
ABSTRACT
Alzheimer disease (AD) is the most common neurodegenerative disease. An imbalance between the production and clearance of Aβ (amyloid beta) is considered to be actively involved in AD pathogenesis. Macroautophagy/autophagy is a major cellular pathway leading to the removal of aggregated proteins, and upregulation of autophagy represents a plausible therapeutic strategy to combat overproduction of neurotoxic Aβ. PPARA/PPARα (peroxisome proliferator activated receptor alpha) is a transcription factor that regulates genes involved in fatty acid metabolism and activates hepatic autophagy. We hypothesized that PPARA regulates autophagy in the nervous system and PPARA-mediated autophagy affects AD. We found that pharmacological activation of PPARA by the PPARA agonists gemfibrozil and Wy14643 induces autophagy in human microglia (HM) cells and U251 human glioma cells stably expressing the human APP (amyloid beta precursor protein) mutant (APP-p.M671L) and this effect is PPARA-dependent. Administration of PPARA agonists decreases amyloid pathology and reverses memory deficits and anxiety symptoms in APP-PSEN1ΔE9 mice. There is a reduced level of soluble Aβ and insoluble Aβ in hippocampus and cortex tissues from APP-PSEN1ΔE9 mice after treatment with either gemfibrozil or Wy14643, which promoted the recruitment of microglia and astrocytes to the vicinity of Aβ plaques and enhanced autophagosome biogenesis. These results indicated that PPARA is an important factor regulating autophagy in the clearance of Aβ and suggested gemfibrozil be assessed as a possible treatment for AD.
Abbreviation: Aβ: amyloid beta; ACTB: actin beta; ADAM10: ADAM metallopeptidase domain 10; AD: Alzheimer disease; AIF1/IBA1: allograft inflammatory factor 1; ANOVA: analysis of variance; APOE: apolipoprotein E; APP: amyloid beta precursor protein; APP-PSEN1ΔE9: APPswe/PSEN1dE9; BAFA1: bafilomycin A1; BDNF: brain derived neurotrophic factor; BECN1: beclin 1; CD68: CD68 molecule; CREB1: cAMP responsive element binding protein 1; DAPI: 4‘,6-diamidino-2-phenylindole; DLG4/PSD-95: discs large MAGUK scaffold protein 4; DMSO: dimethyl sulfoxide; ELISA: enzyme linked immunosorbent assay; FDA: U.S. Food and Drug Administration; FKBP5: FK506 binding protein 5; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; gemfibrozil: 5-(2,5-dimethylphenoxy)-2,2-dimethylpentanoic acid; GFAP: glial fibrillary acidic protein; GLI2/THP1: GLI family zinc finger 2; HM: human microglia; IL6: interleukin 6; LAMP1: lysosomal associated membrane protein 1; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; NC: negative control; OQ: opposite quadrant; PPARA/PPARα, peroxisome proliferator activated receptor alpha; PSEN1/PS1: presenilin 1; SEM: standard error of the mean; SQSTM1: sequestosome 1; SYP: synaptophysin; TFEB: transcription factor EB; TNF/TNF-α: tumor necrosis factor; TQ: target quadrant; WT: wild type; Wy14643: 2-[4-chloro-6-(2,3-dimethylanilino)pyrimidin-2-yl]sulfanylacetic acid
Introduction
Alzheimer disease (AD) is an irreversible, age-associated neurodegenerative disease that is characterized by progressive memory loss and cognitive decline. The finding of extracellular senile plaques comprising Aβ (amyloid beta) is one of the main neuropathological hallmarks of AD [Citation1–Citation3]. It has been suggested that excessive Aβ production and deficits in Aβ clearance play pivotal or causative roles in AD pathogenesis and impaired Aβ clearance in the brain is one of the primary contributing factors [Citation4]. Therefore, the enhancement of brain Aβ clearance is one of the promising strategies for AD prevention and treatment [Citation5–Citation8].
Macroautophagy, herein referred to as autophagy, is a major cellular pathway by which unwanted protein material is aggregated in intracellular autophagosomes, which are then degraded in lysosomes [Citation9]. Autophagy dysfunction has been implicated in the pathogenesis of many neurodegenerative diseases [Citation9–Citation11]. In AD patients, MTOR (mechanistic target of rapamycin kinase) signaling, an important signaling pathway controlling autophagy, is enhanced leading to a lower level of autophagy [Citation12]. Deletion of BECN1/Beclin1, a key protein for the formation of autophagosomes, increases both the intracellular and extracellular Aβ loads in AD cell and animal models [Citation13,Citation14]. TFEB (transcription factor EB), a regulator of the autophagy-lysosome pathway, mediated activation of autophagy in various models and decreased Aβ pathology [Citation15,Citation16]. Moreover, previous studies have shown that autophagy has an essential role against the development of a number of neurodegenerative diseases including AD [Citation17–Citation21]. Therefore, induction of autophagy may serve as a viable therapeutic target for the treatment of AD [Citation11,Citation22–Citation25].
Nuclear receptor PPARA/PPARα (peroxisome proliferator-activated receptor alpha), encoded by the PPARA gene, is a PPAR isoform that regulates the expression of genes involved in glucose and lipid metabolism and inflammatory processes [Citation26–Citation28] by binding to PPAR response elements in the promoter region of the genes [Citation29]. Therefore PPARA acts as a key regulator of energy metabolism, and mitochondrial and peroxisomal function [Citation30]. Enhanced PPARA expression protects against cardiovascular disorders though its role of enhancing PPARG expression [Citation30]. Activation of PPARA reverses the normal suppression of autophagy in the fed state and induces autophagic lipid degradation [Citation31], and mediates innate host defense [Citation32]. Genetic variants in the PPARA gene have been found to be associated with AD [Citation33]. PPARA also regulates neuronal ADAM10 (ADAM metallopeptidase domain 10) expression and stimulates APP (amyloid beta precursor protein) processing towards the α-secretase pathway [Citation34]. Statins, as ligands of PPARA, increase BDNF (brain derived neurotrophic factor) expression and improves memory and learning in AD mice via the PPARA-CREB1 (cAMP responsive element binding protein 1) pathway [Citation35]. Whether PPARA-mediated autophagy is involved in the pathogenesis of AD remains undetermined.
In this study, we hypothesized that activation of PPARA would directly regulate autophagy in the nervous system and reduce the AD-related pathology in a mouse model. By using cellular and animal models, we showed that PPARA is a potential therapeutic target for AD and PPARA-mediated autophagy explains the potential beneficial effect of PPARA agonists in AD.
Results
PPARA agonists induce autophagy in a PPARA-dependent manner
In order to test whether activation of PPARA would affect autophagy in glial cells, we treated human microglia (HM) cells with two known PPARA activators, gemfibrozil, a U.S. Food and Drug Administration (FDA)-approved drug primarily used to treat hyperlipidemia [Citation36,Citation37], and Wy14643 [Citation31]. An increased protein level of the lipidated (PE-conjugated) form of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta; LC3B-II):LC3B-I and a decreased protein level of SQSTM1/p62 (sequestosome 1) in a dose-dependent manner were observed in HM cells treated with gemfibrozil and Wy14643 (). We confirmed these results in human glioma U251 cells stably expressing the human APP mutant (APP-p.M671L) (U251-APP cells) that were created in our previous study [Citation38] (Fig. S1A-D). We found oral administration of gemfibrozil and Wy14643 in drinking water upregulated autophagy in mouse hippocampus and cortex tissues compared with the vehicle group (dimethyl sulfoxide [DMSO] only) (Fig. S1E-H).
Figure 1. PPARA agonists induce autophagy in HM cells and this effect is PPARA-dependent. Dose-dependent increase of LC3B-II:LC3B-I and decrease of SQSTM1 protein levels in HM cells treated with either gemfibrozil (a-b) or Wy14643 (c-d). Knockdown of the PPARA gene reversed the altered protein levels of LC3B-II:LC3B-I and SQSTM1 in response to gemfibrozil (e-f) and Wy14643 (g-h). HM cells were treated with different concentrations of gemfibrozil and Wy14643 for 24 h before harvest for western blotting. Relative protein abundance was normalized to ACTB. Data are representative of 3 independent experiments with similar results. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey’s post-hoc test. Bars represent mean ± SEM.
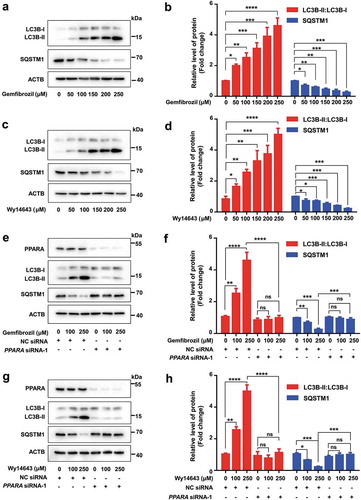
Next, we used siRNAs to knockdown the PPARA gene and characterized the effect of PPARA in gemfibrozil and Wy14643-induced autophagy. We designed 3 siRNAs for PPARA and found that siRNA-1 at a concentration of 25 nM had the best inhibitory effect (Fig. S2). This siRNA was used in the subsequent knockdown assays. The basal level of autophagy and lysosomal biogenesis could be blocked after knockdown of PPARA in HM cells, as demonstrated by the decreased protein levels of autophagy-related proteins BECN1 and LC3B-II:LC3B-I and lysosome-related proteins TFEB and LAMP1 (lysosomal associated membrane protein 1), and increased protein level of SQSTM1 (Fig. S3A-B). In contrast, autophagy and lysosomal biogenesis were enhanced in HM cells with an overexpression of PPARA (Fig. S3C-D). HM cells transfected with the negative control (NC) siRNA showed a pronounced autophagy induction upon gemfibrozil and Wy14643 treatment, whereas knockdown of PPARA reduced the autophagy induced by gemfibrozil and Wy14643 (). These results could be validated in U251-APP cells (Fig. S4). Consistently, the increase of BECN1, TFEB and LAMP1 levels in response to gemfibrozil and Wy14643 treatment could be counteracted by knockdown of PPARA (Fig. S5). Furthermore, the autophagy induced by gemfibrozil and Wy14643 could be abolished by a pretreatment with bafilomycin A1 (BAFA1) (Fig. S6), an inhibitor of the vacuolar (V)-type H+-translocating ATPase that results in blockage of autophagosome-lysosome fusion and accumulation of LC3B-II [Citation39]. These results suggested that PPARA is a positive regulator of autophagy and mediates the autophagy induced by gemfibrozil and Wy14643.
Knockdown of PPARA reverses the increased autophagic flux in response to PPARA agonists in HM cells and U251-APP cells
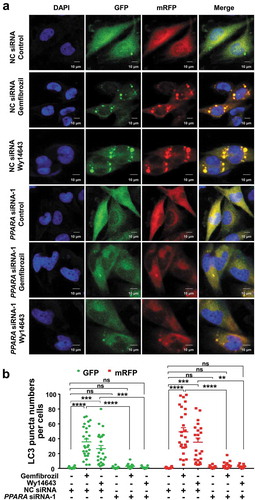
We introduced the tandem monomeric RFP-GFP-tagged LC3 (mRFP-GFP-LC3) reporter into HM and U251-APP cells to determine the effect of gemfibrozil and Wy14643 on autophagic flux. mRFP-GFP-LC3 distribution was evaluated in HM and U251-APP cells treated with rapamycin (an autophagy inducer, as a positive control) or a PPARA agonist. The mRFP-GFP-LC3 in autolysosomes displayed red fluorescence as the GFP signal was sensitive to the acidic condition whereas the mRFP signal was more stable in the lysosome lumen [Citation40]. We observed an increased number of red puncta in the rapamycin-treated HM and U251-APP cells (Fig. S7), indicating increased autophagic flux. Treatment with PPARA agonists gemfibrozil and Wy14643 had a similar effect on the increase of autophagic flux as rapamycin (). Knockdown of PPARA resulted in a few red puncta in HM cells treated with gemfibrozil and Wy14643 (), suggesting that PPARA knockdown blocked the autophagic flux induced by PPARA agonists. Similar results were observed in the U251-APP cells (Fig. S8). Collectively, these results demonstrated the PPARA-mediated autophagy induced by PPARA agonists gemfibrozil and Wy14643.
Figure 2. Knockdown of PPARA reverses the increased autophagic flux in response to gemfibrozil and Wy14643 treatment in HM cells. Cells were transfected with negative control (NC) siRNA (25 nM) and PPARA siRNA-1 (25 nM) for 24 h, followed by an infection with mRFP-GFP-LC3 lentivirus for 24 h. Infected cells were treated with either gemfibrozil or Wy14643 (200 µM) for 24 h. (a) Gemfibrozil and Wy14643 treatment increased the maturation of autolysosomes as shown by the increased red puncta of mRFP-GFP-LC3 in cells, and this effect could be restored by knockdown of the PPARA gene. (b) Quantification of LC3 puncta in (a). DAPI for nucleus, GFP for autophagosome and mRFP for autolysosome. Data shown are representative of 3 experiments. ns, not significant; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey’s post-hoc test. Bars represent mean ± SEM.
PPARA agonists improve memory impairment in APP-PSEN1ΔE9 mice
We used 8-month-old APPswe/PSEN1dE9 (hereafter referred as APP-PSEN1ΔE9), which have the mutated human APP (Swedish mutations K595N/M596L) and also the human PSEN1/PS1 (presenilin 1) deletion [Citation41], to examine the therapeutic efficacy of gemfibrozil and Wy14643 following the procedure described in previous studies for drug delivery in AD mice [Citation42–Citation44]. In brief, the APP-PSEN1ΔE9 mice and age-matched wild-type (WT) littermates were given gemfibrozil (50 µg/mL), Wy14643 (50 µg/mL) or DMSO in drinking water for 2 months, followed by a Morris water maze task. All the APP-PSEN1ΔE9 mice or WT littermates managed the task after a 7-day training period, although there was a significantly impaired learning ability in APP-PSEN1ΔE9 mice as compared to their WT littermates (). We observed a significantly improved learning in the APP-PSEN1ΔE9 mice given a PPARA agonist relative to the vehicle group (). It should be noted that the swim speed was comparable in all groups of the APP-PSEN1ΔE9 mice and their WT littermates regardless of the treatment with a PPARA agonist, which indicated there was no neuromotor differences among the groups ().
Figure 3. Administration of PPARA agonists rescues spatial memory impairment in APP-PSEN1ΔE9 mice. (a) Morris water maze behavioral assessment for APP-PSEN1ΔE9 mice or WT littermates treated with either DMSO (Vehicle) or PPARA agonist (either gemfibrozil or Wy14643) showing differences in escape latency, path length and swim speed during learning session. (b) Probe trial performance at 4 h (short-term memory). In the absence of drug treatment, the APP-PSEN1ΔE9 mice had a poorer performance than the untreated littermates, as shown by no preference for the trained target quadrant (TQ) (as demonstrated by percent of time and percent of distance in the target quadrant) and fewer crosses over the platform location. After the administration of gemfibrozil and Wy14643 for 2 months, APP-PSEN1ΔE9 mice showed a clear preference for the trained target quadrant and increased crosses over the platform location. OQ, opposite quadrant. (c) Probe trial performance at 72 h. Treatment with gemfibrozil and Wy14643 improved the performance of APP-PSEN1ΔE9 mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; one-way ANOVA with the Tukey’s post-hoc test. Bars represent mean ± SEM.
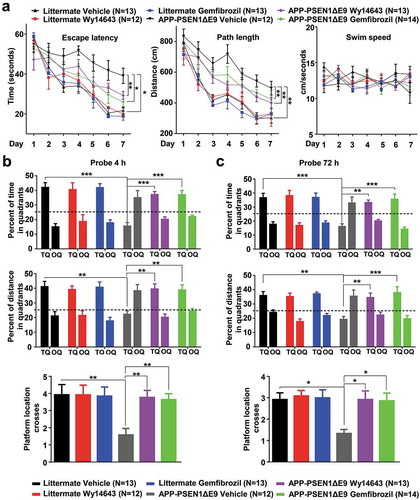
We used a 4-h probe trial and a 72-h probe trial after the last training session to evaluate short-term memory and long-term memory for spatial reference, respectively. The WT littermates treated with vehicle or PPARA agonist showed similar preferences during both tests. There was a strong memory impairment in APP-PSEN1ΔE9 mice compared to WT littermates treated with vehicle (using % time and % distance in the target quadrant as readouts; ). Gemfibrozil and Wy14643-treated APP-PSEN1ΔE9 mice showed a clear preference for the target quadrant compared with the APP-PSEN1ΔE9 vehicle group, with a comparable preference to that of the WT littermates vehicle group for both tests. The APP-PSEN1ΔE9 mice treated with gemfibrozil and Wy14643 showed an increased number of platform location crosses compared to the vehicle group (-)). These results suggested a beneficial effect of PPARA activation by either gemfibrozil or Wy14643 treatment during the memory consolidation phase in APP-PSEN1ΔE9 mice.
PPARA agonists reduce anxiety and enhances structural neuroplasticity in APP-PSEN1ΔE9 mice
Progressive cognitive decline and increased anxiety are two important clinical hallmarks observed in AD patients [Citation45]. In order to see if gemfibrozil and Wy14643 might exert anxiolytic effects in APP-PSEN1ΔE9 mice we used the open field test [Citation46]. Automated video tracking analysis showed no significant difference in locomotor activity between the WT littermates and APP-PSEN1ΔE9 mice with or without a PPARA agonist (). However, gemfibrozil and Wy14643 administration led to reduced anxiety in APP-PSEN1ΔE9 mice, as the treated animals spent significantly more time in the center area when compared to the vehicle group (). This suggested that pharmacological activation of PPARA in APP-PSEN1Δ9 mice leads to a reduction in anxiety. We further verified the effect of gemfibrozil and Wy14643 treatment on the molecular and structural signatures of anxiety and cognition. In line with the behavioral result, we found that gemfibrozil and Wy14643 treatment suppressed the protein level of anxiety-modulating FKBP5 (FK506 binding protein 5) [Citation47] in the cortex, but not in the hippocampus tissues, of APP-PSEN1ΔE9 mice compared to the vehicle group (). Note that there was no obvious effect of PPARA agonists on the protein levels of FKBP5 in cortex and hippocampus tissues of the WT littermates (Fig. S9).
Figure 4. PPARA agonists reverse the signatures of anxiety and enhance structural neuroplasticity in APP-PSEN1ΔE9 mice. (a) Representative paths in the open field for APP-PSEN1ΔE9 mice and WT littermates treated with either DMSO (Vehicle) or a PPARA agonist (either gemfibrozil or Wy14643). (b) Quantification of total distance travelled in the open field (left) and percentage of time stayed in the center area (right). (c-d) Decreased protein levels of anxiety-modulating FKBP5 in the cortex and hippocampus tissues of APP-PSEN1ΔE9 mice treated with vehicle or PPARA agonist (gemfibrozil or Wy14643). (e-f) Increased protein levels of structural plasticity markers SYP (synaptophysin) and DLG4 in the cortex and hippocampus tissues of APP-PSEN1ΔE9 mice treated with vehicle or PPARA agonist (gemfibrozil and Wy14643). (g) Representative images of DLG4 immunostaining in hippocampus from APP-PSEN1ΔE9 mice receiving PPARA agonists or vehicle. (h) Quantification of the DLG4 immunoreactivity in (g). (i) Representative images of Golgi-Cox-impregnated hippocampal neurons and magnified dendritic spines from APP-PSEN1ΔE9 mice and WT littermates treated with vehicle or PPARA agonist (gemfibrozil and Wy14643). (j) Quantification of the spine density in (i). Data are representative of 3 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.001; one-way ANOVA with the Tukey’s post-hoc test. Bars represent mean ± SEM.
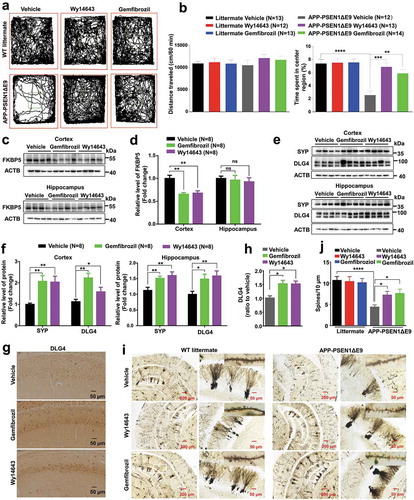
We also assessed the effect of PPARA agonists on presynaptic and postsynaptic integrity, as the APP-PSEN1ΔE9 mice showed improved learning and memory (). The protein levels of structural neuroplasticity markers SYP (synaptophysin) and DLG4/PSD-95 (discs large MAGUK scaffold protein 4) were significantly increased in cortex and hippocampus tissues of APP-PSEN1ΔE9 mice after gemfibrozil and Wy14643 administration (). Accordingly, immunohistochemical analysis also showed an increase of immunoreactivity for DLG4 in hippocampus tissues of APP-PSEN1ΔE9 mice after administration of PPARA agonists (). There was an increase of spine density in Golgi-Cox-impregnated neurons from hippocampus tissues of APP-PSEN1ΔE9 mice treated with gemfibrozil and Wy14643 as compared with the vehicle group, whereas gemfibrozil and Wy14643 treatment had no apparent effect on the alterations of dendritic spines in the hippocampus tissues of WT littermates (-). Taken together, these results demonstrated that PPARA agonists could reduce the synaptic dysfunction, anxiety symptoms and memory deficits of APP-PSEN1ΔE9 mice.
PPARA agonists induce autophagy and alleviate hippocampal and cortex amyloid pathology
As gemfibrozil is a drug for treating hyperlipidemia in adults approved by the FDA, we performed a hematoxylin and eosin staining to show potential hepatotoxic effects of gemfibrozil and Wy14643 on liver tissue. We found no obvious hepatic abnormalities in WT littermates and APP-PSEN1ΔE9 mice administrated with gemfibrozil or Wy14643 compared to the vehicle group (Fig. S10). We then characterized the effect of PPARA agonists on the pathological conditions of AD in APP-PSEN1ΔE9 mice by looking at soluble Aβ levels and amyloid plaque depositions. Administration of PPARA agonists significantly reduced the amounts of soluble total Aβ in the hippocampus and cortex tissues of APP-PSEN1ΔE9 mice (). Specifically, soluble Aβ40 and Aβ42 species, which play major synaptotoxic roles in AD [Citation48], were decreased significantly in hippocampus and cortex tissues of APP-PSEN1ΔE9 mice with treatment of PPARA agonists relative to the vehicle group, although soluble Aβ42:Aβ40 was not changed in these tissues of APP-PSEN1ΔE9 mice with or without treatment of the PPARA agonists (). This result was further confirmed by the ELISA (enzyme linked immunosorbent assay) analysis and we found that administration of PPARA agonists in APP-PSEN1ΔE9 mice significantly reduced the amount of insoluble Aβ42 and Aβ40 in the hippocampus and cortex tissues, though the insolube Aβ42:Aβ40 was not significantly altered ( and Fig. S11). Immunohistochemical staining for coronal brain sections also showed a significant decrease of antibody 4G8-labeled amyloid plaque burden in APP-PSEN1ΔE9 mice after administration of PPARA agonists (Fig. S12).
Figure 5. PPARA agonists induce autophagy and alleviate hippocampal and cortex amyloid pathology in APP-PSEN1ΔE9 mice. (a-b) Levels of soluble total Aβ, Aβ42, Aβ40, and Aβ42:Aβ40 in the hippocampus and cortex tissues in APP-PSEN1ΔE9 mice treated with DMSO (vehicle) or PPARA agonist (either gemfibrozil or Wy14643) as determined by western blotting. (c) Levels of insoluble Aβ40, Aβ42, and Aβ42:Aβ40 in the hippocampus tissues of APP-PSEN1ΔE9 mice treated with DMSO (vehicle) or PPARA agonist (gemfibrozil or Wy14643) determined by ELISA. (d-e) Protein levels of PPARA, the autophagy markers (BECN1, LC3B-II:LC3B-I and SQSTM1) and the lysosome markers (TFEB and LAMP1) in the hippocampus tissues of APP-PSEN1ΔE9 mice. (f-i) Representative double staining image showing an increased colocalization of 4G8-labled amyloid plaques with LC3 (f-g) and 4G8-labled amyloid plaques with LAMP1 (h-i) in coronal brain sections from APP-PSEN1ΔE9 mices treated with or without PPARA agonist (gemfibrozil and Wy14643). Line graphs in (f) and (h) (right) indicated fluorescence intensity across the white dotted lines. (j-k) Western blotting analyse of autophagy markers LC3B-II:LC3B-I and SQSTM1 in cell lysates from U251-APP cells treated with BAFA1 (50 nM), gemfibrozil (200 μM), Wy14643 (200 μM), or both (BAFA1 and gemfibrozil, or BAFA1 and Wy14643). (l) Levels of extracellular Aβ42, Aβ40, and Aβ42:Aβ40 in the supernatant of U251-APP cells treated with BAFA1 (50 nM), gemfibrozil (200 μM), Wy14643 (200 μM), or both (BAFA1 and gemfibrozil, or BAFA1 and Wy14643). Data are representative of 3 independent experiments with similar results. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; one-way ANOVA with the Tukey’s post-hoc test. Bars represent mean ± SEM.
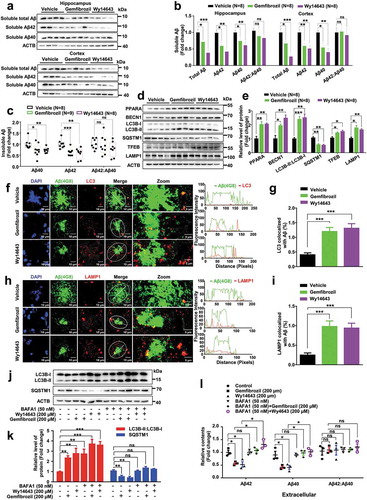
Autophagy was shown to be impaired in brain tissues of AD patients [Citation49] and murine models [Citation19]. To test whether the reduced level of Aβ in APP-PSEN1ΔE9 mice caused by administration of gemfibrozil and Wy14643 ( and Fig. S11–12) was associated with autophagy, we measured the protein levels of autophagy and lysosome marker proteins (BECN1, LC3B, SQSTM1, TFEB, and LAMP1) in hippocampus and cortex tissues of APP-PSEN1ΔE9 mice treated with or without a PPARA agonist. Consistent with the result of the cellular assay ( and Fig. S1A-D), we found a significant increase of autophagy in hippocampus and cortex tissues of APP-PSEN1ΔE9 mice in response to gemfibrozil and Wy14643 ( and Fig. S13). To further assess whether the effect of PPARA agonists on Aβ clearance was caused by activation of autophagy, double immunofluorescence staining of LC3 and Aβ (4G8), LAMP1 and Aβ (4G8), were performed in coronal brain sections of APP-PSEN1ΔE9 mice, respectively. Administration of gemfibrozil and Wy14643 resulted in an increased level of colocalization of LC3 with 4G8 () and LAMP1 with 4G8 (). We used BAFA1, an inhibitor of the vacuolar (V)-type ATPase that results in blockage of autophagosome-lysosome fusion and accumulation of LC3B-II [Citation39], to further show the role of autophagy induced by PPARA agonist in Aβ clearance in U251-APP cells. Autophagy was activated in U251-APP cells by gemfibrozil and Wy14643, as indicated by the increased LC3B-II:LC3B-I and decreased SQSTM1 protein levels (). This effect was associated with a decrease of extracellular Aβ40 and Aβ42 and intracellular Aβ40 and Aβ42 in comparison with untreated cells ( and Fig. S14). Treatment of BAFA1 (50 nM) alone raised the LC3B-II:LC3B-I, but with no effect on extracellular and intracellular Aβ40 and Aβ42 levels relative to untreated cells ( and Fig. S14). However, adding BAFA1 could reverse the decreased levels of Aβ40 and Aβ42 induced by gemfibrozil and Wy14643 treatment ( and Fig. S14). These results supported the key role of autophagy activated by gemfibrozil and Wy14643 in Aβ clearance.
PPARA agonists increase autophagy in microglia and astrocytes in APP-PSEN1ΔE9 mice
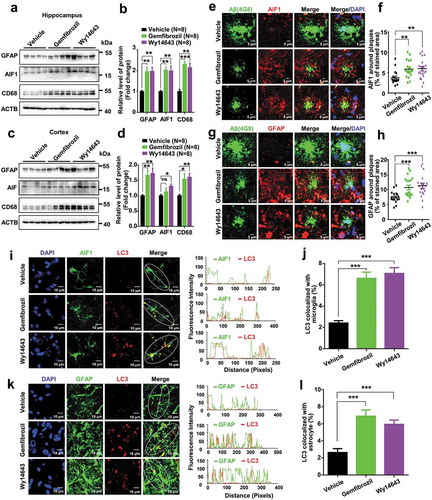
Previous studies have shown that reactive astrocytes surround the senile plaques in AD brains [Citation50], contain intracellular Aβ deposits [Citation51] and degrade Aβ [Citation52]. Activated microglia also accumulated around the senile plaques in AD patients [Citation53] and restricted amyloid plaque formation by phagocytosing Aβ [Citation54–Citation58]. The early stage of the neurodegeneration process was associated with glial dysfunction, resulting in a reduced Aβ clearance and disruption of synaptic connectivity [Citation59]. Consistent with these previous reports [Citation60], we found that amyloid plaques in APP-PSEN1ΔE9 mice were surrounded by microglia and astrocytes at the time of the treatment (8-month old) (Fig. S15). Following administration of a PPARA agonist, we observed an increase of protein levels of the astrocyte marker GFAP (glial fibrillary acidic protein), microglia marker AIF1/IBA1 (allograft inflammatory factor 1) and CD68 (CD68 molecule; which is indicative of phagocytic phenotype [Citation61]) in hippocampus and cortex tissues of APP-PSEN1ΔE9 mice (). Note that AIF1 and GFAP protein levels were not obviously changed in hippocampus and cortex tissues of the WT littermates treated with or without PPARA agonist (Fig. S16). There were significant increases of AIF1 and GFAP immunoreactivity around amyloid plaques in APP-PSEN1ΔE9 mice receiving gemfibrozil and Wy14643 treatment compared to the vehicle group (), suggesting that PPARA activation enhances the recruitment of microglia and astrocytes to the vicinity of amyloid plaques, which was critically for the mediation of Aβ phagocytic uptake [Citation62,Citation63]. Intriguingly, we also observed an increase of double immunofluorescence staining of AIF1 and LC3 and of GFAP and LC3 in APP-PSEN1ΔE9 mice treated with gemfibrozil and Wy14643 relative to the vehicle group (). These results suggested that gemfibrozil and Wy14643 enhance Aβ clearance via autophagy through the participation of microglia and astrocytes.
Figure 6. PPARA agonists induce widespread recruitment of microglia and astrocytes in the vicinity of amyloid plaques and this is associated with autophagy. Western blot showing the protein levels of GFAP, AIF1 and CD68 in the hippocampus (a-b) and cortex tissues (c-d) of APP-PSEN1ΔE9 mice treated with vehicle or PPARA agonist (either gemfibrozil or Wy14643). (e-h) Double staining using the 4G8 (green) and anti-AIF1 (red) antibodies (E) or the 4G8 (green) and the anti-GFAP (red) antibodies (g) in coronal brain sections from APP-PSEN1ΔE9 mice. Scale bar: 5 µm. (e-f) Increased AIF1 immunoreactivity (red) around amyloid plaques (4G8; green) between APP-PSEN1ΔE9 mice administrated with vehicle and PPARA agonist (gemfibrozil and Wy14643). (g-h) Increased GFAP immunoreactivity (red) around amyloid plaques (4G8; green) in APP-PSEN1ΔE9 mice treated with PPARA agonist (gemfibrozil and Wy14643) as compared to APP-PSEN1ΔE9 mice treated with vehicle. (i-l) Double staining using the anti-LC3 (red) and anti-AIF1 (green) antibodies (i) or the anti-LC3 and the anti-GFAP (green) antibodies (k) in coronal brain sections from APP-PSEN1ΔE9 mice. Scale bar:10 µm. There was an increased colocalization of LC3 with AIF1 (i-j) or with GFAP (k-l) in APP-PSEN1ΔE9 mice administrated with PPARA agonist (either gemfibrozil or Wy14643) compared to vehicle. Line graphs in I and K (right) indicate fluorescence intensity across the white dotted lines. All values are mean ± SEM; Student’s t test; ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
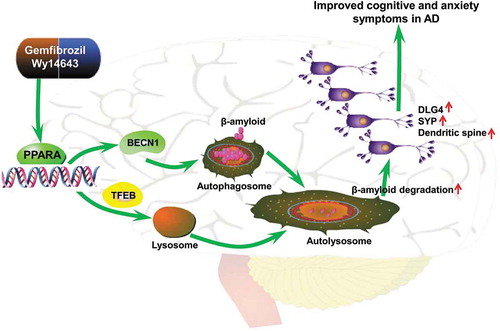
There is no effective therapy for AD at the present time partly because of the limited understanding of its pathophysiological mechanism. Manipulation of the autophagy system has been considered as a promising treatment for AD [Citation11,Citation22–Citation25]. It has been reported that PPARA expression was significantly reduced in AD brains [Citation64] and that PPARA agonists inhibited the Aβ-stimulated expression of TNF/TNF-α (tumor necrosis factor alpha) and IL6 (interleukin 6) reporter genes in GLI2/THP1 (GLI family zinc finger 2) monocytes [Citation65]. In this study, we have provided in vitro and in vivo evidence that upregulation of PPARA mediated-autophagy and lysosomal pathway reduced amyloid pathology and lessened cognitive decline and anxiety symptoms in a murine model. First, we demonstrated that PPARA agonists gemfibrozil and Wy14643 induced autophagy lysosome biogenesis in HM and U251-APP cells and this effect was abolished after knockdown of PPARA. Second, PPARA agonists decreased cognition decline and alleviated anxiety symptoms in APP-PSEN1ΔE9 mice, and this improvement was associated with increased autophagy in the brain tissues of APP-PSEN1ΔE9 mice treated with either gemfibrozil or Wy14643, as shown by a reduction of amyloid deposition and prevention of neuronal dendritic spine loss. This result was consistent with a previous study that showed Wy14643 improved cognitive abilities and reduced neuronal damage in a mouse model of AD although the potential molecular mechanism was undetermined [Citation66]. Moreover, administration of gemfibrozil and Wy14643 reduced Aβ generation in a cellular AD model, which was regulated by autophagy and lysosomal pathway. It is noteworthy that several studies had shown either no abnormalities of APP-PSEN1ΔE9 mice in the open field test, or reduced anxiety at various ages up to 18 months [Citation67–Citation71]. It was also reported that increased anxiety was observed in APP-PSEN1ΔE9 mice at both 18 and 24 months [Citation72]. In our study, we found that APP-PSEN1ΔE9 mice exhibited increased anxiety at an age of 10 months and PPARA agonists alleviated this symptom. The exact reason for the discrepancy between our study and previous reports [Citation67–Citation71] remained to be determined, but different evaluation platforms and detection time points might be an explanation from the technical point of view. In addition, we noticed that a recent study also showed that APP-PSEN1ΔE9 mice had increased anxiety at the age of 24 weeks [Citation73]. Third, administration of gemfibrozil and Wy14643 led to a recruitment of astrocytes and microglia to the vicinity of amyloid plaques in coronal section of APP-PSEN1ΔE9 mouse brains, and enhanced Aβ clearance via autophagy. Based on these lines of evidence, we have suggested that activation of PPARA-mediated autophagy would be a promising therapeutic approach against AD ().
Figure 7. A proposed role of the PPARA-mediated autophagy against AD-like pathology in a murine model. Activation of the nuclear receptor PPARA by gemfibrozil or Wy14643 upregulates autophagy, activates microglia and astrocytes, removes Aβ, increases protein levels of SYP and DLG4, reverses memory deficits and decreases anxiety symptoms.
Autophagy is important for preserving the complex neuronal architecture of hippocampal and cortical circuits, and an abnormal autophagy pathway is increasingly regarded as a contributor for the Aβ-mediated pathogenesis observed in AD [Citation19,Citation20,Citation23,Citation74–Citation77]. Previous studies have reported that activation of PPARA induces lysosomal biogenesis in brain cells [Citation78] and has a neurotrophic role in the nervous system [Citation35]. Our results have shown that PPARA agonists regulate autophagy and lysosome biogenesis in nervous system, leading to a reduction of Aβ accumulations and improved cognition impairment in APP-PSEN1ΔE9 mice, further expanded the active role of PPARA activation in curing neurodegenerative diseases. The identification of PPARA as a new and promising therapeutic target could pave the way for a focused clinical trial of the FDA approved drug gemfibrozil for AD treatment, albeit this drug was initially approved as a lipid-lowering drug to prevent hyperlipidemia [Citation36,Citation37,Citation79].
Evidence has been accumulating to suggest that astrocytes and microglia have played a key role in the development of AD to counteract neurotoxicity of Aβ accumulation [Citation80–Citation89]. For instance, Aβ activated PARP (poly[ADP-ribose] polymerase) in response to oxidative stress generated by the astrocytic nicotinamide adenine dinucleotide phosphate oxidase and cause neuronal death [Citation90]. APOE (apolipoprotein E) promoted colocalization of astrocytes with Aβ plaques and facilitate the degradation of deposited Aβ [Citation80]. Microglia played a vital role in immune regulation and homeostasis of inflammatory environment in the vicinity of Aβ plaques in AD [Citation81]. Microglia directly phagocytosed Aβ plaques [Citation82] and were indirectly involved in Aβ clearance by releasing various cytokines [Citation89]. The phagocytosis of activated microglia maintained brain homeostasis via the clearance of cellular debris and possibly the pruning of synapses [Citation83–Citation85]. Activation of microglia by intracranial delivery of lipopolysaccharide or pro-inflammatory cytokines mitigated Aβ deposition [Citation86–Citation88], and microglial dysfunction led to a defective Aβ clearance in AD mice [Citation91] and accelerated progression of AD-like disease in mice [Citation92]. Genetic defects in different receptors or proteins involved in phagocytosis resulted in neurodegeneration [Citation93,Citation94] and might be responsible for increased amyloidosis in mouse AD models [Citation95]. Conversely, driving microglial activation towards a more phagocytic phenotype reduces Aβ pathology in mouse models of AD [Citation96]. These studies have highlighted the importance of phagocytosis of astrocytes and microglia in brain homeostasis and suggested that identifying key regulators of phagocytosis may represent a therapeutic target for the treatment of AD. In this study, we have found that activation of PPARA could induce a colocalization of astrocytes and microglia to the vicinity of Aβ plaques in APP-PSEN1ΔE9 mice and enhanced Aβ clearance, and this effect was apparently associated with the increased autophagy induced by PPARA agonists. Our current findings not only have given further support to the evidence showing the activation of astrocyte and microglia in Aβ clearance, but have also provided a novel target, PPARA, for activating astrocyte and microglia. Further study should be carried out to elucidate the underlying mechanism for activation of astrocyte and microglia by PPARA agonists. An interesting question arising from this study is that whether gemfibrozil – and Wy14643-induced PPARA-mediated autophagy has any cell-type specificities in the nervous system. A well-designed in vivo experiment using animal models with neuron- or glial cell-specific knockout of PPARA is warranted to comprehensively understand the beneficial role of PPARA-mediated autophagy induced by gemfibrozil and Wy14643 for treating AD.
The current study had several limitations. First, the whole catabolic pathway might be activated by gemfibrozil and Wy14643, and the PPARA-mediated autophagy might not be a selective autophagy pathway. However, we observed no hepatotoxicity in APP-PSEN1ΔE9 mice receiving gemfibrozil and Wy14643 treatment under the dosage used in this study (Fig. S10). Analysis of transcriptomic and proteomic profilings in different tissues might provide further information regarding this important issue. Second, PPARA could also regulate ADAM10 [Citation34], we could not exclude a possibility that the reduction of soluble Aβ was caused by the effect on APP processing rather than autophagy, or at least together with autophagy. Third, we only tested one dosage of gemfibrozil and Wy14643 in APP-PSEN1ΔE9 mice and observed significant amelioration effect. This administration amount was estimated based on the recommended minimum dosage of gemfibrozil for treating hyperlipidemia in adults by the FDA. Future study should be performed to test whether other drug concentrations of gemfibrozil and Wy14643 would have a better efficiency for ameliorating cognitive impairment.
In conclusion, we have demonstrated that the PPARA agonists gemfibrozil and Wy14643 improve spatial learning, memory impairments and anxiety symptoms in APP-PSEN1ΔE9 mice via modulation of the PPARA-mediated autophagy and lysosomal pathway and activation of the astrocytes and microglia, which together contribute to Aβ clearance. These results have shown a novel mechanism behind the therapeutic effect of gemfibrozil and Wy14643 in AD and provide an experimental basis for starting a clinical trial for the FDA approved drug gemfibrozil as a treatment for AD. It would also be rewarding to have a retrospective study for these patients who regularly took gemfibrozil and assessed whether they had a lower occurrence of AD and improved cognition compared to age-matched subjects who did not take this drug during aging.
Materials and methods
Reagents, chemicals and cells
Details of primary antibodies used in the western blotting and immunofluorescence are listed in . The following secondary antibodies and chemicals were used in this study: peroxidase-conjugated anti-rabbit antibody (KPL, 474-1516), peroxidase-conjugated anti-mouse antibody (KPL, 474-1806), Alexa Fluor® 594-conjugated anti-rabbit IgG (Invitrogen, A21207), and Alexa Fluor® 488-conjugated anti-mouse IgG (Invitrogen, A21202). Rapamycin (InvivoGen, trl-rap), bafilomycin A1 (BAFA1, InvivoGen, tlrl-baf), 4',6-diamidino-2-phenylindole (DAPI, Roche, 10236276001), bovine serum albumin (BSA; Biotechnology, 0332-100G), pentobarbital (Sigma, P3761), gemfibrozil (Abcam, ab142883), and Wy14643 (Abcam, ab141142) were purchased as indicated. Vector p3*Flag-CMV-14-PPARA (PPL00582-2a) was purchased from Public Protein/Plasmid library.
Table 1. Primary antibodies used in this study.
Human microglia (HM) cells were obtained from the Kunming Cell Bank, Kunming Institute of Zoology. The U251 cell line, with stable expression of the APP mutant APP-p.M671L (U251–APP cells), was created in our previous study [Citation38]. HM and U251-APP cells were cultured in Dulbecco’s Modified Eagle Medium and Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum (Gibco-BRL, 10099-141) respectively at 37°C in a humidified atmosphere incubator with 5% CO2 and 95% humidity. Drugs were applied directly to the culture medium for treatment.
Animals and drug administration
Eight-month-old APP-PSEN1ΔE9 mice and WT littermates were used in this study. The APP-PSEN1ΔE9 mice overexpress the mutated human APP (Swedish mutations K595N/M596L) and also have the human PSEN1 (presenilin 1) deleted from its exon 9 [Citation41]. APP-PSEN1ΔE9mice and WT littermates were bred and maintained in a temperature-controlled room and maintained on a 12-h light/dark cycle, with ad libitum access to food and water. Mouse genomic DNA was extracted by using the AxyPrep™ Multisource Genomic DNA Miniprep Kit (Axygen, AP-MN-MS-GDNA-50). Genotyping for the APP-PSEN1ΔE9 mice was performed by using PCR using primer pairs oIMR1597 5ʹ-GACTGACCACTCGACCAGGTTCTG-3ʹ/oIMR1598 5ʹ- CTTGTAAGTTGGATTCTCATATCCG-3ʹ and oIMR1644 5ʹ- AATAGAGAACGGCAGGAGCA-3ʹ/oIMR1645 5ʹ- GCCATGAGGGCACTAATCAT-3ʹ for APP and PSEN1, respectively.
Animal experiments were performed on sex- and age-matched groups. Mice of both genders were used for experiments. The APP-PSEN1ΔE9 and WT littermates at an age of 8 months had ad libitum access to gemfibrozil or Wy14643 solution for 2 months, and control mice (vehicle group) had ad libitum access to drinking water containing DMSO. The gemfibrozil (Abcam, ab142883) and Wy14643 (Abcam, ab141142) were dissolved in DMSO at a concentration of 50 mg/mL, and diluted in drinking water at a concentration of 50 µg/mL. All containers with gemfibrozil and Wy14643 were covered with foil to prevent light exposure. Fresh solutions were made every 2 days for the duration of treatment.
The mice were euthanized after the behavioral tests. The brain was removed, gently rinsed in cold saline (0.9% sodium chloride), and immediately dissected into two halves. One half was immediately stored at −80°C for biochemistry assay and the other half was used for Golgi staining or fixed in 4% paraformaldehyde in phosphate-buffered saline (pH 7.4) for immunohistochemistry assays.
All experiments involving animals and the animal care and experimental protocol were approved by the Institutional Animal Care and Use Committee of Kunming Institute of Zoology, Chinese Academy of Sciences.
Morris water maze behavioral assessment
Morris water maze behavioural assessment was performed as previously described [Citation60]. In brief, experiments were performed in a 120-cm diameter, 50-cm deep tank filled with opacified water kept at 20 ± 1°C. The tank was equipped with a 10-cm diameter platform submerged 1 cm under the water surface. Training consisted of daily sessions (3 trials per session) for 7 consecutive days. The start positions varied pseudorandomly among the 4 cardinal points. Mean intertrial interval was 90 min. Each trial ended when the animal reached the platform. A 60 sec cut-off was used, after which mice were gently guided to the platform. Once on the platform, animals were given a rest for 20 s before being returned to their cage. Four h (short-term memory) and 72 h after the last training trial (day 10), retention was assessed during probe trial in which the platform was no longer presented. Animals were video tracked using SMART 3.0 software (Panlab HARVARD, MA, USA) and behavioural parameters (swim speed, travelled distance, latency, percentage of time in each quadrant, percentage of distance in each quadrant) were automatically calculated.
Open-field behavioral assessment
Spontaneous locomotor activity and anxiety of mice in the open-field test were recorded and tracked using the SMART 3.0 software (Panlab HARVARD, MA, USA). In brief, mice were individually placed in the center of an open-top chamber (40 × 40 × 40 inches) with an array of photobeams around the periphery. Animals were allowed to explore the chamber for 60 min for a day. The chamber was cleaned with 70% ethanol after each trial. Locomotor activity in the training trials was recorded by the photobeams, and the distance moved was subsequently determined. The time spent in the center was measured as a potential indicator for anxiolytic behavior. Locomotor activity and anxiety were analyzed according to movement of mice in the open field.
Western blot analysis
Western blotting for target proteins were performed using the common approach as described in our previous study [Citation97]. In brief, cell lysates of different mouse brain tissues (hippocampus and cortex), cultured HM and U251-APP cells were prepared using the protein lysis buffer (Beyotime Institute of Biotechnology, P0013). Protein concentration was determined using the BCA protein assay kit (Beyotime Institute of Biotechnology, P0012). A total of 20 μg protein was separated by 12% or 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and was transferred to a polyvinylidene difluoride membrane (Bio-Rad, L1620177 Rev D). The membrane was soaked with 5% (w:v) skim milk for 2 h at room temperature. The membrane was incubated with primary antibodies overnight at 4°C. The membranes were washed 3 times with TBST (Tris-buffered saline [Cell Signaling Technology, 9997] with Tween 20 [0.1%; Sigma, P1379]), each time 5 min, followed by incubation with the peroxidase-conjugated anti-mouse (474–1806) or anti-rabbit (474–1516) IgG (1:5000; KPL) for 1 h at room temperature. The epitope was visualized using an ECL western blot detection kit (Millipore, WBKLS0500). ImageJ software (National Institutes of Health, Bethesda, Maryland, USA) was used to evaluate the densitometry. Western blot for ACTB (actin beta) or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a loading control to measure the densitometry of target gene. The LC3B-II densitometric signal is determined by the ratio of LC3B-II to LC3B-I before normalized to ACTB or GAPDH.
RNA interference and transfection
Three siRNAs targeting PPARA and the negative control (NC) siRNA were obtained from RiboBio (Guangzhou, China). Cultured HM cells and U251-APP cells were transfected according to the procedure. In brief, cells (2 × 105 per well) were seeded in 6-well plates to grow to 50% confluence. Before transfection, culture medium was removed and washed once with the Opti-MEM medium (Gibco-BRL, 31985-070). The PPARA siRNA or NC siRNA was dissolved in the Opti-MEM medium, and was then mixed with 6 μL LipofectamineTM 3000 (Invitrogen, L3000008) to achieve a final volume of 150 μL. The siRNA-Lipofectamine mixture was incubated at room temperature for 20 min, then the mixture was added to each well together with an additional 850 μL Opti-MEM medium. After an incubation period of 6 h, the medium was removed and 2 mL fresh medium was added to each well for growth. We optimized the siRNA concentration for transfection and the time for cell harvest after transfection by testing 3 different concentrations of siRNA (6.25, 25, or 50 nM) in a 48-h time frame. The efficiency of siRNA to reduce the protein expression of PPARA was determined by western blot analysis.
Tandem mRFP-GFP fluorescence microscopy
A tandem monomeric RFP-GFP-tagged LC3 (mRFP-GFP-LC3) lentivirus (GeneChem, GPL2001) was used to monitor autophagy flux as previously reported [Citation98]. Briefly, HM and U251-APP cells were cultured in Dulbecco’s Modified Eagle Medium and Roswell Park Memorial Institute1640 medium supplemented with 10% fetal bovine serum, respectively. Cells were infected according to the manufacturer’s protocol. For evaluating tandem fluorescent LC3 puncta, 48 h after mRFP-GFP-LC3 lentivirus infection, cells were captured by an Olympus FluoView™ 1000 confocal microscope (Olympus, America).
Immunofluorescence
Mouse brain sections were prepared for immunofluorescence as previously described [Citation97]. The brain was fixed in 4% paraformaldehyde at 4°C and then optimal cutting temperature compound (Leica, 14020108926)-embedded, sectioned (20-μm thick) using a freezing microtome (Leica, CM1850UV-1-1, Amtzell, Germany). Coronal sections were used for immunofluorescence. After antigen retrieval, sections were blocked in phosphate-buffered saline supplemented with 5% bovine serum albumin for 1 h. Sections were then incubated with the respective primary antibodies overnight at 4°C. Secondary antibodies were Alexa Fluor® 594-conjugated anti-rabbit IgG (1:500; Invitrogen, A21207) or Alexa Fluor® 488 anti-mouse IgG (1:500; Invitrogen, A21202). The slides were exposed to DAPI nuclear staining (1:1000; Invitrogen, D1306) for 15 min, before being sealed with antifade mounting medium (Beyotime Institute of Biotechnology, P0128M) and glass covers. Immunofluorescent sections were visualized, and images were captured using an Olympus FluoView™ 1000 confocal microscope (Olympus, USA). For the quantification of staining intensity, total cell and background fluorescence intensity was measured using ImageJ software, respectively. The intensity of specific staining was calculated as previously described [Citation99]. Quantification of the colocalization of LC3 and anti-β-amyloid, LAMP1 and anti-β-amyloid, LC3 and AIF1, or LC3 and GFAP were conducted using ImageJ software.
Immunohistochemistry
Paraffin-embedded brain sections of APP-PSEN1ΔE9 mice were incubated overnight at 4°C with primary antibodies mouse anti-β-amyloid (clone 4G8; 1:500; BioLegend, 800701) or rabbit anti-DLG4 (1:200; Abcam, ab18258). The horseradish peroxidase-labeled secondary antibody was goat anti-mouse (1:200; Servicebio, GB23301) or goat anti-rabbit (1:200; Servicebio, GB23303). The images were captured by CaseViewer (Pannoramic250/MIDI, 3DHISTECH), and staining intensity was quantified using ImageJ software.
Quantification of the microglia and astrocytes around plaques
Aβ plaques, GFAP and AIF1 immunoreactivity were quantified using ImageJ software as previously described [Citation60]. Laser power, numeric gain and magnification were kept constant between animals to avoid potential technical artefacts. Images were first converted to 8-bit gray scale and binary thresholded to highlight a positive staining. At least six sections per mouse were quantified. The average value per structure was calculated for each mouse. For quantification of AIF1 and GFAP immunoreactivity around plaques (4G8 labeled), a region of interest was drawn around the center of the plaque. Mean fluorescence intensity values were measured for either AIF1 or GFAP immunoreactivity and were processed via Icy software (Institut Pasteur). Analysis of data was blind with respect to treatments.
Dendrite and spine analysis in Golgi-Cox stained slices
Golgi-Cox staining (FD NeuroTechnologies, PK401) was used to detect the dendritic spines of neurons. All procedures were performed under dark conditions. Mouse brain tissues were impregnated for 2 weeks and processed according to the manufacturer’s instructions as in our previous study [Citation97]. Approximately 20 neurons were randomly selected from each group and quantified with a double blind, controlled design. ImageJ and Image-Pro Plus (IPP; Media Cybernetics, Inc., Washington, MD, USA) were used to evaluate the number of spines, the total dendritic length and complexity of neurons.
Aβ ELISA analysis
Mouse hippocampus and cortex tissue homogenates and U251-APP cell lysates were extracted using the protein lysis buffer (Beyotime Institute of Biotechnology, P0013) containing protease and phosphatase inhibitors (Abcam, ab201119), and were centrifuged at 13,000 g for 15 min at 4°C. The remaining pellet containing insoluble Aβ was subsequently solubilized in sodium dodecyl sulfate buffer (2% sodium dodecyl sulfate, 25 mM Tris-HCl, pH 7.4) [Citation100]. The levels of Aβ40/Aβ1–40 and Aβ42/Aβ1–42 in APP-PSEN1ΔE9 mouse brain tissues were measured using commercial ELISA kits (Elabscience, E-EL-M0067c to detect mouse Aβ40, and E-EL-M0068c to detect mouse Aβ42). The levels of Aβ40 and Aβ42 in U251-APP cells supernatant and cell lysates were measured using commercial ELISA kits (Elabscience, E-EL-H0542c to detect human Aβ40, and E-EL-H0543c to detect human Aβ42). The ELISA was performed for Aβ40 and Aβ42 according to the manufacturer’s instructions.
Statistical analysis
All statistical analysis was performed using PRISM software (GraphPad Software, Inc., La Jolla, CA, USA). Student’s t test was used for data sets with a single intervention. The one-way ANOVA (analysis of variance) was performed using the Tukey’s post hoc test for multiple comparisons. Data were represented as mean ± SEM (standard error of the mean). A P value of < 0.05 was considered to be statistically significant.
Supplemental Material
Download MS Word (13.2 MB)Acknowledgments
We thank Dr. Nengyin Sheng and the three anonymous reviewers for their helpful comments on the early version of the manuscript. We are grateful to Ian Logan for his comments and language editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Scheltens P, Blennow K, Breteler MM, et al. Alzheimer‘s disease. Lancet. 2016 Jul 30;388(10043):505–517. PubMed PMID: 26921134.
- Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol. 2017 Sep 29;13(10):612–623. PubMed PMID: 28960209.
- Querfurth HW, LaFerla FM. Alzheimer‘s disease. N Engl J Med. 2010 Jan 28;362(4):329–344. PubMed PMID: 20107219.
- Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer‘s disease. Science. 2010 Dec 24;330(6012):1774. PubMed PMID: 21148344; PubMed Central PMCID: PMC3073454.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer‘s disease at 25 years. EMBO Mol Med. 2016 Jun;8(6):595–608. PubMed PMID: 27025652; PubMed Central PMCID: PMC4888851.
- Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016 Sep 1;537(7618):50–56. PubMed PMID: 27582220.
- Bu XL, Xiang Y, Jin WS, et al. Blood-derived amyloid-beta protein induces Alzheimer‘s disease pathologies. Mol Psychiatry. 2017 Oct 31. PubMed PMID: 29086767. DOI:10.1038/mp.2017.204
- Jin WS, Shen LL, Bu XL, et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017 Aug;134(2):207–220. PubMed PMID: 28477083.
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015 Jun;16(6):345–357. PubMed PMID: 25991442.
- Chung KM, Hernandez N, Sproul AA, et al. Alzheimer‘s disease and the autophagic-lysosomal system. Neurosci Lett. 2018 May 18. pii: S0304-3940(18):30344-6. PubMed PMID: 29758300. DOI:10.1016/j.neulet.2018.05.017
- Metaxakis A, Ploumi C, Tavernarakis N. Autophagy in age-associated neurodegeneration. Cells. 2018 May 5;7(5):pii: E37. PubMed PMID: 29734735; PubMed Central PMCID: PMC5981261.
- Caccamo A, Majumder S, Richardson A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010 Apr 23;285(17):13107–13120. PubMed PMID: 20178983; PubMed Central PMCID: PMC2857107.
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun;118(6):2190–2199. PubMed PMID: 18497889; PubMed Central PMCID: PMC2391284.
- Swaminathan G, Zhu W, Plowey ED. BECN1/Beclin 1 sorts cell-surface APP/amyloid β precursor protein for lysosomal degradation. Autophagy. 2016 Dec;12(12):2404–2419. PubMed PMID: 27715386; PubMed Central PMCID: PMC5173276.
- Xiao Q, Yan P, Ma X, et al. Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Abeta generation and amyloid plaque pathogenesis. J Neurosci. 2015 Sep 2;35(35):12137–12151. PubMed PMID: 26338325; PubMed Central PMCID: PMC4556784.
- Zhang YD, Zhao JJ. TFEB participates in the Aβ-induced pathogenesis of Alzheimer‘s disease by regulating the autophagy-lysosome pathway. DNA Cell Biol. 2015 Nov;34(11):661–668. PubMed PMID: 26368054.
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006 Jun 15;441(7095):885–889. PubMed PMID: 16625204.
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006 Jun 15;441(7095):880–884. PubMed PMID: 16625205.
- Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer‘s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011 Jan;134(Pt 1):258–277. PubMed PMID: 21186265; PubMed Central PMCID: PMC3009842.
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2011 Dec 20;8(2):108–117. PubMed PMID: 22187000.
- Schaeffer V, Goedert M. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy. 2012 Nov;8(11):1686–1687. PubMed PMID: 22874558; PubMed Central PMCID: PMC3494601.
- Di Meco A, Li JG, Blass BE, et al. 12/15-Lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and tau pathology by stimulating autophagy in aged triple transgenic mice. Biol Psychiatry. 2017 Jan 15;81(2):92–100. PubMed PMID: 27499089.
- Caccamo A, Ferreira E, Branca C, et al. p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry. 2017 Jun;22(6):865–873. PubMed PMID: 27573878; PubMed Central PMCID: PMC5479312.
- Frake RA, Ricketts T, Menzies FM, et al. Autophagy and neurodegeneration. J Clin Invest. 2015 Jan;125(1):65–74. PubMed PMID: 25654552; PubMed Central PMCID: PMC4382230.
- Chauhan S, Ahmed Z, Bradfute SB, et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat Commun. 2015 Oct 27;6:8620. PubMed PMID: 26503418; PubMed Central PMCID: PMC4624223.
- Keller H, Dreyer C, Medin J, et al. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci U S A. 1993 Mar 15;90(6):2160–2164. PubMed PMID: 8384714; PubMed Central PMCID: PMC46045.
- Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998 Jun 25;393(6687):790–793. PubMed PMID: 9655393; PubMed Central PMCID: PMC 9655393.
- Kersten S. Integrated physiology and systems biology of PPARalpha. Mol Metab. 2014 Jul;3(4):354–371. PubMed PMID: 24944896; PubMed Central PMCID: PMC4060217.
- Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004 Feb;61(4):393–416. PubMed PMID: 14999402.
- Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999 Jul 10;354(9173):141–148. PubMed PMID: 10408502.
- Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014 Dec 4;516(7529):112–115. PubMed PMID: 25383539; PubMed Central PMCID: PMC4267857.
- Kim YS, Lee HM, Kim JK, et al. PPAR-alpha activation mediates innate host defense through induction of TFEB and lipid catabolism. J Immunol. 2017 Apr 15;198(8):3283–3295. PubMed PMID: 28275133.
- Kolsch H, Lehmann DJ, Ibrahim-Verbaas CA, et al. Interaction of insulin and PPAR-alpha genes in Alzheimer‘s disease: the Epistasis Project. J Neural Transm (Vienna). 2012 Apr;119(4):473–479. PubMed PMID: 22065208.
- Corbett GT, Gonzalez FJ, Pahan K. Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci U S A. 2015 Jul 7;112(27):8445–8450. PubMed PMID: 26080426; PubMed Central PMCID: PMC4500265.
- Roy A, Jana M, Kundu M, et al. HMG-CoA reductase inhibitors bind to PPARalpha to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab. 2015 Aug 4;22(2):253–265. PubMed PMID: 26118928; PubMed Central PMCID: PMC4526399.
- Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans affairs high-density lipoprotein cholesterol intervention trial Study group. N Engl J Med. 1999 Aug 5;341(6):410–418. PubMed PMID: 10438259; PubMed Central PMCID: PMC10438259.
- Frick MH, Elo O, Haapa K, et al. Helsinki heart study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987 Nov 12;317(20):1237–1245. PubMed PMID: 3313041; PubMed Central PMCID: PMC3313041.
- Zhang DF, Li J, Wu H, et al. CFH variants affect structural and functional brain changes and genetic risk of Alzheimer’s disease. Neuropsychopharmacol. 2016 Mar;41(4):1034–1045. PubMed PMID: 26243271; PubMed Central PMCID: PMC4748428.
- Rubinsztein DC, Cuervo AM, Ravikumar B, et al. In search of an “autophagomometer”. Autophagy. 2009 Jul;5(5):585–589. PubMed PMID: 19411822.
- Feng L, Ma Y, Sun J, et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy. 2014 Aug;10(8):1442–1453. PubMed PMID: 24991827; PubMed Central PMCID: PMC4203520.
- Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004 Jan 15;13(2):159–170. PubMed PMID: 14645205.
- Giannopoulos PF, Chu J, Joshi YB, et al. Gene knockout of 5-lipoxygenase rescues synaptic dysfunction and improves memory in the triple-transgenic model of Alzheimer‘s disease. Mol Psychiatry. 2014 Apr;19(4):511–518. PubMed PMID: 23478745; PubMed Central PMCID: PMC3688674.
- Giannopoulos PF, Chu J, Sperow M, et al. Pharmacologic inhibition of 5-lipoxygenase improves memory, rescues synaptic dysfunction, and ameliorates tau pathology in a transgenic model of tauopathy. Biol Psychiatry. 2015 Nov 15;78(10):693–701. PubMed PMID: 25802082; PubMed Central PMCID: PMC4529386.
- Di Meco A, Joshi YB, Lauretti E, et al. Maternal dexamethasone exposure ameliorates cognition and tau pathology in the offspring of triple transgenic AD mice. Mol Psychiatry. 2016 Mar;21(3):403–410. PubMed PMID: 26077691.
- Pietrzak RH, Lim YY, Neumeister A, et al. Amyloid-beta, anxiety, and cognitive decline in preclinical Alzheimer disease: a multicenter, prospective cohort study. JAMA Psychiatry. 2015 Mar;72(3):284–291. PubMed PMID: 25629787.
- Seibenhener ML, Wooten MC. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp. 2015 Feb 6;(96):e52434. PubMed PMID: 25742564; PubMed Central PMCID: PMC4354627. DOI:10.3791/52434
- Attwood BK, Bourgognon JM, Patel S, et al. Neuropsin cleaves EphB2 in the amygdala to control anxiety. Nature. 2011 May 19;473(7347):372–375. PubMed PMID: 21508957; PubMed Central PMCID: PMC3145099.
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. CSH Perspect Med. 2012 Jul;2(7):a006338–a006338. PubMed PMID: 22762015; PubMed Central PMCID: PMC3385944
- Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014 Jan;112:24–49. PubMed PMID: 24211851.
- Shao Y, Gearing M, Mirra SS. Astrocyte-apolipoprotein E associations in senile plaques in Alzheimer disease and vascular lesions: a regional immunohistochemical study. J Neuropathol Exp Neurol. 1997 Apr;56(4):376–381. PubMed PMID: 9100668.
- Pihlaja R, Koistinaho J, Malm T, et al. Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer‘s disease. Glia. 2008 Jan 15;56(2):154–163. PubMed PMID: 18004725.
- Nicoll JA, Weller RO. A new role for astrocytes: β-amyloid homeostasis and degradation. Trends Mol Med. 2003 Jul;9(7):281–282. PubMed PMID: 12900213.
- McGeer PL, Itagaki S, Tago H, et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987 Aug 18;79(1–2):195–200. PubMed PMID: 3670729.
- Simard AR, Soulet D, Gowing G, et al. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer‘s disease. Neuron. 2006 Feb 16;49(4):489–502. PubMed PMID: 16476660.
- Gomez-Nicola D, Perry VH. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 2015 Apr;21(2):169–184. PubMed PMID: 24722525; PubMed Central PMCID: PMC4412879.
- Malm TM, Jay TR, Landreth GE. The evolving biology of microglia in Alzheimer‘s disease. Neurotherapeutics. 2014 Jan;12(1):81–93. PubMed PMID: 25404051; PubMed Central PMCID: PMC4322081.
- Liu Z, Condello C, Schain A, et al. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010 Dec 15;30(50):17091–17101. PubMed PMID: 21159979; PubMed Central PMCID: PMC3077120.
- Lee S, Varvel NH, Konerth ME, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer‘s disease mouse models. Am J Pathol. 2010 Nov;177(5):2549–2562. PubMed PMID: 20864679; PubMed Central PMCID: PMC2966811.
- Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer‘s disease. Nat Immunol. 2015 Mar;16(3):229–236. PubMed PMID: 25689443.
- Alves S, Churlaud G, Audrain M, et al. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer‘s disease mice. Brain. 2017 Mar 1;140(3):826–842. PubMed PMID: 28003243.
- Zotova E, Bharambe V, Cheaveau M, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain. 2013 Sep;136(9):2677–2696. PubMed PMID: 23943781.
- Guillot-Sestier MV, Doty KR, Gate D, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015 Feb 4;85(3):534–548. PubMed PMID: 25619654; PubMed Central PMCID: PMC4352138.
- Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer‘s disease model. Cell. 2015 Mar 12;160(6):1061–1071. PubMed PMID: 25728668; PubMed Central PMCID: PMC4477963.
- de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer‘s disease. J Alzheimers Dis. 2006 Jul;9(2):167–181. PubMed PMID: 16873964.
- Combs CK, Bates P, Karlo JC, et al. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Int. 2001 Nov-Dec;39(5–6):449–457. PubMed PMID: 11578780.
- Inestrosa NC, Carvajal FJ, Zolezzi JM, et al. Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer‘s disease. J Alzheimers Dis. 2013;33(4):941–959. PubMed PMID: 23109558.
- Lalonde R, Kim HD, Fukuchi K. Exploratory activity, anxiety, and motor coordination in bigenic APPswe + PS1/ΔE9 mice. Neurosci Lett. 2004 Oct 14;369(2):156–161. PubMed PMID: 15450687.
- Lalonde R, Kim HD, Maxwell JA, et al. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005 Dec 23;390(2):87–92. PubMed PMID: 16169151.
- Reiserer RS, Harrison FE, Syverud DC, et al. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer‘s disease. Genes Brain Behav. 2007 Feb;6(1):54–65. PubMed PMID: 17233641.
- Pugh PL, Richardson JC, Bate ST, et al. Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer‘s disease. Behav Brain Res. 2007 Mar 12;178(1):18–28. PubMed PMID: 17229472.
- Olesen LØ, Bouzinova EV, Severino M, et al. Behavioural phenotyping of APPswe/PS1δE9 mice: age-rrelated changes and effect of long-term paroxetine treatment. PLoS One. 2016 Nov 4;11(11):e0165144. PubMed PMID: 27814403; PubMed Central PMCID: PMC5096719.
- Liu Y, Yoo MJ, Savonenko A, et al. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer‘s disease. J Neurosci. 2008 Dec 17;28(51):13805–13814. PubMed PMID: 19091971.
- Gao JY, Chen Y, Su DY, et al. Anxiety-like but not despair-like behaviors are further aggravated by chronic mild stress in the early stages of APPswe/PS1dE9 transgenic mice. BioRxiv. 2017 Oct 12. DOI:10.1101/202283
- Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer‘s disease. J Cell Biol. 2005 Oct 10;171(1):87–98. PubMed PMID: 16203860; PubMed Central PMCID: PMC2171227.
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007 Dec 1;120(Pt 23):4081–4091. PubMed PMID: 18032783.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013 Aug;19(8):983–997. PubMed PMID: 23921753.
- Lucin KM, O‘Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer‘s disease. Neuron. 2013 Sep 4;79(5):873–886. PubMed PMID: 24012002; PubMed Central PMCID: PMC3779465.
- Ghosh A, Jana M, Modi K, et al. Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: implications for lysosomal storage disorders. J Biol Chem. 2015 Apr 17;290(16):10309–10324. PubMed PMID: 25750174; PubMed Central PMCID: PMC4400343.
- Robins SJ, Collins D, Wittes JT, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. 2001 Mar 28;285(12):1585–1591. PubMed PMID: 11268266.
- Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004 Jul;10(7):719–726. PubMed PMID: 15195085.
- Meyer-Luehmann M, Prinz M. Myeloid cells in Alzheimer‘s disease: culprits, victims or innocent bystanders? Trends Neurosci. 2015 Oct;38(10):659–668. PubMed PMID: 26442698.
- Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010 Aug;117(8):949–960. PubMed PMID: 20552234; PubMed Central PMCID: PMCPMC3653296.
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005 May 27;308(5726):1314–1318. PubMed PMID: 15831717.
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009 Oct 15;64(1):110–122. PubMed PMID: 19840553; PubMed Central PMCID: PMC2834890.
- Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011 Sep 9;333(6048):1456–1458. PubMed PMID: 21778362.
- Herber DL, Roth LM, Wilson D, et al. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004 Nov;190(1):245–253. PubMed PMID: 15473997.
- Herber DL, Mercer M, Roth LM, et al. Microglial activation is required for Aβ clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharm. 2007 Jun;2(2):222–231. PubMed PMID: 18040847.
- DiCarlo G, Wilcock D, Henderson D, et al. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001 Nov-Dec;22(6):1007–1012. PubMed PMID: 11755009.
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer‘s disease. Lancet Neurol. 2015 Apr;14(4):388–405. PubMed PMID: 25792098; PubMed Central PMCID: PMC5909703.
- Abeti R, Abramov AY, Duchen MR. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain. 2011 Jun;134(Pt 6):1658–1672. PubMed PMID: 21616968.
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer‘s disease mice. J Neurosci. 2008 Aug 13;28(33):8354–8360. PubMed PMID: 18701698; PubMed Central PMCID: PMC2597474.
- El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007 Apr;13(4):432–438. PubMed PMID: 17351623.
- Kaifu T, Nakahara J, Inui M, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003 Feb;111(3):323–332. PubMed PMID: 12569157; PubMed Central PMCID: PMC151867.
- Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001 Jul 13;293(5528):306–311. PubMed PMID: 11452127.
- Wyss-Coray T, Yan F, Lin AHT, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer‘s mice. Proc Natl Acad Sci U S A. 2002 Aug 6;99(16):10837–10842. PubMed PMID: 12119423; PubMed Central PMCID: PMC125059.
- Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer‘s disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674–678. PubMed PMID: 23254930; PubMed Central PMCID: PMC3812809.
- Su LY, Luo R, Liu Q, et al. Atg5- and Atg7-dependent autophagy in dopaminergic neurons regulates cellular and behavioral responses to morphine. Autophagy. 2017 Jul 19;13(9):1496–1511. PubMed PMID: 28722508; PubMed Central PMCID: PMC5612517.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMC4835977.
- Burgess A, Vigneron S, Brioudes E, et al. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010 Jul 13;107(28):12564–12569. PubMed PMID: 20538976; PubMed Central PMCID: PMC2906566.
- Krauthausen M, Kummer MP, Zimmermann J, et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer‘s disease model. J Clin Invest. 2015 Jan;125(1):365–378. PubMed PMID: 25500888; PubMed Central PMCID: PMC4382235.