
ABSTRACT
Macroautophagy/autophagy has been implicated in cytoplasmic and viral antigen presentation on major histocompatibility complex (MHC) class II molecules. However, the role of autophagy in the presentation of phagocytized tumor-associated antigens in vivo remains unclear. Following the administration of apoptotic tumor cells and in vivo chemotherapy, mice with a dendritic cell–specific deletion of Atg5, a key autophagy gene, exhibit reduced CD4+ T-cell priming but not CD8+ cytotoxic T-cell priming. Interestingly, Atg5-deficient dendritic cells have an elevated expression of scavenger receptor CD36 and show excessive lipid accumulation. Atg5-deficient dendritic cells increased CD36-dependent phagocytosis of apoptotic tumor cells. CD36 blockade ameliorates elevated phagocytosis and increases CD4+ T-cell priming in dendritic cells; intratumoral CD36 blockade inhibits tumor growth. Our results demonstrate that Atg5 is required for proper antigen phagocytosis and presentation to MHC class II via modulation of CD36 in dendritic cells and may be a future therapeutic target for anti-tumor therapy.
Abbreviations: APC: antigen-presenting cell; ATG: autophagy-related; BMDC: bone marrow–derived dendritic cell; BODIPY: 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene; CSFE: carboxyfluorescein diacetate succinimidyl ester; DAPI: 4ʹ,6-diamidino-2-phenylindole; IFNG/IFN-γ: interferon gamma; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MHC: major histocompatibility complex; NLDC: neonatal liver–derived dendritic cell; PDCD1/PD-1: programmed cell death 1; PI: propidium iodide; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol 3-phosphate; SERPINB/OVA: serine (or cysteine) peptidase inhibitor, clade B; TIMD4/TIM-4: T cell immunoglobulin and mucin domain containing 4
Introduction
Macroautophagy is a catalytic process and conserved pathway found in almost all eukaryotic species that transport cytoplasmic substrates for lysosomal degradation [Citation1]. Autophagy-related (ATG) proteins generate a double-membrane vesicle called the autophagosome [Citation2]. In addition to its function of homeostasis, this autophagic process is used by antigen-presenting cells to present intracellular, endocytic, or phagocytic material on major histocompatibility complex (MHC) class II molecules to CD4+ T cells [Citation3]. Although several studies report the roles of ATGs involved in the presentation of MHC class II antigens such as viral infections or central nervous system diseases, the roles of Atgs in anti-tumor immune responses remain largely unknown [Citation4,Citation5].
Autophagy is involved in the regulation of tumor immunity in both tumor cells and immune cells. Previous studies suggest that tumor cells undergo autophagy to disrupt tumor immune surveillance or anti-tumor immunity [Citation6]. In addition, a recent study revealed that Atg5 in myeloid cell compartments contribute to tumor progression and metastasis [Citation7]. However, little is known about the roles of Atg5 in dendritic cells in the generation of anti-tumor immunity.
Apoptotic tumor cells are the most significant tumor antigens produced by chemotherapy and radiotherapy and help generate the anti-tumor T-cell response [Citation8]. In addition, apoptotic tumor cells are major phagocytic antigens in the generation of anti-tumor T-cell responses [Citation9]. Current studies indicate that ATGs are associated with the regulation of phagocytosis [Citation10]. The ATG12–ATG5-ATG16L1 complex is involved in the conjugation of microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) to the phagophore membrane. Moreover, LC3-associated phagocytosis is a non-canonical autophagy where LC3 is conjugated to phagosomal membranes using the canonical autophagy machinery [Citation11]. In addition, antigens with Toll-like receptor signals and live or apoptotic cell debris were processed in LC3 to phagosomes, resulting in a more rapid degradation in lysosomes [Citation12]. Another study shows that autophagy modulates phagocytosis by regulating the expression of scavenger receptors [Citation13]. Although several studies have demonstrated the link between phagocytosis and the autophagic process, little is known about the roles of ATG5 in the processing and antigen presentation of phagocytic apoptotic tumor cells and the links between scavenger receptors, which are important for apoptotic tumor cell clearance and antigen presentation in dendritic cells.
Here, we used apoptotic tumor cells to investigate the roles of ATG5 in anti-tumor T-cell activation. Our in vivo results showed that ATG5 is required for activation of tumor antigen-specific CD4+ T cells but not for activation of CD8+ cytotoxic T lymphocytes. Furthermore, ATG5 was required for the priming of anti-tumor CD4+ T-cell responses in ITGAX/CD11c+ dendritic cells. Atg5 deficiency promoted increased phagocytosis of apoptotic tumor cells and induced excessive lipid accumulation in dendritic cells. Moreover, increased phagocytosis of apoptotic tumor cells in Atg5-deficient dendritic cells was due to the increased expression of scavenger receptor CD36. The blockade of CD36 ameliorated elevated phagocytosis of apoptotic tumor cells and increased CD4+ T-cell priming in Atg5-deficient dendritic cells. Moreover, intratumoral CD36 blockade remarkably suppressed tumor growth.
Results
Atg5 deficiency reduces anti-tumor CD4+ T-cell priming
The generation of anti-tumor immunity was based on the recognition of dead or apoptotic tumor cells caused by immunological cell death in antigen-presenting cells (APCs) [Citation14]. Tumor-specific T cells induced by APCs in tumor-draining lymph nodes and infiltrated into the tumor mass directly or indirectly killed tumor cells, subsequently promoting tumor mass reduction. To understand the function of Atg5 in anti-tumor immunity, we generated hematopoietic cell-specific Atg5-deficient chimera mice because the lack of the Atg5 gene is known to be neonatally lethal in mice [Citation15]. Reconstitution of atg5 knockout mice in the hematopoietic compartment was successfully established 8 weeks after chimerism was detected (Figure S1). To examine the role of Atg5 in T-cell immune response against tumor cells, we employed the adaptive immune response tumor-bearing mice model induced by oxaliplatin, which is known to induce immunological cell death in tumor cells and induce adaptive T-cell immunity [Citation16]. Oxaliplatin was intraperitoneally injected into the serine (or cysteine) peptidase inhibitor, clade B (SERPINB/OVA)–expressing EL4 (EG7) tumor-bearing Atg5+/- or atg5−/- chimeras to induce apoptosis and degradation of tumor cells in vivo. After 5 days of systemic chemotherapy, isolated CD4+ and CD8+ T cells from tumor-draining lymph nodes were re-stimulated with SERPINB/OVA protein. Our results showed that interferon gamma (IFNG/IFN-γ) production from CD4+ T cells is significantly reduced in atg5−/- chimeras while IFNG/IFN-γ production from CD8+ cytotoxic T cells is not affected ().
Next, to confirm our results, we used another mice model with irradiated tumor cells to monitor anti-tumor T-cell immune responses. Apoptotic or dead tumor cells produced by tumor radiotherapy serve as the major source of tumor-associated antigens for APCs to activate T cells. Apoptotic tumor cells, which are induced by 4000 rad γ-ray irradiation and 48 h incubation, were used as the source of the tumor-associated antigens (Figure S2). CFSE-labeled OT-II T cells (SERPINB/OVA-specific CD4+ T cells) receptor were adoptively transferred into Atg5+/+ or atg5−/- chimeras, apoptotic EG7 cells were injected subcutaneously into the footpad of each chimera, and CD4+ T-cell proliferation was assessed by CFSE dilution. We found that the proliferation of OT-II cells is significantly reduced in atg5−/- chimeras (,d)). To test whether Atg5 is required for tumor antigen presentation by dendritic cells, we crossed Atg5flox/flox mice with ITGAX/CD11c-Cre+ mice (ITGAX/CD11c-atg5−/-), which are selectively defective for autophagy in dendritic cells. We assessed chemotherapy-induced IFNG/IFN-γ production by CD4+ T cells in the tumor-draining lymph nodes after systemic oxaliplatin treatment. Our results demonstrate that IFNG/IFN-γ production of CD4+ T cells is reduced in ITGAX/CD11c-atg5−/- mice ()). After CFSE-labeled OT-II T cells were adoptively transferred into ITGAX/CD11c-atg5−/- mice, apoptotic EG7 cells were injected subcutaneously into each mouse, and T-cell proliferation was monitored by CFSE dilution. The proliferation of OT-II cells was significantly reduced in ITGAX/CD11c-atg5−/- mice ()). However, when cognitive peptide SERPINB/OVA323-339 pulsed to Atg5f/f or ITGAX/CD11c-atg5−/- splenic dendritic cells, OT-II T-cell priming activity was comparable (Figure S3), suggesting that the intracellular MHC class II presentation pathway of apoptotic tumor cells is affected by Atg5 deficiency in dendritic cells.
Figure 1. Impaired anti-tumor CD4+ T-cell priming in Atg5-deficient dendritic cells. (a) Experimental scheme for B. (b) Atg5± or atg5−/- chimera mice were inoculated subcutaneously with live EG7 cells on day 0. The mice were treated with systemic oxaliplatin on day 8. On day 13, CD4+ T cells and CD8+ T cells from draining lymph nodes were co-cultured with wild-type splenocytes as antigen-presenting cells in the presence or absence of SERPINB/OVA protein for 72 h. Concentrations of IFNG/IFN-γ in the culture medium were measured by ELISA. Mean concentrations are presented with standard deviations (ND: not detected; Student’s t-test, **P < 0.01). Data are representative of 3 independent experiments. (c) Experimental scheme for D. (d) CFSE-labeled OT-II CD4+ T cells were adoptively transferred into Atg5+/- or atg5−/- chimeras on day −1, and then apoptotic EG7 cells were injected into the footpads of mice on day 0. After 64 h, CFSE dilution was measured with flow cytometry. Data are representative of 2 independent experiments. (E) Atg5f/f or ITGAX/CD11c-atg5−/- mice were treated with systemic oxaliplatin 8 days after live EG7 subcutaneous inoculation. After 5 days of oxaliplatin treatment, CD4+ T cells from draining lymph nodes were co-cultured with wild-type splenocytes as antigen-presenting cells in the presence or absence of SERPINB/OVA protein for 72 h. Total amount of IFNG/IFN-γ in the culture supernatant was measured by ELISA. Mean concentrations are presented with SDs (Student’s t-test; ***P < 0.001). Data are representative of 3 independent experiments. (F) CFSE-labeled OT-II CD4+ T cells were adoptively transferred into Atg5f/f or ITGAX/CD11c-atg5−/- mice on day −1, and γ-ray–irradiated EG7 were then injected into the footpads of mice on day 0. CFSE dilution was measured by flow cytometry 64 h after irradiated EG7 injection. Data are representative of 3 independent experiments.
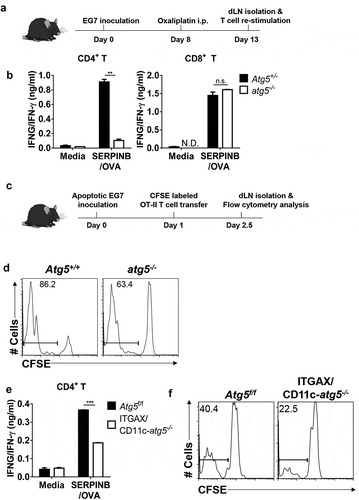
Taken together, these results indicated that Atg5 is essential for the activation of tumor antigen–specific CD4+ T cells but dispensable for CD8+ cytotoxic T lymphocyte activation in vivo. Moreover, these results demonstrate that Atg5 is required for tumor antigen presentation by dendritic cells to prime anti-tumor CD4+ T-cell responses.
Atg5 deficiency does not affect MHC-I antigen presentation
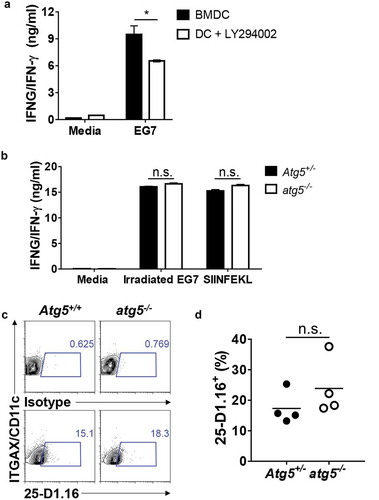
Although the results of our previous experiment indicated that Atg5 is not involved in CD8+ T-cell priming for apoptotic EG7 antigen in dendritic cells, several reports have shown that autophagy is involved in CD8+ T-cell activation through cross-presentation [Citation17]. To test whether autophagy affects CD8+ T-cell priming of MHC class I antigen presentation of the apoptotic tumor cell in dendritic cells, we first generated bone marrow–derived dendritic cells (BMDCs) and treated them with LY294002, a class III phosphatidylinositol 3-kinase (PtdIns3K) inhibitor known to be an autophagy inhibitor. Then, we co-cultured them with apoptotic tumor cells and OT-I T cells, and IFNG/IFN- γ production in the supernatants was measured with ELISA. The results showed that IFNG/IFN-γ production is significantly decreased in LY294002-treated BMDCs (Figure 2(a)). However, we found no difference in IFNG/IFN-γ production between the Atg5+/- or atg5−/- neonatal liver–derived dendritic cells, OT-I T cells, and apoptotic EG7 co-culture ()). Finally, we used a 25-D1.16 antibody, which recognizes SERPINB/OVA-derived peptide SIINFEKL bound to MHC class I molecule H-2Kb, to evaluate APCs. The results showed that SIINFEKL presentation in MHC class I is comparable between Atg5+/- or atg5−/- neonatal liver–derived dendritic cells (NLDCs) and apoptotic EG7 co-culture (,d)). Thus, in MHC class I antigen presentation of apoptotic tumor cells, CD8 + T-cell priming was unimpaired by Atg5 deficiency in dendritic cells.
Figure 2. Effect of Atg5 deficiency and PtdIns3K inhibitor treatment on CD8+ T-cell priming and MHC class I presentation. (a) Wild-type BMDCs were pre-incubated with LY294002 and then co-cultured with irradiated EG7 cells and OT-I CD8+ T cells. Concentrations of IFNG/IFN-γ in the culture medium were measured by ELISA. Mean concentrations are presented with standard deviations (Student’s t-test; *P < 0.05). Data are representative of 3 similar independent experiments. (b) Atg5± or atg5−/- NLDCs were co-cultured with irradiated EG7 cells and OT-I CD8+ T cells. After 72 h, concentrations of IFNG/IFN-γ in supernatant were measured by ELISA. Data are representative of 3 similar experiments. (c) Atg5+/- or atg5−/- NLDCs were co-cultured with irradiated EG7 cells. After 24 h, SERPINB/OVA-specific MHC class I antigen presentation was measured by antibody for SERPINB/OVA257-264 (SIINFEKL) peptide bound to H-2Kb. (d) Dot graph showing data pooled from 4 independent experiments. Error bars indicate the standard error of the mean.
Comparable Atg5-deficient dendritic cell infiltration into tumor mass
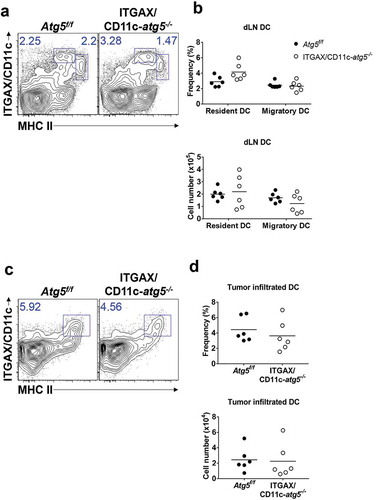
Next, we examined the role of Atg5 in dendritic cell migration and tumor infiltration. Tumor-infiltrated dendritic cells play roles in the regulation of tumor regression or growth and in tumor antigen presentation [Citation18]. In addition, recent research shows that lysosome biogenesis factor transcription factor EB (TFEB), a critical transcription factor for autophagosome generation, controls the migration of dendritic cells [Citation19]. To test whether Atg5 affects the infiltration of dendritic cells into a tumor mass, we subcutaneously inoculated Atg5f/f control mice or ITGAX/CD11c-atg5−/- mice with live EG7 cells. The frequency and number of dendritic cells in tumor-draining lymph nodes and tumor mass were measured by flow cytometry 15 days after tumor injection. The dendritic cells of the lymph nodes were divided into migratory (CD3− CD19− MHCIIhigh ITGAX/CD11c+) and resident (CD3− CD19− MHCII+ ITGAX/CD11c+) dendritic cells. We found no significant difference in the frequency or cell number between migratory or resident dendritic cells in tumor-draining lymph nodes (Figure 3(a,b)) or tumor-infiltrated dendritic cells ()). Although previous studies have suggested the possibility of a link between autophagy and dendritic cell migration, our results suggest that the Atg5 gene deletion in dendritic cells does not affect dendritic cell tumor infiltration.
Figure 3. Comparison of dendritic cell DC infiltration into tumor mass. Atg5f/f or ITGAX/CD11c-atg5−/- mice were inoculated subcutaneously with live EG7 cells. Dendritic cells in the tumor-draining lymph nodes (a) or tumor-infiltrated dendritic cells (c) were analyzed with flow cytometry 15 days after tumor inoculation. (b and d) Dot graph showing the frequency and absolute cell number of dendritic cells. Data are representative of 3 similar experiments.
Increased phagocytosis in Atg5-deficient dendritic cells
Our results indicate that the deletion of the Atg5 gene in dendritic cells reduces the immune response of CD4+ T cells, without affecting the ability of tumor infiltration or migration in dendritic cells. To explain the underlying mechanism of these phenotypes, we examined the possibility that MHC class II presentation is reduced by impaired phagocytosis of tumor-associated antigens. MHC class II presentation and CD4+ T-cell priming can be inappropriately induced when less or more tumor-associated antigens are phagocytosed [Citation20]. To examine phagocytosis ability in Atg5-deficient dendritic cells, Atg5+/- or Atg5−/- BMDCs were co-cultured with DiI-labeled apoptotic EG7 cells, and phagocytosis were measured with flow cytometry. Unexpectedly, our results showed that Atg5 deficiency increases the phagocytosis of apoptotic tumor cells in dendritic cells while the ability to uptake the fluorochrome-labeled latex beads remain intact (). In addition, the kinetics of phagocytosis confirmed that the phagocytosis of apoptotic cells increases from 30 min to 3 h in Atg5-deficient dendritic cells (). However, intracellular degradation activity in Atg5-deficient dendritic cells was comparable to that in wild-type cells ()). To determine the phagocytosis of apoptotic tumor cells also affected by other Atgs such as Atg7 or Atg16l1 in dendritic cells, we transfected Atg7 or Atg16l1 siRNA to BMDCs, and phagocytosis were monitored with flow cytometry. We found that Atg7 or Atg16l1 knockdown does not affect the phagocytosis of apoptotic tumor cells (Figure S4). Our data suggest that regulation of phagocytosis of apoptotic tumor cells is a unique role of Atg5 in dendritic cells.
Figure 4. Increased phagocytosis of apoptotic tumor cells in dendritic cells. (a and b) Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were co-cultured with DiI-labeled apoptotic EG7 cells for 3 h (a) or fluorochrome-labeled latex beads for 1 h (b). Uptake of DiI-labeled apoptotic tumor cells by BMDCs was determined by flow cytometry (c and d). Graph showing the frequency of phagocytosis in DiI-labeled apoptotic tumor cells (c) or latex beads (d) of dendritic cells (Student’s t-test; **P < 0.01). The results shown are representative of 3 similar independent experiments. (e) The footpads of Atg5f/f or ITGAX/CD11c-atg5−/- mice were injected with DiI-labeled apoptotic EG7 cells. After 24 h, the draining lymph node was isolated and phagocytosis of apoptotic tumor cells in migratory (ITGAX/CD11c+ MHC class IIhi) or resident dendritic cells (ITGAX/CD11c+ MHC class IIint) was monitored by flow cytometry. (f) Dot graph showing the frequency of DiI-positive dendritic cells in the draining lymph nodes (4 mice per group; Student’s t-test; **P < 0.01). Data are representative of 3 similar independent experiments.
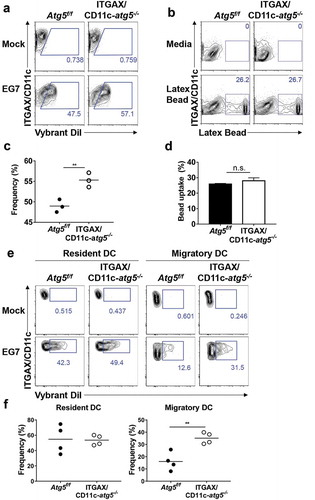
Figure 5. Elevated phagocytosis in Atg5-deficient dendritic cells is not due to change protein degradation activity. (a) Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were co-cultured with DiI-labeled apoptotic EG7 cells. At the indicated time points, phagocytosis of apoptotic tumor cells was monitored with flow cytometry. (b) Dot graph showing the frequency of phagocytosis in DiI-positive dendritic cells over time. (c) Total proteins from Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were isolated with cell lysis buffer. Proteolytic activity in total proteins was detected with an EnzCheK protease assay kit.
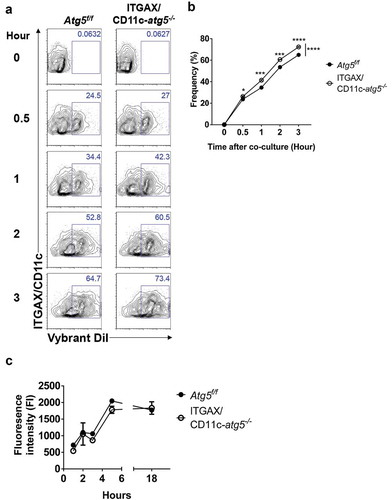
Next, we confirmed these results in vivo. Migratory dendritic cells phagocytose tumor-associated antigens, directly transport them into draining lymph nodes, and present the tumor antigen to the T cells [Citation21]. In addition, tumor-associated antigens are drained via lymphatic flow, are captured by resident dendritic cells in draining lymph nodes, and are presented to the T cells. This suggests that both resident and migratory dendritic cells in the lymph nodes have the ability to phagocytose tumor antigens in vivo. To verify this implication, DiI-labeled apoptotic tumor cells were injected into the footpads of Atg5f/f or ITGAX/CD11c-atg5−/- mice, and after 24 hours, draining lymph nodes was isolated and analyzed with flow cytometry. We found that the phagocytosis of apoptotic tumor cells was increased in migratory dendritic cells but not in resident dendritic cells of ITGAX/CD11c-atg5−/- mice (,f)). In summary, our results show that Atg5 deficiency promotes increased phagocytosis of apoptotic tumor cells in migratory dendritic cells, which are the major dendritic cell type to drive anti-tumor T-cell responses.
Upregulated scavenger receptor in Atg5-deficient dendritic cells
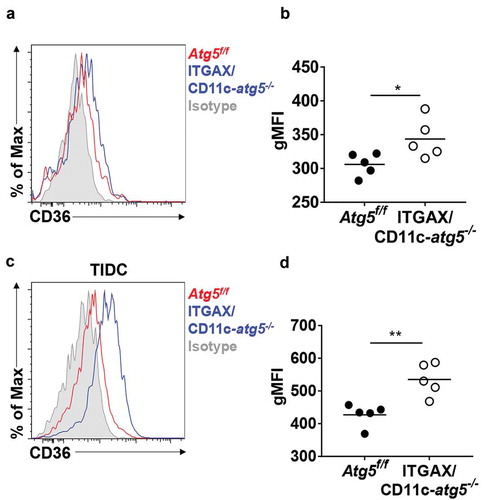
A recent study demonstrated that Atg7 deficiency enhances phagocytic function and increases susceptibility to Mycobacterium tuberculosis infection in macrophages [Citation13]. This research showed that Atg7 regulates phagocytosis by upregulating scavenger receptor expression and increasing the activity of NFE2L2 (nuclear factor, erythroid derived 2, like 2). Scavenger receptors are involved in complex events such as phagocytosis, antigen presentation, and the clearance of apoptotic cells [Citation22]. Among them, CD36 is known to be expressed in dendritic cells and associated with phagocytosis of apoptotic cells [Citation23]. We found that Atg5 deficiency promotes an increase of surface CD36 expression in Atg5f/f or ITGAX/CD11c-atg5−/- mice. Our flow cytometry results showed that surface CD36 expression was elevated in migratory dendritic cells of the lymph nodes (Figure 6(a,b) and in tumor-infiltrated Atg5-deficient dendritic cells in EG7-bearing mice ). Our results suggest that Atg5 controls scavenger receptor CD36 expression in dendritic cells.
Figure 6. Elevated CD36 expression in Atg5-deficient dendritic cells in vivo. (a) Surface CD36 expression in migratory dendritic cells from draining lymph nodes of normal Atg5f/f (red) or ITGAX/CD11c-atg5−/- mice (blue) monitored with flow cytometry. Isotype control used as a negative control (gray). (b) Data shown in dot graph (5 mice per group; *P < 0.05). (c) Surface CD36 expression in tumor-infiltrated dendritic cells from tumor-bearing Atg5f/f (red) or ITGAX/CD11c-atg5−/- mice (blue). (d) Data shown in dot graph (5 mice per group; **P < 0.05). Data are representative of 3 similar independent experiments.
Lipid accumulation in Atg5-deficient dendritic cells
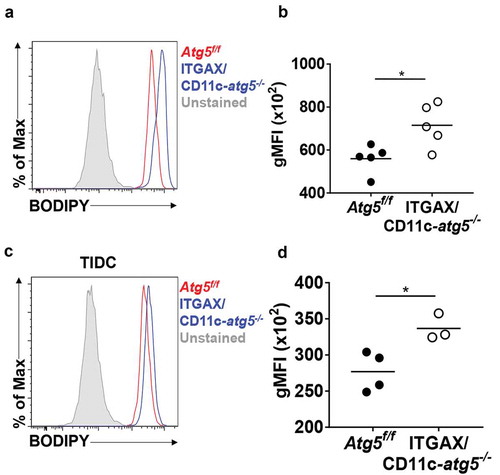
Scavenger receptor CD36 is not only an important receptor for phagocytosis apoptotic cells but also an important receptor for lipid uptake in the steady state in dendritic cells [Citation24]. Excessive accumulation of lipids reportedly impairs cell function, and tumor-infiltrated dendritic cells show higher amounts of intracellular lipids that are induced by endoplasmic reticulum stress and triggered by the tumor microenvironment to inhibit anti-tumor immunity [Citation25]. If the increased expression of CD36 in Atg5-deficient dendritic cells leads to an increase in the amount of lipid in the cells, the function of dendritic cells may become impaired. We examined the total amount of lipids in migratory dendritic cells of the lymph nodes of Atg5f/f or ITGAX/CD11c-atg5−/- mice. Total lipids of dendritic cells were measured by flow cytometry using the lipophilic dye BODIPY. We found that lipids were more accumulated in migratory dendritic cells of ITGAX/CD11c-atg5−/- mice than in those of Atg5f/f mice (Figure 7(a,b)). This higher lipid accumulation was also demonstrated in tumor-infiltrated Atg5-deficient dendritic cells (). Next, we found that mitochondrial lipid usage was affected by Atg5 deficiency in dendritic cells. Mitochondrial fuel usage was assessed in Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs, and our results showed that fatty acid dependency in mitochondria was not affected by Atg5 deficiency in dendritic cells (Figure S5). Thus, our results suggest that increased expression of the scavenger receptor in Atg5 deficient dendritic cells induces excessive lipid accumulation, indicating reduced function of the dendritic cells but no metabolic changes.
Figure 7. Increased lipid accumulation in Atg5-deficient dendritic cells in vivo. (a) Total amount of lipids in migratory dendritic cells from draining lymph nodes of normal Atg5f/f (red) or ITGAX/CD11c-atg5−/- mice (blue) were stained with BODIPY dye and monitored with flow cytometry. Unstained sample was used as negative control (gray). (b) Data shown in dot graph (5 mice per group; *P < 0.05). (c) Total amount of lipids in tumor-infiltrated dendritic cells from tumor-bearing Atg5f/f (red) or ITGAX/CD11c-atg5−/- mice (blue). (d) Data shown in dot graph (3 ~ 4 mice per group; *P < 0.05). Data are representative of 3 similar independent experiments.
CD36 blockade enhances CD4+ T-cell priming with anti-tumor effect
To investigate the association of increased CD36 expression and phagocytosis in Atg5-deficient dendritic cells, we examined whether the phagocytosis of apoptotic tumor cells was affected by CD36 blockade. We blocked the scavenger receptor CD36 using anti-CD36 blocking antibody in Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs and co-cultured with DiI-labeled apoptotic tumor cells. Our results showed that the phagocytosis of apoptotic tumor cells is decreased in both Atg5f/f and ITGAX/CD11c-atg5−/- BMDCs after CD36 blockade. The increased phagocytosis in ITGAX/CD11c-atg5−/- BMDCs compared with Atg5f/f BMDCs was also decreased after CD36 blockade and was comparable with Atg5f/f BMDCs (Figure 8(a,b)). These results indicate that increased phagocytosis caused by Atg5 deficiency in dendritic cells is CD36 dependent.
We also examined whether the priming of CD4 + T cells in dendritic cells could be enhanced by CD36 blocking. Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were co-cultured with apoptotic tumor cells and OT-II T cells with or without CD36 blockade. When CD36 was blocked in Atg5f/f BMDCs, IFNG/IFN-γ production of CD4 + T cells was increased in Atg5f/f BMDCs and Atg5-deficient BMDCs. However, the increased amount of IFNG/IFN-γ production in Atg5-deficient BMDCs was not similar to the increase in Atg5f/f BMDCs ()). Because autophagy is involved in MHC class II antigen presentation in dendritic cells through various mechanisms, it does not seem to increase IFNG/IFN-γ production completely [Citation26].
Figure 8. Blockade of CD36 ameliorates elevated phagocytosis and increases CD4+ T-cell priming. (a) BMDCs from Atg5f/f or ITGAX/CD11c-atg5−/- mice were co-cultured with DiI-labeled apoptotic tumor cells in the presence of anti-mouse CD36 blocking antibody. Phagocytosis of dendritic cells was measured with flow cytometry. (b) Frequency of phagocytosis shown as bar graph (Student t-test, * P < 0.05). Data represent 3 independent experiments. (c) Atg5f/f or ITGAX/CD11c-atg5−/- splenic dendritic cells were co-cultured with apoptotic EG7 and OT-II CD4+ T cells with or without anti-mouse CD36 blocking antibody for 96 h. IFNG/IFN-γ production in the supernatant were measured with ELISA (Student t-test; **P < 0.01; ***P < 0.001; ****P < 0.0001). Data represents 2 independent experiments. (d) EG7 tumor-bearing Atg5f/f or ITGAX/CD11c-atg5−/- mice were injected intratumorally with anti-mouse CD36 blocking antibody at day 11, 13, and 15 after EG7 inoculation. Tumor size was monitored with a digital caliper (5 mice per group; two-way ANOVA; *P < 0.05; ***P < 0.001). Data are representative of 2 independent experiments.
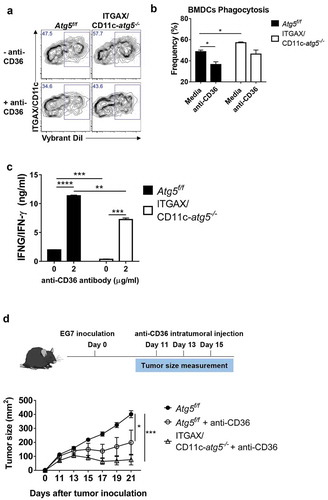
Finally, we examined whether intratumoral blockade of CD36 could promote an anti-tumor effect. In the EG7 subcutaneous model, CD8+ T cells are known to the most important cell type to control tumor size [Citation27,Citation28], and our results showed that Atg5 deficiency does not affect anti-tumor CD8+ T-cell responses. When CD8+ T cells were depleted in mice, tumor size was significantly increased in both Atg5f/f and ITGAX/CD11c-atg5−/- mice (Figure S6C). In addition, we found that tumor growth induced by subcutaneously injected EG7 cells in Atg5f/f or ITGAX/CD11c-atg5−/- mice was not different between the 2 mice groups (Figure S6A and B). However, when EG7 was transplanted into Atg5f/f or ITGAX/CD11c-atg5−/- mice, and then anti-CD36 antibody was injected into the tumor to block CD36, tumor growth was inhibited in Atg5f/f and ITGAX/CD11c-atg5−/- mice ()). Moreover, the anti-tumor effect of CD36 was significantly ameliorated by CD4+ T-cell depletion (Figure S7). Furthermore, we found that EG7 tumor cells and CD8+ T cells do not express CD36 (Figure S8), suggesting that the anti-tumor effect of the anti-CD36 antibody appears predominantly in dendritic cells. Our results suggest that blocking CD36 expression in dendritic cells can regulate phagocytosis and increase reduced MHC class II presentation. They also suggest anti-CD36 antibody as a possible new candidate molecule for anti-tumor therapy.
Discussion
In this study, we investigated the roles of autophagy protein Atg5 in anti-tumor T-cell activation in dendritic cells. Our in vivo results demonstrate that Atg5 is required for the activation of tumor antigen–specific CD4+ T cells but not that of cytotoxic CD8+ T cells. In addition, the results in ITGAX/CD11c-atg5−/- mice indicate that Atg5 is required for the proper priming of anti-tumor CD4+ T-cell responses in dendritic cells. However, Atg5 deficiency did not affect CD8+ T-cell priming and MHC class I presentation in dendritic cells, even though CD8+ T-cell priming was partially affected by PtdIns3K inhibitor treatment. Unexpectedly, Atg5 deficiency promoted increased phagocytosis of apoptotic tumor cells in dendritic cells. Moreover, enhanced phagocytosis in the absence of Atg5 was due to the increased expression of scavenger receptor CD36. Increased phagocytosis of apoptotic tumor cells in Atg5-deficient dendritic cells was reduced by the blockage of CD36. Excessive accumulation of lipids has been reported to impair dendritic cell function, and scavenger receptor CD36 plays an important role in lipid homeostasis. We found lipid accumulation in Atg5-deficient dendritic cells and tumor-infiltrated dendritic cells. In addition, elevated CD36-dependent phagocytosis of apoptotic tumor cells detrimentally affected CD4+ T-cell priming in the apoptotic tumor antigen presentation of dendritic cells. Furthermore, CD36 blockade increased CD4+ T-cell priming in Atg5-deficient dendritic cells. Finally, we evaluated the efficacy of CD36 monoclonal antibodies as therapeutic agents against tumors. Our results show that subcutaneous injection of anti-CD36 antibody effectively inhibits the growth of tumors.
Various autophagy-related proteins have been reportedly associated with MHC class II presentation on intracellular or extracellular antigens in dendritic cells [Citation3]. Especially for extracellular antigens, Atgs contribute to phagocytosis following a process that has been described previously as LC3-associated phagocytosis [Citation11]. A previous study showed that the uptake of dead cells induces LC3-associated phagocytosis and is triggered by engagement of the receptor TIMD4/TIM-4 (T cell immunoglobulin and mucin domain containing 4) [Citation29]. In addition, mice with LC3-associated phagocytosis deficiencies showed defective clearance of dying cells, resulting in enhanced production of pro-inflammatory cytokines [Citation30]. According to our study, Atg5 deficiency does not affect TIMD4 expression in dendritic cells (Figure S10). Future studies are needed to investigate the association of CD36 and TIMD4 in the dying or apoptotic cell processing in dendritic cells.
Specific receptors (such as Toll-like receptors) and the uptake of dectin-1-mediated antigens induce LC3-associated phagosomes, which can fuse with lysosomes, and are sequentially degraded in the phagolysosome with dendritic cells [Citation31]. In addition, LC3-associated phagocytosis phagosomes contribute to the MHC class II presentation of extracellular antigens [Citation32]. According to a previous study, extracellular SERPINB/OVA or phagocytic antigens in Atg5-deficient environments are not proficiently processed and presented to CD4+ T cells [Citation4]. Consistent with previous results, our results showed that apoptotic tumor cells, the major antigen type resulting from chemotherapy or radiotherapy, were processed via autophagic machinery and presented to CD4+ T cells.
Various ATGs are involved in autophagosome formation [Citation33]. For example, in the pre-initiation of autophagosome formation, ATG13, unc-51 like kinase 1/2 (ULK1/2), and FIP200 (FIP200/RB1CC1, RB1-inducible coiled-coil 1) are negatively and positively regulated by mechanistic target of rapamycin kinase (MTOR) or AMP-activated protein kinase (AMPK). In addition, at the initiation stage, ATG14, BECN1 (beclin 1, autophagy related), PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), and PIK3R4/VPS15 (phosphoinositide-3-kinase regulatory subunit 4) are critical proteins for the formation of the PtdIns3K complex and generate phosphatidylinositol 3-phosphate (PtdIns3P) at the site of nucleation in the isolated autophagosome membrane. During the elongation of the autophagosome, LC3, ATG4, ATG3, and ATG12–ATG5-ATG16L1 conjugates associate with the mature autophagosome. However, we could not find a difference in phagocytosis between Atg7 or Atg16l1 knockdown BMDCs. Although additional experiments using genetically knock-out mice models are needed to support our results, we believe that regulation of phagocytosis in dendritic cells is a unique role of ATG5. In addition, recent papers show that ATG5 is the only ATG that is autophagy independent and has unexpected roles in several disease models [Citation34,Citation35]. Although our study has identified a role of ATG5 in anti-tumor immunity of dendritic cells, we recommend that further studies investigate this role at the various stages of MHC class II antigen presentation.
Our results present detailed insight into the role of Atg5 in dendritic cells during apoptotic tumor cell processing and MHC class II presentation. As with the results of the Atg5 chimera, ITGAX/CD11c-specific Atg5-deficient mice failed to induce a CD4+ T-cell response. For tumor antigens, dendritic cells are known to activate CD4+ T cells through MHC class II presentation [Citation36]. In addition, a recent study showed that dendritic cells use ATG-dependent phagocytosis for efficient presentation of myelin antigen by MHC class II during autoimmune central nervous system inflammation [Citation5]. Consistent with previous results, our results showed that apoptotic tumor cells were processed via autophagic machinery and presented to CD4+ T cells. Our results support the previous findings that Atg5 plays an important role in MHC class II presentation in dendritic cells.
By a process called cross-presentation, dendritic cells can present extracellular antigens via MHC class I proteins through several pathways, and several studies show the Atgs contribute to this process [Citation37]. However, only soluble protein, not CD205 dependent-endocytic protein or apoptotic cells, requires Atgs for efficient cross-presentation [Citation38]. Autophagy-related proteins are required for the cross-presentation of soluble antigens, which are produced by macropinocytosis or pinocytosis, but are not required by phagocytosis [Citation3]. Our study confirmed that apoptotic tumor antigen presentation via MHC class I to CD8+ T cells was not affected by Atg5 deficiency in dendritic cells.
Unlike T cells, current studies reported that the deletion of Atgs in dendritic cells does not affect the development or homeostasis of dendritic cells in vivo [Citation38,Citation39]. Although several studies have indicated that autophagy-related genes are related to dendritic cells migration, our results support other previous studies that showed Atg5 deficiency in dendritic cells has no effect on the number and frequency of dendritic cells in tumor-bearing mice. This finding suggests that the deficiency of Atg5 is involved in the presentation of MHC class II antigens through functional changes rather than quantitative changes in dendritic cells.
Efficient phagocytosis of antigens is crucial for an immune response, and macrophages and dendritic cells are professional phagocytes [Citation40]. In contrast to macrophages that degrade phagocytosed antigens rapidly, dendritic cells maintain antigens longer and sustain antigen presentation on MHC class II proteins [Citation41]. As shown by several studies, excessive phagocytosis and proteolysis of antigens is harmful to antigen presentation, and appropriate antigen uptake is a critical factor for efficient antigen presentation [Citation42]. Interestingly, as shown in ), the phagocytosis of apoptotic tumor cells was increased in Atg5-deficient dendritic cells. In vivo results on the phagocytosis capacity of apoptotic tumor cells confirmed that ITGAX/CD11c-specific Atg5 deletion induces the phagocytosis of apoptotic tumor cells in migratory dendritic cells. Our results suggest that excessive phagocytosis in Atg5-deficient dendritic cells affects MHC II antigen presentation and CD4+ T-cell activation.
Scavenger receptor CD36 is a critical receptor for apoptotic tumor cell phagocytosis in dendritic cells [Citation22]. In addition, CD36 contains various ligands such as bacterial cell walls or β-glucans [Citation43,Citation44]. It has been reported that CD36 is involved in a variety of immune responses. In particular, endocytosis via CD36 is required for proper MHC class II antigen pentation while CD8+ T-cell priming normally occurs in CD36-deficient dendritic cells [Citation45,Citation46]. However, a recent study has shown that CD4+ T-cell priming is still successful in CD36-deficient BMDCs [Citation47]. This evidence suggests that CD36-required endocytosis is involved in the presentation of MHC class II antigens; however, this assumption remains controversial. In addition, a current study reported that CD36-mediated phagocytosis of apoptotic medullary thymic epithelial cells is required for allotolerance in dendritic cells in vivo, indicating that CD36-dependent phagocytosis promotes tolerance rather than immunogenicity in dendritic cells [Citation48]. Our results support that the phagocytosis of apoptotic tumor cells through CD36 in dendritic cells is increased by Atg5-deficient dendritic cells. In addition, when CD36 was blocked with antibody, the CD4+ T-cell response significantly increased. This finding indicates that, unlike conventional phagocytosis, CD36-mediated phagocytosis offers the possibility of modulating MHC class II antigen presentation and CD4+ T-cell priming. We have not identified how the CD36-dependent phagosomes are processed in dendritic cells; however, one possible mechanism is that CD36 modulates the endolysosomal pH of apoptotic tumor cell–containing phagosomes, which are phagocytosed by CD36. Two studies show that CD36 is necessary for the action of endosomal pH–elevating agents on long-chain fatty acid uptake and that CD36 trafficking is differentially dependent on endosomal pH [Citation49,Citation50]. In particular, it has been reported that Atg5 deficiency regulates the expression of MHC class II antigens by controlling endolysosomal pH [Citation4]. Therefore, our future research will focus this process, including characteristics of CD36-dependent phagosomes and the intracellular environment of dendritic cells.
We also confirmed the excessive accumulation of lipids in Atg5-deficient dendritic cells due to the increased expression of scavenger receptor CD36, which is an important receptor for cellular lipid uptake [Citation51]. A previous study demonstrated that dendritic cells store more lipids in tumor-bearing mice than those in tumor free mice, causing functional impairment such as the malfunction of CD4+ or CD8+ T-cell priming [Citation25]. In addition, this lipid accumulation in dendritic cells was caused by an increased uptake of extracellular lipids due to elevated scavenger receptor. In the agreement with this study, we also found that Atg5-deficient dendritic cells exhibit elevated CD36 expression and store more lipid contents than wild-type dendritic cells. It has been reported that defects in the autophagy gene cause lipid accumulation. A hepatocyte-specific knock-out of the autophagy gene Atg7 caused hepatic lipid accumulation, and inhibition of autophagy decreased triglycerides β-oxidation and decay [Citation52]. We first elucidated that lipid accumulation in dendritic cells occurs in Atg5 gene defects. However, unlike previous studies, the dependency of lipid as a fuel in mitochondrial oxidative phosphorylation was not significantly changed by Atg5 deficiency in dendritic cells. Our future studies will focus on how CD36-dependent lipid uptake affects the various functions of dendritic cells.
The possibility of CD36-targeted monoclonal antibody as an anti-tumor therapeutic agent has recently been proposed. A previous study showed that tumor metastases were reduced by CD36 blockade in CD44hi cancer stem cells [Citation53]. In effector T cells, large amounts of extracellular fatty acid acquired in a CD36-dependent manner for their proper function and tumor-infiltrating T cells had abnormally high fatty acid uptake and lipid content; moreover, blocking PDCD1/PD-1 (programmed cell death 1) signaling significantly reduced tumor progression and normalized lipid metabolism [Citation54,Citation55]. Our study suggests a new mechanism of tumor therapy using CD36-targeted antibody in dendritic cells. Our study also proposes the level of autophagy in dendritic cells can be used as a marker for predicting the efficacy of an anti-tumor reagent, such as CD36-targeted antibody.
Materials and methods
Mice
Atg5± (B6.129-Atg5tm1Nmz) [Citation15], Atg5flox/flox (B6.129S-Atg5tm1Myok) [Citation15], and ITGAX/CD11c-Cre Tg (B6.Cg-Tg (Itgax-cre)1-1Reiz/J) [Citation56] transgenic mice were generated as described previously and kindly provided by A. Iwasaki (Yale University School of Medicine). OT-1 (C57BL/6-Tg [TcraTcrb]1100Mjb/J) [Citation57] and OT-II (B6.Cg-Tg [TcraTcrb] 425Cbn/J) [Citation58] transgenic mice were purchased from Jackson Laboratories and crossed with CD45.1 (B6.SJL-Ptprca/Pepcb/BoyJ) mice. Mice were bred in a specific pathogen-free facility, and animal care and all animal experiments were approved by KAIST Institutional Animal Care and Use Committee. Male mice matched at 8 and 16 weeks of age were used for each experiment.
Neonatal liver chimera generation
Atg5 chimeras were generated as previously described [Citation59]. Briefly, 2.5 × 106 liver cells from less than 24-h-old pups were confirmed for the Atg5 genotype and transferred into wild-type C57BL/6 mice via tail vein. Before the transfer of the liver cells, recipient mice were lethally irradiated with 475 rad γ-ray 2 times 3 h apart. These mice were maintained with oral antibiotics in the drinking water for 2 weeks. Chimeric mice were used for experiments after 8 weeks post-reconstitution.
Apoptotic tumor cell generation
To induce apoptosis in EG7 cell lines (ATCC, ATCC® CRL-2113™), EG7 cells were irradiated with 4000 rad γ-ray and cultured for 48 h. Apoptosis of EG7 cells were measured with ANXA5/Annexin V (Biolegend, 640,945) and 7-AAD (Biolegend, 420,403) staining and measured with a flow cytometer (FACS Calibur, BD Biosciences). At least 60% of the EG7 cells were ANXA5/Annexin V + and 7-AAD+ double positive.
In vivo t-cell priming by tumor chemotherapy
In vivo T-cell priming was measured as previously described [Citation16,Citation21]. Briefly, indicated mice were inoculated with 1 × 106 live EG7 cells in the shaved right flank on day 0. Each mouse was injected intraperitoneally with 5 mg/kg oxaliplatin (Tocris, 2623) on day 8 to induce tumor degradation and immunological tumor cell death in vivo. Tumor-draining brachial, axillary, and inguinal lymph nodes were removed on day 13. Lymph nodes were minced into small pieces and then digested with a mixture of 2 mg/ml collagenase IV (Worthington Biochemical Corp, LS004189) and 30 µg/ml DNase I (Roche, 10,104,159,001) in Dulbecco’s modified Eagle’s medium (Welgene, LM 001–05) for 30 min at 37°C. Digested lymph nodes were then passed through 70-µm cell strainers. Red blood cells were lysed using an in-house made ACK lysis buffer. Next, CD4+ and CD8+ T cells were enriched with magnetic cell sorting microbeads (Miltenyi Biotec, 130–117-043; 130–117-044). T cells were re-stimulated in vitro with 1 mg/ml SERPINB/OVA protein. IFNG/IFN-γ production in the culture medium at 72 h was determined with a mouse IFNG/IFN-γ ELISA kit (BD Biosciences, 555,138).
In vivo proliferation of OT-II T cells
Anti-tumor CD4+ T-cell response was analyzed as previously described with minor modifications [Citation21]. In brief, SERPINB/OVA-specific OT-II CD4+ T cells from the spleen were negatively enriched with magnetic beads according to the manufacturer’s instruction (Miltenyi Biotec, 130–104-454). These cells were labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the company’s protocol (Invitrogen, C34554) and 2.5 × 106 CD4+ T cells were injected intravenously into mice on day −1. Then, 1 × 107 apoptotic EG7 cells were injected subcutaneously into the footpads of mice on day 0, followed by the isolation of draining popliteal lymph nodes 64 h later. Dilutions of CFSE were assessed using a FACS Calibur.
Dendritic cell generation
Dendritic cells derived from either bone marrow or neonatal liver were differentiated as previously described with slight modifications [Citation60,Citation61]. In brief, liver cells from Atg5± or atg5−/- newborn pups or bone marrow cells from indicated mice were harvested and seeded in tissue culture flasks in RPMI 1640 with 5% in-house prepared CSF2/GM-CSF (colony stimulating factor 2 [granulocyte-macrophage])–containing medium for BMDC generation. Cells were fed every 2 days, and loosely adherent dendritic cells were collected after 7 days.
Phagocytosis assays
For in vitro analysis of phagocytic activity in BMDCs, live EG7 cells were irradiated with 4000 rad γ-ray and incubated in 37°C for 48 h. These cells were labeled with Vybrant DiI (Thermo Fisher Scientific, V22885) according to the manufacturer’s protocol. Next, 2 × 105 Atg5± or atg5−/- BMDCs were incubated with 2 × 105 DiI-labeled apoptotic EG7 cells or 2-μm red fluorescent, carboxylate-modified polystyrene latex beads (Sigma, L3030-1ML). After 3 hours of incubation, cells were washed and analyzed using a FACS Calibur.
For in vivo analysis of phagocytic activity in dendritic cells and macrophages in lymph nodes, 1 × 107 DiI-labeled apoptotic tumor cells were injected into the footpads of the indicated mice. After 24 h of apoptotic tumor cell injection, draining lymph node cells were prepared and pretreated with anti-mouse CD16/32 (BD Biosciences, 553,142) antibody for blocking Fc receptors. Then, they were stained with one of the following antibodies: anti-mouse CD3ε (eBioscience, 25–0031-82), anti-mouse CD19 (eBioscience, 45–0193-82), anti-mouse ITGAX/CD11c (BD Biosciences, 557,400), anti-mouse ITGAX/CD11b (BD Biosciences, 557,397), anti-mouse MHC class II (BD Biosciences, 562,564), or anti-mouse CD45.2 (BD Biosciences, 560,693). DAPI (4ʹ,6-diamidino-2-phenylindole, dihydrochloride) (Invitrogen, D1306) or PI (propidium iodide) (eBioscience, 00–6990-50) was used to exclude dead cells. Gating strategy for flow cytometry analysis was used, as shown in Figure S9. All samples were analyzed on an LSR Fortessa cell analyzer (BD Biosciences).
CD36 expression measurement
To monitor the expression of CD36 in BMDCs, Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were stained with anti-mouse CD36 (BD Biosciences, 562,744). Expression of CD36 was measured with an LSR Fortessa cell analyzer.
To assess CD36 expression in lymph nodes and tumor-infiltrated dendritic cells, Atg5f/f or ITGAX/CD11c-atg5−/- mice were inoculated with 1.5 × 106 live EG7 cells in the shaved right flank on day 0. Fifteen days after EG7 inoculation, tumor-draining lymph nodes and tumor mass were prepared and stained with the previously described method. All samples were analyzed on an LSR Fortessa cell analyzer.
Lipid measurement
Lipid accumulation was measured as previously described [Citation25] with minor modifications. Briefly, after surface staining with fluorescent-labeled antibodies, cells were washed and resuspended in 500 μl BODIPY 493/503 (4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene) (Thermo Fisher Scientific, D3922) at 0.5 μg/ml in Dulbecco’s phosphate-buffered saline for 15 min at room temperature. Samples were then analyzed on an LSR Fortessa cell analyzer.
Tumor growth measurement
Mice were inoculated with 1 × 106 live EG7 cells in the shaved right flank. Tumor growth was measured with a digital caliper (Mitutoyo, 500–181-20) using the following equation: tumor volume = (L × W), where L indicates the length of the long side and W indicates that of the short side.
Cross-presentation assays
Cross-presentation of apoptotic tumor cells was measured as previously described [Citation62] with minor modifications. Briefly, 2 × 105 Atg5± or atg5−/- neonatal liver–derived dendritic cells were co-cultured with 2 × 105 apoptotic EG7 cell cultures for 24 h. Determination of complex quantity of SIINFEKL on H-2Kb (MHC class I) was conducted using anti-mouse SERPINB/OVA257–264 peptide bound to H-2Kb antibody (Ebioscience, 17–5743-82) by FACS Calibur.
To assess IFNG/IFN-γ production in CD8+ OT-I T cells, 2 × 105 Atg5± or atg5−/- neonatal liver–derived dendritic cells were co-cultured with 2 × 105 apoptotic EG7 cells and 2 × 105 splenic CD8+ OT-I T cells from OT-I transgenic mice for 72 h. Then, IFNG/IFN-γ production was measured with a mouse IFNG/IFN-γ ELISA kit.
To address the effect of PtdIns3K in CD8+ T-cell priming, wild-type BMDCs were pretreated with 0.5 mM LY294002 (Selleck Chemicals, S1105) and co-cultured with 2 × 105 apoptotic EG7 cells and 2 × 105 CD8+ OT-I T cells for 72 h. Then, IFNG/IFN-γ production was measured with a mouse IFNG/IFN-γ ELISA kit.
Scavenger receptor blockade
To assess phagocytosis of apoptotic tumor cells after scavenger receptor blockage, 2 × 105 Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were co-cultured with 2 × 105 DiI-labeled apoptotic EG7 cells and 2 µg/ml of anti-mouse CD36 (Abcam, ab23680) blocking antibody for 3 h. Phagocytosis was monitored with a FACS Calibur.
To measure IFN- γ production in OT-II CD4+ T cells after CD36 blockade, 2 × 105 of Atg5f/f or ITGAX/CD11c-atg5−/- splenic dendritic cells enriched with MagniSort mouse ITGAX/CD11c+ cell enrichment kit (eBioscience, 8802–6861-74) were co-cultured with 2 × 105 apoptotic EG7 cells and 2 × 105 CD4+ OT-II T cells for 96 h. Then, IFNG/IFN-γ production was measured with a mouse IFNG/IFN-γ ELISA kit.
To monitor in vivo anti-tumor activity of anti-mouse CD36 antibody, Atg5f/f or ITGAX/CD11c-atg5−/- mice were inoculated with 1 × 106 live EG7 cells in the shaved right flank on day 0. After 11 days, 2 µg anti-CD36 antibody was injected intratumorally every 2 days for the next 6 days. Tumor growth was measured with a digital caliper.
Mitochondria fuel usage
Mito Fuel Flex tests were performed on an XFe96 Bio analyzer (Agilent). Etomoxir (Sigma-Aldrich, E1905), BPTES (Sigma-Aldrich, SML0601), and UK5099 (Sigma-Aldrich PZ0160) were purchased from Sigma-Aldrich, and the assay was processed in accordance with the manufacturer’s protocol. Data were analyzed with Seahorse XF Mito Fuel Flex Test Report Generator.
T-cell depletion experiment
For the T-cell depletion in mice, 150 µl anti-CD4 antibody (for CD4+ T-cell depletion) (Bioxcell, BE0003-1) or 150 µl anti-CD8 antibody (for CD8+ T-cell depletion) (Bioxcell, BE0061) was injected at day −2, day −1, day 7, and day 14 of tumor inoculation. Rat IgG2b antibody (Bioxcell, BE0090) was used as the control.
Measurement of TIMD4 expression in dendritic cells
To measure TIMD4 expression in the dendritic cells of Atg5f/f or ITGAX/CD11c-atg5−/- mice, lymph nodes were isolated and processed as described above. Cells were stained with anti-TIMD4/TIM-4 antibodies (Biolegend, 130,006) and analyzed with an LSR Fortessa cell analyzer.
Sirna knock-down experiment
To knock down the genes Atg7 and Atg16l1 in BMDCs, Atg7 siRNA (Dharmacon, L-049953–00-0005) or Atg16l1 siRNA (Dharmacon, L-051699–01-0005) was transfected with X-tremeGENE siRNA transfection reagent (Roche, 04 476 093 001) according to the manufacturer’s protocol. Negative control siRNA (Qiagen, 1,027,280) was used as the control siRNA.
OT-II cognitive peptide stimulation
To see OT-II CD4+ T-cell priming activity after cognitive peptide stimulation, SERPINB/OVA323-339 peptide was pulsed with Atg5f/f or ITGAX/CD11c-atg5−/- splenic dendritic cells and co-cultured with OT-II CD4+ T cells. After 72 h of co-culture, IFNG/IFN-γ production was measured with a mouse IFNG/IFN-γ ELISA kit.
Protein degradation assays
Total proteins from Atg5f/f or ITGAX/CD11c-atg5−/- BMDCs were isolated with cell lysis buffer (1% Triton X-100 (Bio Basic, TB0198), 25 mM NaCl (Welgene, ML 011–01)) for 15 min on ice. The lysates were centrifuged with 18,409 g of relative centrifugal force 4°C for 15 min, and supernatants were harvested for protein quantitation with Broadford assays (Bio-Rad, 500–0006). Proteolytic activity of 10 µg total proteins was detected in 200 μl phosphate-buffered saline with 0.5% Triton X-100 and 2 mM DTT (Bio Basic DB0058), at pH 5.5 in a 96-flat-well plate using an EnzCheK protease assay kit (Molecular Probes, E6638). Substrate degradation was assayed fluorometrically with a Gemini XPS microplate reader (Molecular Devices).
Statistical analysis
Data are expressed as the means ± SE. Differences between groups were analyzed using Student’s t-tests or two-way ANOVA. All statistical analyses were performed using GraphPad Prism 7 software (GraphPad). Differences were considered statistically significant when P values were greater than 0.05.
Supplemental Material
Download PDF (639.8 KB)Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary materials
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):349–364. 10.1038/s41580-018-0003-4. PubMed PMID: 29618831. .
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. PubMed PMID: 19802558; PubMed Central PMCID: PMCPMC2832191.
- Munz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016 Nov;37(11):755–763. . PubMed PMID: 27667710.
- Lee HK, Mattei LM, Steinberg BE, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010 Feb 26;32(2):227–239. . PubMed PMID: 20171125; PubMed Central PMCID: PMCPMC2996467.
- Keller CW, Sina C, Kotur MB, et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4(+) T cell pathogenicity during CNS inflammation. Proc Natl Acad Sci U S A. 2017 Dec 26;114(52):E11228–E11237. PubMed PMID: 29233943; PubMed Central PMCID: PMCPMC5748192.
- Makris S, Paulsen M, Johansson C. Type I interferons as regulators of lung inflammation. Front Immunol. 2017;8:259.
- Jinushi M, Morita T, Xu Z, et al. Autophagy-dependent regulation of tumor metastasis by myeloid cells. PLoS One. 2017;12(7):e0179357. PubMed PMID: 28686632; PubMed Central PMCID: PMCPMC5501406.
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010 Mar 19;140(6):798–804. . PubMed PMID: 20303871.
- Goldszmid RS, Idoyaga J, Bravo AI, et al. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol. 2003 Dec 1;171(11):5940–5947. PubMed PMID: 14634105.
- Oczypok EA, Oury TD, Chu CT. It’s a cell-eat-cell world: autophagy and phagocytosis. Am J Pathol. 2013 Mar;182(3):612–622. . PubMed PMID: 23369575; PubMed Central PMCID: PMCPMC3589073.
- Lai SC, Devenish RJ. LC3-Associated Phagocytosis (LAP): connections with host autophagy. Cells. 2012 Jul 30;1(3):396–408. . PubMed PMID: 24710482; PubMed Central PMCID: PMCPMC3901117.
- Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007 Dec 20;450(7173):1253–1257. PubMed PMID: 18097414.
- Bonilla DL, Bhattacharya A, Sha Y, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013 Sep 19;39(3):537–547. PubMed PMID: 24035364.
- Caminschi I, Maraskovsky E, Heath WR. Targeting dendritic cells in vivo for cancer therapy. Front Immunol. 2012;3:13. PubMed PMID: 22566899; PubMed Central PMCID: PMCPMC3342351.
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006 Jun 15;441(7095):885–889. PubMed PMID: 16625204.
- Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007 Sep;13(9):1050–1059. PubMed PMID: 17704786.
- Das M, Kaveri SV, Bayry J. Cross-presentation of antigens by dendritic cells: role of autophagy. Oncotarget. 2015 Oct 6;6(30):28527–28528. . PubMed PMID: 26337341; PubMed Central PMCID: PMCPMC4745670.
- Tran Janco JM, Lamichhane P, Karyampudi L, et al. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015 Apr 1;194(7):2985–2991. . PubMed PMID: 25795789; PubMed Central PMCID: PMCPMC4369768.
- Bretou M, Sáez PJ, Sanséau D, et al. Lysosome signaling controls the migration of dendritic cells. Sci Immunol. 2017 Oct 27;2(16):eaak9573. . PubMed PMID: 29079589.
- Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev. 2007 Oct;219:143–156. PubMed PMID: 17850487.
- Asano K, Nabeyama A, Miyake Y, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011 Jan 28;34(1):85–95. PubMed PMID: 21194983.
- Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013 Sep;13(9):621–634. . PubMed PMID: 23928573.
- Albert ML, Pearce SF, Francisco LM, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998 Oct 5;188(7):1359–1368. PubMed PMID: 9763615; PubMed Central PMCID: PMCPMC2212488.
- Martin C, Chevrot M, Poirier H, et al. CD36 as a lipid sensor. Physiol Behav. 2011 Nov 30;105(1):36–42. PubMed PMID: 21354192.
- Herber DL, Cao W, Nefedova Y, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010 Aug;16(8):880–886. PubMed PMID: 20622859; PubMed Central PMCID: PMCPMC2917488.
- Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol. 2018 Mar 26;15:944–952. 10.1038/cmi.2018.2. PubMed PMID: 29578531.
- Lee YH, Martin-Orozco N, Zheng P, et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017 Aug;27(8):1034–1045. PubMed PMID: 28685773; PubMed Central PMCID: PMCPMC5539354.
- Fransen MF, van der Sluis TC, Ossendorp F, et al. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013 Oct 1;19(19):5381–5389. 10.1158/1078-0432.CCR-12-0781. PubMed PMID: 23788581.
- Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011 Oct 18;108(42):17396–17401. PubMed PMID: 21969579; PubMed Central PMCID: PMCPMC3198353.
- Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016 May 5;533(7601):115–119. PubMed PMID: 27096368; PubMed Central PMCID: PMCPMC4860026.
- Tam JM, Mansour MK, Khan NS, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis. 2014 Dec 1;210(11):1844–1854. PubMed PMID: 24842831; PubMed Central PMCID: PMCPMC4271056.
- Loi M, Gannage M, Munz C. ATGs help MHC class II, but inhibit MHC class I antigen presentation. Autophagy. 2016 Sep;12(9):1681–1682. . PubMed PMID: 27439741; PubMed Central PMCID: PMCPMC5082774.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PubMed PMID: 21801009.
- Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015 Dec 24;528(7583):565–569. PubMed PMID: 26649827; PubMed Central PMCID: PMCPMC4842313.
- Zhao Z, Fux B, Goodwin M, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008 Nov 13;4(5):458–469. PubMed PMID: 18996346; PubMed Central PMCID: PMCPMC2682425.
- Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. 2012 Sep 1;1(6):908–916. . PubMed PMID: 23162758; PubMed Central PMCID: PMCPMC3489746.
- Van Kaer L, Parekh VV, Postoak JL, et al. Role of autophagy in MHC class I-restricted antigen presentation. Mol Immunol. 2017 Nov 8 PubMed PMID: 29126597; PubMed Central PMCID: PMCPMC5940586. DOI:10.1016/j.molimm.2017.10.021.
- Mintern JD, Macri C, Chin WJ, et al. Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy. 2015;11(6):906–917. PubMed PMID: 25950899; PubMed Central PMCID: PMCPMC4502655.
- Pua HH, Dzhagalov I, Chuck M, et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007 Jan 22;204(1):25–31. . PubMed PMID: 17190837; PubMed Central PMCID: PMCPMC2118420.
- Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012 Jun 15;12(7):492–502. . PubMed PMID: 22699831; PubMed Central PMCID: PMCPMC5570470.
- Ten Broeke T, Wubbolts R, Stoorvogel W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol. 2013 Dec 1;5(12):a016873. . PubMed PMID: 24296169; PubMed Central PMCID: PMCPMC3839614.
- Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol. 2010 Apr;8(4):296–307. . PubMed PMID: 20234378; PubMed Central PMCID: PMCPMC3037727.
- Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature. 2005 Feb 3;433(7025):523–527. PubMed PMID: 15690042.
- Baranova IN, Kurlander R, Bocharov AV, et al. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol. 2008 Nov 15;181(10):7147–7156. PubMed PMID: 18981136; PubMed Central PMCID: PMCPMC3842223.
- Schulz O, Pennington DJ, Hodivala-Dilke K, et al. CD36 or alphavbeta3 and alphavbeta5 integrins are not essential for MHC class I cross-presentation of cell-associated antigen by CD8 alpha+ murine dendritic cells. J Immunol. 2002 Jun 15;168(12):6057–6065. PubMed PMID: 12055214.
- Belz GT, Vremec D, Febbraio M, et al. CD36 is differentially expressed by CD8+ splenic dendritic cells but is not required for cross-presentation in vivo. J Immunol. 2002 Jun 15;168(12):6066–6070. PubMed PMID: 12055215.
- Biedroń R, Konopiński MK, Marcinkiewicz J, et al. Oxidation by neutrophils-derived HOCl increases immunogenicity of proteins by converting them into ligands of several endocytic receptors involved in antigen uptake by dendritic cells and macrophages. PubMed PMID: 25849867; PubMed Central PMCID: PMCPMC4388828 PLoS One. 2015;104:e0123293.
- Perry JSA, Russler-Germain EV, Zhou YW, et al. CD36 mediates cell-surface antigens to promote thymic development of the regulatory T Cell receptor repertoire and allo-tolerance. Immunity. 2018 May 15;48(5):923–936 e4. PubMed PMID: 29752065; PubMed Central PMCID: PMCPMC5986080.
- Steinbusch LK, Wijnen W, Schwenk RW, et al. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am J Physiol Cell Physiol. 2010 Jun;298(6):C1549–59. PubMed PMID: 20375272.
- Steinbusch LK, Schwenk RW, Ouwens DM, et al. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol Life Sci. 2011 Aug;68(15):2525–2538. PubMed PMID: 21547502; PubMed Central PMCID: PMCPMC3134709.
- Pepino MY, Kuda O, Samovski D, et al. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34:281–303. PubMed PMID: 24850384; PubMed Central PMCID: PMCPMC4329921.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009 Apr 30;458(7242):1131–1135. PubMed PMID: 19339967; PubMed Central PMCID: PMCPMC2676208.
- Pascual G, Avgustinova A, Mejetta S, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017 Jan 5;541(7635):41–45. PubMed PMID: 27974793.
- O’Sullivan D, van der Windt GJ, Huang SC, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014 Jul 17;41(1):75–88. PubMed PMID: 25001241; PubMed Central PMCID: PMCPMC4120664.
- Yin Y, Metzger T, Bailey-Bucktrout S. Tumor infiltrating T cells have abnormal lipid metabolism that can be modulated by PD-L1 blockade. J Immunol. 2016;196(1Supplement):144.21.
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007 Jul 9;204(7):1653–1664. . PubMed PMID: 17591855; PubMed Central PMCID: PMCPMC2118632.
- Hogquist KA, Jameson SC, Heath WR, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994 Jan 14;76(1):17–27. PubMed PMID: 8287475.
- Barnden MJ, Allison J, Heath WR, et al. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998 Feb;76(1):34–40. PubMed PMID: 9553774.
- Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007 Mar 9;315(5817):1398–1401. PubMed PMID: 17272685.
- Ichinohe T, Lee HK, Ogura Y, et al. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009 Jan 16;206(1):79–87. . PubMed PMID: 19139171; PubMed Central PMCID: PMCPMC2626661.
- Zhang Y, Wang Y, Ogata M, et al. Development of dendritic cells in vitro from murine fetal liver-derived lineage phenotype-negative c-kit(+) hematopoietic progenitor cells. Blood. 2000 Jan 1;95(1):138–146. PubMed PMID: 10607696.
- Duewell P, Steger A, Lohr H, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ. 2014 Dec;21(12):1825–1837. PubMed PMID: 25012502; PubMed Central PMCID: PMCPMC4227156.