
ABSTRACT
Extracellular HMGB1 (high mobility group box 1) is considered as a damage-associated molecular pattern protein. However, little is known about its intracellular role. We studied the mechanism whereby intestinal epithelial HMGB1 contributes to host defense, using cell culture, colonoids, conditional intestinal epithelial HMGB1-knockout mice with Salmonella-colitis, il10-/- mice, and human samples. We report that intestinal HMGB1 is an important contributor to host protection from inflammation and infection. We identified a physical interaction between HMGB1 and STAT3. Lacking intestinal epithelial HMGB1 led to redistribution of STAT3 and activation of STAT3 post bacterial infection. Indeed, Salmonella-infected HMGB1-deficient cells exhibited less macroautophagy/autophagy due to decreased expression of autophagy proteins and transcriptional repression by activated STAT3. Then, increased p-STAT3 and extranuclear STAT3 reduced autophagic responses and increased inflammation. STAT3 inhibition restored autophagic responses and reduced bacterial invasion in vitro and in vivo. Moreover, low level of HMGB1 was correlated with reduced nuclear STAT3 and enhanced p-STAT3 in inflamed intestine of il10-/- mice and inflammatory bowel disease (IBD) patients. We revealed that colonic epithelial HMGB1 was directly involved in the suppression of STAT3 activation and the protection of intestine from bacterial infection and injury.
Abbreviations: ATG16L1: autophagy-related 16-like 1 (S. cerevisiae); DAMP: damage-associated molecular pattern; HBSS: Hanks balanced salt solution; HMGB1: high mobility group box 1; IBD: inflammatory bowel disease; IL1B/Il-1β: interleukin 1 beta; IL10: interleukin 10; IL17/IL-17: interleukin 17; MEFs: mouse embryonic fibroblasts; STAT3: signal transducer and activator of transcription 3; TLR: toll-like receptor; TNF/TNF-α: tumor necrosis factor
Introduction
Extracellular HMGB1 (high mobility group box 1) has been studied primarily as a damage-associated molecular pattern (DAMP) molecule [Citation1,Citation2]. It is thought that HMGB1 plays important roles in maintaining nuclear homeostasis and regulating both mitochondrial function and morphology [Citation3]. However, it intracellular role in inflammation and bacterial infection remains poorly understood. Recently, we showed that cytosolic HMGB1 regulates apoptosis by protecting the autophagy proteins BECN1 and ATG5 (autophagy related 5) from calpain-mediated cleavage during inflammation [Citation4]. Our findings suggest that intestinal epithelial HMGB1 is pivotal for limiting tissue injury in inflammatory bowel diseases (IBDs). Intestinal epithelial cells are consistently exposed to bacteria, and these interactions play a key role in development, homeostasis, and immunity [Citation5–Citation9]. Frequent microbial challenges to epithelial cells can trigger discrete signaling pathways that promote molecular changes, such as cytokine and chemokine secretion, as well as alterations to molecules displayed on the surface of epithelial cells. Together, these molecular changes allow both innate and adaptive immune cells to adopt diverse regulatory and inflammatory activities [Citation10]. Our previous studies have shown that HMGB1 is highly expressed in gut epithelial cells where its expression appears to be regulated in part by gut microbes [Citation4]. Because many of these types of host-microbe interactions have shown to play a role in host defense, we investigated the effects and mechanisms of intracellular deficiency of intestinal epithelial HMGB1 in the context of bacterial infection.
Table 1. Real-time PCR primers.
Autophagy plays a vital role in cellular homeostasis and coordinates multiple aspects of the cellular response to pathogens. HMGB1 is known to promote autophagy and limit apoptosis [Citation4,Citation11]. In the context of oxidative stress, HMGB1 acts as a sensor of autophagy. The inhibition of HMGB1 release or its absence reduces the number of autolysosomes and limits autophagic flux in cells under conditions of oxidative stress conditions in vitro [Citation12]. HMGB1 binds with the autophagy protein BECN1 and can act to sustain autophagy [Citation4]. By contrast, the deletion of HMGB1 limits autophagy [Citation11]. STAT3 (Signal transducer and activator of transcription 3) is a key molecular pathway known to be activated by many pathogens and involved in complex immune disorders [Citation13–Citation15]. STAT3 inhibits the expression of multiple autophagy proteins (i.e., BECN1) at the transcriptional level [Citation16,Citation17]. STAT3 is activated via phosphorylation in response to cytokines and growth factors, including interleukin 6 (IL6), interferons, and epidermal growth factor (EGF) [Citation18–Citation21]. HMGB1 has been implicated in autophagy, which is important for pathogenic bacterial invasion and clearance. However, the effects of intracellular HMGB1 in bacterial infection have been scarcely studied. The interplay between HMGB1 and STAT3 in autophagy has not yet been established.
In the present study, we hypothesized that intracellular HMGB1 in intestinal epithelial cells protects tissues from bacterial infection and injury and that intestinal epithelial cell deletion of HMGB1 decreases autophagy. Thus, mice that lack intestinal epithelial cell expression of HMGB1 are susceptible to bacterial infection. Using intestinal epithelial cell HMGB1 conditional knockout (Hmgb1ΔIEC) mice, we investigated the responses of these mice to Salmonella infection at the early and late stages of colonization. We investigated the mechanism of intestinal HMGB1 ablation on STAT activation using the Hmgb1ΔIEC mouse model and colonoids infected with pathogenic Salmonella. Deletion of intestinal epithelial HMGB1 results in high levels of STAT3 activation and suppression of autophagy after Salmonella infection. STAT3 inhibition restores autophagic responses and reduced bacterial invasion. Our findings indicated that colonic epithelial HMGB1 was directly involved in the suppression of bacteria-induced STAT3 activation in intestinal infection and inflammation.
Results
Enhanced susceptibility of Hmgb1ΔIEC mice to Salmonella infection
First, we assessed whether Hmgb1ΔIEC mice were susceptible to Salmonella infection in streptomycin-pretreated mice. Streptomycin treatment is known to reduce the intestinal flora and render mice susceptible to intestinal colonization by various micro-organisms. In previous studies, we established a streptomycin-pretreated Salmonella-colitis mouse model to study host-pathogen interactions [Citation22]. Post-infection 8 h was correlated with the early stages of bacterial colonization; we defined 4 d to represent the late stage of infection. We found that the body weight of the Salmonella-infected Hmgb1ΔIEC female mice dropped much earlier and more rapidly than that of Hmgb1loxP/loxP female mice (n = 7; ). There was no detectable HMGB1 expression in intestinal epithelial cells of Hmgb1ΔIEC mice by immunohistochemistry ()). These data confirmed that the intestinal epithelial HMGB1 KO model was established.
Figure 1. Hmgb1ΔIEC mice were more susceptible to Salmonella infection than Hmgb1loxP/loxP mice. (a) Relative body weight changes in mice infected with Salmonella or not. (n = 7 mice/group, ANOVA test, * p < 0.05). (b) IHC staining of HMGB1 in the colon of Hmgb1loxP/loxP and Hmgb1ΔIEC mice. Data were from a single experiment and were representative of n = 5 mice per group. (c) Cecum shortening was observed in the intestine of mice infected with Salmonella. The cecum was dissected from the indicated mouse and photographed. The cecum length was significantly less in the Hmgb1ΔIEC mice infected with Salmonella, compared to the Hmgb1loxP/loxP mice infected with Salmonella. (n = 7, student’s t-test, *p < 0.05). (d and e) Representative H&E staining and pathology scores of intestine tissues from mice with or without Salmonella infection (n = 5, student’s t-test, * p < 0.05).
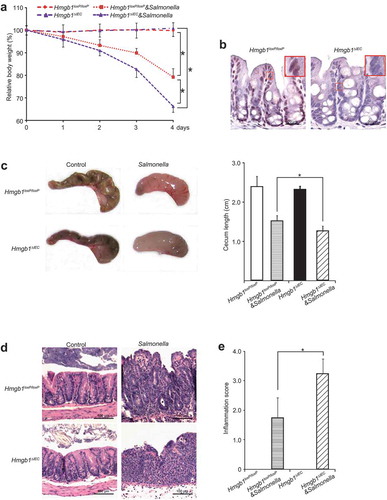
We compared the development of colitis in Hmgb1ΔIEC and Hmgb1loxP/loxP mice infected with Salmonella ()). Cecal inflammation was a key feature in this Salmonella-colitis mouse model [Citation23]. We found that the ceca of Hmgb1ΔIEC mice infected with S. Typhimurium appeared smaller and more red compared with those of Hmgb1loxP/loxP mice infected with S. Typhimurium. The length of ceca in the group of Hmgb1ΔIEC mice infected with Salmonella was significantly shorter compared with that of Hmgb1loxP/loxP mice ()). H&E staining revealed more severe intestinal inflammation in Hmgb1ΔIEC mice infected with S. Typhimurium compared with wild-type mice. ()). Next, the degree of intestinal inflammation was quantified histologically in a blinded fashion by a trained pathologist. The inflammation score in the Salmonella-infected Hmgb1ΔIEC mice was significantly higher than that of Salmonella-infected Hmgb1loxP/loxP mice ()). Thus, we concluded that Hmgb1ΔIEC mice were susceptible to Salmonella infection. Our findings indicate that intestinal HMGB1 contributes to host protection against the pathological consequences of enteric bacterial infection.
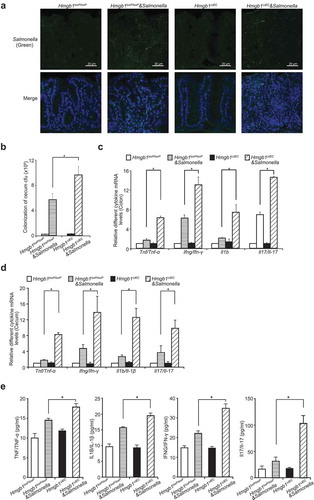
We next examined whether HMGB1 expression acts to protect intestinal cells from Salmonella invasion. An immunofluorescence assay showed more Salmonella staining (green) in the intestine of Hmgb1ΔIEC mice (Figure 2(a)). We also counted the numbers of Salmonella loading in the intestine and found that Hmgb1ΔIEC mice had an increased Salmonella load in the intestine, whereas Hmgb1loxP/loxP mice had significantly less Salmonella ()). Thus, our data indicate that deletion of intestinal HMGB1 led to enhanced Salmonella invasion in the colon.
Figure 2. Deletion of intestinal epithelial HMGB1 led to enhanced Salmonella invasion and increases levels of inflammatory cytokines. (a) Salmonella location (green) in the ceca of Hmgb1loxP/loxP and Hmgb1ΔIEC mice. Hmgb1loxP/loxP and Hmgb1ΔIEC mice were infected with wild-type Salmonella Typhimurium for 4 d. (b) Salmonella concentrations in the cecum of Hmgb1loxP/loxP and Hmgb1ΔIEC mice. (n = 5, student’s t-test, *p < 0.05). (c and d) The mRNA transcript levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 were significantly higher in Salmonella-infected Hmgb1ΔIEC mice compared with Hmgb1loxP/loxP mice in colon and cecum. (Data are shown as means ± SD from 3 replicates; student’s t-test, * p < 0.05). (e) Serum protein levels of TNF/TNF-α, IFNG/IFN-γ, IL1B/IL-1β, and Il17/Il-17 were significantly higher in Salmonella-infected Hmgb1ΔIEC mice compared with Hmgb1loxP/loxP mice. Mice were infected wild-type S. Typhimurium for 8 h (n = 5, student’s t-test, * p < 0.05).
Absence of intestinal HMGB1 is associated with increased levels of inflammatory cytokines post infection
We further hypothesized that the absence of intestinal HMGB1 may be associated with higher levels of cytokines after Salmonella infection. At 8 h post-infection, the mRNA transcript levels of Tnf/Tnf-α, interferon gamma (Ifng/Ifn-γ), Il1b/Il-1β, and Il17/Il-17 in the colon mucosa were significantly higher in the Salmonella-infected Hmgb1ΔIEC mice, compared with Hmgb1loxP/loxP mice ()). In cecum, the mRNA levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 were also significantly higher in the Salmonella-infected Hmgb1ΔIEC mice, compared with Hmgb1loxP/loxP mice ()). The protein levels of inflammatory cytokines (TNF/TNF-α, IFNG/IFN-γ, IL1B/IL-1β, and Il17/Il-17) in the serum were significantly increased in Hmgb1ΔIEC mice compared to the Hmgb1loxP/loxP mice after infection ()). For example, serum Il17/Il-17 was ~3-fold higher Hmgb1ΔIEC mice compared with Hmgb1loxP/loxP mice at 8 h post-infection. Therefore, HMGB1 may be an important contributor to intestinal homeostasis and host protection that guards against bacterial invasion and infection.
No change in the NFΚB, MAPK/JNK, and TLR signaling pathways in the colon of Hmgb1ΔIEC versus Hmgb1loxP/loxP mice
HMGB1 is an intracellular protein that can translocate to the nucleus where can bind to DNA and regulate gene expression [Citation24,Citation25]. However, our data indicated that the gene-targeted deletion of intestinal epithelial HMGB1 did not change the inflammatory NFΚB or MAPK/JNK signaling pathways when compared with the lox-mice (Figure S1(a)). HMGB1 can interact with TLR ligands and cytokines, and can act to activate cells via multiple surface receptors, including TLR2 and TLR4 [Citation26,Citation27]. However, we did not find a difference in the expressions of TLR 2, 4 and 5 at the protein levels between Hmgb1ΔIEC and Hmgb1loxP/loxP mice (Figure S1(b)).
Deletion of intestinal epithelial HMGB1 results in high levels of STAT3 activation after Salmonella infection and STAT3 nuclear translocation
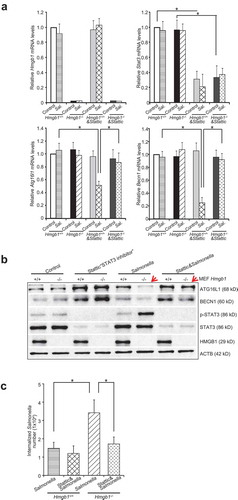
STAT pathways are associated with inflammatory cytokines and autophagy. Therefore, we assessed whether intestinal epithelial HMGB1 deletion leads to changes in STAT activation 8 h post-infection. Interestingly, western blotting revealed that levels of phospho-STAT3, an indicator of STAT3 activation, was much higher in the colon of Hmgb1ΔIEC mice after Salmonella infection compared with Hmgb1loxP/loxP mice. However, there was no difference in the total levels of STAT3 (Figure 3(a)). The expression levels of the other STATs, such STAT5 and p-STAT5, also showed no differences ()).
Figure 3. The absence by gene targeted-deletion of intestinal epithelial HMGB1 increased p-STAT3 and led to total STAT3 nuclear translocation. (a) Western blot analysis of p-STAT3, STAT3, p-STAT5, STAT5 in colonic epithelial cells from Hmgb1loxP/loxP and Hmgb1ΔIEC mice 8 h after Salmonella infection. Relative p-STAT3 protein density expression was normalized with STAT3. (n = 3, student’s t-test, *p < 0.05). (b) Western blot analysis of p-STAT3 and STAT3 levels in cytosolic (c) and nuclear (N) extracts isolated from HCT116 cells that were transfected with HMGB1-siRNA and then colonized with wild-type S. Typhimurium. The nuclear protein SP1, which is absent in the cytosolic fraction, was used as a nuclear protein loading control. The red arrow indicates p-STAT3. (c) The relative intensity of cytoplasmic and nuclear STAT3 from HCT116 cells that were transfected with HMGB1-siRNA and then colonized with wild-type S. typhimurium. Cytosolic and nuclear protein density expression normalized with total protein (n = 3/group. student’s t-test, *p < 0.05).
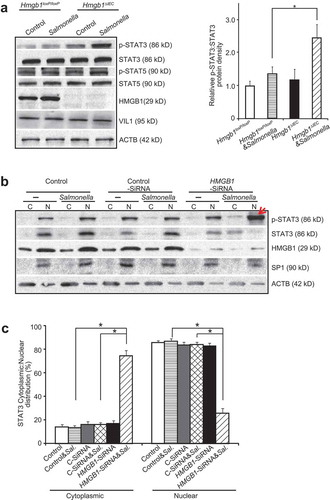
In an experimental loss-of-function experiment in vitro, we further assessed whether knockdown of intestinal HMGB1 would lead to a change in distribution of STAT3. We knocked down HMGB1 protein using a siRNA oligo in human colonic epithelial HCT116 cells and collected both the cell cytoplasmic (C) and nuclear (N) fractions. We found that nuclear levels of p-STAT3 were enhanced in human colonic epithelial cells treated with siHMGB1 RNA and then infected with Salmonella (), indicated with a red arrow; N: nuclei; C: cytoplasm). Under these same conditions, we observed higher amounts of total cytoplasm STAT3 and less total nuclear STAT3 (,c)).
We examined STAT3 phosphorylation in response to Salmonella infection using hmgb1-/- and Hmgb1+/+ mouse embryonic fibroblast (MEF) cells. We found that the complete absence of HMGB1 led to high expression levels of p-STAT3 in Salmonella-infected MEF cells (Figure S2(a) and S2(b), indicated by arrows). In hmgb1-/- MEF cells infected with Salmonella, we also observed increased amounts of total cytoplasm STAT3 and less total nuclear STAT3 (Figure S2(b) and S2(c)). These data suggest that lacking HMGB1 led to enhanced nuclear p-STAT3 and increased cytoplasm STAT3 post infection.
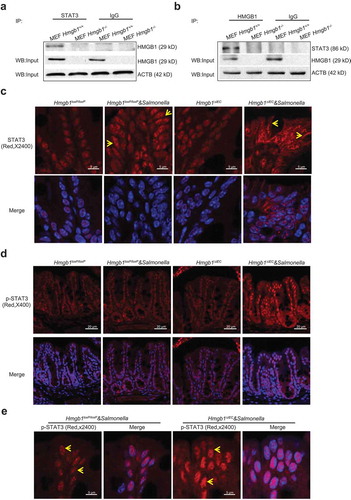
We further hypothesized that HMGB1 can physically interact with STAT3. Our immunoprecipitation data showed that anti-HMGB1 antibody could pull-down total STAT3 protein (), We also used anti-STAT3 antibody for the IP and was able to pull-down total HMGB1 ()). In contrast, we did not detect HMGB1-STAT3 interaction in hmgb1-/- cells. These data indicated the physically interact between HMGB1 with STAT3 in the wild-type cells, whereas, HMGB1 KO abolished this physical interaction. We used Predicting Protein-Protein Interactions (PrePPI) software [Citation28,Citation29] (accessed at https://honiglab.c2b2.columbia.edu/PrePPI/) to study the interaction between proteins HMGB1 (P09429) and STAT3 (P40763). We found that HMGB1 can interact with STAT3: three binding domains in HMGB1 and five binding domains in STAT3 (Figure S3).
Figure 4. HMGB1 physically interacted with STAT3, and Hmgb1 deletion in the intestinal epithelium resulted in STAT redistribution to the cytosol after wild-type S. Typhimurium. (a) STAT3 physically interacted with HMGB1 in vitro. STAT3 physically interacted with HMGB1 in MEF cells, and Hmgb1 deletion abolished this interaction (upper lane HMGB1). MEF cell lysates were immunoprecipitated with anti-STAT3 antibody, and by western blotting the precipitated complex was probed with anti-HMGB1 antibody; HMGB1 was detected in input (lower lane HMGB1, ACTB was used as a protein loading control. (b) HMGB1 physically interacted with STAT3 in MEF cells, and Hmgb1 deletion abolished this interaction. (c) Higher intensity of STAT3 immunostaining in Hmgb1loxP/loxP and Hmgb1ΔIEC mice infected wild-type S. Typhimurium. Arrows indicate the immunostaining of STAT3, HMGBloxP/loxP and Hmgb1ΔIEC mice 8 h post-infection. All experiments were performed in triplicate, and representative data were shown, n = 7 mice/group. (d and e) Higher intensity of p-STAT3 immunostaining in colon of Hmgb1loxP/loxP and Hmgb1ΔIEC mice infected with wild-type S. Typhimurium. Arrows indicate the immunostaining of nuclear p-STAT3, which was punctate in the Hmgb1ΔIEC mice 8 h post-infection. All experiments were performed in triplicate, and representative data are shown; n = 7 mice/group.
HMGB1 deletion in the intestinal epithelium resulted in STAT redistribution after Salmonella infection
We then hypothesized that deletion of colonic epithelial HMGB1 leads to redistribution of STAT3 because of lacking mutual physical interactions. We also investigated whether Salmonella infection changed the distribution of STAT3. In our cellular models of HMGB1 deficiency, levels of nuclear STAT3 were reduced in Salmonella-infected Hmgb1ΔIEC mice ()). We observed increased amounts of cytosolic STAT3 after infection in HMGBIΔIEC intestinal epithelial cells. Increased phospho-STAT3 (Y705) indicates activation of the STAT3 signaling pathway. Similar to our results in Salmonella-infected Hmgb1ΔIEC mice, we found that nuclear STAT3 was decreased and cytosolic STAT3 was increased in Salmonella-infected MEF hmgb1-/- cells (Figure S4). We observed a very strong nuclear phospho-STAT3 signal in the colon of Salmonella-infected Hmgb1ΔIEC mice ()). The p-STAT3 appeared to be mostly nuclear in the infected colonic epithelial Hmgb1ΔIEC cells at the top of the crypts, although there was cytosolic p-STAT3 staining at the bottom of colon crypts. Under high magnification, we observed enhanced phospho-STAT3 staining in nuclei of colonic epithelial cells from Salmonella-infected Hmgb1ΔIEC mice (High magnitude images in ), arrows).
Absence of intestinal epithelial HMGB1 increases STAT3 activity and suppresses autophagic responses to Salmonella infection in vivo
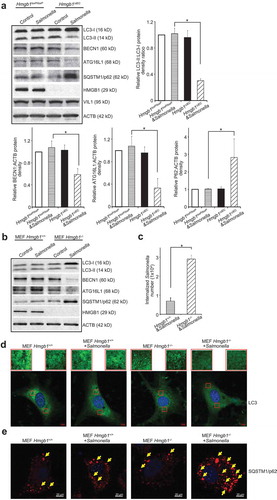
Deletion of intestinal epithelial HMGB1 leads to an abnormal autophagic response in the mouse model of DSS-colitis [Citation4]. We also characterized autophagy in intestinal epithelium HMGB1-knockout mice post Salmonella infection. SQSTM1/p62 is a marker of autophagy which is specifically degraded by autophagy. STAT3 is a transcriptional repressor of BECN1/Beclin 1. We observed significantly reduced expression levels of the autophagy regulators LC3-II:LC3-I ratio, ATG16L1 and BECN1, and an increased level of SQSTM1/p62 in the intestine of Hmgb1ΔIEC mice infected with Salmonella (). We used hmgb1-/- and Hmgb1+/+ mouse MEF cells to test whether the HMGB1 gene was sufficient to a normal autophagy phenotype in vitro. We observed reduced expression of the autophagy markers ATG16L1 and BECN1, and LC3-II:LC3-I ratio in hmgb1-/- MEF cells infected with Salmonella ()) compared with Hmgb1+/+ MEF cells after Salmonella infection. LC3 immunostaining indicated that there was less autophagy occurring in cells that lacked HMGB1 compared with Hmgb1+/+ MEF cells after Salmonella infection ()). Immunostaining of SQSTM1/p62 further showed more SQSTM1/p62 in the hmgb1-/- cells colonized with S. Typhimurium ()). These data matched the changes of SQSTM1/p62 at the protein level in western blots (,b)). We then assessed Salmonella invasion in the infected Hmgb1+/+ and hmgb1-/- MEF cells. As shown in ), HMGB1 expression significantly suppressed Salmonella invasion, when compared with hmgb1-/- MEFs.
Figure 5. The gene targeted-deletion of Hmgb1 suppressed autophagic responses to wild-type S. Typhimurium infection in vivo and in vitro. (a) Western blot analysis of autophagy markers in the colon of Hmgb1loxP/loxP and Hmgb1ΔIEC mice 8 h post-infection. VIL1 was used as a marker for epithelial cells and as an internal control. Additionally, ACTB was used as another internal control. (Data are shown as means ± SD from 3 replicates; student’s t-test, *p < 0.05.) (b) Western blot analysis of autophagy regulators in Hmgb1+/+ and hmgb1-/- MEF cells colonized with wild-type S. Typhimurium. ACTB was used as an internal control. (c) Salmonella invasion in infected Hmgb1+/+ and hmgb1-/- MEFs. The MEFs were stimulated with wild-type S. Typhimurium for 30 min, washed, and incubated in fresh Dulbecco’s Modified Eagle Medium for 30 min. (Data represent means ± SD from 3 replicate experiments, student’s t-test, *p < 0.05). (d) LC3 immunostaining of Hmgb1+/+ and hmgb1-/- cells colonized with wild-type S. Typhimurium. Fewer autophagosomes in the hmgb1-/- colonized with wild-type S. Typhimurium group. Images are each representative of experiments that were performed in triplicate. (e) SQSTM1/p62 immunostaining of Hmgb1+/+ and hmgb1-/- cells colonized with S. Typhimurium. Images are each representative of experiments that were performed in triplicate.
Inhibition of STAT3 activity restored autophagic responses in vitro and in vivo
We next investigated whether increased STAT3 activity would suppress autophagic responses in HMGB1-deficient cells. To test the possibility that autophagy was compromised or that STAT3 activity was increased, we carried out siRNA and inhibitor assays. At the mRNA level, our data showed that Stattic (a STAT3 inhibitor) treatment significantly reduced Stat3 levels in cells, whereas Atg16l1 and Becn1 did not show distinct changes after Stattic treatment in hmgb1-/- MEFs followed by colonization with wild-type S. Typhimurium (Figure 6(a)). At the protein level, we noticed that Salmonella-induced ATG16L1 and BECN1 expression decreased in hmgb1-/- MEFs and Stattic treatment can inhibit this decrease ()). Furthermore, deletion of HMGB1 led to enhanced Salmonella invasion in the hmgb1-/- MEFs, whereas inhibition of STAT3 activity by Stattic treatment significantly reduced the Salmonella invasion ()). Together, these data indicate that inhibition of STAT3 restores expression levels of key autophagy proteins (such as BECN1 and ATG16L1) and the autophagic response in cells infected with bacteria. STAT3 occurs at the upstream of autophagy regulation that leads to an autophagy defect in HMGB1-deficient cells.
Figure 6. Inhibition of STAT3 inactivated autophagy responses to wild-type S. Typhimurium infection. (a) The mRNA levels of Atg16l1 and Becn1, respectively, did not show distinct change after Stattic treatment in hmgb1-/- MEF cells followed by colonization with wild-type S. Typhimurium. (Data represent means ± SD is from 3 replicate experiments; student’s t-test, *p < 0.05.) (b) At the protein level, wild-type S. Typhimurium-induced ATG16L1 and BECN1 expression decreased in hmgb1-/- MEFs and Stattic treatment could inhibit this decrease (Data represent means ± SD is from three replicate experiments; student’s t-test, *p < 0.05). (c) Internalized Salmonella number in Stattic treated Hmgb1+/+ and hmgb1-/- MEF cells followed by colonization with wild-type S. Typhimurium. (Data represent means ± SD from 3 replicate experiments, student’s t-test, *p < 0.05.).
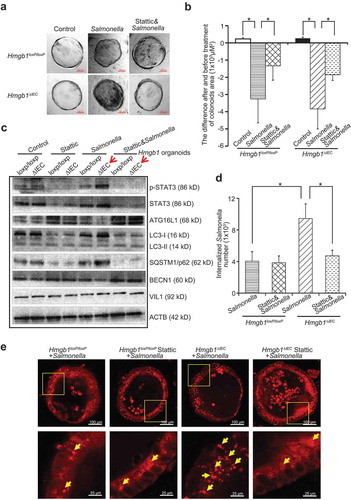
Organoids, including colonoids (large intestine) and enteroids (small intestine), are in vitro primary intestinal cultures derived from the intestine adult stem cells [Citation30]. The system can be used to study the interactions of enteric pathogens – such as Salmonella, C. difficile, E. coli – with the intestinal epithelia [Citation31–Citation33]. We cultured Hmgb1loxP/loxP and Hmgb1ΔIEC mouse colonoids, colonized with Salmonella and then treated with Stattic. The morphology of organoids was deformed and damaged post-Salmonella infection (Figure 7(a)). Furthermore, we quantified the changes of colonoid area in the groups with/without Stattic treatment. Our data showed that Salmonella infection led to reduced area of colonoids in the Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids colonized, whereas the Stattic treated colonoids had less reduction of colonoid area, indicating Stattic protected organoids from Salmonella-induced damage ()). Deletion of HMGB1 led to enhanced Salmonella invasion in the Hmgb1ΔIEC mouse colonoids, whereas inhibition of the STAT3 activity by Stattic treatment reduced bacterial invasion ()). After Salmonella infection, we also observed increased expression of p-STAT3, reduced expression of ATG16L1 and BECN1, and increased expression of SQSTM1/p62 in Hmgb1ΔIEC colonoids infected with Salmonella, compared to the infected Hmgb1loxP/loxP colonoids ()). Immunostaining of SQSTM1/p62 showed Stattic inhibited the STAT3 activation thus restoring the autopgic responses to Salmonella infection ()).
Figure 7. Deletion of Hmgb1 led to increased p-STAT3 and suppressed autophagic responses to S. Typhimurium infection in Hmgb1ΔIEC colonoids. (a) The micrographs showed representative Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without Salmonella infection and stattic treatment. Please note the morphological changes of the Salmonella-infected colonoids. Colonoids were colonized by Salmonella for 30 min, washed, and incubated for 1 h in Mini gut medium with Gentamicin (500 µg/ml). Each condition was examined in triplicates with multiple (>10) organoids in each sample. Each experiment was repeated 3 times. (b) The changes of colonoid size in Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without Stattic treatment. Data represent means ± SD from 3 replicate experiments, student’s t-test, *p < 0.05. (c) Numbers of internalized Salmonella in Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without stattic treatment. Data represent means ± SD from 3 replicate experiments, student’s t-test, *p < 0.05. (d) At the protein level, p-STAT3 expression was increased by wild-type Salmonella. Expressions of ATG16L1, BECN1 and SQSTM1/p62 in Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without Stattic treatment. Experiment was repeated 3 times. (e) Immunostaining of SQSTM1/p62 in Salmonella infected Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without Stattic treatment. Images are each representative of experiments that were performed in triplicate.
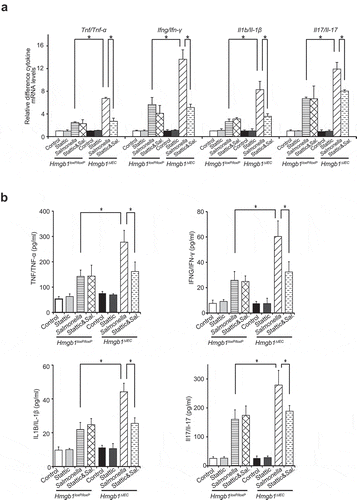
The mRNA levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 were upregulated in Hmgb1ΔIEC colonoids colonized with Salmonella, whereas stattic treatment inhibited the upregulation of the cytokines (Figure 8(a)). Levels of inflammatory cytokines (TNF/TNF-α, IFNG/IFN-γ, IL1B/IL-1β, and Il17/Il-17) in the supernatant were significantly increased in Hmgb1ΔIEC colonoids, compared to the Hmgb1loxP/loxP colonoids after infection, and Stattic treatment inhibited the increased inflammatory cytokines ()). Together, our data in colonoids indicate that deletion HMGB1 led to enhanced bacterial invasion, increased p-STAT3 expression, reduced autophagic responses, and increased inflammatory cytokines; these changes could be restored by inhibiting the STAT3 activity.
Figure 8. Deletion of Hmgb1 led to increased levels of inflammatory cytokines in Hmgb1ΔIEC colonoids with S. Typhimurium infection. (a) The mRNA levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 in Salmonella-infected Hmgb1loxP/loxP and Hmgb1ΔIEC colonoids with or without stattic treatment. (Data are shown as means ± SD from 3 replicates; student’s t-test, *p < 0.05.) (b) TNF/TNF-α, IFNG/IFN-γ, IL1B/IL-1β, and Il17/Il-17 were significantly higher in Salmonella-infected Hmgb1ΔIEC colonoids supernatant and stattic treatment inhibited the increase of cytokines (Data represent means ± SD is from 3 replicate experiments; student’s t-test, *p < 0.05.).
In vivo, we administered 50 mg/kg Stattic to HMGB1loxp and Hmgb1ΔIEC mice by oral gavage, followed by Salmonella infection. Four d post-infection, mice were sacrificed, and tissue samples from the intestinal tracts were harvested for analysis. As shown in Figure 9(a), the Stattic treated Hmgb1ΔIEC mice had less body weight loss post infection than the Hmgb1ΔIEC mice without treatment. The Stattic treated Hmgb1ΔIEC mice had significantly longer cecum than the Hmgb1ΔIEC mice without treatment ()). The histological analysis also supported the protective role of Stattic in the infected Hmgb1ΔIEC mice (,d)). Furthermore, inhibition of STAT3 activity by Stattic treatment significantly reduced Salmonella invasion ()). At the protein level, we noticed that Salmonella-induced ATG16L1 and BECN1 expression decreased in Hmgb1ΔIEC C mice and Stattic treatment can inhibit this decrease, whereas SQSTM1/p62 was reduced after Stattic treatment ()). We tested the cytokines at the mRNA level in colon. Our data showed that Stattic treatment significantly reduced levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 in Hmgb1ΔIEC mice post infection ()). Taken together, our data suggest that inhibition of STAT3 activity restored autophagic responses to Salmonella infection in vitro and in vivo.
Figure 9. The inhibition of STAT3 activity restored autophagic responses and reduces bacterial infection and invasion in vivo. (a) Relative body weight changes in Stattic-treated mice with/without Salmonella infection. (n = 5 mice/group, ANOVA test, *P < 0.05). (b) The cecum length of the Stattic treated Hmgb1ΔIEC mice was significantly longer than the Hmgb1ΔIEC mice without treatment. (n = 5, student’s t-test, *p < 0.05.) (c) Representative H&E staining and (d) pathology scores of intestine tissues from Stattic-treated mice with/without Salmonella infection. (n = 5, student’s t-test, *p < 0.05). (e) Salmonella concentrations in the cecum of in Hmgb1loxP/loxP and Hmgb1ΔIEC Stattic-treated mice with/without Salmonella infection. (n = 5, student’s t-test, *p < 0.05). (f) Changes of p-STAT3 and autophagic responses to Salmonella infection and Stattic treatment. (g) Stattic treatment significantly reduced mRNA levels of Tnf/Tnf-α, Ifng/Ifn-γ, Il1b/Il-1β, and Il17/Il-17 in Hmgb1ΔIEC mice post infection in colon. Data are shown as means ± SD from 3 replicates; student’s t-test, *p < 0.05.
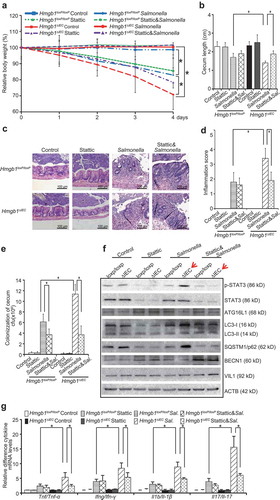
Altered HMGB1 and STAT3 expression in colitis mice and ulcerative colitis (UC) patients
In IBD, a common hallmark is defective processing of intracellular bacteria. A reduction in the expression of HMGB1 was observed in il10-/- mice with colitis symptoms compared with mice without symptoms of colitis. Moreover, we observed correlated changes in p-STAT3 levels in intestinal epithelial cells in an experimental colitis model, whereas the levels of total STAT3 were also increased in the intestine of il10-/- mice (Figure 10(a)). Intestinal HMGB1 expression was reduced in patients with UC, which is consistent with previous reports [Citation4]. Furthermore, in the inflamed human intestine with low HMGB1 expression, intestinal p-STAT3 staining was also very strong in patients with UC. Total STAT3 protein was distributed in both the cytoplasm and nuclei ()). Together, we found a similar trend of HMGB1 and STAT3 expression in UC patients and an experimental colitis model, in which il10-/- mice spontaneously develop IBD [Citation34].
Figure 10. Altered HMGB1 and STAT3 expression was observed in an experimental mouse model of colitis and in ulcerative colitis (UC) patients. (a) Intestinal HMGB1, p-STAT3 and STAT3 staining in an experimental mouse model of colitis (il10-/- mice). HMGB1 staining revealed translocation from the nucleus to the cell membrane in il10-/- mice exhibiting hallmarks of colitis. Levels of intestinal p-STA3 staining were higher in il10-/- mice with symptoms of colitis. Intestinal STAT3 staining revealed translocation from the nucleus to the cell membrane in il10-/- mice with symptoms of colitis. Data are from a single experiment and are representative of 6 mice per group. (b) Intestinal HMGB1, p-STAT3 and STAT3 staining in UC patients. Intestinal HMGB1 staining revealed translocation from the nucleus to the cell membrane in inflamed colon tissues from UC patients. Intestinal p-STA3 staining was higher in inflamed colon tissues from UC patients. Intestinal STAT3 staining revealed translocation from the nucleus to the cell membrane in inflamed colon tissues from UC patients. Images are representative of experiments that we carried out in triplicate; normal, n = 10; UC, n = 16. (c) A working model for intestinal epithelial HMGB1 in bacterial infection. Deletion of Hmgb1 in intestinal epithelial cells abolished the HMGB1-STAT3 binding. Lacking colonic epithelial HMGB1 led to redistribution of STAT3 post bacterial infection. Then, increased p-STAT3 and extranuclear STAT3 reduced autophagic responses, increased inflammation, and enhanced bacterial invasion. These changes could be restored by inhibiting the STAT3 activity.
Discussion
Our present study shows that intestinal epithelial HMGB1 deletion led to a limited ability to prevent bacterial invasion and intestinal inflammation. Our studies fill a gap in knowledge by focusing on changes in autophagy in intestinal epithelial HMGB1 knockout mice and in a Salmonella-colitis model. We found that HMGB1 can physically interact with STAT3 and that the absence of intestinal HMGB1 leads to dysregulation of STAT3, thereby suppressing autophagy [Citation16,Citation35]. Inhibition of STAT3 restores the autophagic response in cells infected with bacteria, indicating that STAT3 activation precedes the inhibition of autophagy ()). This represents the first report of the protective role of intestinal HMGB1 in bacterial infection and invasion. Our present findings demonstrate a novel mode of regulation between STAT3 and HMGB1, with potential therapeutic implications in infectious and inflammatory diseases.
Our data indicate that intestinal epithelial HMGB1 could protect host from bacterial invasion at early stages of infection (8 h post-infection). Extracellular HMGB1 is an important mediator of sepsis lethality. It has been reported that extracellular HMGB1 accumulates in the circulation of patients and mice during sepsis and that its neutralization can improve sepsis survival in experimental animal models [Citation3,Citation36]. HMGB1 could be a late mediator of sepsis lethality. However, the roles of cytosolic HMGB1 are not well understood in the context of infection with alive bacteria, rather than lipopolysaccharide (LPS). In this present study, our well-controlled in vivo model clearly showed that Hmgb1ΔIEC mice had an increased bacterial burden and mortality after Salmonella infection. Levels of p-STAT3 were upregulated and STAT3 target gene Becn1 was suppressed by Salmonella infection. Inflammatory cytokines in the serum were significantly increased in Hmgb1ΔIEC mice compared with Hmgb1loxP/loxP mice after infection.
We have demonstrated a key role of STAT3 in inducing inflammation and suppressing autophagic responses to bacterial infection. We have shown the physical interaction of HMGB1 and STAT3 in the normal cells and intestine. Deletion of intestinal epithelial HMGB1 results in redistribution of STAT3 because of lacking mutual physical interactions in respond to bacterial infection. There are reduced nuclear STAT3, increased extranuclear STAT3, and high levels of p-STAT3 in Salmonella-infected Hmgb1ΔIEC mice. STAT3 acts as a repressor of autophagy. As the downstream of STAT3 activation, we then observed suppressed authophagic responses to infection. Shen et al. reported that overexpression of STAT3 variants, including wild-type, nonphosphorylatable, and extranuclear STAT3, inhibited starvation-induced autophagy [Citation17]. Recent publications have reported a positive effect of HMGB1 on STAT3 expression in other experimental systems [Citation37,Citation38]. To the best of our knowledge the relationship between HMGB1 and STAT3 in the bacterial infection has not yet been established. STAT3 inhibits the expression of multiple autophagy proteins (i.e., BECN1) at the transcriptional level and plays a critical role in infection [Citation39,Citation40]. STAT3 deficiency leads to disruption of mitochondrial ultrastructure and increased autophagy [Citation41]. We showed that BECN1 protein expression is decreased after Salmonella infection, which was associated with high STAT3 activity. However, we did not observe Salmonella-induced changes in Becn1 expression at the mRNA level ()). We observed that HMGB1 deletion leads to significantly reduced autophagy ability via the activity of SATA3 in the intestine of Hmgb1ΔIEC mice infected with Salmonella. Our inhibitor assays also showed that autophagy reductions are downstream of STAT3 activation. STAT3 seems to occur at the upstream of autophagy regulation. If STAT3 activation is inhibited, autophagic responses after infection with Salmonella are restored.
A significant reduction in the expression of HMGB1 was observed in the experimental colitis model of il10-/- mice with colitis symptoms. We found increased levels of p-STAT3 and cytosolic STAT3 in il10-/- mice with colitis symptoms. In the intestine of IBD patients, we observed decreased levels of intestinal HMGB1 with reduced STAT3 staining and STAT3 translocation. Thus, our studies of intestinal HMGB1 expression and Salmonella-colitis add novel insights into the essential roles of HMGB1 in host-bacterial interactions and colitis. We think the HMGB1-dependent inhibition of STAT3 activity and protection of the autophagic flux may play a broader role in the intestinal inflammation.
Interestingly, we did not observe changes in NFΚB, MAPK/JNK, or TLR in the intestine of Hmgb1ΔIEC mice. HMGB1 is an intracellular protein that translocates to the nucleus where it can bind DNA and regulate gene expression. It can also be released from cells. HMGB1 released from cells seems to involve two distinct processes: necrosis, in which cell membranes are permeabilized and intracellular constituents may diffuse out of the cell; and some form of active or facilitated secretion induced by signaling mediated via NFΚB. However, our data indicated that the intestinal deletion of HMGB1 did not change the NFKB or MAPK/JNK signaling pathways compared with loxP-mice (Figure S1(a)). HMGB1 can interact with TLR ligands and cytokines, and can activate cells through multiple surface receptors, including TLR2, TLR4, and AGER/RAGE [Citation27]. However, our data did not reveal differences in TLR signaling pathways between intestinal deletion of HMGB1 mice and loxP-mice (Figure S1(b)). HMGB1 can be secreted by human inflamed intestinal tissues and is abundantly found in the stools of IBD patients [Citation42]. Using a colitis-associated cancer model, neutralizing anti-HMGB1 antibody was shown to reduce both tumor incidence and size [Citation43]. However, it is not clear whether HMGB1 is secreted or released by cell death from the way that the studies were done. Our current data suggest that intracellular HMGB1 is needed to inhibit inflammation induced by bacterial infection. Commensals probably maintain some level of HMGB1 function and expression. In DSS-colitis, BECN1 was expression upregulated at mRNA and protein levels, but cleaved in the intestine of Hmgb1ΔIEC mice [Citation4]. In the Salmonella-colitis, HMGB1 may involve in different pathogenic pathways and mechanisms. Dysbiosis, caused by pathogens, diet, and other perturbation could alter HMGB1 function and expression, rendering the host more susceptible to infection and inflammation.
In summary, our studies indicate that intracellular intestinal HMGB1 is essential for limiting the extent and severity of bacterial inflammation and infection by controlling STAT3 in inflammation and autophagy. Intestinal epithelial HMGB1 is directly involved in the suppression of bacteria-induced STAT3 activation and the protection of intestine tissues from bacterial infection and injury. We reveal that intestinal HMGB1 is essential for reducing tissue injury in infection. Our data suggest the directions in which the work might be built on going forward.
Materials and methods
Human tissue samples
This study was performed in accordance with approval from the University of Rochester Ethics Committee (RSRB00037178). Colorectal tissue samples were obtained with informed consent from the sigmoid colon of patients exhibiting no apparent intestinal pathology and from the mucosa of patients undergoing anterior resection. All pathologic diagnoses were evaluated by pathologists. The UC cohort consisted of 16 patients with previous diagnosis of UC (10:6 female:male, mean age = 66.6 in the range of 51–83), while the age-matched control cohort consists of 10 patients (6:4 female:male, mean age = 65.7 age in the range of 44–85) without previous diseases in gastrointestinal tract. All of patient samples were obtained at the University of Rochester Medical Center from 2008 to 2012.
Animal
HMGB1 flox mice (Hmgb1loxP/loxP) were originally reported by Dr. Chang [Citation4]. Hmgb1ΔIEC mice were obtained by crossing the HMGB1 flox mice with Vil1/villin-Cre mice (Jackson Laboratory, 004586, Bar Harbor, ME, USA), as we previously reported [Citation44]. Experiments were performed on 6–8-wk-old mice. All animal work was approved by the University of Rochester, Rush University Committee on Animal Resources, and UIC Animal Care Committee (ACC).
Bacterial strains and growth condition
Salmonella Typhimurium wild-type strain ATCC14028 is used in this study. Non-agitated microaerophilic bacterial cultures were prepared as previously described [Citation45].
Streptomycin pre-treated mouse model
Animal experiments were performed using specific-pathogen-free female Hmgb1loxP/loxP and Hmgb1ΔIEC that were 6–8-wk old, as previously described [Citation22]. Water and food were withdrawn 4 h before oral gavage with 7.5 mg/mouse of streptomycin (100 µl of sterile solution or 100 µl of sterile water in control). Afterwards, animals were supplied with water and food ad libitum. Twenty h after streptomycin treatment, water and food were withdrawn again for 4 h before the mice were infected with 1 × 107 colony-forming units of S. Typhimurium (100 µl suspension) in Hanks balanced salt solution (HBSS) or treated with sterile HBSS (control). Eight h or 4 d after infection, mice were sacrificed, and tissue samples from the intestinal tracts were removed for analysis.
Stattic-treated mice model
Hmgb1loxP/loxP and Hmgb1ΔIEC (female, 6–8-wks old) mice were randomly assigned to vehicle (sterile phosphate-buffered saline [PBS] with 1% Tween 80 [Sigma-aldrich, P5188]) or 50 mg/kg Stattic (Enzo Life Sciences, BML-EI368) by oral gavage [Citation46]. Stattic or vehicle was administered daily. After 3-d pretreatment, mice were infected with S. Typhimurium using a streptomycin pre-treated mouse model as described above. Four d post-infection, mice were sacrificed, and tissue samples from the intestinal tracts were harvested for analysis.
Cell culture
Human colonic epithelial HCT116 cells, Hmgb1+/+ and HMGB1-/- MEF cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, penicillin-streptomycin (penicillin, 100 I.U./ml, streptomycin, 100 µg/ml), and L-glutamine (4.5 g/L), as previously described [Citation47,Citation48].
Mouse colonoids culture and treatment with Salmonella
Hmgb1loxP/loxP and Hmgb1ΔIEC mouse colonoids were prepared and maintained as previously described [Citation31,Citation49]. Mini gut medium (advanced DMEM/F12 supplemented with HEPES, L-glutamine, N2, and B27) was added to the culture, along with RSPO/R-Spondin (Sigma-Aldrich, SRP3292), NOG/Noggin (Sigma-Aldrich, SRP3227), epidermal growth factor (EGF) (Sigma-Aldrich, SRP3196), and WNT3A (Abcam, ab81484). At day 7 after passage, colonoids were colonized with Salmonella. The colonoid area was measured using ImageJ. Region of interest (ROI) was selected by using a circle tool. Each analysis was performed in a triplicate of each cell. A total of 10 images per sample were analyzed.
Immunofluorescence
Colonic tissues were freshly isolated and embedded in paraffin wax after fixation with 10% neutral buffered formalin. Immunofluorescence was performed on paraffin-embedded sections (4 μm), after preparation of the slides as described previously [Citation48] followed by incubation for 1 h in blocking solution (2% bovine serum albumin [Gemini Bio-products, 700-102P], 1% goat serum [Jackson ImmunoResearch Laboratories, 005–000-121] in HBSS) to reduce nonspecific background. Cultured cells were rinsed 3 times with HBSS, fixed for 20 min in 100% cold ETOH, and permeabilized for 10 min with 0.2% Triton X-100 (Sigma-Aldrich, X100), followed by 3 rinses with HBSS, and incubation for 30 min in 10% BSA in HBSS to reduce non-specific background. The permeabilized cells or tissue samples were incubated with primary antibodies for 90 min at room temperature. Samples were then incubated with goat anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A11034) or goat anti-mouse Alexa Fluor 594 (Thermo Fisher Scientific, A11032) and DAPI (Thermo Fisher Scientific, D1306) for 1 h at room temperature. Tissues or cells were mounted with SlowFade (Thermo Fisher Scientific, S2828), followed by a coverslip, and the edges were sealed to prevent drying. Specimens were examined with a Leica SP5 Laser Scanning confocal microscope.
Analysis of STAT3 distribution
Fluorescence images were analyzed using image analysis software LSM 710 META (Carl Zeiss Inc., Oberkochen, Germany). Each analysis was performed in triplicate from each tissue section on a total of 10 images per mouse sample.
Immunoblotting
Mouse colonic epithelial cells were collected by scraping the tissue from the colon of the mouse, including the proximal and distal regions [Citation22,Citation47]. The cells were sonicated in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, pH 8.0, 1% Triton X-100) with 0.2 mM sodium ortho-vanadate, and protease inhibitor cocktail (Sigma-Aldrich, S8830). The protein concentration was measured using the BioRad Reagent (BioRad, Hercules, CA, USA). Cultured cells were rinsed twice with ice-cold HBSS, lysed in protein loading buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol), and then sonicated. Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and immunoblotted with primary antibodies. The following antibodies were used: anti-p-STAT3 (Cell Signaling Technology, 9145), anti-STAT3 (Cell Signaling Technology, 9132), anti-HMGB1 (Cell Signaling Technology, 3935), anti-LC3B (Cell Signaling Technology, 2775), anti-p-NFKBIA/IΚBΑ (Cell Signaling Technology, 9246), anti-NFKBIA/IΚBΑ (Cell Signaling Technology, 4814), anti-p-NFKB/p65 (Cell Signaling Technology, 3033), anti-NFKB (Cell Signaling Technology, 8242), anti-p-MAPK/JNK (Cell Signaling Technology, 9251), anti-MAPK/JNK (Cell Signaling Technology, 9258), anti-BECN1 (Santa Cruz Biotechnology, SC-10086), anti-VIL1 (Santa Cruz Biotechnology, SC-7672), anti-SP1 (Santa Cruz Biotechnology, SC-14027), anti-TLR2 (Santa Cruz Biotechnology, SC-73181), anti-TLR4 (Santa Cruz Biotechnology, SC-293072), anti-TLR5 (Santa Cruz Biotechnology, SC-30003), anti-SQSTM1/p62 (BD Biosciences, 610832), anti-ATG16L1 (Abgent Biotechology Company, AP1817b) or anti-ACTB/β-ACTIN (Sigma-Aldrich, A5316) antibodies and were visualized with ECL (Thermo Fisher Scientific, 32106). Membranes that were probed with more than one antibody were stripped before re-probing.
HMGB1 siRNA
HCT116 cells were grown in 12-well plates. The cells were transfected with on-Target plus smart pool human HMGB1 siRNA (Dharmacon Inc., L-018981) or scrambled siRNA control (Santa Cruz Biotechnology, SC-37007) using Superfect reagent (QIAGEN Inc., 301305). After transfection for 72 h, cells were colonized by Salmonella for 30 min, washed, and incubated for 30 min in DMEM with Gentamicin (500 µg/ml; Sigma-Aldrich, G1914).
Fractionation
The adherent cells were harvested with trypsin-EDTA and then centrifuged at 500 g for 5 min. The cells were washed with phosphate-buffered Saline (PBS) and pelleted at 500 g for 2–3 min. Nuclear and cytoplasmic fractions of the cells were extracted using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78,833) according to the manufacturer’s instructions.
Coimmunoprecipitation assay
Cells were rinsed twice with ice-cold HBSS and lysed in cold immunoprecipitation buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, pH 8.0, 1% Triton X-100) with 0.2 mM sodium ortho-vanadate, and protease inhibitor cocktail. Samples were precleared with protein A-agarose (Thermo Fisher Scientific, 15,918,014). Pre-cleared lysates were incubated with 2 µg of primary antibodies for 1 h at 4°C. Protein A-agarose was added to the lysate and incubated for 30 min with agitation at 4°C before being washed with cold immunoprecipitation buffer. The pellet was resuspended in 0.1 M glycine, pH 2.5, and incubated with agitation for 10 min at 4°C and then centrifuged at 9,000 g for 2 min. The supernatant was removed and neutralized with 1 M Tris·HCl, pH 8.0. The samples were diluted with concentrated (5×) electrophoresis sample buffer (125 mM Tris, pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue) with 2% β-mercaptoethanol, boiled for 5 min, separated by SDS-polyacrylamide gel electrophoresis, and transferred onto a nitrocellulose membrane. Membrane blots were probed with secondary antibody and visualized by ECL.
S. Typhimurium invasion of Hmgb1+/+ and Hmgb1-/- cells
Infection of MEF cells was performed by a previously described method [Citation48,Citation50,Citation51]. Bacterial solution was added, and bacterial invasion was assessed after 30 min. Cell-associated bacteria, representing bacteria adhered to and/or internalized into the monolayers, were released by incubation with 100 μl of 1% Triton X-100. Internalized bacteria were those obtained from lysis of the epithelial cells with 1% Triton X-100 30 min after the addition of gentamicin (500 μg/ml). For both cell-associated and internalized bacteria, 0.9 ml of LB broth was added, and each sample was vigorously mixed and quantified by plating for colony-forming units on MacConkey agar medium.
Real-time quantitative PCR
Total RNA was extracted from epithelial cell monolayers or mouse colonic epithelial cells using TRIzol reagent (Thermo Fisher Scientific, 15,596,026). The RNA integrity was verified by gel electrophoresis. RNA reverse transcription was done using the iScript cDNA synthesis kit (Bio-Rad Laboratories, 1,708,891) according to the manufacturer’s directions. The RT-cDNA reaction products were subjected to quantitative real-time PCR using the MyiQ single-color real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA) and iTaqTM Universal SYBR Green supermix (Bio-Rad Laboratories, 1,725,121) according to the manufacturer’s directions. All expression levels were normalized to actb levels of the same sample. Percent expression was calculated as the ratio of the normalized value of each sample to that of the corresponding untreated control cells. All real-time PCR reactions were performed in triplicate. All PCR primers () were designed using Lasergene software (DNAStar Inc, Madison, WI, USA).
Statistical analysis
All of the data are expressed as means ± SD. Differences between 2 samples were analyzed by Student’s t-test. Differences among 3 or more groups were analyzed using ANOVA with GraphPad Prism 5. P values of 0.05 or less were considered statistically significant.
Supplemental Material
Download PDF (2.2 MB)Acknowledgments
We would like to acknowledge the NIDDK grants R01 DK105118 and R01DK114126 (JS), the UIC Cancer Center, Crohn’s and Colitis Foundation of America (JSM), and DDRCC (NIDDK P30DK42086) (EBC). The study sponsors play no role in the study design or in the collection, analysis, and interpretation of date.
Disclosure statement
YZ, XZ, RL, JSM, YX, EBC and JS declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342.
- Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162.
- Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251.
- Zhu X, Messer JS, Wang Y, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125(3):1098–1110.
- Mshvildadze M, Neu J. The infant intestinal microbiome: friend or foe? Early Hum Dev. 2010;86(Suppl 1):67–71.
- Hooper LV. Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol. 2009;7(5):367–374.
- Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307.
- Bates JM, Akerlund J, Mittge E, et al. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2(6):371–382.
- Santaolalla R, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2012;28(2):124–129.
- Swamy M, Jamora C, Havran W, et al. Epithelial decision makers: in search of the ‘epimmunome‘. Nat Immunol. 2010;11(8):656–665.
- Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892.
- Kang R, Livesey KM, Zeh HJ 3rd, et al. HMGB1 as an autophagy sensor in oxidative stress. Autophagy. 2011;7(8):904–906.
- Fu XY. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006;16(2):214–219.
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809.
- Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46(8):812–814.
- You L, Wang Z, Li H, et al. The role of STAT3 in autophagy. Autophagy. 2015;11(5):729–739.
- Shen S, Niso-Santano M, Adjemian S, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48(5):667–680.
- Wang YX, van Boxel-Dezaire AHH, Cheon H, et al. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A. 2013;110(42):16975–16980.
- Niemand C, Nimmesgern A, Haan S, et al. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Iimmunol. 2003;170(6):3263–3272.
- Kim HS, Kim DC, Kim HM, et al. STAT1 deficiency redirects IFN signalling toward suppression of TLR response through a feedback activation of STAT3. Sci Rep. 2015;5:13414.
- Kong LY, Gelbard A, Wei J, et al. Inhibition of p-STAT3 enhances IFN-alpha efficacy against metastatic melanoma in a murine model. Clin Cancer Res off J Am Assoc Cancer Res. 2010;16(9):2550–2561.
- Duan Y, Liao AP, Kuppireddi S, et al. Beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest. 2007;87(6):613–624.
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71(5):2839–2858.
- Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255(3):332–343.
- Kalinina N, Agrotis A, Antropova Y, et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol. 2004;24(12):2320–2325.
- Saenz R, Futalan D, Leutenez L, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211.
- Van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11(1):91–99.
- Zhang QC, Petrey D, Deng L, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490(7421):556–560.
- Zhang QC, Petrey D, Garzon JI, et al. PrePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res. 2013;41:828–833.
- Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265.
- Zhang YG, Wu S, Xia Y, et al. Salmonella-infected crypt-derived intestinal organoid culture system for host-bacterial interactions. Physiol Rep. 2014;2(9):e12147.
- Engevik MA, Yacyshyn MB, Engevik KA, et al. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol. 2015;308(6):510–524.
- VanDussen KL, Marinshaw JM, Shaikh N, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64(6):911–920.
- Herfarth HH, Mohanty SP, Rath HC, et al. Interleukin 10 suppresses experimental chronic, granulomatous inflammation induced by bacterial cell wall polymers. Gut. 1996;39(6):836–845.
- Du Toit A. Autophagy: STAT3 maintains order. Nat Rev Mol Cell Biol. 2012;13(12):754.
- Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101(1):296–301.
- Xu YJ, Li L, Chen Y, et al. Role of HMGB1 in regulation of STAT3 expression in CD4(+) T cells from patients with aGVHD after allogeneic hematopoietic stem cell transplantation. Clin Immunol. 2015;161(2):278–283.
- Wang G, Zhang J, Dui D, et al. High mobility group box 1 induces the activation of the Janus kinase 2 and signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway in pancreatic acinar cells in rats, while AG490 and rapamycin inhibit their activation. Bosn J Basic Med Sci. 2016;16(4):307–312.
- Lu R, Wu S, Zhang YG, et al. Salmonella protein AvrA activates the STAT3 signaling pathway in colon cancer. Neoplasia. 2016;18(5):307–316.
- Lu R, Zhang YG, Sun J. STAT3 activation in infection and infection-associated cancer. Mol Cell Endocrinol. 2017;451:80–87.
- Elschami M, Scherr M, Philippens B, et al. Reduction of STAT3 expression induces mitochondrial dysfunction and autophagy in cardiac HL-1 cells. Eur J Cell Biol. 2013;92(1):21–29.
- Vitali R, Stronati L, Negroni A, et al. Fecal HMGB1 is a novel marker of intestinal mucosal inflammation in pediatric inflammatory bowel disease. Am J Gastroenterol. 2011;106(11):2029–2040.
- Maeda S, Hikiba Y, Shibata W, et al. Essential roles of high-mobility group box 1 in the development of murine colitis and colitis-associated cancer. Biochem Biophys Res Commun. 2007;360(2):394–400.
- Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64(7):1082–1094.
- Sun J, Hobert ME, Rao AS, et al. Bacterial activation of beta-catenin signaling in human epithelia. Am J Physiol Gastrointest Liver Physiol. 2004;287(1):220–227.
- Adachi M, Cui C, Dodge CT, et al. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncol. 2012;48(12):1220–1226.
- Sun J, Hobert ME, Duan Y, et al. Crosstalk between NF-kappaB and beta-catenin pathways in bacterial-colonized intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2005;289(1):129–137.
- Lu R, Wu S, Liu X, et al. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PloS one. 2010;5(5):e10505.
- Lu R, Voigt RM, Zhang Y, et al. Alcohol injury damages intestinal stem cells. Alcohol Clin Exp Res. 2017;41(4):727–734.
- McCormick BA, Colgan SP, Delp-Archer C, et al. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123(4):895–907.
- Wu S, Liao AP, Xia Y, et al. Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. Am J Pathol. 2010;177(2):686–697.