128–137.
- Qin S, Parthun MR. Histone H3 and the histone acetyltransferase Hat1p contribute to DNA double-strand break repair. Mol Cell Biol. 2002;22:8353–8365.
- Dong B, Xu XJ, Chen GQ, et al. Autophagy-associated alpha-arrestin signaling is required for conidiogenous cell development in Magnaporthe oryzae. Sci Rep. 2016;6:30963.
- Jiang CJ, Shimono M, Sugano S, et al. Abscisic acid interacts antagonistically with salicylic acid signaling pathway in rice-Magnaporthe grisea interaction. Mol Plant Microbe Interact. 2010;23:791–798.
- Talbot NJ, Foster AJ. Genetics and genomics of the rice blast fungus Magnaporthe grisea: developing an experimental model for understanding fungal diseases of cereals. Adv Bot Res. 2001;34:263–287.
- deJong JC, MCCormack BJ, Smirnoff N, et al. Glycerol generates turgor in rice blast. Nature. 1997;389:244–245.
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552.
- Tucker SL, Talbot NJ. Surface attachment and pre-penetration stage development by plant pathogenic fungi. Annu Rev Phytopathol. 2001;39:385–417.
- Saunders DG, Aves SJ, Talbot NJ. Cell cycle-mediated regulation of plant infection by the rice blast fungus. Plant Cell. 2010;22:497–507.
- Ichimura Y, Imamura Y, Emoto K, et al. In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J Biol Chem. 2004;279:40584–40592.
- Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20:5971–5981.
- Hanada T, Ohsumi Y. Structure-function relationship of Atg12, a ubiquitin-like modifier essential for autophagy. Autophagy. 2005;1:110–118.
- Mizushima N, Sugita H, Yoshimori T, et al. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–33892.
- Yamaguchi M, Noda NN, Nakatogawa H, et al. Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J Biol Chem. 2010;285:29599–29607.
- Lang T, Reiche S, Straub M, et al. Autophagy and the cvt pathway both depend on AUT9. J Bacteriol. 2000;182:2125–2133.
- Noda T, Kim J, Huang WP, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol. 2000;148:465–479.
- Reggiori F, Tucker KA, Stromhaug PE, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90.
- Yang S, Rosenwald AG. Autophagy in Saccharomyces cerevisiae requires the monomeric GTP-binding proteins, Arl1 and Ypt6. Autophagy. 2016;12:1721–1737.
- Davis S, Wang J, Zhu M, et al. Sec24 phosphorylation regulates autophagosome abundance during nutrient deprivation. Elife. 2016;5.
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835.
- Rospert S, Dubaquie Y, Gautschi M. Nascent-polypeptide-associated complex. Cell Mol Life Sci. 2002;59:1632–1639.
- Shulga N, James P, Craig EA, et al. A nuclear export signal prevents Saccharomyces cerevisiae Hsp70 Ssb1p from stimulating nuclear localization signal-directed nuclear transport. J Biol Chem. 1999;274:16501–16507.
- Lopez N, Halladay J, Walter W, et al. SSB, encoding a ribosome-associated chaperone, is coordinately regulated with ribosomal protein genes. J Bacteriol. 1999;181:3136–3143.
- Albanese V, Reissmann S, Frydman J. A ribosome-anchored chaperone network that facilitates eukaryotic ribosome biogenesis. J Cell Biol. 2010;189:69–81.
- Liu X, Yang J, Qian B, et al. MoYvh1 subverts rice defense through functions of ribosomal protein MoMrt4 in Magnaporthe oryzae. PLoS Pathog. 2018;14:e1007016.
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, et al. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757.
- Zhou T, Dagdas YF, Zhu X, et al. The glycogen synthase kinase MoGsk1, regulated by Mps1 MAP kinase, is required for fungal development and pathogenicity in Magnaporthe oryzae. Sci Rep. 2017;7:945.
- Strenkert D, Schmollinger S, Sommer F, et al. Transcription factor-dependent chromatin remodeling at heat shock and copper-responsive promoters in Chlamydomonas reinhardtii. Plant Cell. 2011;23:2285–2301.
- Guo R, Chen J, Mitchell DL, et al. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011;39:1390–1397.
- Zhang Q, Chen L, Yu X, et al. A B-type histone acetyltransferase Hat1 regulates secondary metabolism, conidiation, and cell wall integrity in the taxol-producing fungus Pestalotiopsis microspora. J Basic Microbiol. 2016;56:1380–1391.
- Tscherner M, Zwolanek F, Jenull S, et al. The Candida albicans histone acetyltransferase Hat1 regulates stress resistance and virulence via distinct chromatin assembly pathways. PLoS Pathog. 2015;11:e1005218.
- Chang P, Fan X, Chen J. Function and subcellular localization of Gcn5, a histone acetyltransferase in Candida albicans. Fungal Genet Biol. 2015;81:132–141.
- Gonzalez-Prieto JM, Rosas-Quijano R, Dominguez A, et al. The UmGcn5 gene encoding histone acetyltransferase from Ustilago maydis is involved in dimorphism and virulence. Fungal Genet Biol. 2014;71:86–95.
- Dahlin JL, Chen X, Walters MA, et al. Histone-modifying enzymes, histone modifications and histone chaperones in nucleosome assembly: lessons learned from Rtt109 histone acetyltransferases. Crit Rev Biochem Mol Biol. 2015;50:31–53.
- Haigney A, Ricketts MD, Marmorstein R. Dissecting the molecular roles of histone chaperones in histone acetylation by type B histone acetyltransferases (HAT-B). J Biol Chem. 2015;290:30648–30657.
- Limbeck E, Vanselow JT, Hofmann J, et al. Linking site-specific loss of histone acetylation to repression of gene expression by the mycotoxin ochratoxin A. Arch Toxicol. 2018;92:995–1014.
- Guo X, Niemi NM, Hutchins PD, et al. Ptc7p dephosphorylates select mitochondrial proteins to enhance metabolic function. Cell Rep. 2017;18:307–313.
- Funakoshi T, Matsuura A, Noda T, et al. Analyses of APG13 gene involved in autophagy in yeast, Saccharomyces cerevisiae. Gene. 1997;192:207–213.
- Kamada Y, Funakoshi T, Shintani T, et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513.
- Eisenberg T, Schroeder S, Buttner S, et al. A histone point mutation that switches on autophagy. Autophagy. 2014;10:1143–1145.
- Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398.
- Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530.
- Shintani T, Suzuki K, Kamada Y, et al. Apg2p functions in autophagosome formation on the perivacuolar structure. J Biol Chem. 2001;276:30452–30460.
- Wang CW, Kim J, Huang WP, et al. Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J Biol Chem. 2001;276:30442–30451.
- Barth H, Meiling-Wesse K, Epple UD, et al. Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 2001;508:23–28.
- Li YT, Yi C, Chen CC, et al. A semisynthetic Atg3 reveals that acetylation promotes Atg3 membrane binding and Atg8 lipidation. Nat Commun. 2017;8:14846.
- Reggiori F, Shintani T, Nair U, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1:101–109.
- Yen WL, Legakis JE, Nair U, et al. Atg27 is required for autophagy-dependent cycling of Atg9. Mol Biol Cell. 2007;18:581–593.
- Backues SK, Chen DC, Ruan JS, et al. Estimating the size and number of autophagic bodies by electron microscopy. Autophagy. 2014;10:155–164.
- Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471–483.
- Feng Y, Backues SK, Baba M, et al. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy. 2016;12:648–658.
- Talbot NJ, Ebbole DJ, Hamer JE. Identification and characterization of Mpg1, a gene involved in pathogenicity from the rice blast fungus Magnaporthe-grisea. Plant Cell. 1993;5:1575–1590.
- Qi Z, Wang Q, Dou X, et al. MoSwi6, an APSES family transcription factor, interacts with MoMps1 and is required for hyphal and conidial morphogenesis, appressorial function and pathogenicity of Magnaporthe oryzae. Mol Plant Pathol. 2012;13:677–689.
- Zhang H, Zhao Q, Liu K, et al. MgCRZ1, a transcription factor of Magnaporthe grisea, controls growth, development and is involved in full virulence. FEMS Microbiol Lett. 2009;293:160–169.
- Li X, Gao CY, Li LW, et al. MoEnd3 regulates appressorium formation and virulence through mediating endocytosis in rice blast fungus Magnaporthe oryzae. PLoS Pathog. 2017;13:e1006449.
- Howard RJ, Ferrari MA, Roach DH, et al. Penetration of hard substrates by a fungus employing enormous turgor pressures. Proc Natl Acad Sci USA. 1991;88:11281–11284.
- Qi Z, Liu M, Dong Y, et al. The syntaxin protein (MoSyn8) mediates intracellular trafficking to regulate conidiogenesis and pathogenicity of rice blast fungus. New Phytol. 2016;209:1655–1667.
- Guo M, Guo W, Chen Y, et al. The basic leucine zipper transcription factor Moatf1 mediates oxidative stress responses and is necessary for full virulence of the rice blast fungus Magnaporthe oryzae. Mol Plant Microbe Interact. 2010;23:1053–1068.
- Xu JR, Staiger CJ, Hamer JE. Inactivation of the mitogen-activated protein kinase Mps1 from the rice blast fungus prevents penetration of host cells but allows activation of plant defense responses. Proc Natl Acad Sci USA. 1998;95:12713–12718.
- Zhao X, Mehrabi R, Xu JR. Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot Cell. 2007;6:1701–1714.
- Kudla J, Bock R. Lighting the way to protein-protein interactions: recommendations on best practices for bimolecular fluorescence complementation analyses. Plant Cell. 2016;28:1002–1008.
- Robert T, Vanoli F, Chiolo I, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79.
- Li LW, Chen XL, Zhang SP, et al. MoCAP proteins regulated by MoArk1-mediated phosphorylation coordinate endocytosis and actin dynamics to govern development and virulence of Magnaporthe oryzae. PLoS Genet. 2017;13:e1006814.