
ABSTRACT
Impaired macroautophagy/autophagy is involved in the pathogenesis of hepatic fibrosis. However, how aberrant autophagy promotes fibrosis is far from understood. Here, we aimed to define a previously unrevealed pro-fibrotic mechanism for the stress protein TRIB3 (tribbles pseudokinase 3)-mediated autophagy dysfunction. Human fibrotic liver tissues were obtained from patients with cirrhosis who underwent an open surgical repair process. The functional implications of TRIB3 were evaluated in mouse models of hepatic fibrosis induced by bile duct ligation (BDL) or thioacetamide (TAA) injection. Human fibrotic liver tissues expressed higher levels of TRIB3 and selective autophagic receptor SQSTM1/p62 (sequestosome 1) than nonfibrotic tissues and the elevated expression of TRIB3 and SQSTM1 was positively correlated in the fibrotic tissues. Silencing Trib3 protected against experimentally induced hepatic fibrosis, accompanied by restored autophagy activity in fibrotic liver tissues. Furthermore, TRIB3 interacted with SQSTM1 and hindered its binding to MAP1LC3/LC3, which caused the accumulation of SQSTM1 aggregates and obstructed autophagic flux. The TRIB3-mediated autophagy impairment not only suppressed autophagic degradation of late endosomes but also promoted hepatocellular secretion of INHBA/Activin A-enriched exosomes which caused migration, proliferation and activation of hepatic stellate cells (HSCs), the effector cells of liver fibrosis. Disrupting the TRIB3-SQSTM1 interaction with a specific helical peptide exerted potent protective effects against hepatic fibrosis by restoring autophagic flux in hepatocytes and HSCs. Together, stress-elevated TRIB3 expression promotes hepatic fibrosis by interacting with SQSTM1 and interfering with its functions in liver-parenchymal cells and activating HSCs. Targeting this interaction is a promising strategy for treating fibroproliferative liver diseases.
Abbreviations: 3-MA: 3-methyladenine; AAV: adeno-associated virus; ACTA2/α-SMA: actin, alpha 2, smooth muscle, aorta; BDL: bile duct ligation; BECN1/Beclin 1: beclin 1, autophagy related; CHX: cycloheximide; CQ: chloroquine; Edu: 5-ethynyl-2-deoxyuridine; ESCRT: endosomal sorting complexes required for transport; HSC: hepatic stellate cell; ILV: intralumenal vesicle; LAMP1: lysosomal-associated membrane protein 1; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MVB: multivesicular body; PIK3C3: phosphatidylinositol 3-kinase, catalytic subunit type 3; PPI: protein-protein interaction; SQSTM1/p62: sequestosome 1; TAA: thioacetamide; TEM: transmission electron microscopy; TGFB1/TGFβ1: transforming growth factor, beta 1; TLR2: toll-like receptor 2; TRIB3: tribbles pseudokinase 3
Introduction
Hepatic fibrosis is a critical structural basis of the pathogenesis of a variety of chronic liver diseases, including viral hepatitis, alcoholic and cholestatic liver diseases [Citation1,Citation2]. Although great progress has been achieved in understanding the pathogenesis of hepatic fibrosis in recent years, finding effective antifibrotic agents for hepatic fibrosis and cirrhosis remains an unconquered area in drug development [Citation3]. Macroautophagy/autophagy, a cellular self-digestion process important for maintaining cell and tissue homeostasis during nutrient starvation and diverse cellular stresses [Citation4,Citation5], has been reported to be involved in the regulation of tissue injury repair and fibrosis [Citation6–Citation8]. Unlike the ubiquitin-proteasome system (UPS) which is responsible for the degradation of individual short-lived proteins, autophagy functions as a bulk degradation process for long-lived proteins and organelles such as the mitochondria, endoplasmic reticulum and ribosomes [Citation9]. In addition, autophagy has an important role in the unconventional extracellular release of unwanted or damaged cytoplasmic constituents instead of their degradation, which may be either beneficial or detrimental to neighboring cells [Citation10]. During autophagy, autophagosomes can either fuse with lysosomes or with multivesicular bodies (MVBs), which are late endosomes containing multiple intralumenal vesicles (ILVs), resulting in degradation of their contents in lysosomes. Alternatively, if the autophagy-lysosomal pathway is obstructed, MVBs will fuse with the plasma membrane and release the ILVs as exosomes [Citation11,Citation12].
Exosome secretion is the most common alternative pathway to alleviate stress when autophagic degradation is compromised [Citation13]. In the liver, some exosomes containing miRNAs participate in the pathogenesis of liver fibrosis by modulating hepatic stellate cell (HSC) activation [Citation14,Citation15], and conversely, exosomes derived from umbilical cord mesenchymal cells attenuate liver fibrosis in mice by suppressing the SMAD signaling pathway in hepatocytes [Citation16]. We recently report that hepatocellular autophagy suppression is involved in the pathogenesis of hepatic fibrosis induced by bile duct ligation (BDL) or thioacetamide (TAA) in an IL17A-dependent manner and that blocking IL17A provides therapeutic benefits in mouse models of hepatic fibrosis [Citation17]. However, it has not been determined whether specific exosomes are released from autophagy-impaired hepatocytes and/or whether HSCs, which play vital roles in liver fibrogenesis, directly respond to these exosomes.
Our previous work indicated that the TRIB3 (tribbles pseudokinase 3), a stress protein upregulated in response to multiple stressors, directly interacts with the cargo receptor SQSTM1/p62 (sequestosome 1) and interferes with its binding to MAP1LC3/LC3 and to ubiquitinated substrate proteins, which suppresses autophagic flux and protects several tumor-promoting factors from autophagic degradation in cancer cells [Citation18]. However, it is largely unknown whether elevated TRIB3 expression causes aberrant autophagy activity in parenchymal hepatocytes and HSCs or whether high TRIB3 expression contributes to the progression of hepatic fibrosis. In this study, we examined the expression of TRIB3 in human fibrotic liver tissues and its actions in mouse models of BDL- or TAA-induced hepatic fibrosis. Our study reveals a previously unidentified pro-hepatic fibrosis mechanism for TRIB3 and suggests an attractive therapeutic strategy for treating fibroproliferative liver diseases.
Results
Elevated TRIB3 expression contributes to the development of liver fibrosis
To determine the relationship between TRIB3 and autophagy activity in hepatic fibrosis, we examined the expression levels of TRIB3 and autophagy-associated proteins in human normal and fibrotic liver tissues and found that TRIB3 expression was upregulated in fibrotic tissues compared with normal liver tissues. BECN1/Beclin 1 (beclin 1, autophagy related), PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) and SQSTM1 expression, LC3-II:LC3-I ratio and insoluble SQSTM1 aggregation were also increased in fibrotic tissues, indicating aberrant autophagic flux (). Interestingly, TRIB3-SQSTM1 and SQSTM1-LC3 interactions were observed in fibrotic liver tissues (). Using human tissue microarrays containing 5 normal liver tissue samples and 22 fibrotic liver tissue samples (Table S1), higher levels of TRIB3, SQSTM1 and LC3 were detected in fibrotic tissues than in normal liver tissues (). Moreover, a positive correlation was observed between TRIB3 expression and SQSTM1 or LC3 expression in fibrotic tissues (). Confocal microscopy analysis revealed that fibrotic tissues exhibited increased expression of TRIB3 in hepatocytes (ALB [albumin]) and HSCs (ACTA2/α-SMA [actin, alpha 2, smooth muscle, aorta]) (), as well as enhanced colocalization of TRIB3 with SQSTM1, compared with non-fibrotic tissues (). These data indicate that elevated TRIB3 expression positively correlates with the development of human hepatic fibrosis associated with suppressed autophagy.
Figure 1. High TRIB3 expression positively correlates with SQSTM1 and LC3 expression in human fibrotic liver tissues. (A) Levels of TRIB3, LC3-II:LC3-I, BECN1, PIK3C3, and soluble and insoluble SQSTM1 in human fibrosis and normal liver tissues were detected using western blotting. Representative immunoblots and the ratios of the indicated proteins to ACTB (actin, beta) are presented. Data are the means ± SEM of 3 assays. (B) Human liver extracts were immunoprecipitated with an anti-SQSTM1 antibody (Ab) and blotted with an anti-TRIB3 Ab or anti-LC3-II/I Ab. Representative immunoblots from 3 assays are shown. (C) TRIB3, SQSTM1 and LC3 expression of were detected with immunohistochemical staining in human cirrhotic and normal liver tissues. Representative images of sections from cirrhotic (n = 22) and normal (n = 5) liver tissues are shown (scale bar: 40 μm). The quantitative analysis of expression is indicated in the graphs. Statistical significance was determined using Student’s t-test. (D) Scatter plots reveal the correlation of TRIB3 expression with the level of SQSTM1 or LC3 in cirrhotic tissues. (E and F) Expression of TRIB3 and colocalization of TRIB3 with SQSTM1 in fibrotic and normal human liver tissues was detected by immunostaining. Hepatocytes were indicated by ALB staining and HSCs by ACTA2 staining (scale bar: 20 μm). Data are representative images of 3 assays.
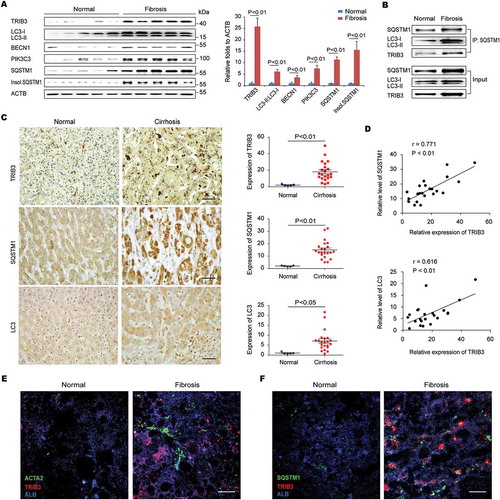
We next assessed the potential roles of TRIB3 in regulating of autophagy during development of BDL-induced hepatic fibrosis. As expected, BDL treatment promoted a time-dependent expression of TRIB3, ACTA2, COL1A1 (collagen type I alpha 1 chain), autophagic proteins as well as the aggregation of insoluble SQSTM1 in the livers. Notably, the levels of both TRIB3 and SQSTM1 were originally upregulated at the early stage and continuously increased from day 1 to day 15 after BDL injury (Fig. S1A). Moreover, treatment with chloroquine (CQ) to inhibit autophagy did not further increase the LC3-II:LC3-I ratio and the aggregation of insoluble SQSTM1 (Fig. S1B), suggesting that there is an obstructed autophagic flux in BDL-injured livers. Using liver-specific Trib3-knockdown (Trib3[LKD]) and WT control mice, we verified the role of TRIB3 in the development of TAA- or BDL-induced hepatic fibrosis. Extensive fibrosis and a large number of dysplastic nodules were observed in the livers of TAA-treated WT mice at the end of the 12 weeks after initial TAA treatment, but Trib3-knockdown mice showed less nodules and jaundice () and a reduction of liver-infiltrating inflammatory cells in liver tissues () compared with WT mice. More importantly, Trib3 knockdown resulted in a remarkable decrease in collagen accumulation in the interstitial non-vessel areas of TAA- or BDL-treated livers ( and Fig. S1C), accompanied with reduced content of hydroxyproline ( and Fig. S1D) and mRNA levels of Col1a1, Timp1 and Tgfb1 in liver tissue ( and Fig. S1E). Additionally, mice challenged with TAA showed a significant upregulation of ACTA2, a marker for HSC activation, which was attenuated in TAA-treated Trib3[LKD] mice (). No significant difference in CYP2E1 (cytochrome P450, family 2, subfamily e, polypeptide 1) content was detected between Trib3[LKD] and WT mice (), indicating that the suppressed hepatic fibrosis in Trib3[LKD] mice was not simply resulted from changes in TAA metabolites. To explore whether silencing Trib3 improved hepatic fibrosis via autophagy, we examined the expression of autophagy signaling proteins in fibrotic liver tissues. Silencing Trib3 not only inhibited the levels of LC3-II:LC3-I, BECN1 and PIK3C3 but also reduced the aggregation of insoluble SQSTM1 in TAA-treated livers without affecting the level of soluble SQSTM1 (). These data suggest that TRIB3 mediates hepatic fibrosis by suppressing autophagy in fibrotic liver tissues.
Figure 2. Trib3 knockdown protects mice from TAA-induced hepatic fibrosis. (A) Representative images of liver morphology showed that the Trib3 knockdown protected mice from TAA-induced hepatic fibrosis (n = 12–15/group, scale bar: 3 mm). (B) Trib3 knockdown repressed the TAA-induced recruitment of inflammatory cells, as indicated by hematoxylin and eosin staining of liver sections (scale bar: 20 μm). Data are representative images of 3 assays (n = 12/group). (C) Sirius red staining of liver sections (scale bar: 20 μm) and a summary of collagen deposition measured with sirius red staining. Data are representative images and means ± SEM of 3 assays (n = 12/group). (D) Hydroxyproline content. Data are means ± SEM of 3 assays (n = 12/group). (E) Hepatic expression of Col1a1, Timp1 and Tgfb1 measured by qRT-PCR. Data are means ± SEM of 3 assays (n = 12/group). (F) ACTA2 expression was detected with immunofluorescence staining in liver sections (scale bar: 20 μm). Data are representative images and means ± SEM of 3 assays (n = 12/group). (G) Content of CYP2E1 in liver tissues. Data are means ± SEM of 3 assays (n = 6/group). (H) Levels of TRIB3, LC3-II:LC3-I, BECN1, PIK3C3, SQSTM1 and insoluble SQSTM1 in TAA-induced fibrotic liver tissues were analyzed by western blotting. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays.
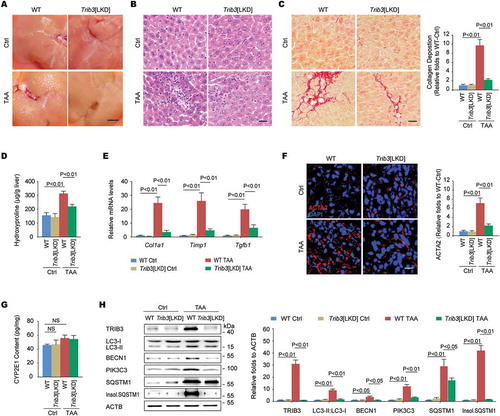
TRIB3 interacts with SQSTM1 to hinder the autophagic degradation of late endosomes in hepatocytes
We found that primary mouse hepatocytes expressing TRIB3 and SQSTM1 showed colocalization and co-IP (co-immunoprecipitation) of SQSTM1 with TRIB3 following starvation (). In hepatocytes co-expressed SQSTM1 and LC3 with or without TRIB3, SQSTM1 was co-immunoprecipitated with LC3, but this interaction was reduced by TRIB3 overexpression (). Overexpressed TRIB3 not only augmented the expression of LC3-II:LC3-I, BECN1, PIK3C3 and SQSTM1 but also resulted in the aggregation of insoluble SQSTM1 in primary hepatocytes (). To confirm the role of TRIB3 in the suppression of autophagic flux, hepatocytes were infected with an RFP-GFP-LC3 expressing adenovirus to measure autophagic flux based on the concept of lysosomal quenching of GFP. Using live-cell imaging, RFP-GFP-LC3 was detected as red speckles after starvation, whereas TRIB3 overexpression increased the presence of yellow puncta and dynamically moving yellow spots, indicating autophagy suppression in cells (). Elevated TRIB3 expression increased the numbers of autophagosomes as well as multivesicular structures, corresponding to late endosomes or MVBs, in hepatocytes (). To quantify MVB distribution, we measured the perinuclear lysobisphosphatidic acid (LBPA) fraction, defined as the LBPA+ area located within 2 μm of the nucleus. Compared with control hepatocytes, both CQ-treated and TRIB3-overexpressed cells exhibited increases in the perinuclear LBPA+ fraction and enlarged LBPA+ puncta size (Fig. S2A and ). As autophagic flux is essential for late endosome degradation, we predicted that high TRIB3 expression hindered SQSTM1-mediated lysosome tracking of late endosomes. Indeed, both SQSTM1 and the late endosomal marker CD63 showed significant aggregation in TRIB3-overexpressed hepatocytes, as indicated by the numbers of SQSTM1- or CD63-positive puncta (Fig. S2B). Furthermore, we performed immunostaining for both CD63 and the lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) to more precisely define the organelle alterations in CQ-treated or TRIB3-overexpressed hepatocytes. Indeed, the colocalization of CD63 and LAMP1 was enhanced in hepatocytes after starvation for 24 h, whereas either CQ treatment or TRIB3 overexpression remarkably reduced this colocalization, indicating that high TRIB3 expression inhibits the fusion of late endosomes with lysosomes (Fig. S2C and ). Next, TAA treated hepatocytes were used to mimic the insults occurred in vivo. We found that TAA stimulated concentration-dependent accumulation (Fig. S2D) and colocalization of TRIB3 and SQSTM1 (Fig. S2E) in hepatocytes. To validate whether the autophagic degradation of late endosomes was impaired by TRIB3, Trib3 depleted hepatocytes expressing RFP-GFP-LC3 were treated with TAA for 12 h. More ectopically expressed RFP-GFP-LC3 was detected as yellow speckles in TAA-treated cells than in untreated control cells. However, dynamically moving red spots with occasional yellow spots were observed in TAA-treated Trib3-KD cells (Fig. S2F), indicating that TRIB3 mediates the accumulation of autophagosomes in TAA-challenged cells. Consistent with these results, silencing Trib3 markedly decreased the levels of TRIB3, LC3-II:LC3-I, SQSTM1, insoluble SQSTM1, LAMP1 and CD63 in TAA-treated hepatocytes (Fig. S2G). Together, these data indicate that TRIB3 controls autophagic flux and the autophagic degradation of late endosomes by interacting with SQSTM1 in hepatocytes.
Figure 3. TRIB3 interacts with SQSTM1 to hinder the cargo functions of SQSTM1 and the autophagic degradation of late endosomes. (A) Primary hepatocytes were co-infected with Ad.Trib3-HA and Ad.Sqstm1-MYC. Colocalization of TRIB3 and SQSTM1 was detected by immunostaining (left panel). Cell extracts were immunoprecipitated with an anti-HA Ab and blotted with an anti-MYC Ab (right panel). Data are representative immunostaining images and immunoblots of 3 assays. (B) The SQSTM1-LC3 interaction was evaluated by an IP assay. Data are representative immunoblots of 3 assays. (C) Levels of autophagy-associated proteins in Ad.Ctrl or Ad.Trib3 treated hepatocytes were detected by western blotting. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays. (D) Hepatocytes expressing RFP-GFP-LC3 were infected with Ad.Ctrl or Ad.Trib3. Autophagic flux was detected with live-cell imaging microscopy. Data are representative images showing red-colored autolysosomes or red/green double-colored autophagosomes of 3 assays. (E) TEM images of autophagosomes and late endosomes (MVBs) in hepatocytes. Red arrows denote autophagosomes; yellow arrows indicate MVBs. The numbers of autophagic vacuoles and MVBs were determined by quantitative analysis. Data are representative images and means ± SEM of 3 TEM assays. (F) Hepatocytes were immunostained with an anti-LBPA Ab and DAPI. The perinuclear LBPA+ fraction was defined as the fraction of LBPA+ area located within 2 μm of the nucleus, and the perinuclear LBPA+ fraction of LBPA+ puncta was quantified (n = 50 cells pooled from 3 assays). (G) Hepatocytes were further immunostained with an anti-CD63 Ab and an anti-LAMP1 Ab to detect the colocalization of late endosomes with lysosomes. Data are representative images and means ± SEM of 3 assays.
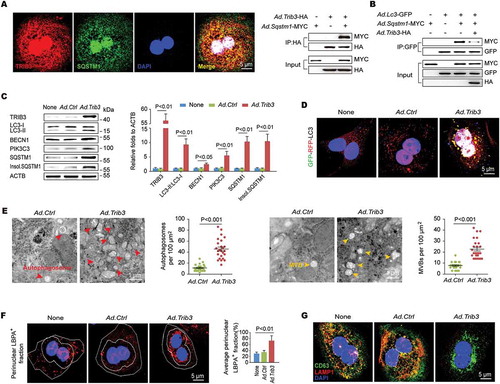
TRIB3 promotes exosome secretion from hepatocytes
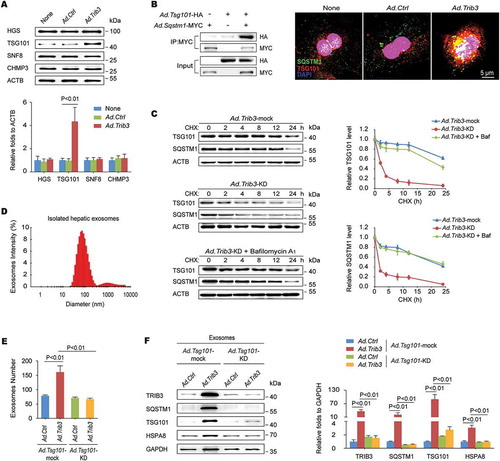
Given that autophagy dysfunction is a critical initiator of exosome release, which acts as an alternative way to alleviate cellular stress, we reasoned that the TRIB3-mediated obstruction of autophagic flux could lead to an augmentation of exosome secretion from hepatocytes. As the ESCRT (endosomal sorting complexes required for transport) machinery is essential for the formation of MVBs and secretion of MVB-derived exosomes [Citation19], we examined the effect of enhanced TRIB3 expression on the levels of typical ESCRT components in hepatocytes, and found that TRIB3-overexpressed hepatocytes exhibited a significant increase in the level of TSG101 (tumor susceptibility 101 [subunit of ESCRT-I]), but not HGS/HRS (hepatocyte growth factor-regulated tyrosine kinase substrate [subunit of ESCRT-0]), SNF8/VPS22 (SNF8 subunit of ESCRT-II) or CHMP3/VPS24 (charged multivesicular body protein 3 [subunit of ESCRT-III]), compared with Ad.Ctrl-infected hepatocytes (). Furthermore, TSG101 co-precipitated with SQSTM1 in hepatocytes co-infected with Ad.Tsg101-HA and Ad.Sqstm1-MYC ( left). In addition, immunofluorescence staining revealed that TSG101 colocalized with SQSTM1 in TRIB3-overexpressed hepatocytes following starvation for 24 h ( right). Silencing Trib3 promoted the degradation of both TSG101 and SQSTM1 in cycloheximide (CHX)-treated hepatocytes, which was delayed by the autolysosome inhibitor bafilomycin A1 (). These data suggest that TRIB3 interferes with SQSTM1-associated autophagic degradation of TSG101. To confirm the involvement of TRIB3 in exosome release, we isolated exosomes using an exosome precipitation kit () and detected protein expression using SDS-PAGE followed by Coomassie staining. As expected, the protein level was increased by 4.9-fold in the exosomal lysates derived from TRIB3-overexpressed hepatocytes, but not in the whole-cell lysates, compared with Ad.Ctrl-infected cells (Fig. S3). Using an optimized sandwich ELISA to quantify CD63, a well-characterized exosome marker, the number of exosomes in the culture supernatant was determined. We observed that the number of exosomes was increased by 2.1-fold in the supernatant of TRIB3-overexpressed hepatocytes compared with control hepatocytes, whereas silencing Tsg101 remarkably eliminated this increase (). To determine whether Tsg101 deficiency impaired exosome biogenesis, we isolated exosomal fractions from the supernatants of Ad.Ctrl- or Ad.Trib3-infected hepatocytes and immunoblotted for TRIB3, SQSTM1, and exosomal markers to measure relative exosome release. Hence, purified exosomes from TRIB3-overexpressed hepatocytes exhibited increased expression of exosomal markers such as TSG101, HSPA8/HSC70 and GAPDH as well as TRIB3 and SQSTM1, which was completely reversed by Tsg101 deletion (). These data indicate that TRIB3 causes SQSTM1-associated accumulation and dysfunction of TSG101, leading to enhanced exosome release from hepatocytes.
Figure 4. Elevated TRIB3 expression promotes TSG101-mediated exosome secretion. (A) Overexpression of TRIB3 enhanced the level of TSG101 but did not affect expression of HGS, SNF8 and CHMP3 in primary hepatocytes. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays. (B) Hepatocytes were co-infected with Ad.Tsg101-HA and Ad.Sqstm1-MYC for 24 h. The extracts were immunoprecipitated with an anti-MYC Ab and blotted with an anti-HA Ab (left panel). Colocalization of TSG101 and SQSTM1 was detected by immunostaining (right panel). Data are representative immunoblots and immunostaining images of 3 assays. (C) Analysis of TSG101 and SQSTM1 degradation kinetics in hepatocytes infected with Ad.Trib3-mock or Ad.Trib3-KD and treated with the protein synthesis inhibitor CHX for the indicated times. Where specified, the medium was supplemented with bafilomycin A1 (Baf) to inhibit the autophagy-lysosome degradation pathway. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays. (D) Exosomes were isolated from the medium of Ad.Trib3-infected hepatocytes and identified with a dynamic light scattering system. Data are representative particle size distribution of 3 assays. (E) Quantitative analysis of hepatocyte-derived exosomes by ELISA. Data are the means ± SEM of 3 assays. (F) The expression of TRIB3, SQSTM1 and exosomal markers TSG101, HSPA8 and GAPDH was detected by western blotting. Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. Data are the means ± SEM of 3 assays.
TRIB3 activates HSCs via hepatocyte-derived exosomes
Based on the findings described above, we wondered whether the increased number of exosomes secreted from TRIB3-overexpressed hepatocytes directly activated HSCs, the major effector cells of liver fibrogenesis. We found that the culture supernatant from TRIB3-overexpressed hepatocytes stimulated HSC migration (Fig. S4A), proliferation (Fig. S4B), morphological alterations (Fig. S4C) and ectopically increased expression of ACTA2 and COL1A1 (Fig. S4D). To exclude the interference of potential HSC activators in the ‘exosome-free’ supernatant, we performed immunostaining for ACTA2 and COL1A1 to precisely analyze the biological function of exosomes from TRIB3-overexpressed hepatocytes (HepaTRIB3-exosomes). After treatment with HepaTRIB3-exosomes for 72 h, HSCs exhibited significant activation, proliferation and migration, as indicated by immunofluorescence staining of ACTA2 and COL1A1 (), Edu staining () and a transwell assay (), respectively. In contrast, treatment of HSCs with a ‘HepaTRIB3-exosome-free’ supernatant fraction did not cause these biological changes (–C). The potential HSC activators in HepaTRIB3-exosomes were identified with an antibody array of 200 cytokines (Table S2). We found that 175 proteins did not exhibit a change in expression or exhibited a low signal intensity, 16 proteins were downregulated, and 9 proteins were upregulated in HepaTRIB3-exosomes compared with control-hepatocyte-derived exosomes (). When HSCs were treated with exosomes isolated from hepatocytes, the bulk of Dil-stained exosomes were engulfed by HSCs (), which acted as mediators for cell-cell communication by delivering ectopic cytokines from hepatocytes to HSCs. Because INHBA/Activin A, a member of TGFB/TGF-β family and a modulator of inflammation and fibrosis, was the most abundant cytokine in HepaTRIB3-exosomes, Inhba-depleted hepatocytes were generated to examine the profibrotic action of HepaTRIB3-exosomes. HepaTRIB3-exosomes secreted from Inhba-depleted hepatocytes decreased the expression of ACTA2 and COL1A1 in HSCs compared with exosomes from Inhba-mock hepatocytes, indicating that INHBA mediated HepaTRIB3-exosome-induced HSC activation ().
Figure 5. Exosomes secreted from TRIB3-overexpressing hepatocytes stimulate HSC activation, migration and proliferation. (A-C) Exosome-free supernatants from TRIB3-overexpressing or control hepatocytes were collected as supernatant’. Freshly isolated mouse primary HSCs were treated with exosomes or supernatant’ for 72 h, and the expression of ACTA2 and COL1A1 (A), proliferation (B) and migration (C) were evaluated by immunostaining, Edu staining and a transwell assay, respectively. Data are representative images and means ± SEM of 3 assays. (D) Heat map of differentially expressed proteins in exosomes secreted from TRIB3-overexpressed or control hepatocytes, as determined using a cytokine antibody array. Nine cytokines were upregulated in TRIB3-overexpressing exosomes vs Control exosomes (means ± SEM, n = 3/group). (E) Isolated exosomes were stained with DiI, and HSCs were treated with exosomes for 24 h. Data are representative images of 3 assays. (F) Primary hepatocytes were infected with Ad.Inhba-mock or Ad.Inhba-KD with or without Ad.Trib3, and the exosomes were isolated and added to HSCs for 72 h. Then, the expression of ACTA2 and COL1A1 in HSCs was evaluated by immunostaining. Data are representative images of 3 assays.
Considering that TRIB3 was detectable in hepatocyte-derived exosomes, we wondered if HepaTRIB3-exosomes suppressed autophagic flux in HSCs. Indeed, HepaTRIB3-exosome-treated HSCs showed remarkable expression of autophagy-associated proteins, as well as ACTA2 and COL1A1, whereas Trib3-depleted HSCs showed significantly reduced levels of these proteins after the treatment of HepaTRIB3-exosomes (Fig. S5A, S5B), indicating that HSC-produced TRIB3 is the main participant in HSC activation. To verify this, we infected primary HSCs with Ad.Ctrl or Ad.Trib3 and observed that enhanced TRIB3 expression significantly promoted HSC activation (Fig. S5C), migration (Fig. S5D) and proliferation (Fig. S5E) as well as the obstruction of autophagic flux (Fig. S5F). To determine how TRIB3 mediated HSC activation, SQSTM1-immunoprecipitated proteins from TRIB3-overexpressed HSCs were subjected to ion-trap mass spectrometry, which revealed that 6 transdifferentiation-associated proteins, namely, SNAI1/SNAIL (snail family transcriptional repressor 1), SNAI2/SLUG (snail family transcriptional repressor 2), FN1 (fibronectin 1), VIM (vimentin), MMP2 (matrix metallopeptidase 2) and MMP9 (matrix metallopeptidase 9), were the major components that interacted with SQSTM1 (Fig. S5G), which was confirmed by IP assay (Fig. S5H). These data suggest that TRIB3 induces HSC activation and transdifferentiation by suppressing autophagic flux.
Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis
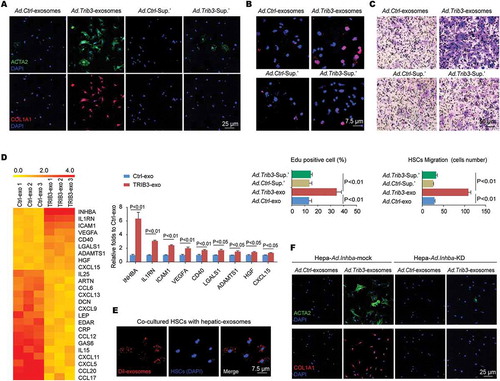
Because the TRIB3-SQSTM1 interaction plays a critical role in fibrotic liver tissues, we speculated that the fusion peptide Pep2-A2, which was generated from the UBA domain of SQSTM1 linked with a cell-penetrating peptide (Pep2, hlyvspwgg) and could interrupt this interaction in tumor cells [Citation18], might exert a therapeutic effect against liver fibrosis. As expected, the Pep2-A2 treatment inhibited the interaction and colocalization of TRIB3 and SQSTM1 in hepatocytes overexpressed TRIB3 and SQSTM1 (). Additionally, pretreatment of hepatocytes with Pep2-A2 restored TRIB3-suppressed autophagic flux (), accompanied by decreased levels of LC3-II:LC3-I, BECN1, PIK3C3, SQSTM1, insoluble SQSTM1, CD63, and TSG101 compared with Pep2-Ctrl-pretreated hepatocytes (), indicating that Pep2-A2 protected hepatocytes from autophagy impairment and late endosome dysfunction. Moreover, pretreating hepatocytes with Pep2-A2 not only reduced the number of exosomes in the medium () but also eliminated the HSC-activating effect of HepaTRIB3-exosomes () compared with Pep2-Ctrl-pretreated hepatocytes. The expression of transdifferentiation signal proteins, MMP2 and MMP9 in HSCs was also attenuated by HepaTRIB3-exosomes from Pep2-A2-pretreated hepatocytes (). In addition, treatment of HSCs with Pep2-A2 inhibited TRIB3-induced HSC activation (Fig. S6A) and the expression of autophagy-associated proteins and pro-transdifferentiation factors (Fig. S6B). Taken together, these data indicate that disrupting the TRIB3-SQSTM1 interaction in either hepatocytes or HSCs can reduce the activation and transdifferentiation of HSCs via restoring impaired autophagic flux.
Figure 6. An α-helical peptide of SQSTM1 (Pep2-A2) interrupts the TRIB3-SQSTM1 interaction and restores autophagic flux. (A) Hepatocytes were co-infected with Ad.Trib3-HA and Ad.Sqstm1-MYC for 24 h and then treated with Pep2-A2 or Pep2-Ctrl (5 mM) for 24 h, and extracts were immunoprecipitated with an anti-HA Ab and blotted with an anti-MYC Ab (left panel). The colocalization of TRIB3 and SQSTM1 was examined by immunostaining (right panel). Data are representative immunoblots and immunostaining images of 3 assays. (B) TRIB3-overexpressing hepatocytes were infected with an adenovirus expressing RFP-GFP-LC3. After 24 h, cells were treated with Pep2-A2 for 12 h, and autophagic flux was detected. Data are representative images showing red-colored autolysosomes or red/green double-colored autophagosomes of 3 assays. (C) Hepatocytes were infected with Ad.Trib3 and treated with Pep2-A2 or Pep2-Ctrl for 24 h, and the expression of autophagy-associated proteins CD63 and TSG101 was detected by immunoblotting. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays. (D) Quantitative analysis of hepatocyte-derived exosomes by ELISA. Data are means ± SEM of 3 assays. (E and F) Hepatocytes were infected with Ad.Trib3, treated with Pep2-A2 or Pep2-Ctrl for 24 h, and the exosomes were isolated and added to HSCs for 72 h. The expression of ACTA2, COL1A1 (E), MMPs and transdifferentiation-associated proteins (F) in HSCs was evaluated by immunostaining or immunoblotting, respectively. Representative immunoblots and the ratio of the indicated protein to ACTB are presented. Data are the means ± SEM of 3 assays.
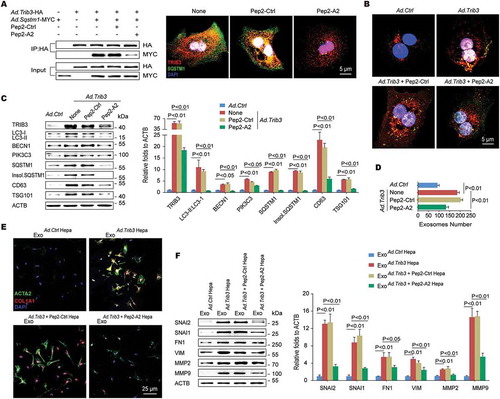
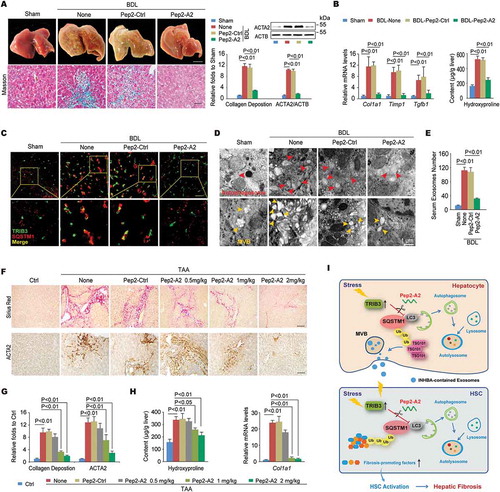
We next evaluated the role of Pep2-A2 in BDL- or TAA-induced mouse models of hepatic fibrosis. Treatment of mice with Pep2-A2, but not Pep2-Ctrl, remarkably reduced BDL-induced hepatic fibrosis, as indicated by improved liver morphology and collagen deposition ( left). A 75% reduction of collagen deposition and 83.3% downregulation of ACTA2 expression were observed in the liver tissues of Pep2-A2-treated mice compared with tissues from untreated mice ( right). In addition, hepatic pro-fibrotic genes expression and hydroxyproline content were also reduced in Pep2-A2-treated mice compared with untreated mice (). Consistent with in vitro results, Pep2-A2 treatment reduced the colocalization of TRIB3 with SQSTM1 () as well as the soluble- and insoluble-levels of TRIB3, SQSTM1 and LC3-II:LC3-I in fibrotic liver tissues (Fig. S7A). Enhanced co-IP of TRIB3, LC3-II/I or ubiquitinated proteins by SQSTM1 was observed in fibrotic liver tissues, which was disrupted by Pep2-A2 treatment (Fig. S7B). The autophagic flux restoring effects of Pep2-A2 were further confirmed in both hepatocytes and HSCs isolated from control and BDL-induced fibrotic livers (Fig. S7C, S7D). More importantly, the mice underwent therapeutic treatment of Pep2-A2 on day 7, 10 and 13 after BDL displayed reduced hepatic fibrosis, which was accompanied by remarkable decreased TRIB3 level, LC3-II:LC3-I ratio and SQSTM1 aggregation in liver tissues, when compared with the untreated mice (Fig. S7E). Moreover, 3-methyladenine (3-MA) and CQ, specific inhibitors of early- and late-stage autophagy, not only reversed the beneficial effects of Pep2-A2 on hepatic fibrosis (Fig. S7F) but also suppressed its autophagy-restoring effect, as indicated by the reversal of the Pep2-A2-induced decrease in the aggregation of insoluble SQSTM1 by 3-MA or CQ (Fig. S7G). Using transmission electron microscopy (TEM) analysis, a large amount of autophagosomes and MVBs were observed in fibrotic tissues from BDL-injured livers of untreated and Pep2-Ctrl-treated mice, which was significantly decreased by Pep2-A2 treatment (), accompanied by lower level of exosomes in serum (). A similar anti-fibrotic effect of Pep2-A2 was observed in TAA-treated mice, as indicated by improved liver morphology (Fig. S8A), reduced collagen deposition and ACTA2 expression (,G), and less hydroxyproline production and Col1a1 mRNA expression () in the livers. We further examined the expression of autophagy-related proteins in primary hepatocytes and HSCs isolated from control and TAA-induced fibrotic livers. Pep2-A2 not only restored the suppressed autophagic flux in both hepatocytes and HSCs but also promoted the degradation of late endosomes in hepatocytes and transdifferentiation-associated proteins in HSCs, both of which were ectopically accumulated in TAA-injured livers (Fig. S8B-D). These data confirm that disrupting the TRIB3-SQSTM1 interaction exerts a potent therapeutic effect against hepatic fibrosis by restoring obstructed autophagic flux ().
Figure 7. Disrupting the TRIB3-SQSTM1 interaction inhibits the progression of hepatic fibrosis. (A) The Pep2-A2 treatment attenuated hepatic fibrosis in a BDL model. Mice were treated with Pep2-A2 or Pep2-Ctrl (5 mg/kg) once per 3 days for 15 days after the BDL procedure (n = 6–12/group). Representative images showing the morphology of the livers (upper left panel, scale bar: 5 mm), Masson’s trichrome staining of liver sections (lower left panel, scale bar: 40 μm), and a summary of collagen deposition and ACTA2 expression (right panel, means ± SEM of 3 assays, n = 6/group). (B) The hepatic hydroxyproline content and expression of the Col1a1, Timp1 and Tgfb1 mRNA are shown. Data are means ± SEM of 3 assays (n = 6/group). (C) Colocalization of TRIB3 and SQSTM1 in frozen liver tissue sections was examined by immunostaining. (scale bar: 20 μm). Data are representative images of 3 assays. (D and E) The Pep2-A2 treatment decreased the number of autophagosomes (D, upper panel) and MVBs (D, lower panel) in hepatocytes as well as the serum-derived exosomes (E) from mice that underwent BDL. Data are representative images of 3 TEM assays and means ± SEM of 3 ELISA assays. (F-H) Pep2-A2 exerted a dose-dependent anti-fibrosis effect on a TAA-induced hepatic fibrosis model (n = 10–12/group). Data are representative photomicrographs of sirius red staining and ACTA2 immunohistochemical staining of liver sections (F, scale bar: 40 μm), as well as a summary of collagen deposition and ACTA2 expression (G) (means ± SEM of 3 assays, n = 10/group), and the quantification of hydroxyproline content and Col1a1 mRNA levels in liver tissues (H) (means ± SEM of 3 assays, n = 10/group). (I) Schematic illustrating the mechanism by which TRIB3 induces hepatocyte exosome secretion and activates HSCs during hepatic fibrosis.
Discussion
TRIB3 is a member of the pseudokinase family and has a scaffold-like function in protein complex assembly [Citation20]. Recent studies indicate that TRIB3 interacts with a variety of crucial signaling factors, such as AKT, NFKB, and MAPK, to regulate multiple biological functions [Citation21,Citation22]. Our current study not only establishes a positive correlation between elevated TRIB3 expression and the development of hepatic fibrosis but also demonstrates that enhanced TRIB3 obstruct the autophagic flux via hindering the cargo functions of SQSTM1 in both hepatocytes and HSCs, resulting in diverse effects on exosome biogenesis in hepatocytes and the activation of HSCs.
During the pathogenesis of liver fibrosis, the role of autophagy in HSCs has been widely studied, and accumulated evidence suggests that activated autophagy is required for the activation of HSC and consequent fibrogenic process in liver tissue. Hernandez-Gea et al. and Thoen et al. reported that increased autophagy provided energy for the HSC activation through digesting of lipid droplets in quiescent HSCs, which had been verified in vitro by inhibition/downregulation of autophagy with either pharmacological or genetic approaches [Citation23,Citation24]. However, there are conflicting studies about the impact of autophagy on the activation of HSCs, since different animal models or experimental conditions reveal quite dissimilar effects on liver fibrosis. For instance, two elegant studies have shown that either rapamycin or carbamazepine, two classic autophagy enhancers, ameliorates experimental hepatic fibrosis by inhibiting the proliferation and activation of HSCs [Citation6,Citation25], which is consistent with our current findings. We agree with Dr. Hernandez-Gea that the autophagy activity is essential for the activation of HSCs, as both ATG5 and ATG7 are widely recognized as early markers of autophagy activation through participating in the elongation of the phagophore. Indeed, in our system, both in vivo and in vitro data showed that the expression of autophagy upstream signals, such as LC3-II:LC3-I, BECN1 and PIK3C3, were markedly upregulated in activated HSCs, implying the initial activation of autophagy. Whereas, as a later-phase marker of autophagy flux, the ‘cargo’ protein SQSTM1, specially its insoluble form was extremely aggregated in the activated HSCs, indicating the autophagic flux is impaired. These contradictory observations remind us that targeting the autophagic flux for treating liver fibrosis is still open and need further investigation. Notably, autophagy plays a protective role against hepatic fibrosis mainly through its powerful clearing function of SQSTM1. There is always a doubt whether SQSTM1 could be used as an anti-fibrotic target, since different animal models or cell types reveal quite dissimilar effects of SQSTM1 on fibrosis [Citation26–Citation28]. As described in two recent papers, ectopic SQSTM1 expression in hepatocytes induces hepatocellular carcinogenesis (HCC) and fibrosis, whereas total body or HSC-specific SQSTM1 depletion activates HSCs and also aggravates the progression of liver fibrosis and HCC [Citation27,Citation28]. As we know, SQSTM1 is a selective ‘cargo’ protein involving in the trafficking of autophagy substrates to the degradation pathway. Using genetic method to completely deplete SQSTM1 from HSCs would certainly damage its physiological functions and interferes with the selective autophagy-mediated clearance of fibrosis-promoting factors such as SNAI2, SNAI1, MMP2 and MMP9, as observed in our study. In contrast, TRIB3 is a stress-responsive protein that is expressed at high levels in activated HSCs but expresses much lower in quiescent HSCs (Fig. S8C). Disrupting the TRIB3-SQSTM1 interaction by restoring the functions of SQSTM1 and autophagic flux could be a more attractive therapeutic strategy for hepatic fibrosis.
The link between autophagy and exosome secretion has been reported previously [Citation29,Citation30]. Biochemical and morphological studies in both hepatocytes and fibroblasts have indicated that autophagosomes can fuse with endosomes and generate hybrid vacuoles called amphisomes [Citation31] that subsequently directly fuse with lysosomes to form autolysosomes and degrade the endosomal contents. Once autophagy is impaired, specific endosomes called MVBs are no longer directed to the autophagy pathway but instead fuse with the plasma membrane and release their contents within exosomes to maintain cellular homeostasis [Citation32]. Indeed, TRIB3 overexpression obstructs autophagic flux by increasing SQSTM1 accumulation, accompanied by abundant MVB formation and exosome secretion from hepatocytes. Studies of multiple components of the endosomal tracking and vesicle fusion machineries, including ESCRT complexes, have revealed a close relationship between the process of autophagy and MVB biogenesis [Citation33,Citation34]. As shown in the present study, the ESCRT protein TSG101 interacts with SQSTM1 and controls MVB degradation and exosome secretion, which can be regulated by TRIB3 as well. These findings are consistent with previous reports that enhanced TSG101 expression significantly promotes MVB formation and alters the size and number of MVBs [Citation35,Citation36]. In addition, we found that TRIB3-induced suppression of autophagy results in the accumulation of fibrosis-promoting factors, including SNAI2, SNAI1, MMP2 and MMP9, which are critical for HSC transdifferentiation [Citation37].
Extracellular vesicles, including exosomes, play important roles in the diagnosis, treatment, and prognosis of liver diseases [Citation38]. For instance, some exosomes exert protective effects against the development of liver disorders by limiting the inappropriate activity of specific effector cells [Citation14,Citation16]. They are also critical in cell-to-cell communication during the progression of liver disease [Citation39]. Almost all types of liver-resident cells, including hepatocytes, HSCs, and Kupffer cells, are exosome-releasing and/or exosome-targeting cells [Citation15,Citation40]. However, because approximately 80% of the liver mass is composed of hepatocytes, it is reasonable to speculate that exosomes derived from TRIB3-overexpressing hepatocytes are the primary initiators of HSC activation, although the possibility that HSCs activate themselves in an autocrine manner cannot be excluded. Several types of exosomes exhibiting profibrotic characteristics have been reported in different animal models of hepatic fibrosis, but few profibrotic factors packaged in exosomes have been identified [Citation15,Citation41]. In the present study, INHBA-enriched exosomes derived from TRIB3-overexpressing hepatocytes directly activated HSCs, which may be the primary factor contributing to the profibrotic actions of TRIB3. Indeed, INHBA was recently shown to function as a positive modulator that is involved in many chronic liver diseases, including liver cirrhosis and fibrosis in response to liver tissue injury via activation of the SMAD signaling pathway [Citation42,Citation43]. Inhibition of INHBA expression or secretion has been proposed as a therapeutic strategy for liver fibrosis. In our in vitro system, little INHBA was detected in the exosome-free supernatant of TRIB3-overexpressing hepatocytes or HSCs, suggesting that most extracellular INHBA is present in exosomes. Although we did not address whether the INHBA packaged in exosomes represents a useful diagnostic marker for patients with hepatic fibrosis, investigating this possibility may provide valuable information and is worthy of further investigation.
As α-helical epitopes are commonly involved in protein-protein interactions (PPIs), hundreds of short α-helical segments have been developed as inhibitors of PPIs and display therapeutic promise against many diseases, including cancers [Citation18,Citation44]. Here, we found that disrupting the TRIB3-SQSTM1 interaction by Pep2-A2, an α-helical peptide derived from the UBA domain of SQSTM1, attenuated the development of hepatic fibrosis by restoring autophagy activity in both hepatocytes and HSCs. Although most PPIs occur inside the cell, only 15% of peptide drugs entering clinical studies are directed toward intracellular targets [Citation45], indicating that disrupting intracellular PPIs is a difficult challenge in the development of therapeutic peptides. We and others have observed that TLR2 (toll-like receptor 2) is significantly upregulated in response to multiple stresses in a variety of liver cells, including hepatocytes, HSCs and hepatocellular carcinoma cells [Citation46,Citation47], implying that TLR2 can be used as a biomarker and potential target for the treatment of liver diseases. Pep2-A2 was designed by linking a cell-penetrating peptide motif that recognizes TLR2 [Citation48]. Our present study verifies the anti-fibrosis activity of Pep2-A2 in vitro and in vivo, and the Pep2-A2 treatment restored SQSTM1 functions and increased the autophagic degradation of MVBs in hepatocytes and of pro-differentiation factors in HSCs. Therefore, TRIB3-mediated impairments in autophagy play a critical role in the pathogenesis of hepatic fibrosis, and our study provides a potential therapeutic strategy for hepatic fibrosis.
Materials and methods
Materials
All primary antibodies against the indicated proteins used for immunoblotting, immunoprecipitation and immunostaining analysis in the present study are listed as follows: rabbit anti-human/mouse TRIB3 (PA5-75683) from Invitrogen; rabbit anti-human/mouse LC3-II/I (L7543), anti-human/mouse SQSTM1 (P0067) from Sigma; rabbit anti-human/mouse PIK3C3 (4263), anti-mouse HGS/HRS (15087), anti-mouse SNAI2/SLUG (9585), anti-mouse SNAI1/SNAIL (3879), anti-mouse VIM (5741), anti-mouse MMP2 (87809), anti-human/mouse ACTB (4970) and mouse anti-mouse Ubiquitin (3936) from Cell Signaling Technology; rabbit anti-human/mouse BECN1 (ab207612), anti-mouse LAMP1 (ab24170), anti-mouse TSG101 (ab125011), anti-mouse SNF8/VPS22 (ab22768), anti-mouse CHMP3/VPS24 (ab175930), anti-mouse HSPA8/HSC70 (ab51052), anti-mouse MMP9 (ab38898) and mouse anti-human/mouse SQSTM1 (ab56416), anti-mouse CD63 (ab213090), anti-mouse FN1 (ab6328) from Abcam; mouse anti-mouse LBPA (Z-PLBPA) from Echelon; goat anti-human ALB (127–10783) from Ray Biotech; mouse anti-human/mouse ACTA2 (BM0002) from BOSTER; rabbit anti-MYC (562), anti-HA (561), anti-GFP (598), mouse anti-HA (M180-3), anti-GFP (M048-3) from MBL. Alexa Fluor 350 (A21081)-, Alexa Fluor 488 (A21202)- and Alexa Fluor 647 (A31573)-conjugated secondary Abs were obtained from Invitrogen. Click-iT™ Edu Alexa Fluor™ 647 Imaging Kit (C10340) was purchased from Invitrogen; Cell Counting Kit-8 (CK04) was obtained from Dojindo.
Human cirrhosis tissue and tissue microarray
Paraffin-embedded human liver tissue microarray slides (BC03117) and surgically removed fibrotic liver tissues were obtained from US Biomax with pathological information. The tissue microarrays were examined by immunohistochemical analysis, and the liver tissues were used in confocal and western blotting analyses. All protocols using human specimens were approved by the Institutional Review Board of the Chinese Academy of Medical Sciences and Peking Union Medical College. Informed consent was obtained from all subjects. The study conformed to the principles outlined in the Declaration of Helsinki.
Generation of liver specific Trib3-knockdown mice
The conditional Trib3-knockdown (Trib3fl/fl) mice were generated by Cyagen Biosciences Inc (Guangzhou, China) in the C57BL/6 background as described previously [Citation49]. After the genotyping identification, adult Trib3fl/fl mice were i.v. injected with AAV (adeno-associated virus) vectors (3x1011 per mouse) expressing GFP-tagged Cre recombinase under control of the Alb (albumin) promoter for 4 weeks. Trib3fl/fl mice injected with an Alb-promoted AAV vector expressing GFP were used as wild type (WT). The efficiency of Cre-induced recombination was detected by western blotting.
Animal model of hepatic fibrosis
Six- to 8-week-old male BALB/c mice and C57BL/6 mice (used as control for the Trib3-knockdown mice) were obtained from Vital River Laboratory Animal Technology (Beijing, China) and maintained in a temperature- and light-controlled facility. Hepatic fibrosis was induced by BDL or treatment with TAA (Sigma, 163678) according to previously published procedures [Citation17]. Briefly, mice were anesthetized with 45 mg/kg of pentobarbital and the abdominal cavity was opened from the abdominal midline. The common bile duct was ligated twice with 1–0 silk suture and the bile duct was cut between the two ligations. To generate a TAA-induced model of hepatic fibrosis, mice were i.p. injected with TAA in PBS (200 mg/kg) at twice/week for 12 weeks. The mice were intraperitoneally (i.p.) injected with Pep2-Ctrl (hlyvspwgg-yllgkedaare) or Pep2-A2 (hlyvspwgg-ggwltrllqtk) on days 0, 3, 6, 9, 12 and 15 after the BDL procedure or twice a week for 7 weeks from the 6th week after the first TAA injection. For therapeutic treatment, the mice were i.p. injected with Pep2-A2 on days 7, 10 and 13 after the BDL procedure. Saline- or peptide-treated hepatic fibrosis mice were i.p. injected with 3-MA (Sigma, M9281) or CQ (Sigma, C6628) at 30 mg/kg/day from day 1 to day 15 after BDL. At the end of the experiments, the mice were sacrificed by using an overdose of pentobarbital (i.p., 100 mg/kg) for the collection of serum and livers. The livers were excised and fixed or frozen for morphological evaluation or western blotting analysis. All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals prepared and approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College.
Isolation of primary hepatic cells
Primary hepatic cells including hepatocytes and HSCs were isolated from normal or fibrotic livers as described previously [Citation50]. Mouse livers were perfused in situ with EGTA solution, followed by pronase (Sigma, P5147) solution and collagenase D (Roche, 11088882001) solution at 37°C for primary hepatic cell isolation. After perfusion, the liver was transferred into a sterile Petri dish containing protease and collagenase D solution. After being gently minced, the liver and the cell suspension was filtered through a 70 μm cell strainer and centrifuged at 300 g for 3 min at 4°C to isolate hepatocytes. The supernatant was transferred to a new tube and centrifuged at 1000 g for 10 min at 4°C. Aspirate the supernatant until 15 ml remains in the tube, then add 50 μL DNase I (Roche, 10104159001) to reduce the viscosity of the cell suspension, and washed with 50 ml GBSS/B once. The cell pellet was resuspended in 12% Nycodenz (AXIS-SHIELD, 1002424) solution, and centrifuged at 1700 g (no brake) for 15 min at 4°C to isolate HSCs. Primary hepatocytes were cultured in DMEM medium (Gibco, 10564029), supplemented with 10% fetal bovine serum (FBS), insulin (Sigma, I9278), dexamethasone (Sigma, D4902) and penicillin-streptomycin (Gibco, 15070063) on collagen-coated dishes (Corning, 354236). HSCs were cultured in RPMI-1640 medium (Gibco, 72400120), supplemented with 10% FBS and penicillin-streptomycin.
Histomorphology
The liver tissues were rapidly harvested and fixed with 4% paraformaldehyde, and embedded in paraffin for histopathological examination. Tissue sections (5 μm thick) were stained with hematoxylin and eosin (Solarbio, G1120), sirius red (Sigma, 365548), or Masson’s trichrome blue (Solarbio, G1345) according to standard procedures. The grade of hepatic inflammation and fibrosis was blindly assessed by a professional pathologist. The average IOD of collagen deposition from 10 randomly chosen regions per sample at a magnification of ×200 was measured using Image-Pro Plus 5.1 image analysis software.
Confocal assay
Hepatocytes or HSCs were seeded on the coverslip-bottom dishes and treated with or without indicated agents. The cells on coverslips or liver sections (5 μm thick) were fixed with 4% paraformaldehyde for 10 min, washed for 3 times and treated with 0.5% Triton X-100 (Sigma, T8787) to penetrate the cell membranes. The samples were stained with indicated primary Abs overnight at 4°C, and incubated with Alexa Fluor 350-, Alexa Fluor 488- or Alexa Fluor 647-conjugated secondary Abs (1:200) for 30 min. Nuclei were visualized by 4ʹ,6-diamidino-2-phenylindole (DAPI) staining. Images were obtained with a FV3000 confocal microscope (Olympus, Japan) and analyzed using Olympus confocal software FV10-ASW.
Transmission electron microscopy
Fresh mouse liver tissues or hepatocytes were fixed with 2% glutaraldehyde and 1% osmium tetroxide, rinsed in 100 mM sodium phosphate buffer (pH 7.2), dehydrated in ethanol and embedded in epon (Sigma, 45347). Ultrathin sections of liver tissues or hepatocytes were collected on formvar-coated grids and stained with 10% uranyl acetate and 1% lead citrate, and then examined with a H600 Transmission Electron Microscope (Hitachi, Japan) operated at 80 KV.
Isolation and tracing of hepatic exosomes
For exosome isolation, the cultured hepatocytes were treated with or without indicated agents. The exosomes were isolated from the supernatants or serum using ExoQuick-TC exosome precipitation solution (System Biosciences, EXOTC50A-1). Size determination of isolated exosomes was measured using Zetasizer Nano ZS particle and molecular size analyzer (Malvern Instruments, U.K.) according to the manufacturer’s instructions. Isolated hepatic exosomes (5X108 particles) which were pre-stained with 1 μM Dil staining solution (Invitrogen, D3911) for 2 h to trace exosomes. Dil stained exosomes were then co-cultured with HSCs for 24 h and exosome engulfed HSCs were observed under a confocal microscope. The cytokines in exosomes secreted from hepatocytes were assayed with an antibody array containing 200 cytokines (Ray Biotech, GSM-CAA-4000) according to the manufacturer’s instructions.
Quantification of hydroxyproline content
Liver tissue hydroxyproline content was measured using a kit (K226) from Biovision. In brief, the livers were hydrolyzed with 2.5 N NaOH at 120°C, 0.1 kPa for 40 min. After neutralization with hydrochloric acid, the hydrolyzation products were diluted with distilled water. The hydroxyproline content of the hydrolyzation products was assessed colorimetrically at 560 nm. The results were represented as micrograms per gram liver.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from the mouse liver tissues using TRIzol reagent (Invitrogen, 15596026) following the manufacturer’s instructions. The concentration of RNA was determined by a NanoDrop ND-1000 Spectrophotometer (NanoDrop Tech, U.S.). Next, the RNA was reverse-transcribed and amplified. FastStart Universal SYBR Green Master (Roche, 4913850001) was used for the real-time PCR analysis of the mRNAs of Col1a1, Timp1, Tgfb1, and the internal control Actb using a GeneAmp PCR 9700 Thermocycler (Applied Biosystems, U.S.). The following specific primer sequences were used: 5ʹ-GGGGCAAGACAGTCATCGAA-3ʹ and 5ʹ-GGG TGGAGGGAGTTTACACG-3ʹ (mouse Col1a1); and 5ʹ- CAGATACCATGATGG CCCCC-3ʹ and 5ʹ-TATGACCAGGTCCGAGTTGC-3ʹ (mouse Timp1); and 5ʹ-TGGCCAGATCCTGTCCAAAC-3ʹ and 5ʹ-GTTGTACAAAGCGAGCACCG-3ʹ (mouse Tgfb1); and 5ʹ-TAACCAACTGGGACGATATG-3ʹ and 5ʹ-AAACAGGGA CAGCACAGCCT-3ʹ (mouse Actb). Relative expression was calculated using the comparative threshold cycle method and determined by the value of 2–ΔΔCt.
Elisa
The concentrations of CYP2E1 or exosomes were detected by ELISA (enzyme-linked immunosorbent assay) using kits from LifeSpan BioSciences (LS-F7860-1) or System Biosciences (EXOCET96A-1) according to the manufacturers’ instructions.
Transwell
HSC migration assays were performed using Transwell chambers with 8-μm pore size filter membranes which were precoated with 10 mg/ml FN1 (fibronectin 1). HSC suspensions were seeded into the upper chamber (5 x104 cells/well in 0.4% FBS in DMEM). After 48 h, non-invaded cells on the upper side of the filter were wiped with a cotton swab. Cells invaded were fixed with 4% paraformaldehyde in PBS, stained with 0.5% crystal violet (Solarbio, C8470) and counted using brightfield microscopy in 8 random fields (×200).
Immunoblotting and immunoprecipitation
ReadyPrep Protein Extraction Kit (Bio-Rad, 163–2100) was used to extract protein samples from liver tissue or cultured primary cells according to the manufacturer’s instructions. Protein concentrations in lysates were determined using a BCA reagent. For insoluble fraction preparation, pellets were washed 4 times with RIPA and resuspended in 2% SDS in Tris-buffered saline (40 mM Tris, 8 M urea, 4% CHAPS [Sigma, C3023]). The sampling volume for the insoluble fraction was adjusted according to the protein concentration in lysates. Equal amounts of proteins were subject to SDS-PAGE. The binding of the primary antibodies was detected by peroxidase-conjugated secondary antibodies (Cell Signaling Technology, 4074 or 7076) and enhanced chemiluminescence, and bands were quantified with the Amersham ECL System (GE Healthcare, U.S.). To perform co-IP assays, cell lysate was IP with the indicated antibodies overnight at 4°C with gentle agitation, followed by incubation with protein A/G Plus-Agarose (Santa Cruz Biotechnology, SC2003) for 2 h at 4°C. The immunocomplex was washed 3 times and then mixed with 2× SDS sample buffer and boiled for 5 min. The precipitates were resolved by SDS-PAGE and detected with immunoblotting.
Statistical analysis
All results are represented as the means ± SEM. Two-group comparisons were analyzed by Student’s t-test as appropriate, while multiple comparisons among three or more groups were performed using one-way ANOVA; p < 0.05 or p < 0.01, were considered statistically significant. All statistical results were analyzed using SPSS 17.0 software.
Supplemental Material
Download MS Word (7.3 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet. 2014;383:1749–1761.
- Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436.
- Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901.
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232.
- Mi S, Li Z, Yang HZ, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. 2011;187:3003–3014.
- Liu H, Mi S, Li Z, et al. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B-mediated degradation of BCL2 in lung epithelial cells. Autophagy. 2013;9:730–742.
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472.
- Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116.
- Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289.
- Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–125.
- Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403.
- Chen L, Charrier A, Zhou Y, et al. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology. 2014;59:1118–1129.
- Seo W, Eun HS, Kim SY, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology. 2016;64:616–631.
- Li T, Yan Y, Wang B, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013;22:845–854.
- Zhang XW, Mi S, Li Z, et al. Antagonism of Interleukin-17A ameliorates experimental hepatic fibrosis by restoring the IL-10/STAT3-suppressed autophagy in hepatocytes. Oncotarget. 2017;8:9922–9934.
- Hua F, Li K, Yu JJ, et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat Commun. 2015;6:7951.
- Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–869.
- Boudeau J, Miranda-Saavedra D, Barton GJ, et al. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452.
- Du K, Herzig S, Kulkarni RN, et al. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577.
- Kiss-Toth E, Bagstaff SM, Sung HY, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279:42703–42708.
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946.
- Thoen LF, Guimarães EL, Dollé L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360.
- Zhu J, Wu J, Frizell E, et al. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–1204.
- Ni HM, Woolbright BL, Williams J, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625.
- Duran A, Hernandez ED, Reina-Campos M, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016;30:595–609.
- Umemura A, He F, Taniguchi K, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29:935–948.
- Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol. 2015;17:300–310.
- Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163.
- Berg TO, Fengsrud M, Stromhaug PE, et al. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892.
- Fader CM, Sanchez D, Furlan M, et al. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250.
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452.
- Rusten TE, Vaccari T, Lindmo K, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–1825.
- Villarroya-Beltri C, Baixauli F, Mittelbrunn M, et al. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat Commun. 2016;7:13588.
- Razi M, Futter CE. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell. 2006;17:3469–3483.
- Gorrell MD. Liver fibrosis: the hepatocyte revisited. Hepatology. 2007;46:1659–1661.
- Masyuk AI, Masyuk TV, Larusso NF. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol. 2013;59:621–625.
- Hirsova P, Ibrahim SH, Verma VK, et al. Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology. 2016;64:2219–2233.
- Conde-Vancells J, Rodriguez-Suarez E, Embade N, et al. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res. 2008;7:5157–5166.
- Witek RP, Yang L, Liu R, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–330 e322.
- Kreidl E, Ozturk D, Metzner T, et al. Activins and follistatins: emerging roles in liver physiology and cancer. World J Hepatol. 2009;1:17–27.
- Ding ZY, Jin GN, Wang W, et al. Activin A-smad signaling mediates connective tissue growth factor synthesis in liver progenitor cells. Int J Mol Sci. 2016;17:408.
- Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci. 2013;34:393–400.
- Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur J Med Chem. 2015;94:459–470.
- Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–1900.
- Lin H, Yan J, Wang Z, et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology. 2013;57:171–182.
- Li K, Lv XX, Hua F, et al. Targeting acute myeloid leukemia with a proapoptotic peptide conjugated to a Toll-like receptor 2-mediated cell-penetrating peptide. Int J Cancer. 2014;134:692–702.
- Li K, Wang F, Cao WB, et al. TRIB3 Promotes APL Progression through Stabilization of the Oncoprotein PML-RARalpha and Inhibition of p53-Mediated Senescence. Cancer Cell. 2017;31:697–710 e697.
- Mederacke I, Dapito DH, Affo S, et al. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305–315.