
ABSTRACT
Multiple sources contribute membrane and protein machineries to construct functional macroautophagic/autophagic structures. However, the underlying molecular mechanisms remain elusive. Here, we show that RAB2 connects the Golgi network to autophagy pathway by delivering membrane and by sequentially engaging distinct autophagy machineries. In unstressed cells, RAB2 resides primarily in the Golgi apparatus, as evidenced by its interaction and colocalization with GOLGA2/GM130. Importantly, autophagy stimuli dissociate RAB2 from GOLGA2 to interact with ULK1 complex, which facilitates the recruitment of ULK1 complex to form phagophores. Intriguingly, RAB2 appears to modulate ULK1 kinase activity to propagate signals for autophagosome formation. Subsequently, RAB2 switches to interact with autophagosomal RUBCNL/PACER and STX17 to further specify the recruitment of HOPS complex for autolysosome formation. Together, our study reveals a multivalent pathway in bulk autophagy regulation, and provides mechanistic insights into how the Golgi apparatus contributes to the formation of different autophagic structures.
Abbreviations
ACTB: actin beta; ATG9: autophagy related 9A; ATG14: autophagy related 14; ATG16L1: autophagy related 16 like 1; BCAP31: B cell receptor associated protein 31; BECN1: beclin 1; Ctrl: control; CQ: chloroquine; CTSD: cathepsin D; DMSO: dimethyl sulfoxide; EBSS: Earle’s balanced salt solution; EEA1: early endosome antigen 1; GDI: guanine nucleotide dissociation inhibitor; GFP: green fluorescent protein; GOLGA2: golgin A2; HOPS: homotypic fusion and protein sorting complex; IP: immunoprecipitation; KD: knockdown; KO: knockout; LAMP1: lysosomal associated membrane protein 1; LC3: microtubule-associated protein 1 light chain 3; OE: overexpression; PtdIns3K: class III phosphatidylinositol 3-kinase; SQSTM1/p62: sequestosome 1; RAB2: RAB2A, member RAS oncogene family; RAB7: RAB7A, member RAS oncogene family; RAB11: RAB11A, member RAS oncogene family; RUBCNL/PACER: rubicon like autophagy enhancer; STX17: syntaxin 17; TBC1D14: TBC1 domain family member 14; TFRC: transferrin receptor; TGOLN2: trans-golgi network protein 2; TUBB: tubulin beta class I; ULK1: unc-51 like autophagy activating kinase 1; VPS41: VPS41, HOPS complex subunit; WB: western blot; WT: wild type; YPT1: GTP-binding protein YPT1.
Introduction
Macroautophagy (hereafter referred to as autophagy) is a lysosomal degradative pathway, which is essential to development and homeostasis [Citation1–Citation3]. The deregulation of autophagy is tightly associated with a variety of human diseases [Citation4,Citation5]. Morphologically, autophagy is initiated from phagophores in mammalian cells. After nucleation, the phagophore membrane expands and ultimately seals to generate an autophagosome, which then fuses with a lysosome or vacuole leading to the degradation of autophagy cargoes [Citation6–Citation9]. How autophagosome forms and matures into autolysosome remain to be the fundamental unresolved questions in autophagy field.
Independent studies have shown that the biogenesis of autophagosomes needs membranes from multiple sources, including ER [Citation10,Citation11], Golgi network [Citation12–Citation15], mitochondria [Citation16], the plasma membrane [Citation8], the endosomes [Citation17–Citation19] and ER-Golgi intermediate [Citation20–Citation23]. It is unresolved that how the membranes from different resources are directionally delivered for the formation and expansion of a phagophore, which eventually seals to form an autophagosome. Due to its widespread distribution and characteristic transmembrane structure, ATG9A (hereafter referred to as ATG9) is considered to be the good candidate to deliver membranes from different sources [Citation7,Citation8,Citation17,Citation24–Citation37], but it remains unclear how Golgi-derived ATG9-positive (ATG9+) vesicles are transported and integrated into early autophagic structures in mammalian cells.
RAB small GTPases are critical molecular switches in trafficking pathways [Citation38,Citation39]. Several RAB GTPases in autophagy regulation have been identified [Citation40,Citation41], but how these RAB GTPases relay signals sequentially to fulfill the entire autophagy process remains poorly understood. It has been proposed that endocytosis or phagocytosis involves a RAB-conversion mechanism in which signals are seamlessly transduced to promote the maturation of endosomes or phagosomes [Citation42,Citation43], but it is not known whether an analogous regulation also exists in the autophagy pathway.
In previous study, we identified RUBCNL as a vertebrate-specific autophagy regulator [Citation44], and we showed that RUBCNL antagonizes RUBCN/Rubicon to activate the class III phosphatidylinositol 3-kinase (PtdIns3K) during late steps of autophagy. In addition, RUBCNL interacts with STX17 on autophagosomes to promote autolysosome formation. Here, we reported that RAB2A (hereafter referred to as RAB2), another RUBCNL-interactor, unexpectedly regulated both autophagy initiation and termination in mammalian cells. We observed that the Golgi apparatus contributed to autophagy initiation by donating RAB2, which participated in the formation of phagophores by further recruiting and activating ULK1. Next, RAB2 switched to interact with RUBCNL and STX17 to become an autophagosomal GTPase, which further specified the recruitment of HOPS (homotypic fusion and protein sorting) complex to autophagosome to facilitate the fusion with lysosomes. Our study provides mechanistic insights into the regulatory mechanisms underlying the roles of the Golgi apparatus in autophagosome biogenesis and maturation.
Results
Golgi-resident RAB2 relocates to autophagic membrane structures through microtubule-based vesicular transport
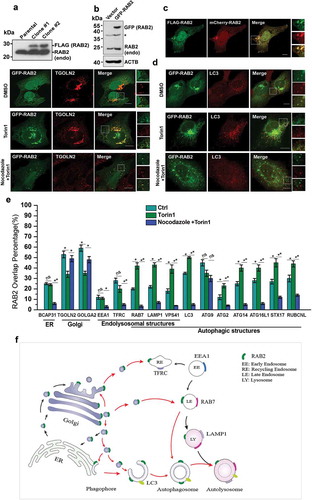
In our previous study, Mass spectrometry (MS) analysis of the proteins showed that co-immunoprecipitated (co-IP) with RUBCNL resulted in the identification of RAB2 as another potential RUBCNL-binding partner [Citation44]. Therefore, RAB2 might be an autophagy regulator. To test this notion, we first investigated its subcellular localization. Because the reliable antibodies for imaging of endogenous RAB2 were not available, we established stable cell lines expressing FLAG-RAB2 at a level that was close to its endogenous counterpart (Figure 1(a)). Next, we employed this stable cell line to optimize the imaging conditions, which enabled transiently expressed GFP-RAB2 or mCherry-RAB2 to behave similarly to FLAG-RAB2 (). Subsequently, we applied these conditions for following confocal microscopy analysis. In unstressed cells, RAB2 predominantly resided on the Golgi apparatus, as exhibited by extensive colocalization with GOLGA2 or TGOLN2/TGN46 ( and S1). In addition, the localization of RAB2 on ER, late endosomal and autophagic membrane structures was apparent, albeit at relatively lower levels. In contrast, the localization of RAB2 on early endosomes and mitochondria was limited. Interestingly, autophagy stimulation by Torin1 treatment significantly enhanced the colocalization of RAB2 with autophagic markers, except for ATG9 ( and S1). Meanwhile, the overlay percentage of RAB2 with Golgi markers was reduced. These results showed that RAB2 is a Golgi-resident small GTPase, and implied that RAB2, upon autophagy induction, may relocate from the Golgi network to autophagic structures. To test this hypothesis, we used nocodazole, a microtubule polymerization inhibitor in cell culture and performed confocal microscopy analysis, knowing that intracellular vesicular trafficking depends on microtubules. Nocodazole treatment largely abolished the colocalization of RAB2 with autophagic markers, except for ATG9, while the overlay between RAB2 and Golgi markers remained largely unaltered ( and S1). These results demonstrated that RAB2 largely co-existed with a portion of ATG9 on Golgi apparatus-related membrane structures, and that Golgi-resided RAB2 relocated to autophagic structures through microtubule-based vesicle trafficking. These observations allowed us to outline the RAB2 trafficking routes in mammalian cells, as shown in .
Figure 1. Golgi-derived RAB2+ vesicles fuse into autophagic membrane structures by vesicular trafficking. (a) Western-blot (WB) analysis of FLAG-RAB2 stable cell lines using anti-RAB2 antibody. (b) WB analysis of GFP-RAB2 and endogenous RAB2 level using anti-RAB2 antibody. (v) The subcellular localization of mCherry-RAB2 was similar to the stably expressed FLAG-RAB2. (d) Confocal microscopy analysis of GFP-RAB2 with TGOLN2 and LC3 as indicated under untreated, Torin1- or Torin1 plus nocodazole-treated conditions. Scale bars: 10 µm. (e) Quantification of colocalization presented in and S1. Data were shown as mean ± SD, *p < 0.05, **p < 0.01; ‘ns’ indicates no statistical significance. (f) Schematic representation of the trafficking routes of RAB2. Red arrow heads indicated the routes uncovered in this work.
RAB2 regulates autophagy initiation
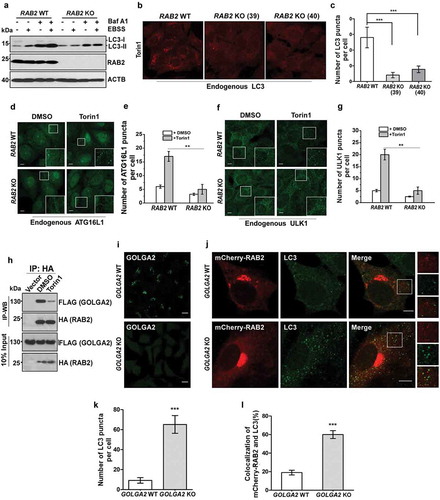
The colocalization pattern of RAB2 suggested a potential function of RAB2 in autophagy. To test this idea, we generated RAB2-knockout (KO) U2OS cell lines (Figure 2(a)), and we found that the levels of LC3-II were significantly reduced in the absence of RAB2 in both autophagy-stimulated and -unstimulated cells, indicating that RAB2 KO resulted in a defect in LC3 lipidation. Consistently, RAB2 depletion significantly diminished cytosolic LC3 puncta (), and this defect could be rescued by the re-expression of wild-type (WT) RAB2 (Fig. S2A and S2B). LC3 lipidation is mainly catalyzed by ATG12–ATG5-ATG16L1 on the elongating phagophore membrane [Citation45]. Indeed, membrane recruitment of endogenous ATG16L1 was abolished in RAB2 KO cells (). In addition, RAB2 knockdown (KD) in mouse livers led to SQSTM1/p62 accumulation and the defects in the biogenesis of autophagic membrane structures in vivo (Fig. S2C, S2D and S2E). More importantly, RAB2 KO eliminated the formation of the earliest autophagic structures labelled by endogenous ULK1 or GFP-ATG13 (, S2F and S2G). Collectively, these data indicated that Golgi-derived RAB2+ vesicles participated in autophagy initiation. The observation that autophagy stimuli decreased the colocalization of GOLGA2/GM130 and RAB2 ( and S1) led us to hypothesize that there might be functional correlation between GOLGA2 and RAB2 in autophagy initiation. Indeed, RAB2 was able to co-IP with GOLGA2, which was consistent with previous study [Citation46], and their interaction was decreased in autophagy-stimulated cells indicating that autophagy stimuli dissociated RAB2 from GOLGA2 (). Consistently, GOLGA2 depletion by either shRNA knockdown (KD) (Fig. S2H, S2I and S2J) or Crispr-Cas9-mediated knockout () was able to elevate LC3 lipidation levels and to increase the colocalization of RAB2 and LC3. Together, these data suggested that autophagy stimuli liberate a population of RAB2+ vesicles from the Golgi network for autophagy initiation.
Figure 2. RAB2 is required for autophagy initiation in mammalian cells. (a) Measurement of LC3 lipidation. Control or clonal RAB2 KO U2OS cell line were untreated and treated with EBSS and/or bafilomycin A1 (Baf A1) for 2 h, and then analyzed by WB. (b) Control and clonal RAB2 KO U2OS (#39 and #40) were treated with Torin1 for 2 h, which was followed by fixation, anti-LC3 immunostaining and confocal microscopy analysis. Scale bars: 10 µm. (c) Quantification of LC3 puncta described in (B). Data are shown as mean ± SD, ***p < 0.001. (d) Measurement of early autophagic membrane structures positive for endogenous ATG16L1, which was quantified in (e). Scale bars: 10 µm. Data are shown as mean ± SD, **p < 0.01. (f) Measurement of early autophagic membrane structures positive for endogenous ULK1, which was quantified in (g). Scale bars: 10 µm. Data are shown as mean ± SD, **p < 0.01. (h) Co-IP of HA-RAB2 and FLAG-GOLGA2 under uninduced and autophagy-induced conditions. (l) Immunostaining analysis of endogenous GOLGA2 in GOLGA2 knockout (KO) and control HEK293 cells (j) Measurement of LC3 puncta and RAB2-LC3 colocalization in GOLGA2 KO U2OS cells, which was quantified in (k and l). Scale bars: 10 µm. Data are shown as mean ± SD, ***p < 0.001.
Autophagy stimuli trigger RAB2-mediated ULK1 acquisition and activation to facilitate the formation of a phagophore
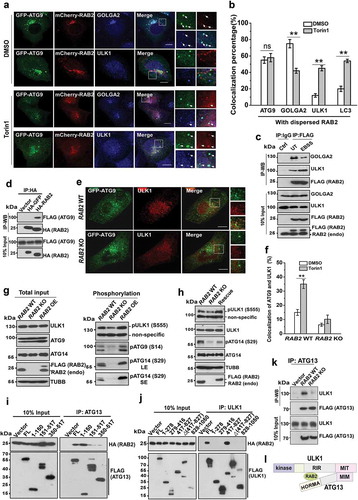
The fact that RAB2 KO abolished the formation of the earliest autophagic structures marked by ULK1 implied that the Golgi-derived RAB2+ATG9+ vesicles participate in the formation of phagophores. In yeast, both ATG9 and ATG1 are required for the formation of phagophore assembly site [Citation47,Citation48]. Therefore, we envisioned that RAB2 might regulate ULK1 and ATG9 to facilitate the formation of the phagophore in mammalian cells. We optimized the conditions by intentionally expressing GFP-ATG9 at very low levels which enabled faithful resembling of endogenous ATG9 (Fig. S3A). We observed that in unstimulated cells majority of RAB2+ATG9+ vesicles were positive for GOLGA2 but not for ULK1, however, this pattern was largely reversed when autophagy was stimulated (Figure 3(a,b) and S3B). Because reliable antibodies for the immunoprecipitation of endogenous RAB2 were lacking, we used the cell line that stably expressed FLAG-RAB2 at a level close to its endogenous counterpart (). Consistently, autophagy stimuli-triggered dissociation of RAB2 from GOLGA2 was accompanied by the increased interaction of RAB2 with ULK1 (), and the colocalization of RAB2 and ULK1 were enriched by wortmannin, which is capable of accumulating early autophagic structures (Fig. S3C and S3D). In addition, we did not detect the interaction between GOLGA2 and ULK1 (data not shown), which indicated that they may bind to RAB2 in a sequential manner. Furthermore, RAB2 interacted with ATG9 and facilitated the colocalization of ATG9 and ULK1 (). In contrast, RAB2 KO did not affect the colocalization of ATG9 and TFRC (transferrin receptor), and ATG9 depletion decreased the colocalization of RAB2 and TFRC (Fig. S3E, S3F, S3G and S3H). Importantly, RAB2 appeared to regulate ULK1 activation (), which was further confirmed by the alteration of ULK1-mediated phosphorylation of ATG14 and ATG9 in the RAB2 KO, OE and RAB2 WT -rescued KO cells. Biochemical dissection showed that RAB2 directly interacted with the HORMA domain of ATG13 and the RIR (417-827aa, RAB2-Interaction Region, which was named in this study) of ULK1 (, S3I and S3J), and RAB2 was required for the integrity ULK1-ATG13 complex (). Since the phosphorylation of various of autophagy machineries including ATG9 and ATG14 by ATG1/ULK1 is critical for autophagy initiation [Citation25,Citation49–Citation51], these results indicated that RAB2 regulates ULK1 recruitment and activation to further propagate signals for autophagy initiation.
Figure 3. RAB2 regulates ULK1 recruitment and activation for autophagy initiation. (a) Confocal microscopy analysis of the colocalization of GFP-ATG9, mCherry-RAB2 and ULK1 or GOLGA2, and quantification was shown in (b). Scale bars: 10 µm. Data are shown as mean ± SD, **p < 0.01; ‘ns’ indicates no significance. (c) Co-IP of RAB2 and GOLGA2 or ULK1 under unstressed and starved conditions. (d) Co-IP of HA-RAB2 and FLAG-ATG9 using HA-GFP as a negative control. (e) Confocal microscopy analysis of the colocalization of GFP-ATG9 and ULK1 in RAB2 WT and KO U2OS cells under Torin1 treatment, and quantification was shown in (f). Scale bars: 10 µm. Data are shown as mean ± SD, **p < 0.01. (g) WB analysis of ULK1 phosphorylation at Serine555, ATG14 phosphorylation at Serine29 and ATG9 phosphorylation at Serine14 in RAB2 WT, KO or OE U2OS cells cultured in complete medium. Long exposure (LE), short exposure (SE). (h) WB analysis of phosphorylation of ULK1 and ATG14 in rescued RAB2 KO cells. (i) Dissection of RAB2 and ATG13 interaction by co-IP assay. (j) Dissection of RAB2 and ULK1 interaction by co-IP assay. (k) Co-IP of FLAG-ATG13 and ULK1 in RAB2 WT or KO cells. (l) Schematic representation of RAB2 interaction with the HORMA domain of ATG13 and the RIR (RAB2-Interaction Region, named in this study) domain of ULK1.
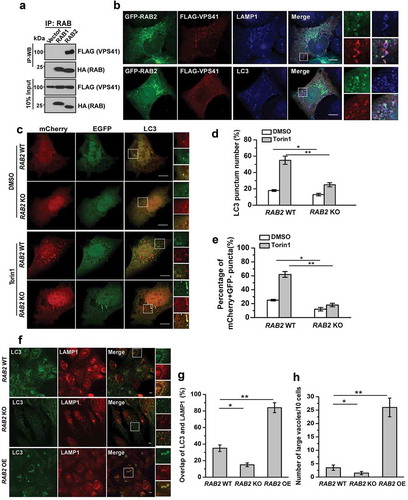
RAB2 interacts with RUBCNL and STX17 to become an autophagosomal GTPase
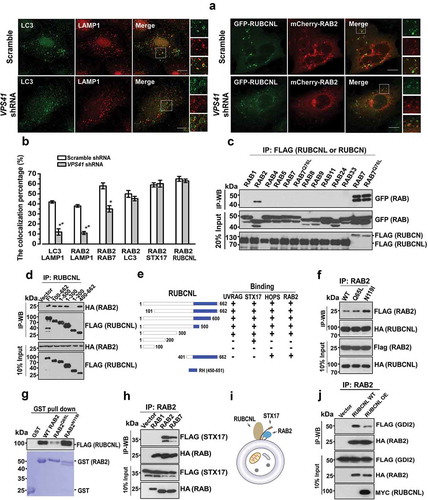
RAB2 also colocalized with autophagosomal markers, including RUBCNL and STX17 (), implying that RAB2 was probably retained on autophagosomes after the closure step of autophagosome formation. Therefore, we further dissected RAB2 subcellular localization after the disruption of autophagosome maturation by silencing VPS41, the essential functional subunit of HOPS complex, and we observed the colocalization of LC3 and LAMP1 was significantly reduced (Figure 4(a,b) and Fig. S4A), confirming that the loss of the tethering function of HOPS led to a defect in autolysosome formation. Similarly, RAB2 showed impaired colocalization with RAB7 or LAMP1 in the absence of functional HOPS. In contrast, HOPS inactivation did not affect colocalization of RAB2 with STX17 and RUBCNL ( and S4A), indicating that RAB2 colocalized with RUBCNL and STX17 on autophagosomes rather than with LAMP1 or RAB7 on lysosomes or late endosomes. Consistent with our previous Mass spectrometry (MS) analysis, RUBCNL selectively interacted with RAB2 (), and their interaction was mapped to both the N- and C-termini of RUBCNL, which partially overlapped with the HOPS-binding regions of RUBCNL () [Citation44]. Further co-IP assays and in vitro pulldown assay using purified recombinant proteins showed that RUBCNL and STX17 directly interacted with three forms of RAB2, with a preference for the GDP-bound form (). It is well-established that membrane-associated RAB GTPases are recycled constantly to the cytoplasm via GDI-mediated membrane extraction [Citation38] and that GDI2 is responsible for detachment of RAB2 from membrane structures [Citation52]. Therefore, we hypothesized that RUBCNL or STX17 may inhibit GDI2-mediated membrane extraction of RAB2. Indeed, the interaction between RAB2 and GDI2 was largely disrupted in RUBCNL OE cells (). Moreover, RUBCNL KD significantly reduced the colocalization of RAB2 with STX17 and LC3 (Fig. S4B). Because autophagosome-targeting of RUBCNL is partially dependent on STX17 [Citation44], we concluded that RUBCNL and STX17 prevent GDI2-mediated membrane extraction to maintain RAB2 on autophagosomes.
Figure 4. RAB2 directly interacts with RUBCNL and STX17 to become an autophagosomal GTPase. (a) Confocal microscopy analysis of the colocalization of the organelle markers as indicated in VPS41 KD and control cells, and quantification was shown in (b). Scale bars: 10 µm. Data are shown as mean ± SD, **p < 0.01, *p < 0.05. (c) Co-IP of FLAG-RUBCNL with GFP-tagged RAB GTPases, and co-IP of FLAG-RUBCN with GFP-WT RAB7 or GFP-RAB7Q76L were applied as positive controls. (d) Co-IP of FLAG-WT RUBCNL or mutants with HA-RAB2. (e) Summary of the interaction of RUBCNL with different binding partners. (f) Co-IP of HA-RUBCNL with FLAG-WT RAB2, FLAG-RAB2Q65L or FLAG-RAB2N119I. (g) In vitro GST-pulldown assay using purified recombinant proteins for FLAG-RUBCNL and GST-WT RAB2, RAB2Q65L or RAB2N119I as indicated. (h) Co-IP of FLAG-STX17 with HA-RAB1, HA-RAB2 or HA-RAB7. (i) Schematic representation of the trimeric complex of RUBCNL, STX17 and RAB2 on autophagosome. (j) Co-IP of FLAG-GDI2 and HA-RAB2 in RUBCNL OE and control cells.
The autophagosomal trimeric complex containing RAB2, RUBCNL and STX17 recruits HOPS complex for autophagosome maturation
We then postulated that the autophagosomal trimeric complex containing RAB2, STX17 and RUBCNL further specifies the recruitment of HOPS complex to facilitate autophagosome maturation. Indeed, RAB2 but not RAB1 interacted and colocalized with HOPS complex (Figure 5(a,b) and data not shown). Next, we performed autophagosome maturation assays using the tandem mCherry-GFP-LC3 construct [Citation53,Citation54] (), and we observed that RAB2 KO significantly reduced autophagosome maturation, as measured by the percentage of mCherry+ GFP− puncta in U2OS cells. Furthermore, the colocalization of LC3 and LAMP1 puncta, the indicator for autophagosome-lysosome fusion, was reduced in RAB2 KO cells but increased in RAB2 OE cells (). Notably, large vesicles positive for both LAMP1 and LC3 only appeared in RAB2 OE cells, which further demonstrated the accelerated autolysosome formation driven by RAB2 OE (). Overexpression of RAB2 mutants was able to inhibits autophagy flux (Fig. S5A). Furthermore, we monitored the delivery of autophagosomal membranes to lysosomes by GFP-LC3 processing assays [Citation54,Citation55]. The appearance of free GFP, the indicator of the lysis of the inner autophagosomal membrane was blocked in RAB2 KO cells (Fig. S5B and S5C). RAB2 appeared to regulate endocytic pathway as well (Fig. S5D), which was consistent with the observations by other groups [Citation56,Citation57]. However, its dual role in both endocytosis and autophagy was not due to a direct effect on lysosome biogenesis or maturation, because cathepsin D maturation was not altered by RAB2 KO (Fig. S5E). Overall, our data showed that the autophagosomal tripartite complex containing RAB2, STX17 and RUBCNL recruited HOPS for autophagosome maturation (Fig. S5F).
Figure 5. The autophagosomal trimeric complex containing RAB2, RUBCNL and STX17 recruits HOPS complex to promote autophagosome maturation. (a) Co-IP of FLAG-VPS41 with HA-RAB1 or HA-RAB2. (b) Confocal microscopy analysis of the colocalization of GFP-RAB2, FLAG-VPS41 and LAMP1 or LC3. (c) Confocal microscopy analysis of mCherry-GFP-LC3 expressed in RAB2 WT or KO U2OS cells in the presence of DMSO or Torin1. mCherry-positive GFP-negative (mCherry+ GFP−) puncta, which indicate autolysosomes, were quantified and summarized in (d and e). Scale bars: 10 µm. Data are shown as mean ± SD, *p < 0.05, **p < 0.01. (f) Confocal microscopy analysis of the colocalization of LC3 and LAMP1 in RAB2 WT, KO or OE U2OS cells, which was quantified and summarized in (G and H). Scale bars: 10 µm. Data are shown as mean ± SD, *p < 0.05, **p < 0.01.
Discussion
In this study, we suggest a model in which RAB2 links the Golgi apparatus to autophagosome formation and maturation. It should be noted that RAB2 interacts with several Golgins, including GOLGA2, to maintain the homeostasis of the Golgi apparatus [Citation58–Citation61]. Whether other Golgins also have a similar role in autophagy initiation requires further investigation. In addition, it seems that GOLGA2 may inhibit autophagy through other mechanisms [Citation62]. Furthermore, it is not known what are the upstream signals in dissociating RAB2 from GOLGA2 upon autophagy stimulation. We recently noticed that starvation altered the phosphorylation pattern of GOLGA2 (unpublished data), and the characterization of the putative kinases involved is currently undertaken in the lab.
RAB2 may regulate autophagy initiation through three different mechanisms. First, RAB2 transports Golgi-derived ATG9+ vesicles to the phagophore assembly sites. Previous works have shown that plasma membrane- and recycling endosome-derived ATG9+ vesicles contribute to autophagosome biogenesis [Citation17,Citation18,Citation28]. Our work indicates that the Golgi-derived ATG9+ vesicles are delivered by RAB2 for the construction of early autophagic structures, because the colocalization of ULK1 and ATG9, but not that of ATG9 and TFRC, was significantly reduced in RAB2 KO cells, and the translocation of RAB2, but not that of ATG9, to recycling endosomes was abolished by nocodazole treatment. Second, RAB2 recruits ULK1 to phagophore assembly sites, as ULK1 appears to be soluble and forms a diffused cytosolic pattern in the absence of RAB2. Previous studies have demonstrated that ULK1 may have a tethering function, which is independent of its kinase activity [Citation29,Citation63–Citation65]. It has been indicated that ATG9 is self-interacted [Citation66], and it requires at least three ATG9 vesicles to mark a PAS in yeast [Citation7]. Therefore, it is conceivable that RAB2-ULK1 interactions may directly contribute to the initial tethering of ATG9+ vesicles prior to fusion, which enables the small donating vesicles to mature into a phagophore. Third, RAB2 facilitates ULK1 activation to propagate signals for autophagy initiation. The acquisition of ULK1 to RAB2+ vesicles may result in clustering of the ULK1 complex, which is essential for ULK1 activation [Citation47,Citation65,Citation67,Citation68]. Next, activated ULK1 phosphorylates ATG9 to enable the ATG9+ vesicles to fuse into phagophores [Citation25,Citation49,Citation50,Citation69], and phosphorylates ATG14 and BECN1 to activate PtdIns3K for nucleation [Citation51,Citation70].
Studies in yeast have shown that YPT1 is recruited and activated by its GEF, TRAPIII, at PAS, which is followed by YPT1-mediated ATG1 recruitment [Citation35,Citation71]. RAB1, the mammalian orthologue of YPT1, also regulates autophagy [Citation28,Citation72–Citation77]. In addition, RAB1, together with TRAPPIII and TBC1D14, maintains an ATG9 pool on the Golgi apparatus by acquiring the vesicles from recycling endosomes [Citation28]. Moreover, a circuit involving TBC1D14, RAB11 and ULK1 on the recycling endosome also contributes to autophagy initiation [Citation18]. Although RAB2 homolog has not been detected in yeast [Citation78], RAB2 appears to be conserved in model organisms ranging from C. elegans and Drosophila to humans. We demonstrated here that the Golgi network contributes membranes and protein machineries including RAB2 and ATG9 to autophagy, which is different from RAB1- or RAB11-mediated pathways linking recycling endosomes to autophagy in mammalian cells.
A series of studies have shown that RAB2 is essential for endocytic pathways [Citation79–Citation81] in which C. elegans GOP1 and its mammalian homolog CLEC16A activates RAB2 as GDF (GDI displacement factor) [Citation38,Citation81]. RUBCNL appears to have a similar GDF function, because RUBCNL preferentially interacts with the GDP-bound form of RAB2, and RUBCNL inhibits RAB2-GDI2 interactions. In addition, because both STX17 and RAB2 localize on different compartments [Citation82,Citation83], to avoid disadvantageous mistargeting of HOPS complex, it might be necessary to form a tripartite complex with RUBCNL on autophagosomes to further specify the recruitment of HOPS complex for autophagosome maturation. Whether the complex containing RUBCNL, STX17 and RAB2 also functions in endocytic pathway is currently not known. During the preparation of this manuscript, two groups reported that the Drosophila RAB2 regulates autophagosome maturation by recruiting HOPS complex [Citation57,Citation84], which is in accordance with second part of our findings in this work. However, this work provides further insights into the mechanisms of how RAB2 recruits HOPS to promote autophagosome maturation by introducing RUBCNL and STX17 as the cofactors. In addition, a possible function of RAB2 in autophagy initiation in Drosophila was not addressed, which may worth further investigation.
In conclusion, this study not only identifies RAB2 as a unique GTPase participating in the formation of both autophagosome and autolysosome in mammalian cells, but also provides further mechanistic insights into the mechanisms of how the Golgi apparatus contributes to bulk autophagy pathway.
Materials and methods
Antibodies
Anti-SQSTM1 (MBL, PM045), anti-ATG16L1 (MBL, PM040), anti-GOLGA2 (MBL, PM179-3), anti-LC3 (MBL, PM036), anti-AIFM2 (Cell Signaling Technology, 5318), anti-CTSD (Santa Cruz Biotechnology, sc-6486), anti-EGFR (Santa Cruz Biotechnology, sc-120), anti-RAB2 (BBI Life Sciences, D122959-0200), anti-RAB1A (Protein tech, 11671–1-AP), anti-VPS41 (Santa Cruz Biotechnology, sc-377118), anti-ULK1 (Santa Cruz Biotechnology, sc-390904), anti-LAMP1 (Santa Cruz Biotechnology, sc-20011), anti-TFRC (Santa Cruz Biotechnology, sc-32272), anti-BCAP31 (Santa Cruz Biotechnology, sc-393810), anti-TOMM20 (Santa Cruz Biotechnology, sc-17764), anti-SQSTM1 (pSer403) (Genetex, GTX128171), anti-ATG14 (Cell Signaling Technology, 5504), anti-LAMP1 (Cell Signaling Technology, D2D11), anti-EEA1 (Cell Signaling Technology, C45B10), anti-p-ATG14 (S29) (Cell Signaling Technology, 13155S), anti-LC3 (Sigma, L8918), anti-RAB2 (Abcam, GR188995-4), anti-HA-Tag-HRP (MBL, M180-7), anti-FLAG-Tag-HRP (MBL, M185-7), anti-HA (Biolegend, 16B12), anti-GFP (Santa Cruz Biotechnology, M048-3), anti-MYC (Santa Cruz Biotechnology, 9E10), anti-ubiquitin (Cell Signaling Technology, 3936T), anti-LAMP1 (D2D11) (Cell Signaling Technology, 9091S) anti-ULK1 (D8H5) (Cell Signaling Technology, 8054S), anti-p-ULK1 (S555) (Cell Signaling Technology, 5869S), anti-ATG9 (Cell Signaling Technology, 13509S), anti-p-ATG9 (S14) (Donated by Prof. Chen Quan), Alexa Fluor 488 (Abcam, GR238847-1), Alexa Fluor 546 (Thermo Fisher Scientific, A11003), Alexa Fluor 546 (Thermo Fisher Scientific, A11010), Alexa Fluor 405 (Thermo Fisher Scientific, A31556), Alexa Fluor 405 (Thermo Fisher Scientific, A81553), Alexa Fluor 488 (Thermo Fisher Scientific, A11008), Alexa Fluor 488 (Thermo Fisher Scientific, A11001).
Chemicals and reagents
Torin1 (Selleck Chemicals, S2827), bafilomycin A1 (Selleck Chemicals, S1413), chloroquine (Sigma, C6628), EGF (PeproTech incorporated, AF-100–15), nocodazole (Sigma, M1404), puromycin (Sigma, P7255), Lipofectamine 3000 (Thermo Fisher Scientific, L3000015), Earle’s basic salt solution (Thermo Fisher Scientific, 1816327), restriction enzymes (Thermo Fisher Scientific), GST agarose (Probegene, PC014), 2× Taq Master Mix (Probegene, ME013), 2 × Ultra-Pfu Master Mix (Probegene, ME026), ClonExpress II One Step Cloning Kit (Vazyme Biotech, C112-01), 2 × Phanta Master Mix (Vazyme Biotech, P511).
Cell lines
U2OS (ATCC), HEK293T (ATCC), HeLa (ATCC), HEK293 (ATCC), RAB2−/- U2OS (constructed in our lab), FLAG-RAB2 U2OS (Constructed in our lab), FLAG-RAB2 HEK293T (constructed in our lab), HA-RUBCNL HEK293T (constructed in our lab), MYC-RUBCNL HEK293T (constructed in our lab).
Oligonucleotides
VPS41 shRNA (GCTTTGACAGTCAGAGGCTTT), GOLGA2 shRNA 1 (CGAGAATGATGAGGTGAAGAT), GOLGA2 shRNA 2 (GCGGATTTGTAAAGCTGACTA), ATG9 shRNA (GTGGACTATGACATCCTATTT).
Recombinant DNA
pEGFP-C1 (Clonetech, PT32595), pEGFP-N1 (Clonetech, PT3027-5), pmCherry-N1 (Clonetech, PT3974-5), pCDNA5-FRT-TO-3 × FLAG (Invitrogen, V6010-20), pcDNA3.1-HA (Invitrogen, V709-20), pmCherry-EGFP (Addgene, 86639; Ivan Yudushkin lab), pOG44 (Stratagene, 1141), pMRX-IP-GFP-LC3-RFP-LC3ΔG (Addgene, 84572; Noboru Mizushima lab).
Software and algorithms
LSM 800 Browser (ZEISS), DNA STAR sequence assay, SPSS 17 Measurement data analysis (IBM).
Cell culture
U2OS, HEK293 and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1% penicillin-streptomycin.
Stable cell lines construction
FLAG-RAB2/293T and FLAG-RAB2/U2OS were obtained by Flp-In™ System. The pOG44 plasmid and the pcDNA5-FRT-RAB2 vector were co-transfected into the Flp-In™ 293T and Flp-In™ U2OS cells. Cells were passed into 2 to 4 10-cm plates 48 h after transfection. Hygromycin was added one day after re-plating. Replace medium every other day, always adding hygromycin. Colonies were visible after approximately 15 days and should be picked. Transfer colonies to 12-well plates and grow in presence of hygromycin. WB was performed to verify FLAG-tagged protein expression.
Generation knockout cell lines for GOLGA2 and RAB2
pLKO-cas9-RAB2 and pLKO-cas9-GOLGA2 sgRNA vectors with designed gDNA sequence 5ʹGCTCGAATGATAACTATTGA3ʹ and 5ʹTGCTGATATTCTCTCAACTG3ʹ were cloned. U2OS or HEK293 cells were seeded in a 6-well plate with 50% confluency one day before transfection. Cells were transfected with 2 μg pLKO-cas9-RAB2 sgRNA or pLKO-cas9-GOLGA2 sgRNA vector by Lipofectamine 3000. Twenty-four h later, regular medium was replaced with medium containing 1 μg/ml puromycin. After 2 days incubation, cells were diluted and seeded into 15cm dishes. Two weeks later, single cell clones were trypsinized and seeded into 96-well plates. WB was performed to screen single cell clones with anti-RAB2 or anti-GOLGA2 antibodies. Then, WB verified KO cell clones were sent for sequencing verification.
Immunoprecipitation and western blot
Cell pellets were homogenized in TAP buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40 [Sigma, 56741], 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 10 nM MG132 [Selleck Chemicals, S2619], protease inhibitor cocktail [Bimake, B14001], phosphatase inhibitor cocktail [Bimake, B15001]) and incubated on ice for 30 min. The cell lysate was cleared by centrifugation at 16,873 g for 30 min. The supernatant was incubated with antibody-conjugated beads (Bimake, B23102) and rotated for 2 h at 4°C. After incubation, the beads were washed 3 times with TAP buffer. WB was performed following standard procedures.
Immunofluorescence
Cells grown on coverslips were transfected with different plasmids, then fixed in 4% paraformaldehyde in PBS (Thermo Fisher Scientific, 10010023) for 20 min at room temperature and permeabilized with 0.1% Triton X-100 (Sangon Biotech, T0694) in PBS for 20 min. Following permeabilization, cells were treated with block buffer (1% BSA (BBI Life Sciences, A600332), 0.1% Triton X-100 in PBS) for 1 h at room temperature. Cells were incubated with primary antibodies diluted in block buffer overnight at 4°C. Cells were washed 3 times with PBS, each for 10 min, followed by incubation with Alexa Fluor-conjugated secondary antibody (Life Technologies) in block buffer for 1 h at room temperature. Slides were examined by using a laser scanning confocal microscope (Zeiss LSM 800).
EGFR degradation assay
Cells cultured in 12-well plates were grown to approximately 80% confluency. Cells were serum-starved overnight (12–18 h). EGFR endocytosis was stimulated by adding of 200ng/ml EGF (peprotech, AF-100–15) in DMEM containing 20 mM HEPES and 0.2% BSA. Four different time lapse after EGF stimulation, the cells were boiled in 100 μl 1× SDS loading buffer. Samples were analyzed by WB of EGFR.
RAB2 knockdown in mouse liver by AAV-mediated shRNA expression in vivo
All animal experiments were performed under the guidelines of the institutional Animal Care and Use Committee at Zhejiang University. Mice were maintained in a barrier facility, at normal room temperatures, on a regular 12 h light and 12 h dark cycle. For Rab2 KD in mouse liver, eight-week-old male C57BL/6J were applied for studies. shRNA-Rab2 sequence: GCCTATCTCTTCAAGTACATCTTCAAGAGAGATGTACTTGAAGAGATAGGCTTTTTT. pAV-U6-GFP inserted with nonsense sequence were used as control. Male mice were used for the AAV gene transfer studies. The mice in each experiment were randomized. pAV-U6-shRNA-Rab2-GFP and pAV-U6-GFP AAV particles were injected with 5 × 1011 vg into each mouse. Four weeks later, protein samples isolated from mouse liver, which were equalized with BCA kit, were performed with WB using anti-SQSTM1 antibody. The other group of mice, which were injected with pAV-U6-shRNA-Rab2-GFP and pAV-U6-GFP AAV particles, were starved for two days. Livers from normal feeding and starved mice were prepared for transmission electron microscopy (TEM).
Protein extraction from tissue
Liver samples (200 mg) were homogenized in 1 ml TAP buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 10 nM MG132, supplemented with protease, phosphatase inhibitors and deacetylase inhibitors (Selleck Chemicals, S1045)) using a homogenizer at 4°C for 45 s, and the homogenates were cleared by centrifugation at 16,873 g for 20 min and 200,000 g for 60 min, respectively. The supernatants were used for WB analysis or immunoprecipitation.
Autophagy analysis
For LC3-II degradation assay, U2OS cells with RAB2 KO or WT were treated with 250 nM Torin1 or vehicle at different time points, and whole cells lysates were briefly sonicated in 1× SDS loading buffer, and incubated at 100°C for 5 min, then subjected to WB analysis with antibodies against LC3. For autophagosome maturation assays, U2OS with RAB2 KO or WT were transfected with GFP-mCherry-LC3, 16 h post-transfection, the cells were treated with EBSS at 37°C for 1 h, and analyzed by fluorescence microscopy. To determine how RAB2 mutants affect autophagic flux, stable cell lines of flag-tagged WT RAB2, RAB2Q65L and RAB2N119I were treated with Torin1 treatment for different times. Cell lysates were analyzed by WB using SQSTM1 and LC3 antibodies. For GFP-LC3-RFP-LC3ΔG cleavage assay, RAB2 KO or WT U2OS cells were seeded in 12-well plates. When reaching 70% confluency, cells were transfected with GFP-LC3-RFP-LC3ΔG. Twenty-four h later, cells were treated with Torin1 for inducing autophagy for different times. Cells were boiled in 100 μl 1× SDS loading buffer. Samples were analyzed by WB using GFP, RFP and LC3 antibodies. For Electron microscopy, RAB2 KO and WT cells were treated with 250 nM Torin1 or vehicle (DMSO) for 2 h. The cells were harvested and washed with PBS at room temperature. The cell pellets were fixed in 0.1 M PBS buffer containing 2.5% (w:v) glutaraldehyde at 4°C for 24 h. The cells were washed with 0.1 M PBS three times for 15 min each time. The cells were post-fixed in 0.1 M PBS buffer containing 1% osmium tetroxide for 1 h at 4°C and then washed with water three times for 15 min each time. When processing resumed, the cells were dehydrated in graded alcohols, embedded in Epon 812 (SPI, 660-AB), sectioned with ultramicrotome (Leica, Germany), and then stained with uranyl acetate and lead citrate. The sections were examined with a transmission electron microscope (JEOL-1230, Japan). For each representative figure which was shown, and at least three different experiments were performed.
Quantification and statistical analysis
Statistical analyses were performed using the Student’s t test in SPSS 17.0 software. Values are expressed as mean ± SD of at least three independent experiments, unless otherwise noted. A P value < 0.05 was considered statistically significant.
Supplemental Material
Download Zip (4.5 MB)Acknowledgments
We are grateful to Prof. Tianhua Zhou from Zhejiang University School of Medicine for supporting this work. We thank Prof. Jiahuai Han from Xiamen University for cDNA and Prof. Quan Chen from Nankai University for ATG9 phosphor-antibody and ATG9 cell lines. We thank Shu Jia and the Imaging Center of Zhejiang University School of Medicine for assistance with confocal microscopy and electron microscopy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. PubMed PMID: 19653858; PubMed Central PMCID: PMC2831538.
- Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009 Jul;10(7):458–467. PubMed PMID: 19491929.
- Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016 Aug;26(8):624–635. PubMed PMID: 27050762.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008 Feb 28;451(7182):1069–1075. PubMed PMID: 18305538; PubMed Central PMCID: PMC2670399.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 Jan 11;132(1):27–42. PubMed PMID: 18191218; PubMed Central PMCID: PMC2696814.
- Bernard A, Klionsky DJ. Autophagosome formation: tracing the source. Dev Cell. 2013 Apr 29;25(2):116–117. PubMed PMID: 23639440; PubMed Central PMCID: PMC3668556.
- Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012 Jul 23;198(2):219–233. PubMed PMID: 22826123; PubMed Central PMCID: PMCPMC3410421.
- Ravikumar B, Moreau K, Jahreiss L, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010 Aug;12(8):747–757. PubMed PMID: 20639872; PubMed Central PMCID: PMC2923063.
- Moreau K, Ravikumar B, Renna M, et al. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011 Jul 22;146(2):303–317. PubMed PMID: 21784250; PubMed Central PMCID: PMC3171170.
- Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009 Dec;11(12):1433–1437. PubMed PMID: 19898463.
- Yla-Anttila P, Vihinen H, Jokitalo E, et al. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009 Nov;5(8):1180–1185. PubMed PMID: 19855179.
- Geng J, Nair U, Yasumura-Yorimitsu K, et al. Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2010 Jul 01;21(13):2257–2269. PubMed PMID: 20444978; PubMed Central PMCID: PMCPMC2893989.
- van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi Is Required for the Expansion of the Autophagosomal Phagophore in Yeast Saccharomyces cerevisiae. Mol Biol Cell. 2010;21(13):2270–2284.
- Yen WL, Shintani T, Nair U, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J cell Biol. 2010 Jan 11;188(1):101–114. PubMed PMID: WOS:000273507300011; English.
- Guo Y, Chang C, Huang R, et al. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci. 2012 Apr 01;125(Pt 7):1706–1715. PubMed PMID: 22328508; eng.
- Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010 May 14;141(4):656–667. PubMed PMID: 20478256; PubMed Central PMCID: PMC3059894.
- Puri C, Renna M, Bento CF, et al. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell. 2013 Sep 12;154(6):1285–1299. PubMed PMID: 24034251; PubMed Central PMCID: PMC3791395.
- Longatti A, Lamb CA, Razi M, et al. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012 May 28;197(5):659–675. PubMed PMID: 22613832; PubMed Central PMCID: PMC3365497.
- Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009 Apr 20;185(2):305–321. PubMed PMID: 19364919; PubMed Central PMCID: PMC2700373.
- Dongyan Tana YC, Wangd J, Zhangd J, et al. The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc Natl Acad Sci U S A. 2013 Nov 26;110(48):19432–19437.
- Lemus L, Ribas JL, Sikorska N, et al. An ER-Localized SNARE Protein Is Exported in Specific COPII Vesicles for Autophagosome Biogenesis. Cell Rep. 2016 Feb 23;14(7):1710–1722. PubMed PMID: 26876173.
- Ge L, Melville D, Zhang M, et al. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife. 2013 Aug 06;2:e00947. PubMed PMID: 23930225; PubMed Central PMCID: PMCPMC3736544.
- Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. eLife. 2014 Nov 28;3:e04135.
- Orsi A, Razi M, Dooley HC, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012 May;23(10):1860–1873. PubMed PMID: 22456507; PubMed Central PMCID: PMCPMC3350551.
- Zhou C, Ma K, Gao R, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2016 Dec 9. PubMed PMID: 27934868. DOI:10.1038/cr.2016.146.
- Feng Y, Klionsky DJ. Autophagic membrane delivery through ATG9. Cell Res. 2017 Feb;27(2):161–162. PubMed PMID: 28072404; PubMed Central PMCID: PMCPMC5339853.
- Mari M, Griffith J, Rieter E, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190(6):1005–1022.
- Lamb CA, Nuhlen S, Judith D, et al. TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 2016 Feb 1;35(3):281–301. PubMed PMID: 26711178; PubMed Central PMCID: PMC4741301.
- Rao Y, Perna MG, Hofmann B, et al. The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat Commun. 2016 Jan 12;7:10338. PubMed PMID: 26753620; PubMed Central PMCID: PMCPMC4729957.
- He C, Klionsky DJ. Atg9 trafficking in autophagy-related pathways. Autophagy. 2007 May-Jun;3(3):271–274. PubMed PMID: 17329962.
- Reggiori F, Shintani T, Nair U, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005 Jul;1(2):101–109. PubMed PMID: 16874040; PubMed Central PMCID: PMCPMC1762033.
- Webber JL, Young AR, Tooze SA. Atg9 trafficking in Mammalian cells. Autophagy. 2007 Jan-Feb;3(1):54–56. PubMed PMID: 17102588.
- Bader CA, Shandala T, Ng YS, et al. Atg9 is required for intraluminal vesicles in amphisomes and autolysosomes. Biol Open. 2015 Sep 09;4(11):1345–1355. PubMed PMID: 26353861; PubMed Central PMCID: PMCPMC4728360.
- He S, Ni D, Ma B, et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat Cell Biol. 2013 Oct;15(10):1206–1219. PubMed PMID: 24056303; PubMed Central PMCID: PMCPMC3805255.
- Kakuta S, Yamamoto H, Negishi L, et al. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J Biol Chem. 2012 Dec 28;287(53):44261–44269. PubMed PMID: 23129774; PubMed Central PMCID: PMCPMC3531741.
- Mari M, Reggiori F. Atg9 reservoirs, a new organelle of the yeast endomembrane system? Autophagy. 2010 Nov 6;(8):1221–1223. PubMed PMID: 20962573. Doi:10.1083/jcb.200912089
- Mari M, Reggiori F. Atg9 trafficking in the yeast Saccharomyces cerevisiae. Autophagy. 2007 Mar-Apr;3(2):145–148. PubMed PMID: 17204846.
- Pfeffer S, Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol. 2004 Nov;5(11):886–896. PubMed PMID: 15520808.
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009 Aug;10(8):513–525. PubMed PMID: 19603039.
- Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014 Mar;21(3):348–358. PubMed PMID: 24440914; PubMed Central PMCID: PMCPMC3921601.
- Lamb CA, Longatti A, Tooze SA. Rabs and GAPs in starvation-induced autophagy. Small GTPases. 2016 Oct;7(4):265–269. PubMed PMID: 27669114; PubMed Central PMCID: PMC5129906.
- Rink J, Ghigo E, Kalaidzidis Y, et al. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005 Sep 09;122(5):735–749. PubMed PMID: 16143105.
- Poteryaev D, Datta S, Ackema K, et al. Identification of the switch in early-to-late endosome transition. Cell. 2010 Apr 30;141(3):497–508. PubMed PMID: 20434987.
- Cheng X, Ma X, Ding X, et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol Cell. 2017 Mar 16;65(6):1029–1043 e5. PubMed PMID: 28306502.
- Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008 May;19(5):2092–2100. PubMed PMID: 18321988; PubMed Central PMCID: PMCPMC2366860.
- Tisdale EJ. A Rab2 mutant with impaired GTPase activity stimulates vesicle formation from pre-Golgi intermediates. Mol Biol Cell. 1999 Jun;10(6):1837–1849. PubMed PMID: 10359600; PubMed Central PMCID: PMC25379.
- Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017 Mar 15;86:225–244.
- Suzuki K, Kubota Y, Sekito T, et al. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007 Feb;12(2):209–218. PubMed PMID: 17295840.
- Feng Y, Backues SK, Baba M, et al. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy. 2016;12(4):648–658. PubMed PMID: 27050455; PubMed Central PMCID: PMC4835963.
- Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014 Feb 06;53(3):471–483. PubMed PMID: 24440502; PubMed Central PMCID: PMCPMC3978657.
- Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12(3):547–564. PubMed PMID: 27046250; PubMed Central PMCID: PMCPMC4835982.
- Shisheva A, Chinni SR, DeMarco C. General role of GDP dissociation inhibitor 2 in membrane release of Rab proteins: modulations of its functional interactions by in vitro and in vivo structural modifications. Biochemistry. 1999 Sep 07;38(36):11711–11721. PubMed PMID: 10512627.
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007 Sep-Oct;3(5):452–460. PubMed PMID: 17534139.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMC4835977.
- Morishita H, Kaizuka T, Hama Y, et al. A new probe to measure autophagic flux in vitro and in vivo. Autophagy. 2017 Apr 3;13(4):757–758. PubMed PMID: 28121224; PubMed Central PMCID: PMCPMC5388228.
- Lund VK, Madsen KL, Kjaerulff O. Drosophila Rab2 controls endosome-lysosome fusion and LAMP delivery to late endosomes. Autophagy. 2018 Aug 13:1–23 PubMed PMID: 29940804. DOI:10.1080/15548627.2018.1458170.
- Lorincz P, Toth S, Benko P, et al. Rab2 promotes autophagic and endocytic lysosomal degradation. J Cell Biol. 2017 May 08;216:1937–1947. PubMed PMID: 28483915.
- Barr FA. Purification and functional interactions of GRASP55 with Rab2. Methods Enzymol. 2005;403:391–401. PubMed PMID: 16473605.
- Saraste J. Spatial and Functional Aspects of ER-Golgi Rabs and Tethers. Front Cell Dev Biol. 2016;4:28. PubMed PMID: 27148530; PubMed Central PMCID: PMCPMC4834429.
- Wong M, Gillingham AK, Munro S. The golgin coiled-coil proteins capture different types of transport carriers via distinct N-terminal motifs. BMC Biol. 2017 Jan 26;15(1):3. PubMed PMID: 28122620; PubMed Central PMCID: PMCPMC5267433.
- Sato K, Roboti P, Mironov AA, et al. Coupling of vesicle tethering and Rab binding is required for in vivo functionality of the golgin GMAP-210. Mol Biol Cell. 2015 Feb 01;26(3):537–553. PubMed PMID: 25473115; PubMed Central PMCID: PMCPMC4310744.
- Joachim J, Jefferies HB, Razi M, et al. Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol Cell. 2015 Dec 17;60(6):899–913. PubMed PMID: 26687599; PubMed Central PMCID: PMC4691241.
- Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell. 2012 Dec 21;151(7):1501–1512. PubMed PMID: 23219485; PubMed Central PMCID: PMCPMC3806636.
- Stjepanovic G, Davies CW, Stanley RE, et al. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc Natl Acad Sci U S A. 2014 Sep 02;111(35):12793–12798. PubMed PMID: 25139988; PubMed Central PMCID: PMCPMC4156731.
- Torggler R, Papinski D, Brach T, et al. Two independent pathways within selective autophagy converge to activate Atg1 kinase at the vacuole. Mol Cell. 2016 Oct 20;64(2):221–235. PubMed PMID: 27768871.
- He C, Baba M, Cao Y, et al. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008 Dec;19(12):5506–5516. PubMed PMID: 18829864; PubMed Central PMCID: PMC2592676.
- Kamber RA, Shoemaker CJ, Denic V. Receptor-bound targets of selective autophagy use a scaffold protein to activate the Atg1 kinase. Mol Cell. 2015 Aug 6;59(3):372–381. PubMed PMID: 26166702.
- Yeh YY, Shah KH, Herman PK. An Atg13 protein-mediated self-association of the Atg1 protein kinase is important for the induction of autophagy. J Biol Chem. 2011 Aug 19;286(33):28931–28939. PubMed PMID: 21712380; PubMed Central PMCID: PMCPMC3190700.
- Papinski D, Kraft C. Atg1 kinase organizes autophagosome formation by phosphorylating Atg9. Autophagy. 2014 Jul;10(7):1338–1340. PubMed PMID: 24905091; PubMed Central PMCID: PMCPMC4203558.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013 Jul;15(7):741–750. PubMed PMID: 23685627; PubMed Central PMCID: PMC3885611.
- Wang J, Menon S, Yamasaki A, et al. Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9800–9805. PubMed PMID: 23716696; PubMed Central PMCID: PMC3683756.
- Winslow AR, Chen CW, Corrochano S, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010 Sep 20;190(6):1023–1037. PubMed PMID: 20855506; PubMed Central PMCID: PMCPMC3101586.
- Zoppino FC, Militello RD, Slavin I, et al. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010 Sep;11(9):1246–1261. PubMed PMID: 20545908.
- Mochizuki Y, Ohashi R, Kawamura T, et al. Phosphatidylinositol 3-phosphatase myotubularin-related protein 6 (MTMR6) is regulated by small GTPase Rab1B in the early secretory and autophagic pathways. J Biol Chem. 2013 Jan 11;288(2):1009–1021. PubMed PMID: 23188820; PubMed Central PMCID: PMC3542987.
- Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014 Jul 1;23(13):3579–3595. PubMed PMID: 24549040; PubMed Central PMCID: PMC4049310.
- Kakuta S, Yamaguchi J, Suzuki C, et al. Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J. 2017 May 18;31:3757–3773. PubMed PMID: 28522593.
- Lipatova Z, Belogortseva N, Zhang XQ, et al. Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proc Natl Acad Sci U S A. 2012 May 1;109(18):6981–6986. PubMed PMID: 22509044; PubMed Central PMCID: PMC3344974.
- Tisdale EJ, Bourne JR, Khosravi-Far R, et al. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol. 1992 Nov;119(4):749–761. PubMed PMID: 1429835; PubMed Central PMCID: PMC2289685.
- Mangahas PM, Yu X, Miller KG, et al. The small GTPase Rab2 functions in the removal of apoptotic cells in Caenorhabditis elegans. J Cell Biol. 2008 Jan 28;180(2):357–373. PubMed PMID: 18227280; PubMed Central PMCID: PMC2213587.
- Lu Q, Zhang Y, Hu T, et al. C. elegans Rab GTPase 2 is required for the degradation of apoptotic cells. Development. 2008 Mar;135(6):1069–1080. PubMed PMID: 18256195.
- Yin J, Huang Y, Guo P, et al. GOP-1 promotes apoptotic cell degradation by activating the small GTPase Rab2 in C. elegans. J Cell Biol. 2017 Apr 19;216:1775–1794. PubMed PMID: 28424218.
- Arasaki K, Shimizu H, Mogari H, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. 2015 Feb 9;32(3):304–317. PubMed PMID: 25619926.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 Mar 21;495(7441):389–393. PubMed PMID: 23455425.
- Fujita N 1, Huang W 1, Lin T-H 1, et al. Genetic screen in Drosophila muscle identifies autophagy-mediated T-tubule 2 remodeling and a Rab2 role in autophagy. eLife. 2017;6.