
ABSTRACT
Imbalance in production and clearance of amyloid beta (Aβ) is the primary reason for its deposition in Alzheimer disease. Macroautophagy/autophagy is one of the important mechanisms for clearance of both intracellular and extracellular Aβ. Here, through screening, we identified alborixin, an ionophore, as a potent inducer of autophagy. We found that autophagy induced by alborixin substantially cleared Aβ in microglia and primary neuronal cells. Induction of autophagy was accompanied by up regulation of autophagy proteins BECN1/Beclin 1, ATG5, ATG7 and increased lysosomal activities. Autophagy induced by alborixin was associated with inhibition of the phosphoinositide 3-kinase (PI3K)-AKT pathway. A knock down of PTEN and consistent, constitutive activation of AKT inhibited alborixin-induced autophagy and consequent clearance of Aβ. Furthermore, clearance of Aβ by alborixin led to significant reduction of Aβ-mediated cytotoxicity in primary neurons and differentiated N2a cells. Thus, our findings put forward alborixin as a potential anti-Alzheimer therapeutic lead.
Abbreviations
Aβ: amyloid beta; ALB: alborixin; ATG: autophagy-related; BECN1: beclin 1; DAPI: 4, 6-diamidino-2-phenylindole; DCFH-DA: 2,7-dichlorodihydrofluorescein diacetate; fAβ: fibrillary form of amyloid beta; GFAP: glial fibrillary acidic protein; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MAP2: microtubule-associated protein 2; MTOR: mechanistic target of rapamycin kinase; PTEN: phosphatase and tensin homolog; ROS: reactive oxygen species; SQSTM1: sequestosome 1; TMRE: tetramethylrhodamine, ethyl ester
Introduction
Alzheimer disease is the most common neurodegenerative disease impacting millions of peoples worldwide. Several studies have linked deposition of amyloid-β (Aβ) to the development of Alzheimer disease. Recently some of the clinical trials targeting Aβ have failed to show improvement in the clinical outcome [Citation1,Citation2]. However, post-trial analysis has suggested that the therapies targeting Aβ may be useful if the intervention is done at earlier stages of AD or in those individuals who do not have symptoms of AD but are at high risk of developing the disease. The preventive clinical trials in risk populations, including dominantly inherited or sporadic AD will take some time.
Several studies have established that the production of Aβ stays normal, however, its clearance gets severely reduced in case of sporadic AD [Citation3–Citation5]. Among many mechanisms for clearance of Aβ, defective autophagy is an important contributing factor to AD, and it is related to Aβ pathology [Citation6,Citation7]. Autophagy is a natural process for clearing the aggregated proteins and worn-out organelles from the cell [Citation8–Citation11]. It also plays a similar role in Alzheimer disease, where, it is engaged in the clearance of intracellular and extracellular Aβ [Citation12,Citation13]. Few recent reports have also suggested that the intracellular Aβ can cause greater harm to the cell health [Citation14,Citation15]. Interestingly, autophagy has also been implicated to mediate the process of secretion of Aβ in the transgenic mouse model, where, deficient autophagy reduced the extracellular Aβ plaques [Citation16]. Further, the expression level of BECN1/Beclin 1 has been found to be low in Alzheimer brain, whereas, its genetic reduction leads to reduced autophagy and accumulation of Aβ and neurodegeneration in the brain of the transgenic mouse model, furthermore, this pathology was reduced by administration of BECN1 [Citation17]. Several more studies have indicated that pharmacological induction of autophagy in cells of central nervous system may help in reducing the load of Aβ [Citation12,Citation18–Citation20].
With this view, we planned this study to explore new autophagy inducing pharmacophores. During the screening program, we came across alborixin, a very potent inducer of autophagy, belonging to a polyether group of ionophores [Citation21]. Alborixin is reported to have strong antimicrobial activity against gram-positive bacteria [Citation22]. It has also been shown to possess anti-cancer activity against human colon cancer cells [Citation23]. Literature survey revealed that some of the ionophore antibiotics have already been known to induce autophagy [Citation24]. However, there was no report related to induction of autophagy by alborixin. Therefore, through this study, we decided to explore its autophagy inducing potential and usefulness for clearance of Aβ.
For all the autophagy-related experiments in this study, we used microglial cells, which are resident macrophages of the brain. Microglial cells have been shown to surround the Aβ plaques in the AD patients. These cells have also been reported to play an important role in the clearance of extracellular Aβ fibrils by autophagy in the mouse model [Citation25]. Additionally, we have also used primary neuronal cells to demonstrate the autophagy induction and clearance of Aβ by alborixin. Overall, in this study, we have demonstrated that alborixin appears to induce autophagy in different types of cells found in the brain and have established the mechanism of autophagy induction by alborixin, We further show that the autophagic activity of alborixin can be utilized to clear Aβ and to provide neuroprotection.
Results
Alborixin induces autophagy in microglial N9 cells
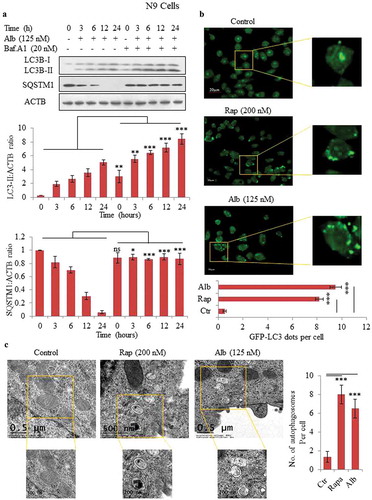
A screening program was run for autophagy inducers in mouse microglial N9 cells to find new autophagy inducers. Through this screening, we identified alborixin as an inducer of autophagy (Figure S1). Our results showed that alborixin induced autophagy in N9 cells in a concentration-dependent manner. Alborixin could induce autophagy even at a low concentration of about 30 nM in 12 h as shown by increased levels of LC3B-II and decreased expression of SQSTM1 (Figure S2). On the basis of this experiment, we chose 125 nM as a concentration for further studies. To assess the effect of alborixin on autophagy flux, cells were further treated with alborixin in presence or absence of bafilomycin A1, which blocks fusion between autophagosomes and lysosomes [Citation26]. On the basis of autophagic flux data in N9 cells, 12-h treatment was chosen to be the appropriate time for further studies (). Confocal microscopy showed increased LC3 puncta in N9 cells treated with alborixin (125 nM), which was comparable to that of rapamycin, 200 nM in 12 h (). Further, we used transmission electron microscopy (TEM) to visualize the formation of autophagosomes. The similar treatments of N9 cells with alborixin and rapamycin led to significant increase in the number of autophagosomes, which could be easily observed by TEM ().
Figure 1. Alborixin induced autophagy in N9 cells. (A) Western blot analysis for LC3B-II and SQSTM1 and calculation of autophagy flux after treatment with alborixin at indicated time periods in the absence and presence of bafilomycin A1 in mouse microglial N9 cells. Autophagic flux was calculated through ratio of LC3B-II:ACTB in the absence and presence of bafilomycin A1. (B) Confocal microscopy analysis of N9 cells for GFP-LC3 puncta. LC3 puncta were visualized and counted after 12-h treatment of N9 cells with alborixin (125 nM) and rapamycin (200 nM). The Images shown here are representative of 1 of 3 similar experiments and were taken at 40x. The scale bar in zoomed-out images: 20 μm. The average number of puncta per cells were calculated after analyzing 200 cells from 3 independent experiments (3n). (C) Transmission electron microscopy was used to analyze the formation of autophagosomes in N9 cells, which were treated similarly as in . The scale bars used in zoomed-out and zoomed-in images are shown in the figure. A total of 45 cells were analyzed for counting average number of autophagosomes per cell. Densitometry of western blots was done by using ImageJ software and statistical comparisons were made as shown in the figure by using Bonferroni test. p value<0.05 was considered to be significant with ***p < 0.001, **p < 0.01, *p < 0.05. The images presented in this figure are only representative in nature.
Alborixin induces autophagy by inhibiting the PI3K-AKT-MTOR pathway in N9 cells
To explore molecular events involved in autophagy induced by alborixin, we analyzed the level of autophagy-related proteins BECN1, ATG7, ATG5 and ATG12. Interestingly, alborixin upregulated the level of all these proteins in N9 cells in a time-dependent manner (). Level of another important autophagy regulator, BECN1, was significantly enhanced through 24 h of treatment ().
Figure 2. Inhibition of the PI3K-AKT-MTOR pathway via upregulation of PTEN in N9 cells. Alborixin at 125 nM, enhanced the protein level of PTEN in a time-dependent manner to inhibit several downstream proteins viz. p-AKT (S473, T308), p-MTOR (S2448) and RPTOR of AKT pathway in N9 cells. Alborixin also upregulated key autophagic proteins BECN1, ATG7, ATG5 and ATG12 in a time-dependent manner in N9 cells. Western blots of all proteins from 3 independent experiment were quantified by using Quantity One and ImageJ softwares and normalized by dividing with ACTB as shown in the bar graphs.
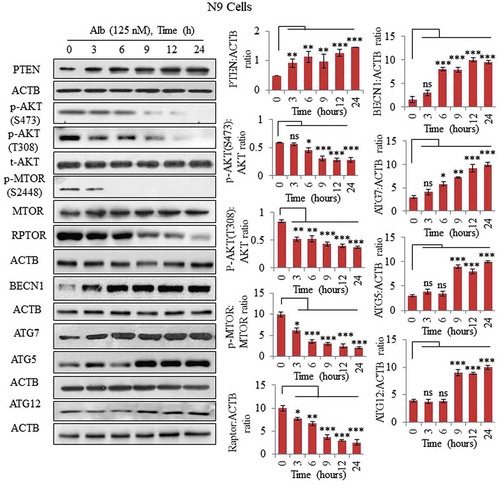
Mechanistically, alborixin was found to inhibit the PI3K-AKT-MTOR pathway, which plays an important role in the process of autophagy. We used 125 nM of alborixin through 24 h, to analyze its effect on major proteins of this pathway. Alborixin strongly reduced the level of several proteins of this pathway, including p-AKT (S473), p-AKT (T308), p-MTOR (S2448) and RPTOR in N9 cells (). Furthermore, inhibition of the PI3K -AKT pathway by alborixin was associated with an upregulated level of the native inhibitor PTEN () [Citation27].
Alborixin induces autophagy in primary neuronal cells by inhibiting PI3K-AKT pathway through upregulation of PTEN
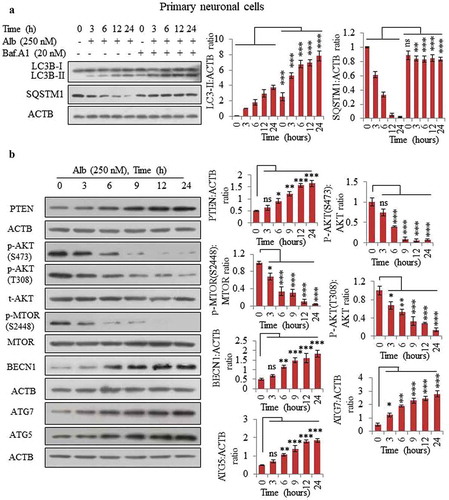
After confirming the autophagy mechanism in N9 cells, we further wanted to know if it can also induce autophagy in a similar way in primary neuronal cells. For this purpose, we cultured the primary neuronal cells obtained from 18-day-old embryo of C57BL/6 mice and treated them with 250 nM of alborixin to calculate the autophagic flux through 24 h. Similar to its effect on N9 cells, alborixin was able to induce the autophagy flux in primary neuronal cells (). Interestingly, just as in N9 cells, an LC3B-II band started to appear even after 3 h of treatment with alborixin in primary neuronal cells as well. Further, inhibition of the AKT pathway appears to play an important role in autophagy induced by alborixin in primary neuronal cells. Primary cells treated with alborixin displayed strong inhibition of p-AKT (S473), p-AKT (T308) and p-MTOR (S2448), which was further reflected in the activation of autophagy proteins such as BECN1, ATG5 and ATG7 (). More interestingly, PTEN activation also seemed to be the main force behind the inhibition of AKT pathway (). Primary neuronal cells used in this study (wherever required) were tested for expression of neuronal marker RBFOX3/NeuN and astroglial marker GFAP by western blotting before experiments (Figure S3A). We also stained the neuronal cultures with neuronal marker MAP2 and astroglial marker GFAP for analyzing the purity of neurons (Figure S3B). Measurable expression of GFAP was not found in any of the primary cultures (Figure S3A and B). Additionally, different concentrations of alborixin used in this study for distinct cell lines were also tested for its toxicity in these cell lines/types. However, alborixin did not induce toxicity in these cell lines (Figure S3C-E).
Figure 3. Alborixin induced autophagy in primary neuronal cells by upregulating PTEN. (A) Calculation of autophagy flux in primary neuronal cells. (B) Alborixin treatment upregulated the expression of PTEN and reduced the level of p-AKT (S473, T308), p-MTOR (S2448) along with enhanced level of BECN1, ATG5 and ATG7. Blots from 3 independent experiments were quantified by using quantity one and imageJ softwares and normalized by dividing by ACTB as shown in the bar graphs. The blots shown in this figure are representative images.
Abrogation of PTEN and upregulation of AKT inhibits autophagy induced by alborixin
To confirm the role of PTEN in autophagy induced by alborixin, we knocked-down PTEN in immortalized human primary microglial HMC3 cells by siPTEN, and treated the cells with alborixin 125 nM in presence and absence of bafilomycin A1 through 24 h and analyzed the level of the autophagy marker LC3B-II. There was no change in level of LC3B-II and SQSTM1 when PTEN was knocked down ( and Figure S4A), suggesting that PTEN is indispensable for autophagy induced by alborixin. However, HMC3 cells treated under similar conditions with alborixin elicited strong autophagic response (). Additionally, SQSTM1 showed decreasing trend with increasing treatment time ( and Figure S4B).
Figure 4. Knocking down of PTEN led to abrogation of alborixin-induced autophagy in HMC3 cells. (A) Western blot analysis of LC3B-II and SQSTM1 in PTEN knocked down HMC3 cells after treatment with alborixin (125 nM) through 24 h. (B) Western blot analysis of LC3B-II and SQSTM1 in wild-type HMC3 cells after treatment with alborixin under similar conditions. Autophagic flux was calculated by using ratio of LC3B-II:ACTB in the absence and presence of bafilomycin A1. (C) Effect of overexpressed WT PTEN and its inactive mutant on autophagy flux in HMC3 cells. Overexpression of PTEN was induced by transfecting the pCMV Flag WT-PTEN plasmid into HMC3 cells, whereas the PTEN mutant was generated by PCR based site-directed mutagenesis. (D) Overexpression of AKT led to abrogation of alborixin-induced autophagy flux in HMC3 cells. For overexpression of AKT, the HMC3 cells were transfected with the Myr-AKT plasmid before treatment with alborixin. Blots presented here are representative only and the quantitative graphs quantified by using ImageJ software shown are mean±SD of 3 independent experiments (3n). Statistical comparisons were made between different samples by using the Bonferroni test as shown in the figure. p value<0.05 was considered to be significant with ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
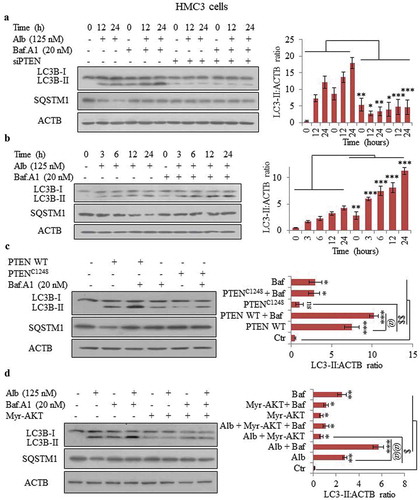
After confirming the role of PTEN in alborixin-induced autophagy, we wanted to know if overexpression of PTEN, independent of alborixin, can induce autophagy. Therefore, we overexpressed PTEN in HMC3 cells by PTEN wild-type plasmid. Interestingly, we observed a clear increase in the autophagic flux of LC3B-II with increased expression of PTEN (). Overexpression of PTEN induced LC3B-II, which was further increased when treatment with bafilomycin A1 was given (). PTEN overexpression also led to decreased expression of SQSTM1 ( and Figure S4D). On the contrary, when we generated a catalytically inactive mutant, PTENC124S by using site-directed mutagenesis, the transfected HMC3 cells did not show any autophagy flux (). Before going for autophagic flux analysis, cells were evaluated for enhanced expression of PTEN after plasmid transfection (Figure S4E).
Furthermore, autophagic flux induced by alborixin was hampered when AKT was overexpressed in HMC3 cells by the Myr-AKT-delta4-129 mutant plasmid (). Inhibition of autophagy flux was also demonstrated by reduced degradation of SQSTM1 in AKT-overexpressing cells (Figure S4F). The HMC3 cells transfected with Myr-AKT-delta4-129 mutant plasmid were checked for the expression of AKT before analyzing the effect of alborixin on autophagy in these cells (Figure S4G).
Alborixin enhances autophagy induced by both soluble and fibrillary forms of amyloid beta in N9 cells
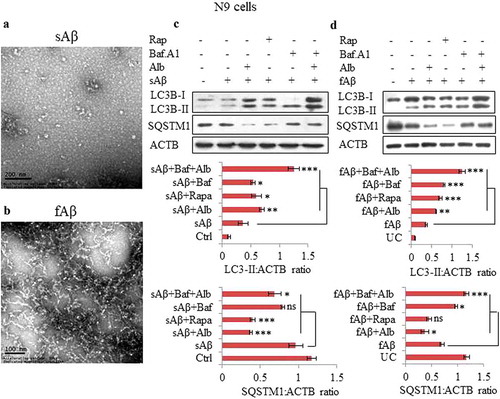
In Alzheimer disease, Aβ induces autophagy; however, the autophagic response of the cells to Aβ challenge may not be strong enough to clear Aβ. Therefore, we intended to explore if alborixin can enhance the autophagic response in cells. In addition, we also asked if soluble and fibrillary forms of Aβ elicit similar kinds of autophagic responses in cells. We prepared both the soluble and fibrillar forms of Aβ and checked them with the help of the transmission electron microscope for their consistency before using them for treatment of cells (). Interestingly, the cells co-treated for 12 h with Aβ (both soluble and fibrillary) and alborixin displayed enhanced autophagy as compared to the cells treated with Aβ alone, this was evident by increased autophagy flux ().
Figure 5. Alborixin enhanced the basal level of autophagy induced by soluble amyloid beta (sAβ) and fAβ. Representative images of transmission electron microscopy (TEM), (A) sAβ and (B) fibrillar amyloid beta (fAβ). Scale bars: (A) 200 nm, (B) 100 nm. (C and D) Level of LC3B-II was increased and that of SQSTM1 was reduced after co-treatment of N9 cells with either soluble or fibrillar forms of Aβ and alborixin 125 nM. Bafilomycin A1 (20 nM) reversed the effect of alborixin. Rapamycin (200 nM) in this experiment was used as a standard. Autophagic flux was calculated by using LC3B-II:ACTB in the absence and presence of bafilomycin A1. Ratio of values obtained through densitometry of western blots were quantified by using ImageJ software. Data presented here are mean±SD of 3 independent experiments and the blots shown in this figure are representative images. Statistical comparisons were made between samples treated with either sAβ or fAβ and all other samples by using Bonferroni test. p values ***p < 0.001, **p < 0.01, *p < 0.05.
Alborixin facilitates clearance of both soluble and fibrillary forms of amyloid beta in microglia N9 and primary neuronal cells
Based on our results, we hypothesized that alborixin may be a good candidate for promoting the clearance of Aβ for treatment of Alzheimer disease. To prove this hypothesis in vitro, we co-treated the N9 cells with fluorescently tagged Aβ1–42-HiLyte Fluor 555 and alborixin for 12 h. Confocal microscopy showed significantly reduced red fluorescence intensity of Aβ, indicating the enhanced clearance of Aβ by alborixin ( and B and Figure S5). Conversely, inhibiting autophagy using bafilomycin A1 strongly enhanced the red fluorescence in the cells, indicating increased accumulation of Aβ due to inhibition of autophagy induced by alborixin and Aβ ( and Figure S5). These results were further corroborated by ELISA analysis of intracellular and extracellular (in the supernatant) Aβ1–42 from the cells treated under similar conditions as in . The intracellular level of Aβ showed similar results () as observed in . The supernatant of alborixin-treated cells displayed a lower level of Aβ as compared to control cells. Interestingly, cells treated with alborixin in presence of bafilomycin A1, showed the increased amount of Aβ left in the media, indicating reduced phagocytosis by the cells ().
Figure 6. Alborixin facilitated the clearance of Aβ in N9 and primary neuronal cells. (A) Confocal images of N9 cells treated with Aβ and alborixin (125 nM) for 12 h. (B) Relative fluorescence intensity (RFI) of confocal images. Scale bars: 5 μm (Figure 6A), and μm (). (C and D) Confocal microscopy and calculation of RFI in primary neuronal cells treated with alborixin (250 nM) for 24 h, whereas, Aβ was added 12 h before termination of the experiment. Rapamycin (200 nM) in all the experiments was used as a standard. Red fluorescence of Aβ1-42-HiLyte Fluor 555 was considerably reduced in cells treated with alborixin. Bafilomycin A1 (20 nM) on the contrary reduced the clearance of Aβ. For each sample, average RFI of at least 500 cells was taken and then final average was calculated for the same sample from 3 independent experiments (3n) by using FV-10-ASW (Version 1.7) software. (E) ELISA of N9 cells treated under similar conditions to quantify Aβ present intracellularly and in the supernatant. (F) Flow cytometric analysis for clearance of Aβ in N9 cells treated under similar conditions as in Figure 6A. However, for flow cytometry, fluorochrome tagged Aβ1–42-HiLyte Fluor 488 was used. Flow cytometric histograms shown here are representative images from 1 of 3 similar experiments. Average of 3 experiments (3n) is depicted in the graph as Figure 6G. Samples were compared statistically by using Bonferroni test. p value ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
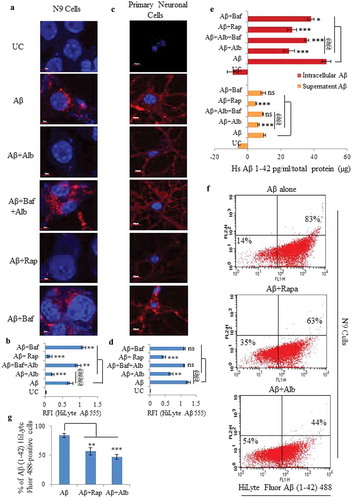
We also confirmed these findings in primary neuronal cells. We observed reduced red fluorescence intensity suggesting activation of autophagy by alborixin in primary neuronal cells ( and Figure S6). Further, fluorimetric quantification of the amount of Aβ1-42-HiLyte Fluor 555 left in the media of N9 cells used in the , displayed results similar to confocal analysis in , the data are shown in the supplementary section Table S1.
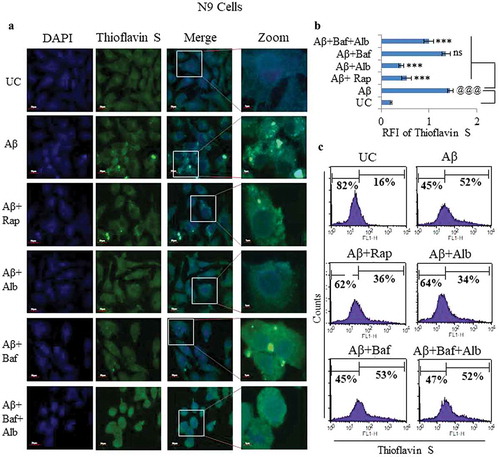
We also analyzed the effect of alborixin on N9 cells to phagocytose Aβ. We used fluorescently tagged Aβ1-42-HiLyte Fluor 488 to treat the N9 cells under similar conditions as in , and measured the intracellular Aβ fluorescence by flow cytometry. The reduced intracellular Aβ fluorescence after treatment with alborixin or rapamycin in comparison to the cells treated with Aβ alone (), strongly corroborated our results found in . We further attempted to analyze the effect of alborixin on the clearance of the fibrillary form of Aβ in N9 cells by using thioflavin S (a dye used to bind fibrillary form of Aβ). Cells treated with a fibrillary form of amyloid beta (fAβ; 10 µM) displayed significantly higher fluorescence intensity than the cells treated with either alborixin (125 nM) or rapamycin (200 nM) (). Thus, implying the involvement of autophagic effect of alborixin in the clearance of fAβ. Flowcytometric analysis for intracellular fAβ of the thioflavin S stained cells, treated under similar conditions also showed comparable results ().
Figure 7. Effect of alborixin on clearance of fibrillar form of Aβ in N9 cells. (A and B) Cells after treatment with fAβ (10 μM) and alborixin (125 nM) or rapamycin (200 nM), were stained with thioflavin S (0.1%) for 10 min before analysis on a confocal microscope at 60x and (C) flow cytometry. Cells treated with both rapamycin and alborixin displayed reduced green fluorescence of stained fAβ. Scale bar for confocal images: 5 µm for zoomed-in and 20 µm for zoomed-out images. A total of 500 cells were analyzed for calculating average fluorescence intensity (RFI) from 3 independent experiments (3n). Statistical comparisons in all the experiments were made as shown in the figures by using Bonferroni test. p values ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
PTEN knockdown and AKT overexpression hampers alborixin induced clearance of Aβ1-42
We further asked, if knocking down PTEN could alter the clearance of Aβ induced by alborixin. To address this, we knocked down PTEN in HMC3 cells and analyzed the clearance of Aβ through confocal microscopy, flow cytometry and ELISA. Our results showed that alborixin-mediated clearance of Aβ was hampered in cells knocked down with siPTEN ( and Figure S7). Constitutive activation of AKT using Myr-AKT-delta4-129 also blocked alborixin-mediated autophagic clearance of Aβ in HMC3 cells ( and Figure S8). These results supported our assumption that alborixin clears Aβ by inhibition of AKT pathway though upregulation of PTEN.
Figure 8. Knockdown of PTEN and overexpression of AKT reversed the clearance of Aβ caused by alborixin. (A) Confocal microscopy and flow cytometric analysis for clearance of amyloid beta (Aβ1-42-HiLyte Fluor 555) in siPTEN-transfected and alborixin-treated HMC3 cells. (B) Analysis for Aβ clearance in Myr-AKT delta4-129-transfected HMC3 cells after treatment with alborixin. (C) WT PTEN overexpression independent of alborixin caused clearance of Aβ, whereas, the catalytically inactive mutant PTENC124S did not have any effect on the clearance of Aβ in HMC3 cells. Confocal images of HMC3 cells were taken after treatment with Aβ and alborixin (125 nM) for 12 h. Average RFI of at least 500 cells was used to calculate final average for the same sample from 3 independent experiments (3n). Scale bar used in confocal images: 10 µm. The X axis of dot plots in Figure 8A-C represents Aβ1-42-HiLyte Fluor 555 fluorescence. Statistical comparisons for Figure 8A-C were made by using Bonferroni test. p values ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
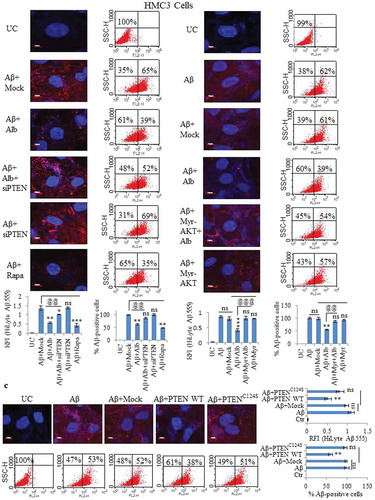
Furthermore, overexpression of PTEN enhanced clearance of Aβ after 12 h of treatment with Aβ1-42-HiLyte Fluor 555. Conversely, Aβ red fluorescence was restored when catalytically inactive mutant of PTENC124S was transfected into HMC3 cells ( and Figure S9).
Additionally, The ELISA results indicated a similar pattern, wherein inhibition of PTEN and overexpression of AKT reduced the clearance of Aβ, whereas, PTEN overexpression independent of alborixin enhanced the clearance of Aβ (Figure S7B, S8B and S9B).
Alborixin induces formation of lysosomes to facilitate the clearance of Aβ1-42
N9 cells treated with Aβ1-42-HiLyte Fluor 488 in the presence of alborixin were stained with LysoTracker DND Red to test, whether, alborixin leads the formation of lysosomes. Interestingly, large number of cells treated with alborixin showed the formation of lysosomes displaying enhanced red fluorescence intensity, thus, leading to reduced green Aβ fluorescence intensity, indicating its clearance from the cell ( and C and Figure S10). Rapamycin treated cells in a similar way displayed increased red fluoresce and decreased green Aβ fluorescence ( and C and Figure S10). Addition of bafilomycin A1 to alborixin-treated cells reversed the autophagic effect of alborixin and reduced red fluorescence and increased green Aβ fluorescence, further confirming the role of alborixin-induced autophagy in clearance of Aβ (, C and Figure S10).
Figure 9. Alborixin caused clearance of Aβ through formation of autolysosomes in N9 cells. (A) Double staining of cells with LysoTracker DND Red (80 nM) and Aβ1-42-HiLyte fluor 488 (2 μg/ml) showed reduced level of Aβ with increasing LysoTracker DND Red fluorescence after treatment with alborixin. However, red fluorescence was highly reduced, when, cells were co-treated with alborixin and bafilomycin A1 (20 nM). (B) Immunofluorescence staining of N9 cells against LAMP1 in presence of Aβ1-42-Hilyte Fluor 488 and DAPI. For both representative Figure 9A and B, RFI was calculated from at least 500 cells from each sample and then final average RFI of each sample was calculated from 3 independent experiments (3n), the data are given in Figure 9C & D. Scale bar for confocal images: 5 µm for zoomed-in images. Statistical comparisons in all the experiments were made as shown in the figures by using Bonferroni test. p values ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
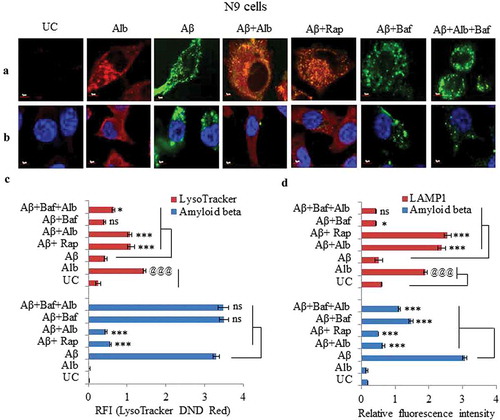
Furthermore, alborixin-treated cells displayed strongly enhanced red fluorescence of LAMP1 which was accompanied by decrease in Aβ1-42-HiLyte Fluor 488 green fluorescence (, D and Figure S11). Treatment with bafilomycin A1 reversed the effect of alborixin effect, which was evident by the increased Aβ1-42-hilyte fluor 488 fluorescence ( and D and Figure S11).
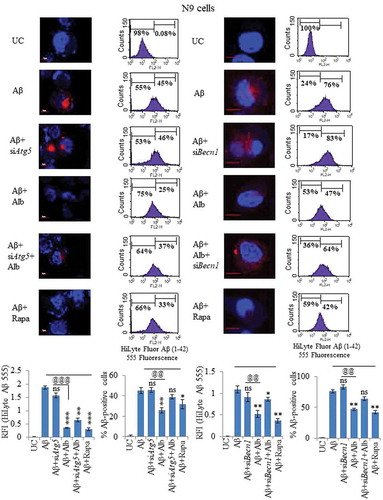
siRNA-mediated suppression of Atg5 and Becn1 significantly reversed the clearance of Aβ caused by alborixin in N9 cells
Our earlier data clearly indicated that alborixin-mediated induction of autophagy is accompanied by upregulation of several autophagy regulating proteins. A knockdown of Atg5 by siRNA displayed significantly increased fluorescence intensity of Aβ1-42-HiLyte Fluor 555, pointing towards partial reversal of alborixin effect on Aβ clearance ( and Figure S12A). Using flow cytometry, we observed that about 45% of the cells treated Aβ1-42-hilyte fluor 555 showed Aβ fluorescence, which declined to 25% after treatment with alborixin suggesting clearance of Aβ (). However, knockdown of Atg5 reversed the effect of alborixin ().
Because knockdown of Atg5 could partially reverse the effect of alborixin on clearance of Aβ, therefore, we knocked down Becn1, another important autophagic protein. Knockdown of Becn1 significantly reversed alborixin-induced clearance of Aβ ( and Figure S13A). These results were further confirmed by flow cytometric analysis of similarly treated cells, where 76% of the cells treated with Aβ1-42-hilyte fluor 555 showed fluorescence, whereas treatment with alborixin reduced this number to 47%, which was significantly restored to 64% when the siBecn1-treated cells were exposed to alborixin ().
Figure 10. Knocking down of Atg5 and Becn1 led to reduced clearance of Aβ in N9 cells. (A) Confocal microscopy images after various treatments of N9 cells with siRNA-mediated knocked down of Atg5. Histogram shows the average RFI of Aβ1-42-HiLyte Fluor 555 for each sample from 3 independent experiments. (B) Flow cytometric analysis of N9 cells treated under similar conditions as in Figure 10A. Histogram represents average of % Aβ1-42-hilyte fluor 555-positive cells from 3 separate experiments (3n). (C) Confocal microscopy and average RFI of Aβ1-42-hilyte fluor 555 in Becn1-knocked down N9 cells after treatment with alborixin. Average RFI of Aβ1-42-hilyte fluor 555 was measured from 3 independent experiments (3n) is shown in the histogram. (D) Flow cytometric analysis for clearance of Aβ in siBecn1-transfected N9 cells. Average number of Aβ1-42-hilyte fluor 555-positive cells from 3 independent experiments are shown in the histogram. Scale bar for confocal images: 20 µm. Bonferroni test was applied for statistical comparisons between samples as shown in the figures. p values ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
Alborixin provides neuroprotection to primary neuronal cells and differentiated N2a cells against Aβ
Aβ causes several toxic effects in neurons, however, mitochondria are one of the main targets of Aβ [Citation28]. Therefore, we assumed that the enhanced ability of cells to clear Aβ should provide protection to the nerve cells against Aβ toxicity. To check this assumption, we analyzed the effect of alborixin (250 nM) on mitochondrial membrane potential in N2a cells treated for 48 h with fibrillary fAβ1-42. The further treatment of cells with alborixin for 24 h clearly rescued the cells from mitochondrial toxicity of Aβ, which was indicated by the increased number of TMRE fluorescent cells treated with Aβ and alborixin as compared to cells treated with Aβ alone ().
Figure 11. Alborixin provided neuroprotection against Aβ1-42 in differentiated neuroblastoma and primary neuronal cells. (A) Flow cytometric analysis for mitochondrial membrane potential in differentiated N2a cells by using TMRE dye. Alborixin rescued the cells from Aβ induced mitochondrial stress as indicated by increased number of TMRE fluorescent cells in comparison to cells treated with Aβ alone. (B) Analysis for ROS generation by flow cytometry. N2a cells were treated similarly as for mitochondrial potential analysis. ROS generation was captured by using DCFH2-DA, which produced green fluorescence after interaction with ROS. As indicated by dot plot, alborixin considerably reduced the ROS generation induced by Aβ. The histograms in Figure 11A and B represent the average number of TMRE- or DCF-positive cells from 3 independent experiments for each sample. (C) Alborixin rescued primary neuronal and differentiated N2a cells from Aβ cytotoxicity. Cells treated with Aβ1-42 for 48 h were checked for their viability through an SRB assay. Alborixin (250 nM) was added to cells 24 h before termination of the experiment. Bonferroni test was applied for statistical comparisons between different samples as shown in the figures. p values ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
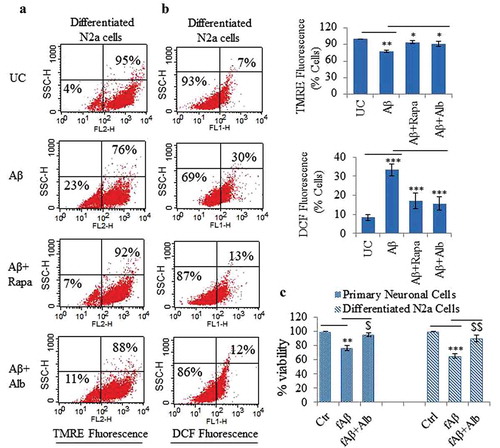
The second most prominent feature of Aβ toxicity is the production of ROS, which is also related to mitochondrial toxicity. Alborixin treatment reduced ROS generation in N2a cells treated with Aβ (). After having these results, we further attempted to analyze if alborixin can have some protective effect on the cell viability in cells treated with of Aβ. Therefore, we treated primary neuronal and differentiated N2a cells with fAβ1-42 for 48 h in the presence and absence of alborixin for 24 h. Interestingly, cells treated with alborixin showed significantly enhanced viability when compared with the cells treated with fAβ1-42 alone ().
Discussion
Defective autophagic clearance is one of the important mechanisms responsible for the deposition of Aβ in Alzheimer disease [Citation7]. Therefore, pharmacological induction of autophagy may enhance the clearance of Aβ and hence alleviate its pathological effects like generation of ROS, mitochondrial stress, activation of NLRP3 inflammasome and hyperphosphorylation of tau protein, etc. Autophagy is a general mechanism in most types of mammalian cells for clearance of aggregated or misfolded proteins [Citation29]. However, brain has specialized resident macrophages; the microglial cells, which actively participate in the autophagic clearance of Aβ along with other types of cells like neurons and astrocytes [Citation25,Citation30,Citation31]. A recent study has shown that old microglial cells, which are relatively non responsive to clearance of Aβ, can be activated to phagocytose and clear Aβ plaques [Citation32]. This study, therefore, was planned to assess the potential of alborixin to induce autophagy in mouse microglial N9 and primary neuronal cells. Alborixin is a polyether ionophore antibiotic isolated from various Streptomyces sp., however, for this study alborixin was isolated from Streptomyces scabrisporus. Ionophore antibiotics have several pharmacological activities; however, this is the first study where the potential and mechanism of induction of autophagy by this class of compounds have been explored. Our initial studies showed that alborixin potently induces autophagy at nano molar concentrations in N9 and primary neuronal cells. At the same time, alborixin did not show any significant toxicity at the concentrations and time points used for treatment of different cell types used in this study. Autophagy is a multistage process, where series of proteins play a role to degrade the unwanted peptides [Citation33]. Therefore, with a view to establish the molecular mechanism of autophagy induced by alborixin, we further analyzed its effect on several autophagic proteins. Interestingly, level of all the major proteins, including BECN1, ATG7, ATG5 and ATG12 observed a sharp increase in N9 cells in a time-dependent manner with a peak reaching at 12 h. Therefore, we planned all the subsequent experiments with 12h treatment with alborixin. However, the autophagy in neuronal cells may be induced differently from cancer cells [Citation34]. Therefore, we also confirmed the upregulated level of BECN1, ATG5 and ATG7 in primary neuronal cells, indicating that alborixin is a universal inducer of autophagy with a similar type of mechanism in different cell types.
Further, we focused our attention on MTOR, the major regulator of autophagy in the cell [Citation35]. Alborixin treatment of different cell types showed a clear decrease in the level of p-MTOR. Thus, indicating that MTOR may be a direct target of alborixin like that of rapamycin. However, looking upstream of MTOR showed a sharp decrease in the expression of both the active AKT forms p-AKT (S473) and p-AKT (T308). These observations led us to believe that the downregulation of this pathway may be targeted through PTEN, the natural inhibitor of PI3K-AKT pathway. PTEN is a tumor suppressor gene, which can induce autophagy through PI3K-AKT pathway [Citation27]; on the contrary, its suppression can inhibit autophagy [Citation36]. Our data also suggested major involvement of PTEN in alborixin induced autophagy in N9 and primary neuronal cells. Wherein, alborixin treatment led to overtly enhanced expression of PTEN in both these cell types to suppress the AKT pathway for induction of autophagy. This finding, further consolidated our observation that alborixin displayed similar mechanism of action of autophagy induction in different cell types.
To confirm the involvement of PTEN in alborixin-induced autophagy, we used HMC3 cells and attempted to knock down the expression of PTEN with siRNA. Interestingly, alborixin could induce autophagy in wild-type HMC3 cells as indicated by the sharp increase in autophagic flux. However, due to knockdown of PTEN, alborixin was unable to elicit the autophagic response in siPTEN HMC3 cells. After confirming the involvement of PTEN in alborixin-mediated autophagy, it was interesting to observe that overexpression of PTEN could induce autophagy in the absence of alborixin and overexpression of its inactive mutant did not induce any autophagic flux. Moving down further, the upregulation of AKT by the Myr-AKT plasmid in HMC3 cells was also able to inhibit the autophagy induced by alborixin. Thus, proving the involvement of PTEN mediated inhibition of AKT pathway in the induction of autophagy by alborixin.
Aβ accumulation can induce autophagy in different types of cells, including microglia and neurons [Citation25,Citation37,Citation38]. However, in AD autophagic response to Aβ is severely hampered due to different reasons [Citation14,Citation39,Citation40]. Therefore, pharmacological induction of autophagy may help in the clearance of Aβ. With this view, we checked, whether, alborixin can enhance the autophagic response to soluble and fibrillar forms of Aβ. Surprisingly, alborixin just like rapamycin, was able to increase the accumulation of LC3B-II and degradation of SQSTM1 in N9 cells, when compared to the treatment of cells with Aβ alone. These data, thus, suggested the utility of alborixin as a pharmacological agent for clearance of Aβ.
In order to investigate if the autophagy induction property of alborixin can be utilized to clear the Aβ, we incubated N9 and primary neuronal cells with fluorochrome-tagged Aβ in the presence or absence of alborixin before analysis on a confocal microscope. Surprisingly, alborixin treatment led to almost complete clearance of Aβ in both N9 and primary neuronal cells as indicated by the reduced intracellular fluorescence intensity of Aβ, although rapamycin treatment had a similar effect. Interestingly, the autophagy inhibitor bafilomycin A1 caused Aβ fluorescence to reappear in the cells treated with both Aβ alone and in combination with alborixin. Thus, suggesting the involvement of autophagy induction by alborixin in the clearance of Aβ. These results were also confirmed through flow cytometry. Alborixin just like rapamycin was as well effective in reducing the intracellular load of fibrillar Aβ in N9 cells, which were stained by thioflavin S for fibrillar Aβ.
Further analysis with LysoTracker DND Red dye showed that the intracellular concentration of fluorochrome tagged Aβ was inversely related with LysoTracker DND Red fluorescence after treatment with alborixin. However, inhibition of alborixin-induced autophagy by bafilomycin A1 led to reversal of this phenomenon. Similar results were obtained when LAMP1 staining was done instead of using LysoTracker DND Red as a marker for lysosomes. Additionally, knocking down of Atg5 and Becn1 also reduced the effect of alborixin on the clearance of Aβ. Therefore, these 2 experiments evidently implicated the process of autophagy for clearance of Aβ by alborixin. Additionally, autophagy can be induced by both BECN1- and ATG5-dependent and -independent pathways [Citation41]. Our data also indicated that knocking down of either Atg5 or Becn1 did not completely reverse the Aβ clearance, thus indicating that alborixin may utilize both BECN1 and ATG5 pathways for induction of autophagy and clearance of Aβ. We further evaluated if knocking down of PTEN or upregulation of AKT could reverse the effect of alborixin on Aβ clearance. Interestingly, the alborixin induced clearance of Aβ was significantly reversed in both the cases. These experiments further proved the cascade of events that involved the upregulation of PTEN by alborixin leading to induction of autophagy and clearance of Aβ.
Based on these observations, we hypothesized that if alborixin is able to clear the Aβ from the cells, it should also be able to reduce the deleterious pathological effects of Aβ on the cells. Aβ is known to induce mitochondrial stress and production of ROS in the cells [Citation42–Citation46], which ultimately leads to the death of neurons. Therefore, we attempted to analyze the effect of alborixin on these stress signals produced by Aβ in differentiated neuroblastoma cells. Our assumption was found to be true, when treatment with alborixin significantly reduced the Aβ induced loss of mitochondrial potential and production of ROS in differentiated N2a cells. It further led to appreciable reversal of cytotoxicity caused by Aβ, which was evident by increased viability of primary neuronal and differentiated N2a cells.
In conclusion, we have explored the potential of alborixin for induction of autophagy, established the molecular mechanism of autophagy induction by alborixin; and demonstrated that alborixin through autophagy can help in the Aβ clearance in different types of brain cells. Based on these data, we have planned to study the therapeutic effects of alborixin in the genetic model of Alzheimer disease. This work may help in catalysing the research work on alborixin and other ionophores for their possible use as autophagy inducers. We believe that the data presented in this study will strengthen the idea of targeting autophagy for treatment of Alzheimer disease.
Materials and methods
Reagents and antibodies
Alborixin was isolated from streptomyces scabrisporus [Citation23]. Reagents include the following: DMEM (Sigma-Aldrich, D1152), RPM1-1640 (Sigma Aldrich, R6504), MEM (Sigma-Aldrich, M0894) and F12 (Sigma-Aldrich, N3520), non-essential amino acids (Sigma, M7145), streptomycin (Sigma-Aldrich, S6501), penicillin (Sigma-Aldrich, P3032), rapamycin (Sigma-Aldrich, R8781), bafilomycin A1 (Sigma-Aldrich, B1793), sodium bicarbonate (Sigma-Aldrich, S5761), sodium pyruvate (Sigma-Aldrich, P2256), PBS (Sigma-Aldrich, D5652), 4, 6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, D9542), 2,7-dichlorodihydrofluorescein diacetate (DCFH2ַ-DA) (Sigma-Aldrich, D6883), dimethylsulfoxide (DMSO; Sigma-Aldrich, D2650), thioflavin S (Sigma-Aldrich, T1892), papain (Sigma-Aldrich, 76220), TMRE (Sigma-Aldrich, 87917), poly-L-lysine (Sigma-Aldrich, P4707), RIPA buffer (Sigma-Aldrich, R0278), BSA (Sigma-Aldrich, A2153), skimmed milk (Sigma-Aldrich, 70166), acrylamide (Sigma-Aldrich, A9099), N,N′-Methylenebis acrylamide (Sigma-Aldrich, M7279), sodium orthovanadate (Sigma-Aldrich, 450243), sodium fluoride (Sigma-Aldrich, 215309), glycine (Sigma-Aldrich, G8898), EDTA (Sigma-Aldrich, EDS), triton X-100 (Sigma-Aldrich, T8787), tween 20 (Sigma-Aldrich, P7949), trizma (Sigma-Aldrich, T6066), sodium dodecyl sulphate (Sigma-Aldrich, L3771), sodium chloride (Sigma-Aldrich, S7653), ammonium persulfate (Sigma-Aldrich, A3678), TEMED (Sigma-Aldrich, T7024), glycerol (Sigma-Aldrich, G5516), HEPES (Sigma-Aldrich, H3375), paraformaldehyde (Sigma-Aldrich, P6148), osmium tetroxide solution (Sigma-Aldrich, 75632), glutaraldehyde solution (Sigma-Aldrich, G5882), uranyl acetate (British Drug Houses, 6159-44-0), protease inhibitor cocktail (Sigma-Aldrich, P8340), Aβ1-42 peptide (Sigma-Aldrich, A9810), anti-LC3B-II (Sigma-Aldrich, L7543), anti-SQSTM1 (Sigma-Aldrich, P0067), anti-GFAP (Sigma-Aldrich, G3893), anti-ACTB (Sigma-Aldrich, A3854), sulforhodamine B dye (Sigma-Aldrich, S1402), hibernated media (Gibco, A12476), neurobasal media (Gibco, 21103049), OPTI-MEM media (Gibco, 11058-021), B-27™ Supplement (Gibco, 17504044), glutamax (Gibco, 35050-061), trypsin (Gibco, 25300-054), fetal bovine serum (Gibco, 10270), LysoTracker® Red DND-99 (Thermo Fisher Scientific, L7528), and lipofectamine 3000 (Thermo Fisher Scientific, L3000-001), 3-(4, 5, -dimethylthiazole-2-yl)-2, 5 diphenyltetrazolium bromide (MTT) (Alpha Aesar, L11939), Aβ1-42-HiLyte FluorTM 555 (Anaspec, 60480) and Aβ1-42-HiLyte FluorTM 488 (Anaspec, AS-60479). ELISA kit for human Aβ1-42 (Immuno-Biological Laboratories, 27712). Antibodies anti-ATG5 (Cell Signalling Technology, 2630), anti-BECN1 (Cell Signalling Technology, 3495), anti-AKT (Cell Signalling Technology, 9272), anti-p-AKT (S473) (Cell Signalling Technology, 4051), anti-p-AKT (T308) (Cell Signalling Technology, 9275), anti-RPTOR (Cell Signalling Technology, 2280), anti-PTEN (Cell Signalling Technology, 9188), anti-RBFOX3/NeuN (Cell Signalling Technology, 12943), HRP-linked rabbit IgG (Cell Signalling Technology,7074), mouse IgG (Cell Signalling Technology, 7076), goat anti-mouse IgG alexa fluor 555 secondary antibody (Invitrogen, A32727), siAtg5 (Cell Signalling Technology, 6345), siPTEN (Cell Signalling Technology, 6251). anti-ATG7 (Santa Cruz Biotechnology, Sc-33211), anti-ATG12 (Santa Cruz Biotechnology, Sc-68884), anti-LAMP1 (Santa Cruz Biotechnology, Sc-20011), anti-p-MTOR (S2448) (Santa Cruz Biotechnology, 293133), anti-MAP2 (Santa Cruz Biotechnology, Sc-20172), mouse scrambled siRNA (Santa Cruz Biotechnology, Sc-37007), and siBecn1 (Santa Cruz Biotechnology, Sc-29798). PVDF Membrane (Millipore, ISEQ00010), ECL-kit (Millipore, WBKLS0500), fugene-6 (Promega, E2312). QuikChange™ Site-Directed Mutagenesis Kit (Stratagene, 200518), protein assay dye (Bio-Rad, 161-0737), protein molecular weight markers (Bio-Rad, 161-0375), Bradford reagent (Bio-Rad, 500-0006).
Cell culture
The microglia (N9) cell line was provided by Dr. Anirban Basu (National Brain Research Centre, Gurgaon, India) and was cultured in RPMI-1640 media containing NaHCO3 (3.7 g/L). HMC3 cells were purchased from ATCC (CRL-3304) and were maintained in MEM medium. Mouse neuroblastoma N2a cells were obtained from the National Centre for Cell Sciences (Pune, India) and were cultured in DMEM high-glucose medium. Media of all types of cells except primary neurons was supplemented with 10% fetal bovine serum, penicillin G (70 mg/L) and streptomycin (100 mg/L). Cells were maintained in a humidified environment in an incubator (Thermocon Electron Corporation, Houston, TX, USA) with 95% humidity and 5% CO2. Alborixin, rapamycin, and bafilomycin A1 were dissolved in dimethylsulfoxide (DMSO). Control cells in all the experiments received the similar concentration of DMSO (˂0.2%) as treated samples. All types of cells used in this study were tested for mycoplasma contamination before use in experiments.
Primary neuron culture
Primary neurons were cultured from C57BL/6 mouse at embryonic day 18 in compliance with Institutional Animals Ethics Committee (IAEC), IIIM, Jammu, India. Cortices were dissected in ice-cold hibernate E media and digested in papain (1 mg/mL; Sigma-Aldrich, 76220) for 10 min at 37°C. Desired neuron concentrations were seeded in poly-L-lysine coated 12-mm round coverslips and culture dishes for immunofluorescence and immunoblotting respectively. Neurons were maintained in neurobasal media supplemented with 2% B27 and 0.5 mM GlutaMax for 15 days at 37°C temperature and 5% CO2.
Transient transfection with the GFP- LC3 plasmid
The GFP-LC3 plasmid was a kind of gift from Alec Kimmelman (Radiation Oncology, Harvard Medical School). For transfection of GFP- LC3, N9 cells were grown on coverslips at a density of 0.7 × 106 in OPTI-MEM media. Transient transfection was done by preparing transfection mix (1 μg of plasmid and 5 μl of Fugene-6) and kept for 20 min before adding this mixture to N9 cells. After 24 h of transfection, cells were treated with 125 nM of alborixin for 12 h. Treatment with rapamycin at 200 nM was taken as a standard. Cells were washed twice with PBS and fixed with 4% of paraformaldehyde. Samples were analyzed for GFP-LC3 puncta on confocal microscope at 40x.
Transmission electron microscopy
For the transmission electron microscopy (TEM) to analyze autophagosomes, N9 cells after treatments were fixed in glutaraldehyde (2.5%):paraformaldehyde (1%) mixture for 1 h at 4°C in 0.1 M phosphate buffer, pH 7.2, samples were post-fixed in 1% osmium tetroxide for 2 h at 4°C, dehydrated in graded acetone and embedded in Araldite (Fluka, 10951). Ultrathin sections were prepared by using a diamond knife, collected on copper grids (EMS, G 300 Cu). TEM was also used for analyzing the fibrinogenesis of Aβ1-42. Briefly, 10 µL of each protein sample was deposited onto carbon grids. The loaded carbon coated grids were stained using 2% uranyl acetate and allowed to dry before analysis. For both these studies, JEOL JEM 1400 Plus electron microscope was used at 80 kV.
Western blot
After various treatments, different cell types (N9, HMC3, differentiated N2a and primary neuronal cells) were lysed with RIPA lysis buffer (2 mM PMSF, 0.5 mM NAOV4, 50 mM NaF, protease inhibitor cocktail [Sigma-Aldrich, P8340]). Proteins (70 μg) were subjected to SDS-PAGE, followed by transfer to PVDF membrane at 100V for 2 h under cold conditions. Protein blots were blocked with 5% skimmed milk for 1 h at room temperature, followed by treatment with different primary antibodies overnight at 4°C and further incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Blots were incubated with Millipore Immobilon western chemiluminescent HRP substrate, before being analyzed for signal on either x-ray film or chemidoc system.
Preparation of fibrillar amyloid beta
Aβ1–42 peptide was dissolved in water containing 0.1% NH3 to prepare a 1 mM stock. The Aβ peptide solution was further diluted with the same volume of PBS and distributed into aliquots and saved at −80°C. Fibrillation of Aβ was induced by subjecting the aliquot vials to 37°C for 3 days [Citation47].
Analysis of autophagy flux
For all the experiments depicting autophagy, the autophagic flux was analyzed according to guidelines published by Klionsky et al [Citation48].
Preparation of HiLyte FluorTM 555 and 488-labeled β-Amyloid1-42 peptide
HiLyte FluorTM 555- and 488-labeled Aβ1–42 peptides was dissolved according to instructions of the manufacturer (Anaspec Inc.). Briefly, the peptide was dissolved in 1% NH3OH to prepare a 2 mg/ml stock. Aβ peptide solution was further diluted with the same volume of PBS to make 1mg/ml stock and stored at −20°C. Aβ peptide solution prepared by this method was treated as a soluble form.
Confocal microscopy to analyze the clearance of amyloid beta peptide
Clearance of Aβ1-42 peptide was analyzed in 3 different cell types viz. N9, primary neurons and HMC3 cells. For confocal microscopy cells were grown on coverslips for 24 h. All cell types were treated with alborixin at the indicated concentrations for 12 h. Fluorochrome-tagged Aβ1-42 HiLyte Fluor tm 555 (2 μg/ml) was added to cells for 12 h in the absence or presence of alborixin. Rapamycin at 200 nM was used as a standard and bafilomycin A1 (20 nM) was used as a negative control for this experiment. At the end of treatment, cells were washed 3 times with PBS and fixed with 4% paraformaldehyde for 15–20 min at room temperature. Cells were again washed with PBS and slides were prepared by using PBS and glycerol (1:9) as a mounting media. Images were taken at Olympus Fluoview, FV-1000 confocal laser scanning microscope at 60x.
Thioflavin S staining of fibrillary amyloid beta
N9 cells were seeded at a density of 0.7 × 106/well of 6 well plate for 24 h and treated with fAβ (10 µM) and alborixin (125 nM) for 12 h. Rapamycin at 200 nM was used as a standard in this experiment. At the end of treatment, cells were fixed in 4% paraformaldehyde for 15 min at room temperature. Cells were further incubated in 0.2% Triton X-100 for 5 min and washed with PBS before staining with thioflavine S (0.01%; Sigma-Aldrich, T1892) for 10 min. Cells were washed 3 times with 50% ethanol followed by washing twice with distilled water. Images were taken on confocal microscope at 60x.
Enzyme-linked immunosorbent assay (ELISA)
N9 cells were treated with a variety of stimuli as indicated and the concentrations of human Aβ1-42 in the culture media and lysate prepared from the lysis of cell pellet were evaluated with a sandwich ELISA kit in accordance with the manufacturer’s recommendations. Quantification of Aβ was done by standard curve generated by using recombinant human Aβ1-42.
Flow cytometric analysis of intracellular Aβ1-42
Cells after various treatments were washed and resuspended in PBS for analysis by flow cytometry for the intracellular fluorescence of Aβ1-42-HiLyte Fluor 488 or Aβ1-42-HiLyte Fluor 555 in FL-1 and FL-2 channels respectively at BD FACS Calibur flow cytometer.
Transfection of Myr-AKT delta4-129 and pCMV Flag WT-PTEN into HMC3 cells
Myr-AKT delta4-129 was procured from Addgene (10841; Richard Roth) [Citation49]. pCMV Flag WT-PTEN was obtained from Addgene (22231; Hong Wu) [Citation50]. Briefly, HMC3 cells were seeded in 6-well plates at a density of 0.3 × 106 per well. After 24 h, transient transfection was done by adding 2 μg of plasmid and 5 μl of Lipofectamine 3000 into each well of a plate for 24 h and transiently transfected cells were given various treatments before analysis by confocal microscopy, western blotting and ELISA.
Generation of catalytically inactive mutant of PTEN
The catalytically inactive point mutant C124S was generated by using PCR-based site-directed mutagenesis in which the catalytically active cysteine was replaced by serine using the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene, 200518) as per the manufacturer’s protocol. The primers with the following sequences were purchased from Sigma Aldrich: C124S, forward primer (GTCCCTTTCCAGCTTACTGTGAATTGCTGCAACATG) and reverse primer (CATGTTGCAGCAATTCACAGTAAAGCTGGAAAGGGAC).
Lysosome and amyloid beta double staining in microglial N9 cells
Interaction of Aβ and lysosomes was imaged through confocal microscopy by using fluorescent tagged Aβ1-42 and staining lysosomes with LysoTracker DND Red dye in N9 cells. For the assay, N9 cells were grown on coverslips in a 6-well plates and co-treated with alborixin (125 nM) and 2 μg/ml of fluorochrome tagged Aβ1-42-HiLyte Fluor 488 for 12 h. LysoTracker DND Red (80 nM) was added 90 min before the termination of the experiment. Cells were washed 2 times with incomplete media and live cell images were taken on a confocal microscope at 60x.
Immunofluorescence staining for lysosomal localization of amyloid beta in microglia N9 cells
Interaction of Aβ and lysosomes was imaged through confocal microscopy by using fluorescent tagged Aβ1-42 and lysosomes with LAMP1 antibody in N9 cells. For this assay, N9 cells were grown on coverslip in a six-well plate and co-treated with alborixin (125 nM) and 2 μg/ml of fluorochrome tagged Aβ1-42-HiLyte Fluor 488 for 12 h. Media was aspirated and cells were washed 3 times with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were washed 3 times with PBS and blocked by blocking buffer (containing 5% normal goat serum [Cell Signalling Technology, 5425] and 0.3% Triton X-100 in PBS) for 60 min followed by the primary antibody (LAMP1) overnight at 4°C and further incubated with secondary antibody for 1 h at room temperature. Then cells were washed 3 times with PBS for 5 min each. Images were taken on a confocal microscope at 60X.
Transfection of cells with Atg5, Becn1 and PTEN siRNA
N9 cells were grown at a density of 0.7 × 106 on coverslips placed in the 6-well plates. Cell media was replaced with OPTI-MEM media and transfection with siAtg5 and siBecn1 was done by using Lipofectamine 3000 (Thermo Fisher Scientific, L3000-001) in the ratio (1:200). For PTEN transfection 0.3 × 106 HMC3 cells were used, the rest of the transfection protocol was same as that for N9 cells. After 24 h of transfection, cells were co-treated with Aβ1-42- HiLyte Fluor 555 (2 µg/ml) and 125 nM of alborixin for 12 h. Transfected cells after the treatments were analyzed through confocal microscopy, whereas the cells left in each well of the plate were analyzed by flow cytometry as described above. Additionally, PTEN-transfected HMC3 cells were treated with recombinant human Aβ 1-42 in the presence and absence of alborixin for analysis of the intracellular and extracellular Aβ 1-42 through ELISA.
Mitochondrial membrane potential (MMP)
Mitochondrial membrane potential was measured by using TMRE dye in N2a cells, which were differentiated with retinoic acid (10 µM; Sigma-Aldrich, R2625) with 1% FCS for 4 days. Cells were pre-treated with Aβ for 48 h followed by treatment with alborixin, 250 nM and rapamycin, 200 nM for 24 h. TMRE, 300 nM was added 30 min prior to termination of the experiment. Cells were washed with PBS and analyzed by flow cytometry for change in mitochondrial membrane potential (∆Ψm) through TMRE red fluorescence.
Reactive oxygen species (ROS)
ROS production was monitored by flow cytometry using 2ʹ, 7ʹ- dichlorodihydrofluorescein diacetate (DCFH2-DA). Briefly, differentiated N2a cells were pre-treated with Aβ for 48 h followed by treatment with alborixin, 250 nM and rapamycin, 200 nM for 24 h. DCFH2-DA (10 µM) was added 30 min before termination of the experiment. Cells were washed with PBS and were analyzed by flow cytometry.
Neuroprotection assay
Neuroprotection assay was done on primary neurons and N2a cells. In brief, primary neurons were seeded into poly-L-lysine coated 96 well plates whereas, N2a cells were seeded into a normal cell culture 96-well plate. The differentiation of N2a cells was done by treating with retinoic acid (10 µM) and 1% FBS in DMEM media for 4 days. Primary neurons and differentiated N2a cells were incubated with Aβ for 48 h, whereas alborixin was added to the cells for a period of 24 h. At the end of treatment, cells were fixed with ice-cold trichloroacetic acid (TCA) and kept at 40°C for 1 h. After 1 h, the samples were washed thrice with water and allowed to air dry following which 100 µl of 0.4% SRB dye was added at room temperature for 30 min. To remove the unbound SRB plates were again washed thrice with 1% v:v acetic acid and then allowed to dry at room temperature. After drying, the bound dye was solubilized by adding 100 µl of 10 mM TRIS buffer to each well. Finally to solubilize the protein the plate was kept on a shaker for 5 min and OD was taken at 540 nm. Cell viability was calculated by comparing optical density OD of untreated control cells with that of treated groups. The viability of control cells was taken as 100%.
Statistical analysis
Data presented here are means of three similar experiments and the error bars represent the standard deviation (SD) between the experiments. Statistical analysis was done by using Instat-3 software and Bonferroni method was applied to calculate statistical significance, p value<0.05 was considered to be significant with ***p < 0.001, **p < 0.01, *p < 0.05 or @@@p < 0.001, @@p < 0.01, @p < 0.05.
Declaration of interest statement
No potential conflict of interest was reported by the authors.
Supplemental Material
Download MS Word (2.4 MB)Acknowledgments
The financial assistance for this study was provided by Council of Scientific and Industrial Research (CSIR), India, through the project BSC-0205. Authors are also thankful to CSIR for providing SRF fellowship to Abubakar Wani. Thanks are also due to University Grants Commission (UGC) and Indian Council of Medical Research (ICMR), India, for providing research fellowships to Mehak Gupta and Aabid Manzoor Shah respectively. We also acknowledge the help provided by Mr. Ashok Kumar from CSIR-IIIM for providing help in performing confocal microscopy. We are thankful to Dr. Suresh Kumar of Autophagy Inflammation and Metabolism Centre of Biomedical Research Excellence, University of New Mexico Health Sciences Centre, USA for critically analyzing the manuscript. This manuscript has been assigned the institutional MS no. IIIM/2248/2018.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014 Jan 23;370(4):311–321. PubMed PMID: 24450890.
- Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014 Jan 23;370(4):322–333. PubMed PMID: 24450891; PubMed Central PMCID: PMC4159618.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353–356. PubMed PMID: 12130773.
- Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010 Dec 24;330(6012):1774. PubMed PMID: 21148344; PubMed Central PMCID: PMC3073454.
- Bohm C, Chen F, Sevalle J, et al. Current and future implications of basic and translational research on amyloid-beta peptide production and removal pathways. Mol Cell Neurosci. 2015 May;66(Pt A):3–11. PubMed PMID: 25748120; PubMed Central PMCID: PMC4503820.
- Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005 Oct 10;171(1):87–98. PubMed PMID: 16203860; PubMed Central PMCID: PMC2171227.
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007 Dec 1;120(Pt 23):4081–4091. PubMed PMID: 18032783.
- Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992 Feb 15;48(2):158–172. PubMed PMID: 1740188.
- Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem. 2000;69:303–342. PubMed PMID: 10966461.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011 Nov 11;147(4):728–741. PubMed PMID: 22078875.
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012 Feb;8(2):108–117. PubMed PMID: 22187000; PubMed Central PMCID: PMC22187000.
- Caccamo A, Majumder S, Richardson A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010 Apr 23;285(17):13107–13120. PubMed PMID: 20178983; PubMed Central PMCID: PMC2857107.
- Majumder S, Richardson A, Strong R, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. PubMed PMID: 21980451; PubMed Central PMCID: PMC3182203.
- McGowan E, Pickford F, Kim J, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005 Jul 21;47(2):191–199. PubMed PMID: 16039562; PubMed Central PMCID: PMC1373682.
- Lopez EM, Bell KF, Ribeiro-da-Silva A, et al. Early changes in neurons of the hippocampus and neocortex in transgenic rats expressing intracellular human a-beta. J Alzheimers dis. 2004 Aug;6(4):421–431. discussion 443-9. PubMed PMID: 15345813.
- Nilsson P, Loganathan K, Sekiguchi M, et al. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013 Oct 17;5(1):61–69. PubMed PMID: 24095740.
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun;118(6):2190–2199. PubMed PMID: 18497889; PubMed Central PMCID: PMC2391284.
- Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011 Jul 20;12(8):437–452. PubMed PMID: 21772323.
- Ravikumar B, Berger Z, Vacher C, et al. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006 Apr 1;15(7):1209–1216. PubMed PMID: 16497721.
- Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PloS one. 2010 Apr 1;5(4):e9979. PubMed PMID: 20376313; PubMed Central PMCID: PMC2848616.
- Gachon P, Farges C, Kergomard A. Alborixin, a new antibiotic ionophore: isolation, structure, physical and chemical properties. J Antibiot (Tokyo). 1976 Jun;29(6):603–610. PubMed PMID: 950314.
- Delhomme C, Kergomard A, Kergomard G, et al. Alborixin, a new antibiotic ionophore: taxonomy, isolation and biological properties. J Antibiot (Tokyo). 1976 Jul;29(7):692–695. PubMed PMID: 956055.
- Shah AM, Wani A, Qazi PH, et al. Isolation and characterization of alborixin from streptomyces scabrisporus: a potent cytotoxic agent against human colon (HCT-116) cancer cells. Chem Biol Interact. 2016 Aug 25;256:198–208. PubMed PMID: 27378626.
- Park MH, Lee SJ, Byun HR, et al. Clioquinol induces autophagy in cultured astrocytes and neurons by acting as a zinc ionophore. Neurobiol Dis. 2011 Jun;42(3):242–251. b09. PubMed PMID: 21220021.
- Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014 Oct 1;10(10):1761–1775. PubMed PMID: 25126727; PubMed Central PMCID: PMC4198361.
- Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998 Feb;23(1):33–42. PubMed PMID: 9639028.
- Arico S, Petiot A, Bauvy C, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001 Sep 21;276(38):35243–35246. PubMed PMID: 11477064.
- Chen JX, Yan SD. Amyloid-beta-induced mitochondrial dysfunction. J Alzheimers dis. 2007 Sep;12(2):177–184. PubMed PMID: 17917162; PubMed Central PMCID: PMC3687350.
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015 Mar 13;47:e147. PubMed PMID: 25766616; PubMed Central PMCID: PMC4351408.
- Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008 Jul 2;28(27):6926–6937. PubMed PMID: 18596167; PubMed Central PMCID: PMC2676733.
- Xiao Q, Yan P, Ma X, et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014 Jul 16;34(29):9607–9620. PubMed PMID: 25031402; PubMed Central PMCID: PMC4099542.
- Daria A, Colombo A, Llovera G, et al. Young microglia restore amyloid plaque clearance of aged microglia. Embo J. 2017 Mar 1;36(5):583–603. PubMed PMID: 28007893; PubMed Central PMCID: PMC5331757.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007 Nov 15;21(22):2861–2873. PubMed PMID: 18006683.
- Maday S, Holzbaur EL. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016 Jun 1;36(22):5933–5945. PubMed PMID: 27251616; PubMed Central PMCID: PMC4887563.
- Wong PM, Feng Y, Wang J, et al. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015 Aug 27;6:8048. PubMed PMID: 26310906; PubMed Central PMCID: PMC4552084.
- Ueno T, Sato W, Horie Y, et al. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy. 2008 Jul;4(5):692–700. PubMed PMID: 18424911.
- Hung SY, Huang WP, Liou HC, et al. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009 5;May(4):502–510. PubMed PMID: 19270530.
- Li W, Tang Y, Fan Z, et al. Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol Neurodegener. 2013 Aug 10;8:27. PubMed PMID: 23938027; PubMed Central PMCID: PMC3751621.
- Lucin KM, O’Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013 Sep 4;79(5):873–886. PubMed PMID: 24012002; PubMed Central PMCID: PMC3779465.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013 Aug;19(8):983–997. PubMed PMID: 23921753.
- Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009 Oct 1;461(7264):654–658. PubMed PMID: 19794493.
- Butterfield DA, Drake J, Pocernich C, et al. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001 Dec;7(12):548–554. PubMed PMID: 11733217.
- Smith MA, Perry G, Richey PL, et al. Oxidative damage in Alzheimer’s. Nature. 1996 Jul 11;382(6587):120–121. PubMed PMID: 8700201.
- Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001 Aug;60(8):759–767. PubMed PMID: 11487050.
- Mungarro-Menchaca X, Ferrera P, Moran J, et al. beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res. 2002 Apr 1;68(1):89–96. PubMed PMID: 11933053.
- Hauptmann S, Keil U, Scherping I, et al. Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp Gerontol. 2006 Jul;41(7):668–673. PubMed PMID: 16677790.
- Piechotta A, Parthier C, Kleinschmidt M, et al. Structural and functional analyses of pyroglutamate-amyloid-beta-specific antibodies as a basis for Alzheimer immunotherapy. J Biol Chem. 2017 Jul 28;292(30):12713–12724. PubMed PMID: 28623233; PubMed Central PMCID: PMC5535044.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMC4835977.
- Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996 Sep 6;271(36):21920–21926. PubMed PMID: 8702995.
- Mosessian S, Avliyakulov NK, Mulholland DJ, et al. Analysis of PTEN complex assembly and identification of heterogeneous nuclear ribonucleoprotein C as a component of the PTEN-associated complex. J Biol Chem. 2009 Oct 30;284(44):30159–30166. PubMed PMID: 19740742; PubMed Central PMCID: PMC2781571.