
ABSTRACT
Macroautophagy/autophagy occurs at basal levels in all eukaryotic cells and plays an important role in maintaining bio-energetic homeostasis through the control of molecule degradation and organelle turnover. It can be induced by environmental conditions such as starvation, and is deregulated in many diseases including autoimmune diseases, neurodegenerative disorders, and cancer. Interestingly, the modulation of autophagy in mesenchymal stem cells (MSCs) represents a possible mechanism which, affecting MSC properties, may have an impact on their regenerative, therapeutic potential. Furthermore, the ability of MSCs to modulate autophagy of cells in injured tissues/organs has been recently proposed to be involved in the regeneration of damaged tissues and organs. In particular, MSCs can affect autophagy in immune cells involved in injury-induced inflammation reducing their survival, proliferation, and function and favoring the resolution of inflammation. In addition, MSCs can affect autophagy in endogenous adult or progenitor cells, promoting their survival, proliferation and differentiation supporting the restoration of functional tissue. This review provides, for the first time, an overview of the studies which highlight a possible link between the therapeutic properties of MSCs and their ability to modulate autophagy, and it summarizes examples of disorders where these therapeutic properties have been correlated with such modulation. A better elucidation of the mechanism(s) through which MSCs can modulate the autophagy of target cells and how autophagy can affect MSCs therapeutic properties, can provide a wider perspective for the clinical application of MSCs in the treatment of many diseases.
Abbreviations: 3-MA: 3-methyladenine; AD: Alzheimer disease; ATG: autophagy-related; BECN1: beclin 1; BM: bone marrow; CD: cluster of differentiation; EAE: experimental autoimmune encephalomyelitis; IL: interleukin; INF: interferon; LAP: LC3-associated phagocytosis; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MSCs: mesenchymal stem cells; MTOR: mechanistic target of rapamycin kinase; PD: Parkinson disease; PtdIns3K: class III phosphatidylinositol 3-kinase; ROS: reactive oxygen species; SLE: systemic lupus erythematosus; SQSTM1: sequestosome 1; TBI: traumatic brain injury; TGF: transforming growth factor; TNF: tumor necrosis factor
Autophagy
Definition and state of the art
Autophagy is the main cellular mechanism for degrading and recycling intracellular proteins and organelles under different physiological and pathological conditions. Three different general types of autophagy have been identified in mammals: microautophagy, chaperone-mediated autophagy, and macroautophagy, each depending on the mechanism that mediates the delivery of cytoplasmic cargo to lysosomes for degradation [Citation1]. Microautophagy degrades cytosolic cargo incorporated into lysosomes through small vesicles formed from invagination of the lysosomal membrane. In chaperone-mediated autophagy, proteins that expose a targeting pentapeptide motif in their amino acid sequence are selectively recognized by cytosolic chaperones and translocated into the lysosome through LAMP2 (lysosomal associated membrane protein 2), where they are rapidly degraded. Macroautophagy (referred to hereafter as autophagy), is the best-known autophagy process and has been suggested to be the one that contributes more efficiently to lysosomal degradation [Citation2]. In the canonical form of macroautophagy, cytoplasmic components are sequestered within double-membrane vesicles, named autophagosomes. The formation of the autophagosome is a multistep process that includes the nucleation of the vesicle or phagophore, followed by its elongation and closure. Previous studies identify different membrane pools that contribute to the formation of the phagophore such as the plasma membrane, mitochondria, endoplasmic reticulum, and Golgi complex [Citation3]. After autophagosome formation, the outer membrane of the autophagosome fuses with a lysosome membrane, forming the autolysosome, where digestion takes place and the cargo contents are recycled [Citation4] ().
Figure 1. The steps of autophagy. The figure depicts the multistep process leading to autolysosome formation and the main proteins/enzymes involved. It also reports defective autophagy in specific diseases. Abbreviations: AD: Alzheimer disease; ATG: autophagy-related; BECN1: beclin 1; HD: Huntington disease; LAMP: lysosomal associated membrane protein; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; PD: Parkinson disease; PE: phosphatidylethanolamine; PtdInsK3: class III phosphatidylinositol 3-kinase; SLE: systemic lupus erythematosus; TBI: traumatic brain injury.
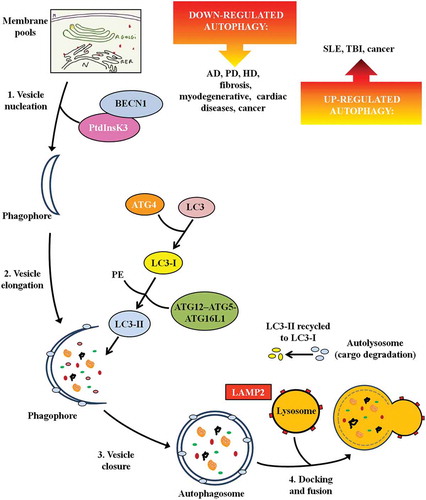
ATG (autophagy-related) genes and ATG proteins are identified as the core machinery for autophagosome biogenesis. They were initially discovered and characterized in yeast [Citation5] and their homologs were subsequently identified in mammals and shown to possess similar mechanisms [Citation6]. During autophagosome nucleation, the macromolecular complex comprised of the class III phosphatidylinositol 3-kinase (PtdIns3K) and BECN1 (beclin 1), involved also in the localization of many of the other autophagy-related proteins to the phagophore membrane [Citation7], is recruited. Successively, in the elongation of phagophore membrane, 2 ubiquitin-like systems are involved, including the ATG12–ATG5-ATG16L1 complex and MAP1LC3/LC3 (microtubule associated protein 1 light chain 3). LC3 is cleaved by ATG4 to form cytosolic LC3-I. LC3-I is covalently bound to phosphatidylethanolamine through the action of ATG7, ATG3 and the ATG12–ATG5-ATG16L1 complex generating LC3-II (). LC3-II is tightly associated with the phagophore and autophagosome membrane and it serves as a typical marker of the completed autophagosome, therefore LC3-II protein is widely used as an indicator of autophagy [Citation8,Citation9].
Non-canonical autophagy pathways have also been described that lead to autophagosomal degradation through variants of the canonical pathway. Different forms of non-canonical autophagy have been identified such as BECN1-independent autophagy that does not involve proteins involved in phagophore nucleation (such as BECN1) or others that do not involve proteins used in phagophore elongation and closure (such as ATG5, ATG7 and LC3). Among the forms of non-canonical autophagy some authors described LC3-associated phagocytosis (LAP) [Citation10]. In LAP, differently from canonical autophagy, LC3 is conjugated to phosphatidylethanolamine directly on the phagosome-sealed membrane using only a part of the canonical autophagy machinery (such as ATG5, ATG7, ATG12 and ATG16L1 for LC3 lipidation). Lipidated LC3-II then facilitates lysosomal fusion and cargo destruction. LAP also affects immune cells; it is present in phagocytic cells, including macrophages, where it plays a crucial role in the clearance of extracellular particles (such as cellular debris) and pathogens.
Physiological and pathological role of autophagy
Autophagy occurs at basal levels in all eukaryotic cells and plays an important role in maintaining bio-energetic homeostasis through the control of molecule degradation and organelle turnover [Citation11]. In this scenario, autophagy targets misfolded proteins and dysfunctional organelles for degradation, preventing the accumulation of detrimental components that can lead to cell damage and death. It is not surprising that defects in the autophagy signaling pathway are associated with many human diseases. Numerous studies demonstrate that deregulated autophagy is linked to neurodegenerative disorders [Citation12], cystic fibrosis [Citation13], myopathies [Citation14] and cardiomyopathy [Citation15]. Interestingly, most of these pathologies are associated with reduced BECN1 levels that could cause an impairment of the early phases of autophagy.
Conversely, autophagy can be rapidly upregulated in response to environmental stress, such as oxidative stress, starvation, hypoxia, inflammation, and infection, each of which has the potential to cause or aggravate cell injury. In this context, activated autophagy constitutes a stress adaptation pathway that promotes cell health and survival; however, paradoxically, excessive stimulation of autophagy can contribute to cell damage [Citation16,Citation17]. More specifically, exacerbated autophagy can lead to a non-apoptotic form of programmed cell death, also called type 2 cell death or autophagic cell death. Autophagic cell death has been reported in Kaposi sarcoma cells treated with imatinib [Citation18] and in prostate cancer cells treated with the proteasome inhibitor MG132 [Citation19]. It is of note that, in the literature, many of the original affirmations of ‘autophagic cell death’ have been revised and reformulated as ‘cell death with autophagy’ because the main support for autophagy involvement in many of these cases was the observation of autophagic vacuoles in the dying cell, which often result in a block of the autophagic flux [Citation20]. Autophagic flux is a measure of autophagic activity, and a number of methods are currently utilized to assess it. Autophagic activity is usually investigated by measuring LC3-II levels, but this should be associated with the analysis of the degradation of substrates (e.g., SQSTM1/p62 [sequestosome 1]). In addition, genetic manipulations (e.g., short interfering RNA for ATG genes), pharmacological inhibitors such as 3-methyladenine (3-MA) and chloroquine, and/or inducers such as rapamycin, could be used to further confirm alterations in autophagic flux [Citation21].
Examples of altered autophagy in diseases
It is now clear that alterations in the autophagic process can contribute to disease pathogenesis [Citation22] (). Among the disorders with autophagic dysfunction, neurodegenerative disorders have received particular attention. Autophagy dysfunction due to reduced lysosomal function or autophagosome-lysosome fusion and consequent autophagosome accumulation (with increased LC3-II levels), has been observed in Alzheimer disease (AD), Parkinson disease (PD), and Huntington disease [Citation23–Citation25]. Autophagic impairment causes the aggregation of abnormal and misfolded proteins (such as amyloid-beta in AD, SNCA/alpha-synuclein in PD, and mutant HTT [huntingtin] in Huntington disease) leading to neuronal toxicity and dysfunctions such as deregulated transcription and altered axonal transport [Citation26]. Similar to neurodegenerative disorders, the pathogenesis of myodegenerative [Citation27] and cardiac [Citation28] diseases may also involve either the failure of autophagosomes to fuse with lysosomes or protein aggregation that exceeds autophagic clearance capacity.
Insufficient autophagy activation (with decreased LC3-II expression and other autophagy-linked proteins) is involved in fibrogenic tissues [Citation29] leading to a reduction of degradation of misfolded proteins and defective organelles. For example, accumulated dysfunctional mitochondria that trigger reactive oxygen species (ROS) production and oxidative stress are observed in fibrotic hepatocytes [Citation30]. Furthermore, in vitro experiments on idiopathic pulmonary fibrosis models demonstrate that the profibrotic mediator TGFB (transforming growth factor beta) is likely responsible for decreased autophagy [Citation31].
Upregulated expression levels of BECN1 and LC3-II are observed in conditions of autophagic stress associated with exacerbated autophagy in murine lupus T and B cells [Citation32] and in human systemic lupus erythematosus (SLE) T lymphocytes [Citation33] notoriously characterized by energy deficit and oxidative stress [Citation34,Citation35]. A link between LAP defects and the establishment of a lupus-like autoimmune disease in mice has recently been reported [Citation36]. More specifically, macrophages from LAP-deficient animals (lacking Cybb/Nox2, or Rubcn/Rubicon), fail to digest engulfed dying cells, leading to elevated inflammatory cytokine production and high levels of anti-double-stranded DNA and anti-nuclear antibodies, and other antibodies against autoantigens commonly associated with SLE [Citation36].
Excessive autophagy is considered a hot topic involved in cell death after traumatic brain injury (TBI). Numerous autophagosomal vacuoles and secondary lysosomes are ultrastructurally detected in mice post-TBI [Citation37] and, furthermore, it is also demonstrated that inhibition of autophagy can attenuate TBI damage and neuronal functional deficits [Citation38].
Interestingly, autophagy activation has been recognized to have a dual role in cancer: on the one hand it diminishes malignant transformation [Citation39], but on the other hand favors tumor progression [Citation40–Citation42] in a disease- and stage-specific manner. It has been proposed, indeed, that in the early stages of cancer development, quality control by autophagy inhibits tumorigenesis conferring a pathway with anti-oncogenic functions. In contrast, in the late stages of oncogenesis, autophagy is a crucial process for cancer cell survival. Autophagy ameliorates the energy state for rapidly dividing cancer cells and contributes to quality control to eliminate intracellular damage caused by the aggressive tumor microenvironment and by anti-oncogenic activities [Citation43].
Role of autophagy in the therapeutic potential of mscs
MSC therapeutic potential
MSCs, defined as an adherent fibroblast-like population with the ability to self-renew [Citation44], were first identified in human bone marrow [Citation45] and have been the most widely studied MSC population. MSCs can be isolated from many other human tissues, including adipose tissue [Citation46], liver [Citation47], synovial membrane [Citation48], umbilical cord blood and Wharton’s jelly [Citation49,Citation50], periosteum [Citation51], dental pulp [Citation52], salivary glands [Citation53], peripheral blood [Citation54], placental tissue [Citation55,Citation56], amniotic fluid [Citation57], tendon [Citation58], and menstrual blood [Citation59]. MSCs express mesenchymal markers such as ENG/CD105, NT5E/CD73, and THY1/CD90, and lack hematopoietic markers such as PTPRC/CD45, CD34, CD14, ITGAM/CD11b, CD79A, CD19, and HLA-DR (major histocompatibility complex, class II, DR) [Citation56,Citation60].
Many in vitro and preclinical studies robustly document the therapeutic potential of MSCs when applied as a treatment for different pathological conditions such as inflammatory/autoimmune disorders, diabetes, neurodegenerative diseases, ischemia-reperfusion injuries, and kidney, liver and lung fibrosis [Citation61,Citation62]. It was initially hypothesized that MSC therapeutic action depends on their ability to differentiate toward a variety of cell lineages and replace damaged cells [Citation60]. However, the controversial ability of MSCs to differentiate in vivo [Citation63,Citation64], as well as the low survival and engraftment in host tissues after transplantation [Citation65], have strongly suggested that MSCs could exert trophic actions on tissue resident cells present in the injured area, or on immune cells recruited to the injury site. Specifically, through cell-to-cell contact and the secretion of growth factors, cytokines, and the release of extracellular vesicles (containing peptides/proteins, mRNA, and micro RNA) [Citation64], MSCs create a reparative environment, which suppresses the activation/function of inflammatory cells, prevents apoptosis and promotes survival of damaged tissue cells [Citation66–Citation69], reduces oxidative injury often involved in tissue damage [Citation70], and favors angiogenesis/arteriogenesis [Citation71].
In detail, MSCs exert immunosuppressive action by: 1) inhibiting the activation/proliferation of immune cells such as T and B cells [Citation61,Citation72–Citation76]; 2) inhibiting maturation toward effector immune cells (cells which actively respond to an immunogenic stimuli) both in vitro [Citation77–Citation81] and in vivo [Citation82,Citation83]; 3) favoring the expansion of immune cells with regulatory activities (which act as suppressors of the immune response) in vitro [Citation84,Citation85] and in vivo [Citation82,Citation83,Citation86–Citation90]; 4) limiting the secretion of pro-inflammatory cytokines and chemokines (IL1B [interleukin 1 beta], IL2, TNF [tumor necrosis factor], and IFNG [interferon gamma]) involved in the propagation of the immune/inflammatory response, and 5) promoting the production of anti-inflammatory factors (IL10, TGFB) [Citation89,Citation91].
The promising findings obtained from preclinical studies provide a strong rationale for investigating MSCs as a potential innovative therapy in humans. Many clinical trials, carried out by academic groups and industry, have indeed investigated and are currently investigating the feasibility and efficacy of MSCs for the treatment of a variety of diseases (https://clinicaltrials.gov). These include hematological diseases, graft-versus-host disease, diabetes, autoimmune diseases (such as Crohn disease, rheumatoid arthritis), neurodegenerative diseases (such as PD and AD), and diseases in the liver (including liver fibrosis), kidneys, and lungs, as well as ischemic diseases (such as myocardial ischemia, ischemic stroke, and critical limb ischemia) [Citation92–Citation94]. The therapeutic use of MSCs and MSC-based products could receive a strong advancement through an improved understanding of the mechanisms through which transplanted MSCs can promote tissue repair under different pathological conditions. In this regard, an underexplored mechanism is the role of autophagy in MSC actions; its understanding could establish a new therapeutic action of MSCs and expand the spectrum of their clinical applications.
Evidence for the involvement of autophagy in MSC therapeutic potential
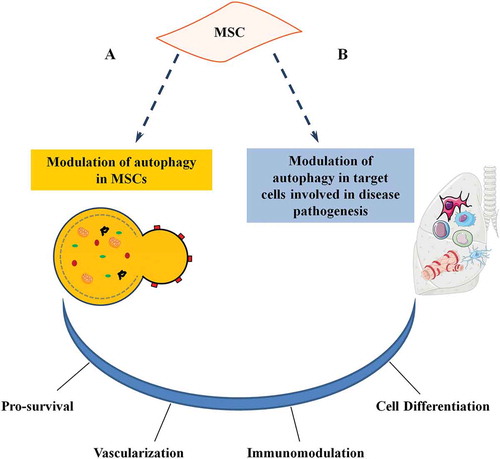
Recently it has been proposed that autophagy may be associated with MSC activities, playing a dual role: i) modulation of autophagy in MSCs may consequently affect MSC functions; ii) MSCs may modulate the autophagy of immune and other cells involved in disease pathogenesis. Both of these mechanisms ultimately affect the therapeutic properties of MSCs.
Autophagy modulation in MSCs affects MSC functions
Autophagy modulation in MSCs may affect their ability to differentiate toward a variety of cell lineages and to affect the proliferation, activation, and functions of immune cells.
At first, we will illustrate the evidence supporting a functional interplay between MSC autophagy and differentiation potential. The first observation suggesting a pivotal role of autophagy in MSC differentiation was that undifferentiated MSCs contain more autophagosomes than differentiated cells [Citation95]. More recently, Nuschke and collaborators [Citation96] confirmed the autophagosome accumulation in undifferentiated bone marrow (BM)-MSCs and, by using tandem fluorescent reporters that allow for the assessment of the fusion of autophagosomes with lysosomes, they observe an impairment of autophagosome-lysosome fusion leading to the blockage of autophagosome degradation. When they expose MSCs to osteogenic differentiating conditions, they observe a transient autophagy activation leading to a rapid degradation of accumulated autophagosomes that are likely to supply cells with the energy and anabolic precursors needed to support the morphological, structural and metabolic remodeling requested by cellular differentiation.
Transient induction of autophagy (detected as increased expression of autophagosome-bound LC3-II and of the pro-autophagic protein BECN1, and reduced SQSTM1/p62 levels), is also observed in dental pulp-derived MSCs under osteogenic differentiation [Citation97] and in BM-MSCs under adipogenic differentiation [Citation98].
Other authors investigated the interplay between autophagy and other types of MSC differentiation. For example, Li and collaborators [Citation99] observe that the expression of neuron-specific markers in BM-MSCs cultured in neuronal differentiation conditions increases in rapamycin-treated MSCs (inducer of autophagy), whereas neuronal markers are reduced when BM-MSCs are treated with autophagy inhibitors.
Activation of the autophagic machinery (increased expression of BECN1, ATG5 and LC3-II with the concomitant decrease of the expression of BCL2) was also observed by Park and collaborators [Citation100] in human tonsil-derived MSCs upon stimulation of skeletal muscle differentiation. However, whereas the autophagy inhibitor bafilomycin A1 decreases the expression of myogenic markers, the induction of myogenic differentiation by 5-azacytidine, although associated with increased expression of myogenic markers, is not associated with increased expression of LC3-II.
To our knowledge, only one study examined the pathway(s) involved in the activation of autophagy after differentiation stimuli [Citation97]. In this study, the authors use gene knockdown and pharmacological inhibition to demonstrate that differentiation (in this case osteogenic)-induced autophagy correlates with the early MTOR (mechanistic target of rapamycin kinase) inhibition mediated by 5ʹ AMP-activated protein kinase (AMPK) activation and the late activation of AKT/PKB-MTOR signaling [Citation97].
Recent findings also outline the important role of mitochondria in MSC differentiation, showing that there are fewer, active mitochondria in undifferentiated MSCs compared to MSCs cultured in adipogenic and osteogenic differentiation stimuli [Citation101]. Some authors even propose a link between mitochondria-selective autophagy (mitophagy) and higher cellular mitochondrial turnover [Citation102]. Although the findings reported above lack in demonstrating a mechanistic link between the activation of autophagy and MSC differentiation, and it remains to be elucidated whether these pathways are different among MSC sources and/or differentiation stimuli applied, these data strongly suggest an involvement of autophagy activation in MSC differentiation.
Very few studies instead explored a possible link between MSC autophagy and MSC ability in modulating the functions of immune cells. The scarce studies lead to controversial results and are limited to examine how the modulation of MSC autophagy may change the suppressive action of MSCs on T-cell proliferation.
A study performed by Dang and collaborators [Citation103] suggests that inhibition of autophagy increases the immunosuppressive action of mouse BM-MSCs transplanted in an autoimmune disease model of experimental autoimmune encephalomyelitis (EAE). The authors observed that Becn1 knockdown in MSCs (short hairpin Becn1-MSCs) improves their therapeutic and immunomodulatory effect in EAE mice with respect to control MSCs. Indeed, after treatment with short hairpin Becn1-MSCs, the authors find a stronger reduction in both CD4+ and CD8+ T cells and in the proliferation of MOG (myelin oligodendrocyte glycoprotein)-specific CD4+ T cells, without changes in T cell polarization. Similar results are found when EAE mice are transplanted with MSCs that have been pre-treated with the autophagy inhibitor 3-MA [Citation103].
In contrast, other authors propose that MSC immunosuppressive actions correlate with MSC autophagy levels. The study from Gao and collaborators [Citation104] compares MSCs with activated autophagy (rapamycin pretreated), and MSCs with inhibited autophagy (3-MA pretreated), for their ability to suppress the proliferation of peripheral blood mononuclear cells stimulated in vitro with anti-CD3 and anti-CD28 antibodies. The authors, by evaluating autophagy flux either by western blot analysis of LC3-II:LC3-I and SQSTM1/p62 expression or by the presence of green puncta in MSCs transfected with green fluorescent protein-LC3-II, find that co-culture with rapamycin-pretreated MSCs significantly reduces T-cell proliferation with respect to co-culture with untreated-MSCs, whereas T-cell proliferation is enhanced by co-culture with 3-MA-pretreated MSCs. The authors associate the higher ability of rapamycin-treated MSCs to suppress T-cell proliferation with increased secretion of TGFB1.
Other authors instead suggest a lack of association between autophagy modulation and MSC immunosuppressive properties. In their study, Chinnadurai and collaborators [Citation105] demonstrate that pre-treatment with INFG increases MSC ability to inhibit T-cell proliferation with concomitant upregulation of the mRNA expression of BECN1, and of other genes involved in autophagy such as ATG12, ATG5, ATG16L1, ATG7, and LC3. Although these findings suggest a mechanistic link between autophagy activation and potentiation of MSC immunosuppressive activity, when the authors applied a commercial fluorescent-based technique to monitor autophagy, they did not observe any increased fluorescent staining (indicative of autophagosome formation) nor in MSCs cultured in the presence of IFNG, nor in MSCs treated with the autophagy inducer rapamycin. Furthermore, the authors did not observe any changes in MSC immunosuppressive ability when they pre-treated MSCs with an autophagy inhibitor (3-MA). All together, these data suggest the absence of a functional link between MSC autophagy and MSC immunosuppression.
Modulation of MSC autophagy is also proposed as a possible strategy to favor MSC-induced T cell polarization toward regulatory cells. MSCs derived from adipose tissue pre-treated with rapamycin are shown to be more effective than untreated MSCs in suppressing in vitro expansion of T helper 17 cells and in reducing the mortality and clinical severity of acute graft versus host disease induced in mice. In vivo benefits are associated with a reduction of T helper 17 cells and an increase in regulatory T cells [Citation106]. The authors correlate the potentiation of immunoregulatory MSC function with the activation of the autophagic machinery, given that they observe increased mRNA expression of some autophagy genes such as ATG5, and LC3; increased protein expression of BECN1, ATG5, ATG7, and LC3-II; and concomitant suppression of the expression of MTOR and MTOR components (RICTOR and RPTOR) [Citation106].
A very recent study suggests that the modulation of autophagy may also affect the secretion capacity of MSCs consequently affecting their functions. Indeed, the subcutaneous injection of MSCs pre-treated with the autophagy inducer rapamycin enhances the wound healing ability of MSCs and, in contrast, MSCs silenced for BECN1 (i.e., early blockage of the autophagic machinery) display a decreased therapeutic action [Citation107]. Because the authors observe higher secretion of VEGF (vascular endothelial growth factor) from MSCs with rapamycin-induced autophagy, they propose that MSC wound healing properties could be related to promotion of angiogenesis driven by autophagy-induced secretion of VEGF.
The discrepancies observed in the outcomes from these few studies can be in part attributable to differences in species from which MSCs were obtained (mice and humans), in cell culture conditions, and also in the inflammatory microenvironment surrounding MSCs. Therefore, the conclusion that some authors have proposed that the modulation of autophagy can represent a strategy to increase the therapeutic properties of MSCs, still has to be convincingly demonstrated and still represents a stimulating field of research.
MSCs modulate autophagy of immune and other cells involved in disease pathogenesis
Several studies strongly indicate the involvement of autophagy in the regulation of proliferation, survival, antigen-presentation, maturation/polarization, and cytokine production of cells of innate and adaptive immunity [Citation108].
For example, autophagy machinery participates in eliminating pathogens, a process driven by autophagic macrophages [Citation109]. The activation of autophagy is highly relevant for the proliferation of activated T lymphocytes. It has been suggested to assure an efficient ATP production for rapid cellular proliferation and cytokine production by switching the energetic metabolism from mitochondrial oxidative phosphorylation (typical of naïve, memory, and regulatory T cells) to glycolysis [Citation110,Citation111]. Autophagy induction is also required for the survival of activated T cells by increasing the rate of degradation of specific pro-apoptotic proteins, such as BCL2L11/BIM and cysteinyl aspartate proteinases CASP3 (caspase 3) and CASP8 [Citation112].
Given the key role played by autophagy in regulating survival and function of innate and adaptive immune cells, it is plausible that MSCs may affect immune cell functions through the modulation of the autophagic machinery. Very few studies have been performed to investigate this topic. One of these shows that treatment with MSCs from umbilical cord reduces exacerbated autophagy and apoptosis observed in T cells from SLE patients [Citation113]. The authors suggest that umbilical cord derived-MSCs regulate T cell autophagy by transferring functioning mitochondria to T cells with a positive impact on their survival. However, the connection between T cell apoptosis and overregulated autophagy, and their relevance in lupus pathology, remain to be fully elucidated [Citation33].
Furthermore, the involvement of autophagy in the ability of MSCs to affect macrophage functions and macrophage autophagy was investigated in an experimental model of silicosis [Citation114]. Treatment of silicotic animals with BM-MSCs is reported to decrease the expression of autophagy-related proteins (LC3 and BECN1) in alveolar macrophages and also mitigate the cellular damage associated with this disorder. The authors suggest a connection between these findings; however, the main challenge is to establish whether the changes/alterations observed in the autophagic flux of immune cells observed after MSC treatment are instrumental to immunosuppressive MSC ability or rather are an indirect consequence of MSC benefits exerted on other pathways associated with disease pathogenesis.
Other studies suggest that MSCs exert their therapeutic actions through managing the autophagic machinery of tissue cells involved in the pathogenesis of disease/injury. To this regard, the anti-apoptotic and pro-survival effects exerted by MSCs in various experimental models has been recently proposed, but not always convincingly demonstrated, to be correlated with their ability to activate autophagy in tissue resident cells. For example, BM-MSC treatment reduces apoptosis and promotes survival of pulmonary cells in in vitro and in vivo models of ischemia-reperfusion-injury [Citation115]. The authors observe that in both models, BM-MSC treatment activates the autophagic machinery, monitored through increased LC3-II:LC3-I, decreased SQSTM1/p62 levels and autophagosomes detection using transmission electron microscopy.
The observed increase of class III phosphoinositide 3-kinase (PI3K) and decrease of class I PI3K in lung tissues treated with BM-MSCs, along with the in vitro observation of a reversion of BM-MSC-induced autophagy in presence of PI3K inhibitor altogether brought the authors of this study to suggest a role of PI3K-AKT signaling in BM-MSC pro-autophagy activity. However, given that they used a pan-PI3K inhibitor, which may also inhibit the class III PtdIns3K, the implication of PI3K-AKT pathway remains to be demonstrated. In addition, the authors did not investigate how modulation of autophagy could affect cell apoptosis.
In a model of hydrogen peroxide-induced injury in H9C2 cardiomyocytes, induction of apoptosis concomitant with induction of autophagy is observed in cardiomyocytes [Citation116]. The treatment with MSC-derived exosomes reduces hydrogen peroxide-induced production of ROS and cell apoptosis with a concomitant further enhancement of autophagy. Although the authors associate the anti-apoptotic and anti-oxidative stress effects of MSC-derived exosomes with the upregulation of autophagy, in the presence of the autophagy inhibitor bafilomycin A1, the effect of exosomes on autophagy is not reversed as expected, but rather potentiated [Citation116].
The possible connection between induction of autophagy and decline of apoptosis following MSC treatment has also been investigated in in vitro and in vivo models of diabetes. In this study, beta-pancreatic cells cultured under chronic high-glucose conditions show increased autophagy [Citation117]. The co-culture with BM-MSCs produces a further increase in autophagosome and autolysosome formation with a concomitant decrease in beta-cell apoptosis and improvement of their function. The addition of autophagy inhibitors (3-MA and chloroquine) to the culture decreases cell survival and partially abolishes the suppressing effects of BM-MSCs on ROS levels. Considering that impaired mitochondria lead to significant production of ROS, the authors suggest that enhanced autophagy, induced by BM-MSC treatment in beta-cells, contributes to the clearance of dysfunctional mitochondria [Citation117]. The same authors find a preservation of the number and functionality of beta-cells, in diabetic rats transplanted with BM-MSCs, which they associate with the enhanced autophagic activity of these cells.
BM-MSCs are also shown to stimulate autophagy in diseased neurons exposed to toxic protein aggregates, increasing neuronal survival. In an in vitro model of AD generated by treating SH-SY5Y cells (a human neuroblastoma cell line) with amyloid beta peptides, the amyloid beta-treated cells show accumulation of autophagic vacuoles not fused with lysosomes, indicating a blockage of the autophagic machinery [Citation118]. The treatment with MSCs markedly increases the number of autophagosomes fused with lysosomes leading to a re-activation of the autophagic machinery, and degradation of intracellular amyloid beta aggregates with a consequent higher neuronal survival. Similar findings on the activation of autophagy, enhancement of aggregate degradation, and higher neuronal survival are also confirmed in an in vivo model of amyloid beta-treated animals [Citation118].
A very recent study uses red fluorescent protein-green fluorescent protein-LC3 transgenic mice (which allows the monitoring of autophagosome and autolysosome formation [Citation119]), to demonstrate that MSC-derived exosomes reduce cell death in ischemic cardiomyocytes, and this is associated with the reduction of autophagy induced by ischemia [Citation120]. The authors observe in vitro that MSC exosomes inhibit autophagy independently of MTOR and AMPK activity, while stimulating TRP53/p53 and BNIP3 (BCL2 interacting protein 3) signaling (increased mRNA and protein levels of TRP53 and BNIP3) via the exosomal transfer of Mir125b-5p from MSCs to cardiomyocytes. This study is the first to investigate autophagy in transgenic mice, and further studies that use these models [Citation119] to monitor autophagy during different stages of disease will be of crucial importance to advance our understanding on the involvement of autophagy in the therapeutic potential of MSCs.
Conclusions
The evidence reported herein strongly indicates that modulation of autophagy in MSCs and by MSCs may represent a strategy that can play an important role in reducing inflammation, apoptosis, and oxidative stress of cells involved in disease pathology, ultimately contributing to the therapeutic action exerted by MSCs.
Many open questions remain concerning the ability of MSCs to respond to the tissue microenvironment, and to either induce or inhibit autophagy in other cells in order to contribute to amelioration of the disease. Among these, what are the sensors that guide MSCs toward autophagy-enhancing or -inhibiting pathways, and how do these ultimately contribute to efficacy? What are the mechanisms through which MSCs modulate autophagy, and is the modulation of autophagy a direct effect by MSCs, or is it an indirect consequence whereby MSCs induce other mechanisms that ultimately affect autophagy?
A better elucidation of the mechanism(s) through which MSCs can modulate autophagy of target cells and how autophagy can affect MSC therapeutic properties, can provide a wider perspective for the clinical application of MSCs in the treatment of many diseases. New MSC-based therapies designed to modulate autophagy signaling pathways might represent an innovative and attractive strategy in regenerative medicine.
Acknowledgments
This work was supported in part by Università Cattolica del Sacro Cuore “linea D1-2017”, “linea D1-2018” to OP, by the Regenerative Medicine Research Center (CROME) of Università Cattolica del Sacro Cuore, Rome, and by Fondazione Poliambulanza Istituto Ospedaliero, Brescia, Italy, Contributo MIUR 5 × 1000 (2015). Part of the scheme in has been produced using Servier Medical Art (www.servier.com).
Figure 2. Role of autophagy on MSC functions. Autophagy plays a dual role in MSC activities. (a) Stress signals or pharmacological agents can modulate autophagy in MSCs. (b) MSCs may modulate autophagy of tissue-resident and recruited cells (target cells) involved in disease pathogenesis. Both actions affect MSC functions and have an impact on the therapeutic potential (either directly a or indirectly b) by influencing survival, vascularization, immunomodulation, and cell differentiation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Wirawan E, Van Den Berghe T, Lippens S, et al. Autophagy: for better or for worse. Cell Res. 2012;22:43–61.
- Bejarano E, Cuervo AM. Chaperone-Mediated Autophagy. Proc Am Thorac Soc. 2010;7:29–39.
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34.
- Klionsky DJ, Emr S. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721.
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS. 1993;333:169–174.
- Mizushima N, Yoshimor T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132.
- Cao Y, Klionsky D. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2017;17:839–849.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologuen of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728.
- Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812.
- Kim JY, Zhao H, Martinez J, et al. Non-canonical autophagy promotes the visual cycle. Cell. 2013;154:365–376.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873.
- Lee JA, Yue Z, Gao F. Autophagy in neurodegenerative diseases. Brain Res. 2016;1649:141–142.
- Luciani A, Villella V, Esposito S, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–875.
- Lai C, Tsai C, Kuo W, et al. Multi-strain probiotics inhibit cardiac myopathies and autophagy to prevent heart injury in high-fat diet-fed rats. Int J Med Sci. 2016;13:277–285.
- Bartlett J, Trivedi P, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;14:1–8.
- Shintani T, Klionsky D. Autophagy in health and disease: a double-edged sword. Science. 2004;306:90–95.
- Sever O, Demir O. Autophagy: cell death or survive mechanism. J Oncol Sci. 2017;3:37–44.
- Basciani S, Vona R, Matarrese P, et al. Imatinib interferes with survival of multi drug resistant Kaposi’s sarcoma cells. FEBS Lett. 2007;581:5897–5903.
- Yang W, Monroe J, Zhang Y, et al. Proteasome inhibition induces both pro- and anti-cell death pathways in prostate cancer cells. Cancer Lett. 2006;243:217–227.
- Nishiyama J, Matsuda K, Kakegawa W, et al. Reevaluation of neurodegeneration in lurcher mice: constitutive ion fluxes cause cell death with, not by, autophagy. J Neurosci. 2010;30:2177–2187.
- Klionsky D, Abeliovich A, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175.
- Murrow L, Debnath J. Autophagy as a stress response and quality control mechanism—implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137.
- Yu W, Cuervo A, Kumar A, et al. Macroautophagy-a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98.
- Chu Y, Morfini G, Langhamer L, et al. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 2012;35:2058–2073.
- Nagata E, Sawa A, Ross CA, et al. Autophagosome-like vacuole formation in Huntington’s disease lymphoblasts. Neuroreport. 2004;15:1325–1328.
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176.
- Bolaños-Meade J, Zhou L, Hoke A, et al. Wagner KR Hydroxychloroquine causes severe vacuolar myopathy in a patient with chronic graft-versus-host disease. Am J Hematol. 2005;78:306–309.
- Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–365.
- Del Principe D, Lista P, Malorni W, et al. Fibroblast autophagy in fibrotic disorders. J Pathol. 2013;229:208–220.
- Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785.
- Patel AS, Lin L, Geyer A, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7:1–9.
- Wu D, Adamopoulos I. Autophagy and autoimmunity. Clin Immunol. 2017;176:55–62.
- Gros F, Arnold J, Page N, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8:1113–1123.
- Gergely P, Grossman C, Niland B, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:175–190.
- Caza TN, Fernandez DR, Talaber G, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis. 2014;73:1888–1897.
- Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–119.
- Lai Y, Hickey R, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550.
- Luo C, Li B, Li Q, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63.
- White E, Di Paola R. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316.
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967.
- Fujii S, Mitsunaga S, Yamazaki M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008;99:1813–1819.
- Song J, Qu Z, Guo X, et al. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy. 2009;5:1131–1144.
- Sridhar S, Botbol Y, Macian F, et al. Autophagy and Disease: always two sides to a problem. J Pathol. 2012;226:255–273.
- Prockop D. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74.
- Pittenger M, Mackay A, Beck S, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147.
- Zuk P, Zhu M, Mizuno H, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–228.
- Campagnoli C, Roberts I, Kumar S, et al. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98:2396–2402.
- De Bari C, Dell’Accio F, Tylzanowski P, et al. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44:1928–1942.
- Erices A, Conget P, Minguell J. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. 2000;109:235–242.
- Anzalone R, Lo Iacono M, Corrao S, et al. New emerging potentials for human Wharton’s jelly mesenchymal stem cells: immunological features and hepatocyte-like differentiative capacity. Stem Cells Dev. 2010;19:423–438.
- Fukumoto T, Sperling J, Sanyal A, et al. Combined effects of insulin-like growth factor-1 and transforming growth factor-beta1 on periosteal mesenchymal cells during chondrogenesis in vitro. Osteoarthr Cartil. 2003;11:55–64.
- Shi S, Gronthos S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Min Res. 2003;18:696–704.
- Rotter N, Oder J, Schlenke P, et al. Isolation and characterization of adult stem cells from human salivary glands. Stem Cells Dev. 2008;17:509–518.
- Villaron E, Almeida J, Lopez-Holgado N, et al. Mesenchymal stem cells are present in peripheral blood and can engraft after allogeneic hematopoietic stem cell transplantation. Haematologica. 2004;89:1421–1427.
- Bailo M, Soncini M, Vertua E, et al. Engraftment potential of human amnion and chorion cells derived from term placenta. Transplantation. 2004;78:1439–1448.
- Parolini O, Alviano F, Bagnara G, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international workshop on placenta derived stem cells. Stem Cells. 2008;26:300–311.
- In’t Anker P, Scherjon S, Kleijburg-van der Keur C, et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. 2004;22:1338–1345.
- Salingcarnboriboon R, Yoshitake H, Tsuji K, et al. Establishment of tendon-derived cell lines exhibiting pluripotent mesenchymal stem cell-like property. Exp Cell Res. 2003;287:289–300.
- Rossignoli F, Caselli A, Grisendi G, et al. Isolation, characterization, and transduction of endometrial decidual tissue multipotent mesenchymal stromal/stem cells from menstrual blood. BioMed Res Int. 2013;2013:1–14.
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317.
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736.
- Bernardo M, Locatelli F, Fibbe W. Mesenchymal stromal cells. Ann N Y Acad Sci. 2009;1176:101–117.
- Phinney D, Prockop D. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair-current views. Stem Cells. 2007;25:2896–2902.
- Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther. 2016;7:1–13.
- Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol Ther. 2009;17:939–946.
- Lai R, Arslan F, Lee M, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–222.
- Kanazawa H, Fujimoto Y, Teratani T, et al. Bone marrow-derived mesenchymal stem cells ameliorate hepatic ischemia reperfusion injury in a rat model. PLoS One. 2011;6:e19195.
- Gatti S, Bruno S, Deregibus M, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transpl. 2011;26:1474–1483.
- Bruno S, Grange C, Deregibus M, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–1067.
- Calió M, Marinho D, Ko G, et al. Transplantation of bone marrow mesenchymal stem cells decreases oxidative stress, apoptosis, and hippocampal damage in brain of a spontaneous stroke model. Free Radic Biol Med. 2014;70:141–154.
- Nagaya N, Fujii T, Iwase T, et al. Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infarction through angiogenesis and myogenesis. Am J Physiol Hear Circ Physiol. 2004;287:H2670–H2676.
- Magatti M, De Munari S, Vertua E, et al. Human amnion mesenchyme harbors cells with allogeneic T-cell suppression and stimulation capabilities. Stem Cells. 2008;26:182–192.
- Tse W, Pendleton J, Beyer W, et al. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75:389–397.
- Augello A, Tasso R, Negrini S, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482–1490.
- Rossi D, Pianta S, Magatti M, et al. Characterization of the conditioned medium from amniotic membrane cells: prostaglandins as key effectors of its immunomodulatory activity. PLoS One. 2012;7:e46956.
- Corcione A, Benvenuto F, Ferretti E, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107:367–372.
- Comoli P, Ginevri F, Maccario R, et al. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol Dial Transpl. 2008;23:1196–1202.
- Asari S, Itakura S, Ferrer K, et al. Mesenchymal stem cells suppress B-cell terminal differentiation. Exp Hematol. 2009;37:604–615.
- Tabera S, Perez-Simon J, Diez-Campelo M, et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica. 2008;93:1301–1309.
- Magatti M, De Munari S, Vertua E, et al. Amniotic mesenchymal tissue cells inhibit dendritic cell differentiation of peripheral blood and amnion resident monocytes. Cell Transpl. 2009;18:899–914.
- Magatti M, Caruso M, De Munari S, et al. Human amniotic membrane-derived mesenchymal and epithelial cells exert different effects on monocyte-derived dendritic cell differentiation and function. Cell Transpl. 2015;24:1733–1752.
- Choi E, Shin I, Park S, et al. Reversal of serologic, immunologic, and histologic dysfunction in mice with systemic lupus erythematosus by long-term serial adipose tissue-derived mesenchymal stem cell transplantation. Arthritis Rheum. 2012;64:243–253.
- Parolini O, Souza-Moreira L, O’Valle F, et al. Therapeutic effect of human amniotic membrane-derived cells on experimental arthritis and other inflammatory disorders. Arthritis Rheumatol. 2014;66:327–339.
- Pianta S, Bonassi Signoroni P, Muradore I, et al. Amniotic membrane mesenchymal cells-derived factors skew T cell polarization toward Treg and downregulate Th1 and Th17 cells subsets. Stem Cell Rev. 2015;11:394–407.
- Uccelli A, de Rosbo N. The immunomodulatory function of mesenchymal stem cells: mode of action and pathways. Ann N Y Acad Sci. 2015;1351:114–126.
- Akiyama K, Chen C, Wang D, et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10:544–555.
- Madec A, Mallone R, Afonso G, et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia. 2009;52:1391–1399.
- Duijvestein M, Wildenberg M, Welling M, et al. Pretreatment with interferon-gamma enhances the therapeutic activity of mesenchymal stromal cells in animal models of colitis. Stem Cells. 2011;29:1549–1558.
- Magatti M, Vertua E, De Munari S, et al. Human amnion favours tissue repair by inducing the M1-to-M2 switch and enhancing M2 macrophage features. J Tissue Eng Regen Med. 2016;11:2895–2911.
- Yan M, Liu X, Dang Q, et al. Intra-articular injection of human synovial membrane-derived mesenchymal stem cells in murine collagen-induced arthritis: assessment of immunomodulatory capacity in vivo. Stem Cells Int. 2017;2017:1–12.
- Nemeth K, Leelahavanichkul A, Yuen P, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49.
- Silini AR, Cargnoni A, Magatti M, et al. The long path of human placenta, and its derivatives, in regenerative medicine. Front Bioeng Biotechnol. 2015;3:1–16.
- Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25:829–848.
- Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22:824–833.
- Oliver L, Hue E, Priault M, et al. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012;21:2779–2788.
- Nuschke A, Rodrigues M, Stolz DB, et al. Human mesenchymal stem cells/multipotent stromal cells consume accumulated autophagosomes early in differentiation. Stem Cell Res Ther. 2014;5:1–14.
- Pantovic A, Krstic A, Janjetovic K, et al. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone. 2013;52:524–531.
- Song B, Chi Y, Li X, et al. Inhibition of notch signaling promotes the adipogenic differentiation of mesenchymal stem cells through autophagy activation and PTEN-PI3K/AKT/mTOR pathway. Cell Physiol Biochem. 2015;36:1991–2002.
- Li B, Duan P, Li C, et al. Role of autophagy on bone marrow mesenchymal stem cell proliferation and differentiation into neurons. Mol Med Rep. 2016;13:1413–1419.
- Park S, Choi Y, Jung N, et al. Autophagy induction in the skeletal myogenic differentiation of human tonsil-derived mesenchymal stem cells. Int J Mol Med. 2017;39:831–840.
- Li Q, Gao Z, Chen Y, et al. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell. 2017;8:439–445.
- Forni MF, Peloggia J, Trudeau K, et al. Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells. 2016;34:743–755.
- Dang S, Xu H, Xu C, et al. Autophagy regulates the therapeutic potential of mesenchymal stem cells in experimental autoimmune encephalomyelitis. Autophagy. 2014;10:1301–1315.
- Gao L, Cen S, Wang P, et al. Autophagy improves the immunosuppression of CD4+ T cells by mesenchymal stem cells through transforming growth factor-beta1. Stem Cells Transl Med. 2016;5:1496–1505.
- Chinnadurai R, Copland I, Ng S, et al. Mesenchymal stromal cells derived from Crohn’s patients deploy indoleamine 2,3-dioxygenase-mediated immune suppression, independent of autophagy. Mol Ther. 2015;23:1248–1261.
- Kim KW, Moon SJ, Park MJ, et al. Optimization of adipose tissue-derived mesenchymal stem cells by rapamycin in a murine model of acute graft-versus-host disease. Stem Cell Res Ther. 2015;6:1–15.
- An Y, Liu W, Xue P. Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death Dis. 2018;9:58.
- Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. 2019;19(3):170–183.
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841.
- Hubbard VM, Valdor R, Patel B, et al. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357.
- Riffelmacher T, Richter FC, Simon AK. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy. 2018;14:199–206.
- Botbol Y, Guerrero-Ros I, Macian F. Key roles of autophagy in regulating T-cell function. Eur J Immunol. 2016;46:1326–1334.
- Chen J, Wang Q, Feng X, et al. Umbilical cord-derived mesenchymal stem cells suppress autophagy of t cells in patients with systemic lupus erythematosus via transfer of mitochondria. Stem Cells Int. 2016;2016:1–13.
- Zhu H, Gao J, Zhao M, et al. Effects of bone marrow-derived mesenchymal stem cells on the autophagic activity of alveolar macrophages in a rat model of silicosis. Exp Ther Med. 2016;11:2577–2582.
- Li J, Zhou J, Zhang D, et al. Bone marrow-derived mesenchymal stem cells enhance autophagy via PI3K/AKT signalling to reduce the severity of ischaemia/reperfusion-induced lung injury. J Cell Mol Med. 2015;19:2341–2351.
- Liu L, Jin X, Hu C, et al. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways. Cell Physiol Biochem. 2017;43:52–68.
- Zhao K, Hao H, Liu J, et al. Bone marrow-derived mesenchymal stem cells ameliorate chronic high glucose-induced β-cell injury through modulation of autophagy. Cell Death Dis. 2015;6:1–13.
- Shin J, Park H, Kim H, et al. Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy. 2014;10:32–44.
- Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13:1619–1628.
- Xiao C, Wang K, Xu Y, et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ Res. 2018;123:564–578.