
ABSTRACT
Autophagosome and lysosome fusion is an important macroautophagy/autophagy process for cargo degradation, and SNARE proteins, including STX17, SNAP29, VAMP7 and VAMP8, are key players in this process. However, the manner in which this process is precisely regulated is poorly understood. Here, we show that VAMP7B, a SNARE domain-disrupted isoform of R-SNARE protein VAMP7, competes with SNARE domain functional isoform VAMP7A to bind to STX17 and inhibits autophagosome-lysosome fusion. Moreover, we show that DIPK2A, a late endosome- and lysosome-localized protein, binds to VAMP7B, which inhibits the interaction of VAMP7B with STX17 and enhances the binding of STX17 to VAMP7A, thus enhancing autophagosome-lysosome fusion. Furthermore, DIPK2A participates in autophagic degradation of mitochondria proteins and alleviates apoptosis. Thus, we reveal a new aspect of autophagosome-lysosome fusion in which different isoforms of VAMP7 compete with STX17 and their regulation by DIPK2A.
Abbreviations: DIPK2A: divergent protein kinase domain 2A; EEA1: early endosome antigen 1; GOLGA2: golgin A2; LAMP1: lysosomal associated membrane protein 1; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3 beta; MFN2: mitofusin 2; MT-CO2: mitochondrially encoded cytochrome c oxidase II; PARP1: poly(ADP-ribose) polymerase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; RAB5A: RAB5A, member RAS oncogene family; RAB7A: RAB7A, member RAS oncogene family; REEP: receptor accessory protein; RTN4: reticulon 4; SNARE: SNAP receptor; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; TOMM20: translocase of outer mitochondrial membrane 20; VAMP7: vesicle associated membrane protein 7; VAMP8: vesicle associated membrane protein 8.
Introduction
Autophagy, a constitutive process by which non-specific substrates are degraded and recycled by macroautophagy or, in specific organelles, by mitophagy, pexophagy and reticulophagy, is crucial for maintaining energy homeostasis and for cellular survival during environmental stresses [Citation1]. During autophagy, the targeted cytoplasmic contents are isolated inside of a double-membraned vesicle, the autophagosome, which is delivered to the lysosome to form the autolysosome, after which the targeted contents are degraded by the lumenal contents of the lysosome. The process of autophagosome-lysosome fusion is regulated by SNARE protein complexes, including the STX17 (syntaxin 17), SNAP29 (synaptosome associated protein 29), VAMP7 (vesicle associated membrane protein 7) and VAMP8 complexes [Citation2,Citation3], as well as the recently reported YKT6 (YKT6 v-SNARE homolog)-containing complexes [Citation4]. However, the manner in which SNARE protein-mediated fusion is regulated remains poorly understood.
VAMP7 is a longin family R-SNARE protein involved in various cellular processes such as autophagosome biosynthesis, cell migration, membrane repair and neurite outgrowth [Citation5]. Like other longin family proteins, such as YKT6 and SEC22, VAMP7 consists of a conserved N terminal regulatory longin domain, followed by a SNARE domain and a C terminal transmembrane domain [Citation6]. Importantly, VAMP7 is required for autophagosome-lysosome fusion as its deletion results in accumulation of autophagosomes and blockage of autolysosomal degradation [Citation2]. In Drosophila, Vamp7 competes with another R-SNARE protein, Ykt6, to bind to the Syx17-Snap29 complex and is directly involved in membrane fusion [Citation7]. Human VAMP7 has several isoforms produced by alternative splicing, among which VAMP7A, that is localized on the lysosome and late endosome, and regulates autophagosome-lysosome fusion, is the main type [Citation2,Citation8]. VAMP7C, VAMP7D and VAMP7H are missing either all or parts of exons 1–4 and lack an intact longin domain [Citation8]. They function similarly to VAMP7A in SNARE complexes, but with different localization [Citation9]. In contrast, VAMP7B, VAMP7I and VAMP7J contain a longin domain, but they lack a SNARE domain [Citation8]. Specifically, VAMP7B, which originates from exon 6 skipping and possesses a frameshifted C terminal sequence, has a sequence totally different from that of VAMP7A and was reported to have a widespread distribution in the cytosol [Citation8]. However, the function of VAMP7B has not been characterized. The manner in which various VAMP7 isoforms cooperate to accomplish the membrane fusion function of VAMP7 remains poorly understood.
STX17 is a well-studied SNARE protein that mediates autophagosome-lysosome fusion. Upon starvation stimulation, STX17 translocates to autophagosomes, where it forms a complex with SNAP29, VAMP8 [Citation3] or VAMP7 [Citation2,Citation10]. STX17 knockdown was shown to prevent mitophagy in an effect that was thought to be a result of defective autophagosome-lysosome fusion [Citation11]. STX17 was not considered to be required for PINK1 (PTEN induced kinase 1)- and PRKN (parkin RBR E3 ubiquitin protein ligase)-mediated mitophagy [Citation10,Citation12], until recent study revealed that the STX17-PGAM5 (PGAM family member 5, mitochondrial serine/threonine protein phosphatase) axis is involved in this process [Citation13]. In addition, STX17 is enriched in stress-induced mitochondrial-derived vesicles (MDVs) and regulates MDV degradation by interacting with VAMP7, thus mediating MDV-endolysosome fusion [Citation10]. Therefore, the role of STX17 in mitophagy has not been comprehensively explored.
DIPK2A/DIA1 (divergent protein kinase domain 2A) has been identified as being lost in cases of familial autism [Citation14]. However, the role of DIPK2A in cognitive disorders is not well understood [Citation15]. It has been reported that DIPK2A is localized to the Golgi apparatus [Citation16] and the ER membrane [Citation17], suggesting that it is involved in the secretion pathway. DIPK2A has been reported to possess kinase activity similar to that of FAM69 family proteins, and it has been predicted to function as an ER kinase [Citation17,Citation18]. DIPK2A also promotes cardiac repair and cell proliferation by regulating the phosphoinositide 3-kinase-AKT/protein kinase B-CDK7 (cyclin dependent kinase 7) pathway [Citation19] and interacting with the IGF1R (insulin like growth factor 1 receptor) [Citation20,Citation21]. In addition, DIPK2A protects cardiomyocytes from ischemia-induced cell death by activating PRKCE (protein kinase C epsilon) [Citation22]. These findings suggest that DIPK2A performs functions related to cellular protection. However, the mechanisms through which DIPK2A is involved in regulating cellular proliferation and cellular protection remain unknown. In this study, we show that DIPK2A is localized on late endosomes and lysosomes, where it promotes autophagosome-lysosome fusion by inhibiting the interaction between STX17 and VAMP7B and enhancing the interaction between STX17 and VAMP7A, thus promoting autophagic degradation of mitochondria, resulting in inhibition of apoptosis and promotion of cell survival.
Results
DIPK2A is localized on lysosomes and late endosomes
We first produced an anti-DIPK2A polyclonal antibody by immunizing rabbits with a peptide from the C terminus of DIPK2A (Figure S1A). This antibody was used to detect a band with an expected molecular weight of ~49 kDa in lysates from HeLa cells (Figure S1A). Consistent with a previous study [Citation17], endogenous DIPK2A was detected in the endomembrane fraction (Figure S1B). Subcellular fractionation showed that DIPK2A was detected in the fractions positive for lysosome marker LAMP1 and Golgi apparatus marker GOLGA2/GM130 (golgin A2), but not in fractions positive for early endosome marker EEA1 ().
Figure 1. DIPK2A is localized to the lysosomes and late endosomes. (A) Sucrose gradient separation analysis of DIPK2A in HeLa cells by western blotting (WB). (B) HeLa cells were co-transfected with DIPK2A-mCherry (red) and GFP-RAB7A (late endosome marker, green), GFP-LAMP1 (lysosome marker, green) or GFP-RAB5A (early endosome marker, green). The localization of the DIPK2A (red) with endosomes (green) or lysosomes (green) was analyzed by fluorescence microscopy. The boxed areas are magnified. Colocalized puncta are marked by arrowheads. Scale bar: 10 μm. (C) HeLa cells were transfected with DIPK2A-mCherry (red) and immunostained for LAMP1 (green). The localization of DIPK2A (red) and lysosomes (green) was analyzed by super-resolution fluorescence microscopy, structure illumination microscopy (SIM). The boxed areas are magnified. Scale bar: 2 μm. (D) DIPK2A-mCherry knock-in HEK293T cells were transfected with GFP-LAMP1, and the localization of DIPK2A (red) and LAMP1 (green) was analyzed by fluorescence microscopy. The boxed areas are magnified. Colocalized puncta are marked by arrowheads. Scale bar: 10 μm. See also Figure S1.
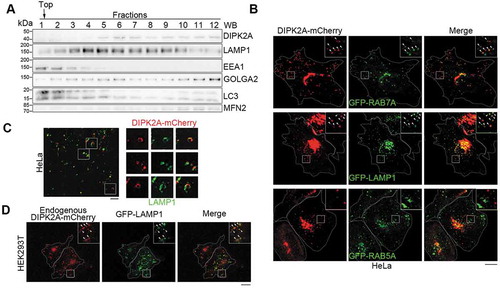
To explore the localization of DIPK2A, we overexpressed DIPK2A-mCherry in HeLa cells. DIPK2A was partially colocalized with the Golgi apparatus (marked by GOLGA2) and the ER (marked by ER-GFP) [Citation23] (Figure S1C), in agreement with previous studies [Citation15,Citation17,Citation24]. Meanwhile, consistent with the results of the subcellular fractionation (), some DIPK2A puncta were also colocalized with late endosomes and lysosomes, as indicated by GFP-RAB7A (RAB7A, member RAS oncogene family) and GFP-LAMP1 (lysosomal associated membrane protein 1), respectively, but not with GFP-RAB5A (RAB5A, member RAS oncogene family)-labeled early endosomes (). Super-resolution microscopic technology, structure illumination microscopy (SIM), revealed that DIPK2A-mCherry puncta were hollow and localized to vesicular structures positive for LAMP1 (). Furthermore, we added mCherry to the C terminus of the endogenous DIPK2A using the CRISPR-Cas9 approach [Citation25] (Figure S1D and S1E). Endogenous DIPK2A-mCherry was also partially colocalized with GFP-LAMP1 (). Taken together, these findings show that DIPK2A is localized on lysosomes and late endosomes.
DIPK2A regulates autophagy, mitophagy and autophagosome-lysosome fusion
To explore the function of DIPK2A, we constructed DIPK2A knockout HeLa (Figure S2A) and U2OS (Figure S2B) cells using the CRISPR-Cas9 approach and found that the protein levels of autophagy marker SQSTM1/p62 (sequestosome 1) and MAP1LC3B/LC3-II (microtubule associated protein 1 light chain 3 beta-II) in DIPK2A knockout cells were higher than those in wild-type cells upon treatment with Earle’s balanced salt solution (EBSS), and autophagy inhibitor chloroquine diphosphate salt (CQ) blocked this effect (–). These results suggest that DIPK2A promotes autophagy.
Figure 2. DIPK2A promotes autophagy and mitophagy. (A-C) Wild-type (WT) or DIPK2A knockout (KO) HeLa cells were cultured in normal medium, EBSS, or EBSS plus CQ for 2 h, harvested, and subjected to western blotting (WB) analysis. ACTB was used as a loading control (A). The relative band intensity of LC3-II (B) and SQSTM1 (C) was compared with the loading control based on 3 independent experiments. (D and E) Wild-type (WT) or DIPK2A knockout (KO) HeLa cells transfected with GFP-PRKN were treated with DMSO or 10 μM CCCP for 24 h, harvested, and subjected to western blotting (WB) analysis. ACTB was used as a loading control (D). The relative protein levels were compared with the loading control based on 3 independent experiments (E). (F-H) HeLa cells co-transfected with mRFP-GFP-LC3 and DIPK2A-BFP (blue) or the control vector were cultured under normal or EBSS (2 h) conditions and imaged by fluorescence microscopy (F). Scale bar: 10 μm. The percentage of GFP+ mRFP+ puncta (G), and the numbers of GFP+ mRFP+ and GFP– mRFP+ puncta (H) were determined (n ≥ 75 cells). (I-K) Wild-type (WT) or DIPK2A knockout (KO) HeLa cells were transfected with mRFP-GFP-LC3, cultured under normal or EBSS (2 h) conditions, and imaged by fluorescence microscopy (I). Scale bar: 10 μm. The percentage of GFP+ mRFP+ puncta (J), and the numbers of GFP+ mRFP+ and GFP– mRFP+ puncta (K) were determined (n ≥ 40 cells). For (G, H, J and K), the results are expressed as mean ± S.D. Statistical significance (p-value) was determined by one-way ANOVA (listed at the top). See also Figure S2.
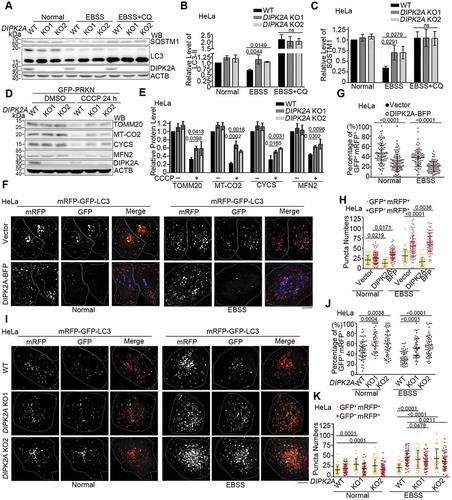
Interestingly, we observed that some DIPK2A puncta were localized on or in the vicinity of mitochondria (Figure S2C), and DIPK2A interacted with MFN2 (mitofusin 2) when they were both overexpressed in HEK293T cells (Figure S2D and E). Indeed, in DIPK2A knockout cells, the expression levels of mitochondrial proteins such as TOMM20 (translocase of outer mitochondrial membrane 20), MT-CO2 (mitochondrially encoded cytochrome c oxidase II), CYCS (cytochrome c, somatic) and MFN2 were higher than those in wild-type cells upon mitophagic stimulation by carbonyl cyanide 3-chlorophenylhydrazone (CCCP) under PRKN-expressing condition ( and ). These results suggest that DIPK2A regulates mitophagy.
Next, we tested whether DIPK2A localization is altered in response to starvation. We divided the cells into 3 groups according to the localization of DIPK2A: (1) ER, apparent ER localization as revealed by high colocalization with RTN4 (reticulon 4); (2) Endo+Lyso, apparent endosome and lysosome localization as revealed by high colocalization with LAMP1; (3) ER+Endo+Lyso, both ER and Endo+Lyso localization (Figure S2F). Next, changes in the ratio of cells were measured following EBSS treatment. The proportion of cells with endosome- and lysosome-localized DIPK2A significantly increased after starvation treatment (Figure S2F and G). Next, we examined whether DIPK2A functions in the autophagosome-lysosome fusion process using tandem fluorescent-tagged LC3 (mRFP-GFP-LC3). As the GFP signal is quenched under acidic conditions, autolysosomes are labeled only by mRFP (GFP– mRFP+ puncta), while yellow puncta (GFP+ mRFP+ puncta) indicate autophagosomes. The percentage of GFP+ mRFP+ puncta reflects the autophagosome-lysosome fusion rate [Citation26,Citation27]. Overexpression of DIPK2A-BFP significantly decreased the percentage of GFP+ mRFP+ puncta in HeLa cells ( and ), as the number of autophagosomes (GFP+ mRFP+ puncta) decreased upon DIPK2A overexpression, while the number of autolysosomes (GFP– mRFP+ puncta) increased (). In contrast, DIPK2A knockout significantly increased the percentage of GFP+ mRFP+ puncta and the number of autophagosomes (–). The difference in the number of autolysosomes was not obvious under normal condition, while DIPK2A knockout cells contained fewer autolysosomes under EBSS treatment (). These results suggest that DIPK2A promotes autophagosome-lysosome fusion.
VAMP7B interacts with DIPK2A and with STX17
To achieve autophagosome-lysosome fusion, autophagosome-localized Q-SNARE STX17 must interact with VAMP7 or VAMP8, which are lysosome-localized R-SNARE proteins [Citation3,Citation10,Citation28]. We next examined whether DIPK2A, which is localized on the lysosome and late endosome (–), interacts with VAMP7 and VAMP8 by immunoprecipitation. DIPK2A did not interact with VAMP8 when they were overexpressed in HEK293T cells (Figure S3A and B). Interestingly, DIPK2A interacted specifically with VAMP7B, but not VAMP7A ( and ). Consistently, detailed mapping showed that the C terminus (145–260 aa), but not the N terminus (1–144 aa; identical to VAMP7A), of VAMP7B interacted with DIPK2A ( and S3C).
Figure 3. VAMP7B interacts with DIPK2A and STX17. (A and B) HEK293T cells transfected with the indicated vectors were immunoprecipitated (IP) by the indicated antibodies and subjected to western blotting (WB) analysis. (C) HEK293T cells transfected with DIPK2A-HA and the indicated VAMP7B mutants were immunoprecipitated (IP) by anti-FLAG antibody and subjected to western blotting (WB) analysis. FL, full length; numbers indicate amino acids. (D) HEK293T cells co-transfected with MYC-STX17 and the indicated vectors were immunoprecipitated (IP) by anti-FLAG antibody and subjected to western blotting (WB) analysis. (E and F) HEK293T cells co-transfected with V5-STX17 and FLAG-tagged VAMP7 truncates (E) or VAMP7A deletion mutants (F) were immunoprecipitated (IP) by anti-FLAG antibody and subjected to western blotting (WB) analysis. FL, full length; numbers indicate amino acids. (G and H) HEK293T cells co-transfected with MYC-tagged STX17 mutants and 3× FLAG-VAMP7A (G) or 3× FLAG-VAMP7B (H) were immunoprecipitated (IP) with anti-MYC antibody and subjected to western blotting (WB) analysis. FL, full length; numbers indicate amino acids. (I) Diagram of the interaction among DIPK2A, STX17, VAMP7A and VAMP7B. See also Figure S3.
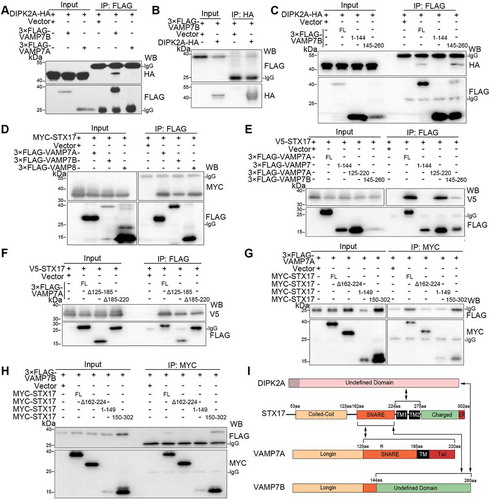
VAMP7B also interacted with STX17, as were observed for VAMP7A and VAMP8 (). The C termini of VAMP7A (125–220 aa) and VAMP7B (145–260 aa) interacted with STX17, but their common N terminus (1–144 aa) did not (). Deletion of either the SNARE domain (Δ125–185 aa) or the 185–220 aa region (Δ185–220 aa) of VAMP7A weakened, but did not eliminate, its interaction with STX17 (). Therefore, in addition to the SNARE domain, the 185–220 aa region of VAMP7A is also required for it to bind to STX17. VAMP7A was reported to adopt a closed conformation depending on the interaction between its longin domain (7–110 aa) and the SNARE domain (125–185 aa) [Citation29]. However, the N terminus of VAMP7A (VAMP7 1–144 aa) and the full-length form of VAMP7B did not interact with VAMP7A (Figure S3D). In contrast, VAMP7A was found to interact with itself through its 185–220 aa region (Figure S3C-E).
Next, we mapped the interaction region of STX17 to VAMP7A and VAMP7B (Figure S3F). Full-length STX17 and its C terminus (150–302 aa) bound to both VAMP7A () and VAMP7B (), while the N terminus (1–149 aa) of STX17 and deletion mutants without the SNARE domain (Δ162–224 aa) did not (, and S3F). Thus, the SNARE domain of STX17 is required for interaction with both VAMP7A and VAMP7B. Normally, protein interactions via SNARE domains could be resolvable by NSF (N-ethylmaleimide sensitive factor, vesicle fusing ATPase) and strengthened by N-ethylmaleimide (NEM) [Citation30]. Interestingly, the interaction between STX17 and both VAMP7A and VAMP7B were enhanced by NEM treatment (Figure S3G).
In summary, the SNARE domain of STX17 interacts with both VAMP7A and VAMP7B, while the C terminus of VAMP7B interacts with both STX17 and DIPK2A ().
VAMP7B inhibits STX17- and VAMP7A-mediated autophagosome-lysosome fusion
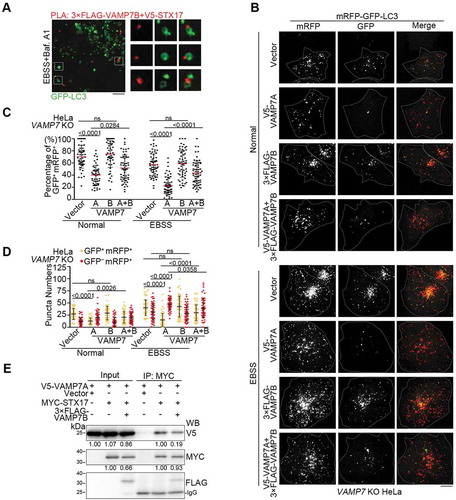
The function of VAMP7B has not been explored yet. Although a previous report indicated cytoplasmic dispersed localization of VAMP7B [Citation8], we noticed that VAMP7B was clearly enriched on autophagosomes marked by endogenous LC3 (Figure S4A). Furthermore, VAMP7B was colocalized with STX17 on LC3-positive autophagosomes in EBSS and bafilomycin A1 (Baf. A1) co-treated cells (Figure S4B), and proximity ligation assay (PLA) by probing both VAMP7B and STX17 further showed that PLA-positive puncta were localized on LC3-labeled vesicles (), suggesting that the interaction between STX17 and VAMP7B occurs on autophagosomes before fusion with lysosomes. VAMP7B overexpression inhibited autophagosome-lysosome fusion, as shown by the increased percentage of GFP+ mRFP+ puncta of LC3 (Figure S4C-E), increased number of autophagosomes and decreased number of autolysosomes (Figure S4F) under starvation condition. Although VAMP7A overexpression promoted autophagosome-lysosome fusion in VAMP7 knockout cells (– and S4G), co-overexpression of VAMP7B and VAMP7A ameliorated the enhancement of autophagosome-lysosome fusion caused by VAMP7A overexpression alone (– and S4G), while VAMP7B alone failed to inhibit autophagosome-lysosome fusion in the absence of VAMP7A (– and S4G). These data suggest that VAMP7A and VAMP7B have opposite function in autophagosome-lysosome fusion. Importantly, VAMP7B weakened the interaction between STX17 and VAMP7A (), indicating that VAMP7B inhibited the interaction between VAMP7A and STX17.
Figure 4. VAMP7B inhibits autophagosome-lysosome fusion by ameliorating interaction between VAMP7A and STX17. (A) HeLa cells transfected with GFP-LC3 (green), 3× FLAG-VAMP7B and V5-SXT17 were treated with EBSS in the presence of bafilomycin A1 (Baf. A1) for 2 h before probed with anti-FLAG and anti-V5 antibodies, and subjected to PLA (red). Samples were observed by structure illumination microscopy (SIM). The boxed areas are magnified. Scale bar: 2 μm. (B-D) VAMP7 knockout (KO) HeLa cells co-transfected with mRFP-GFP-LC3 and the indicated vectors were cultured under normal or EBSS (2 h) conditions and imaged by fluorescence microscopy (B). Scale bar: 10 μm. The percentage of GFP+ mRFP+ puncta (C) and the numbers of GFP+ mRFP+ and GFP– mRFP+ puncta (D) were determined (n ≥ 50 cells). (E) HEK293T cells transfected with the indicated vectors were immunoprecipitated (IP) using an anti-MYC antibody and subjected to western blotting (WB) analysis. The relative band intensities are marked below. For (C and D), the results are expressed as mean ± S.D. The statistical significance (p-value) was determined by one-way ANOVA (listed at the top). See also Figure S4.
DIPK2A promotes autophagosome-lysosome fusion through STX17 and VAMP7
As the C terminus of VAMP7B (145–260 aa) interacts with both DIPK2A ( and ) and STX17 ( and ), we hypothesized that DIPK2A and STX17 compete to interact with VAMP7B. Once DIPK2A was added, DIPK2A bound to VAMP7B, while the interaction between VAMP7B and STX17 was alleviated (). DIPK2A overexpression decreased the number of PLA-positive puncta probing VAMP7B and STX17 interaction ( and ), while DIPK2A knockout cells contained more PLA-positive puncta than wild-type cells (Figure S4H and I), indicating that DIPK2A inhibits the interaction between VAMP7B and STX17. Considering that VAMP7B inhibited the interaction between VAMP7A and STX17 (), we wondered whether DIPK2A promotes the interaction between VAMP7A and STX17. Consistent with a previous report [Citation10], endogenous VAMP7A was immunoprecipitated by STX17 (Figure S4J), and this interaction was enhanced upon DIPK2A overexpression (). Therefore, DIPK2A binds to VAMP7B and inhibits the binding of STX17 to VAMP7B, thus enhancing the interaction between STX17 and VAMP7A.
Figure 5. DIPK2A attenuates the interaction between STX17 and VAMP7B while enhancing the interaction between SXT17 and VAMP7A. (A) HEK293T cells transfected with the indicated vectors were immunoprecipitated (IP) using anti-MYC and anti-FLAG antibodies and subjected to western blotting (WB) analysis. The relative band intensities are marked below. (B and C) HeLa cells were transfected with 3× FLAG-VAMP7B and V5-SXT17 in the presence of DIPK2A-BFP (blue) or not. PLA (red) was performed by probing with anti-FLAG and anti-V5 antibodies (B). Scale bar: 10 μm. The number of PLA-positive puncta were quantified (n ≥ 50 cells) (C). (D) HeLa cells transfected with DIPK2A-3× FLAG or the control vector for 36 h were immunoprecipitated by an anti-STX17 antibody or rabbit IgG, and subjected to western blotting (WB) analysis. The relative band intensities are marked below. (E-G) Wild-type (WT), STX17 knockout (KO) and VAMP7 knockout (KO) HeLa cells co-transfected with mRFP-GFP-LC3 and the indicated vectors were cultured under normal or EBSS (2 h) conditions and imaged by fluorescence microscopy (E). Scale bar: 10 μm. The percentage of GFP+ mRFP+ puncta (F), and the numbers of GFP+ mRFP+ and GFP– mRFP+ puncta (G) were determined (n ≥ 40 cells). For (C, F and G), the results are expressed as mean ± S.D. The statistical significance (p-value) was determined by unpaired two-tailed Student’s t-tests (C) or two-way ANOVA (F and G) (listed at the top). See also Figure S4.
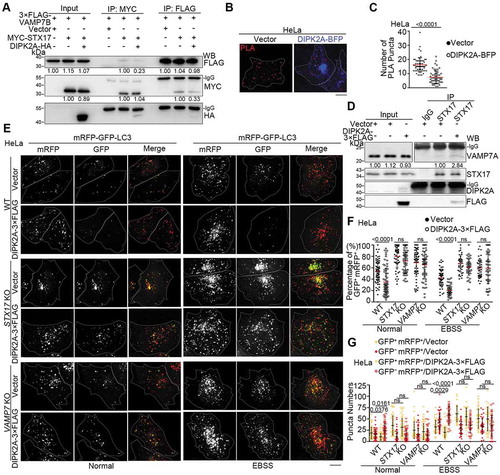
To test whether DIPK2A promotes autophagosome-lysosome fusion through STX17 and VAMP7, we overexpressed DIPK2A in wild-type, STX17 knockout and VAMP7 knockout cells ( and S4K). Although DIPK2A overexpression promoted autophagosome-lysosome fusion in wild-type cells, this effect was attenuated in either STX17 knockout cells or VAMP7 knockout cells (–). YKT6 was recently shown also to mediate autophagosome-lysosome fusion [Citation4], while DIPK2A still promoted autophagosome-lysosome fusion in YKT6 knockdown cells (Figure S4L-N). These findings indicate that DIPK2A promotes autophagosome-lysosome fusion through STX17 and VAMP7.
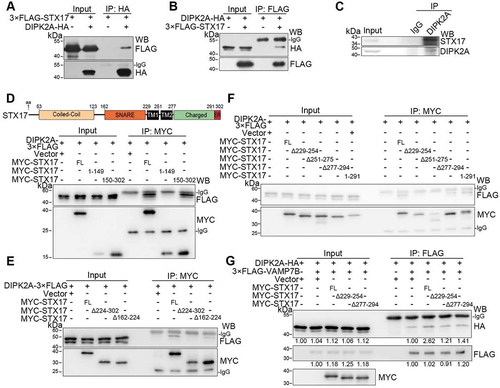
STX17 interacts with DIPK2A to facilitate VAMP7B binding to DIPK2A
To explore how DIPK2A regulates STX17-VAMP7 binding, we next tested whether DIPK2A interacts with STX17 by immunoprecipitation assay. 3× FLAG-STX17 and DIPK2A-HA interacted with each other when they were overexpressed in HEK293T cells ( and ). Endogenous STX17 was also immunoprecipitated with the anti-DIPK2A antibody (). The C terminus of STX17 (150–302 aa), but not the N terminus (1–149 aa), interacted with DIPK2A (). Furthermore, STX17 deletion mutants lacking the 224–302 aa region (Δ224–302 aa) did not interact with DIPK2A, while STX17 without the SNARE domain (Δ162–224 aa) interacted with DIPK2A (). Detailed mapping showed that only STX17 mutants without either of the two transmembrane domains (Δ229–255 aa or Δ251–275 aa) failed to interact with DIPK2A (), suggesting that the two transmembrane domains of STX17 are indispensable for its interaction with DIPK2A. Considering that STX17 interacts with both VAMP7B and DIPK2A, yet it does so through different domains (), we surmised that the interaction between STX17 and DIPK2A reduces the distance between DIPK2A and VAMP7B, thus facilitating the binding of VAMP7B to DIPK2A. Indeed, STX17 overexpression increased the interaction between DIPK2A and VAMP7B, while overexpression of STX17 mutants (Δ229–255 aa or Δ251–275 aa) which did not interact with DIPK2A, did not ().
Figure 6. STX17 interacts with DIPK2A via two transmembrane domains. (A and B) HEK293T cells co-transfected with 3× FLAG-STX17 and DIPK2A-HA were immunoprecipitated (IP) by the indicated antibodies and subjected to western blotting (WB) analysis. (C) HeLa cells were collected and lysed. Endogenous proteins were immunoprecipitated (IP) by an anti-DIPK2A antibody or rabbit IgG (as a control). (D-F) HEK293T cells co-transfected with DIPK2A-3× FLAG and MYC-tagged STX17 truncates were immunoprecipitated (IP) by an anti-MYC antibody and subjected to western blotting (WB) analysis. FL, full length; numbers indicate amino acids. (G) HEK293T cells transfected with the indicated vectors were immunoprecipitated (IP) by an anti-FLAG antibody and subjected to western blotting (WB) analysis. FL, full length; numbers indicate amino acids. The relative band intensities are marked below.
DIPK2A inhibits apoptosis and enhances cell survival
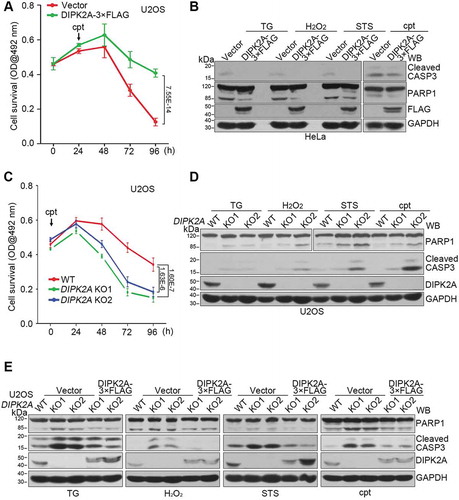
Consistent with a previous study [Citation19], DIPK2A overexpression increased the cell survival rate with () or without (Figure S5A) camptothecin (cpt) treatment, as revealed by MTT assay, indicating the involvement of DIPK2A in cell proliferation. As DIPK2A promoted autophagy (–) and mitophagy (–), and autophagy and mitophagy play important roles in cell fate determination by influencing apoptosis [Citation31], we assessed the role of DIPK2A in apoptosis. We overexpressed DIPK2A in HeLa cells, after which the cells were treated with apoptosis-inducing drugs, including thapsigargin (TG), H2O2, staurosporine (STS), or cpt. The apoptosis level was determined by western blotting analysis of the cleavage of CASP3 (caspase 3) and PARP1 (poly[ADP-ribose] polymerase 1). DIPK2A overexpression inhibited apoptosis induced by all of the selected drugs (), indicating that DIPK2A is a potential negative regulator of apoptosis. However, under normal conditions, the cell survival rate of DIPK2A knockout cells was not different from that of wild-type cells, as revealed by MTT assay (Figure S5B). DIPK2A knockout cells showed enhanced sensitivity to the pro-apoptotic effect of cpt treatment and had a decreased survival rate (). Upon treatment with TG, H2O2, STS, or cpt, DIPK2A knockout cells had a higher apoptosis level than that of wild-type cells, as shown by the cleavage of CASP3 and PARP1 (). Moreover, re-introduction of DIPK2A into DIPK2A knockout U2OS cells rescued the increase in apoptosis level (), suggesting that DIPK2A plays a role in regulating apoptosis. Collectively, DIPK2A functions as a negative regulator of apoptosis to promote cell survival.
Figure 7. DIPK2A inhibits apoptosis. (A) U2OS cells were seeded in 96-well cell plates and transfected with the control vector or DIPK2A-3× FLAG. 1 μM cpt was added 24 h after transfection. The cells were analyzed by MTT assay. (B) HeLa cells were transfected with the control vector or DIPK2A-3× FLAG and treated with 500 ng/mL TG, 2 mM H2O2, 10 nM STS, or 1 μM cpt for 24 h. Cell lysates were subjected to western blotting (WB) analysis using the indicated antibodies. GAPDH was used as a loading control. (C) Wild-type (WT) or DIPK2A knockout (KO) U2OS cells were seeded in 96-well cell plates and treated with 1 μM cpt. The cells were analyzed by MTT assay. (D) Wild-type (WT) and DIPK2A knockout (KO) U2OS cells were treated with 500 ng/mL TG, 2 mM H2O2, 10 nM STS, or 1 μM cpt for 24 h and subjected to western blotting (WB) analysis. GAPDH was used as a loading control. (E) Wild-type (WT) or DIPK2A knockout (KO) U2OS cells were transfected with the indicated vectors and treated with 500 ng/mL TG, 2 mM H2O2, 10 nM STS or 1 μM cpt for 24 h. Cell lysates were subjected to western blotting (WB) analysis. GAPDH was used as a loading control.
The apoptosis-inhibition function of DIPK2A is mediated through STX17-VAMP7
To investigate the relationship between the apoptosis inhibition function of DIPK2A and its autophagy- and mitophagy-promoting ability, we first assessed alterations in the protein levels of mitochondrial proteins during apoptosis. First, consistent with previous studies, the expression levels of mitochondrial proteins such as MT-CO2, CYCS [Citation32] and MFN2 [Citation33,Citation34] increased with time during apoptosis (Figure S5C). DIPK2A overexpression in HeLa cells reduced the levels of these mitochondrial proteins under both normal condition and cpt treatment (), whereas the protein levels of MFN2 and TOMM20 in DIPK2A knockout cells were higher than those in wild-type cells upon cpt treatment (), suggesting that DIPK2A negatively regulates the protein levels of mitochondrial proteins during apoptosis.
Figure 8. Inhibition of apoptosis by DIPK2A depends on its ability to regulate autophagy and on STX17-VAMP7. (A) HeLa cells transfected with the control vector or DIPK2A-3× FLAG were treated with 1 μM cpt for 24 h. The cell lysates were subjected to western blotting (WB) analysis. ACTB was used as a loading control. (B) Wild-type (WT) and DIPK2A knockout (KO) cells were treated with 1 μM cpt for 24 h and subjected to western blotting (WB) analysis. GAPDH was used as a loading control. (C) HeLa cells transfected with the control vector or DIPK2A-3× FLAG were treated with 1 μM cpt, 500 ng/mL TG or 10 nM STS. DMSO or CQ was added at the same time. The cell lysates were subjected to western blotting (WB) analysis. GAPDH was used as a loading control. (D) Wild-type or DIPK2A knockout (KO) HeLa cells transfected with the control vector or DIPK2A-3× FLAG were treated with 1 μM cpt, and with or without 20 nM rapamycin (Rap) for 12 h. The cell lysates were subjected to western blotting (WB) analysis. ACTB was used as a loading control. (E) Wild-type (WT) or STX17-constitutively-knockdown (STX17 KD) HeLa cells were transfected with the indicated vectors and treated with 1 μM cpt for 24 h. The cell lysates were subjected to western blotting (WB) analysis. TUBA4A was used as a loading control. (F and G) Wild-type (WT) or STX17 knockout (KO) HeLa cells were transfected with the indicated vectors, and treated with or without 1 μM cpt for 24 h. The cell lysates were subjected to western blotting (WB) analysis. ACTB was used as a loading control (F). Inhibition of the relative apoptosis level (cleaved PARP1) by DIPK2A was determined based on 3 independent experiments and normalized based on the control groups (without cpt treatment) (G). (H and I) Wild-type (WT) or VAMP7 knockout (KO) HeLa cells were transfected with the indicated vectors, and treated with or without 1 μM cpt for 24 h. The cell lysates were subjected to western blotting (WB) analysis using the indicated antibodies. GAPDH was used as a loading control (H). Inhibition of the relative apoptosis level (cleaved PARP1) by DIPK2A was determined based on 3 independent experiments and normalized based on the control groups (without cpt treatment) (I). (J) HeLa cells transfected with the control vector or 3× FLAG-VAMP7B were treated with DMSO, 1 μM cpt or 10 nM STS for 24 h, after which they were subjected to western blotting (WB) analysis. ACTB was used as a loading control. (K) Model of STX17-, VAMP7- and DIPK2A-mediated autophagosome-lysosome fusion. VAMP7B binds to the SNARE domain of STX17 through its C terminus and inhibits binding of VAMP7A. Once DIPK2A binds to the TM domain of STX17, VAMP7B detaches from the SNARE domain of STX17, while VAMP7A binds to this domain. The simultaneous binding of VAMP7B and DIPK2A to different domains of STX17 enhances the binding between VAMP7B and DIPK2A. Some unidentified factors may also participate in this process.
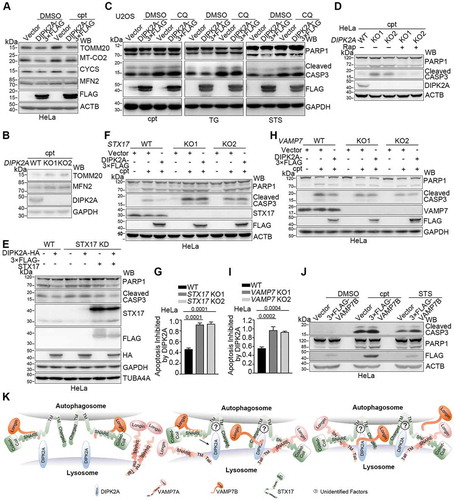
Next, we investigated whether the apoptosis inhibition function of DIPK2A depends on its autophagy- and mitophagy-promoting ability. Although DIPK2A inhibited apoptosis induced by cpt, TG and STS, inhibition of autophagy by CQ blocked this effect (), while the enhanced apoptosis level in DIPK2A knockout cells was reversed by autophagy induction by rapamycin (). These data suggest that DIPK2A inhibits apoptosis by promoting autophagy.
Finally, we examined whether the apoptosis inhibition ability of DIPK2A depends on STX17 and VAMP7. DIPK2A overexpression did not inhibit apoptosis in STX17 knockdown cells, and this effect was rescued by re-introduction of STX17 (). Similarly, the inhibitory effect of DIPK2A on apoptosis was attenuated in either STX17 knockout cells ( and ) or VAMP7 knockout cells ( and ). Considering that DIPK2A regulates autophagy by sequestering VAMP7B from STX17 (), then VAMP7B itself should enhance apoptosis. Indeed, VAMP7B overexpression promoted apoptosis under apoptosis-inducing conditions (). Taken together, these results suggest that the apoptosis inhibition function of DIPK2A is mediated through STX17-VAMP7 and by sequestering VAMP7B from inhibitory interaction with STX17.
Discussion
In this study, we show that the SNARE-domain-deleted VAMP7 isoform, VAMP7B, inhibits autophagosome-lysosome fusion by competing with the functional isoform, VAMP7A, for the SNARE domain of STX17 (). We also show that a new late endosome- and lysosome-localized regulator, DIPK2A, binds to VAMP7B, inhibiting the interaction between VAMP7B and STX17, while enhancing the interaction between VAMP7A and STX17, thus promoting autophagosome-lysosome fusion (). Thus, DIPK2A regulates apoptosis via autophagic degradation of mitochondria by sequestering VAMP7B from inhibitory interaction with STX17.
Q-SNARE STX17 and R-SNARE VAMP7 (VAMP7A) have been reported to regulate autophagosome and lysosome fusion [Citation2]. VAMP7 has several isoforms, and VAMP7A is a late endosome- and lysosome-localized isoform with a functional SNARE domain [Citation2]. VAMP7B is another isoform with no SNARE domain [Citation8]. The mRNA transcript encoding VAMP7B is expressed in various cells [Citation8]. Although we were unable to detect endogenous VAMP7B due to the lack of an available antibody, we showed that exogenously expressed VAMP7B was localized on the autophagosome (Figure S4A). In our study, both VAMP7A and VAMP7B were found to bind to the same domain of STX17. Thus, VAMP7B inhibits the interaction between STX17 and VAMP7A by competition, while the binding of VAMP7B to STX17 leads to autophagosome-lysosome fusion failure because VAMP7B lacks a SNARE domain. Accordingly, VAMP7B overexpression inhibited autophagosome-lysosome fusion (Figure S4C-F) by disturbing the binding of VAMP7A to STX17, which formed a valid combination for SNARE fusion (). The spatiotemporal regulation of VAMP7A activity by VAMP7B may prevent inappropriate binding of VAMP7A to STX17, finely tuning the STX17- and VAMP7A-mediated membrane fusion specificity during autophagosome-lysosome fusion [Citation5]. On the other hand, both VAMP7A and VAMP7B share the same longin domain in the N terminus [Citation8]. It seems that VAMP7B has a similar function as the longin domain alone, as the longin domain was reported to inhibit the function of VAMP7A [Citation35–Citation37]. It is predicted by resonance assignments that the longin domain binds to the SNARE domain to auto-inhibit the SNARE function of VAMP7A [Citation29]. However, consistent with a previous report [Citation38], interaction between the longin domain and SNARE domain of VAMP7A was not detected in our co-immunoprecipitation assays (Figure S3D), which also did not detect interaction between VAMP7B and VAMP7A (Figure S3D). Thus, the detailed mechanisms underlying the auto-inhibition of VAMP7A by the longin domain require further exploration. Besides, considering that NEM treatment enhanced the interaction of STX17 and VAMP7B despite of a lack of SNARE domain in VAMP7B (Figure S3G), it is possible that the association between them is indirect and mediated by another NSF resolvable SNARE interaction. Therefore, the exact interaction mode between STX17 and VAMP7B also merits further study.
Our data show that DIPK2A inhibits the interaction between VAMP7B and STX17. VAMP7B binds to both STX17 and DIPK2A via its C terminal domain, and its binding to STX17 is inhibited in the presence of DIPK2A (). On the other hand, DIPK2A and VAMP7B bind to separate domains of STX17, and the interaction between STX17 and DIPK2A may facilitate the delivery of VAMP7B to DIPK2A by shortening the distance between these proteins (). DIPK2A then binds to the C terminus of VAMP7B, and once VAMP7B is detached from STX17, VAMP7A binds to the SNARE domain of STX17, which is previously occupied by VAMP7B (). In this sense, DIPK2A enhances the interaction of STX17 with VAMP7A and promotes autophagosome-lysosome fusion (). As DIPK2A binds to the transmembrane domain of STX17, it is reasonable to suggest that other regulatory factors probably exist to facilitate this process ().
DIPK2A has been identified as being related with familial autism [Citation14]. Autism spectrum disorders (ASDs) are common neurodevelopmental disorders involving impaired social communication ability, restrictive interests and repetitive patterns of behavior [Citation39]. Impaired autophagy and dysfunctional mitochondrial clearance in dendritic spines have been observed in patients with ASDs [Citation40,Citation41]. Mutations in several autism risk genes, such as TSC1/2 [Citation42], FMR1 (fragile X mental retardation 1) [Citation43] and WDFY3 (WD repeat and FYVE domain-containing 3) [Citation44] have been reported to reduce synapse turnover by disrupting autophagy or mitophagy [Citation45], whereas MTOR hyperactivity, which impairs autophagy, leads to increased spine density, developmental spine pruning deficits and ASD-like social behavior [Citation41,Citation46]. Given the role of DIPK2A in autophagosome-lysosome fusion and cell protection, a thorough understanding of its molecular mechanism will refine our comprehension of autism pathogenesis and provide novel drug targets.
Materials and methods
Reagents and antibodies
Reagents used in this study were as follows: camptothecin (cpt; Selleck, S1288), rapamycin (Selleck, S1039), bafilomycin A1 (Baf. A1; Selleck, S1413), N-ethylmaleimide (NEM; Selleck, S3692), staurosporine (STS; Selleck, S1421), Earle’s balanced salt solution (EBSS; Sigma-Aldrich, E7510), carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich, C2759), chloroquine diphosphate salt (CQ; Sigma-Aldrich, C6628) and thapsigargin (TG; Sigma-Aldrich, T9033).
The primary antibodies used in this study were as follows: anti-ACTB (actin beta; Abclonal, AC026; 1:100,000), anti-cleaved CASP3 (Cell Signaling Technology, 9664; 1:300), anti-CYCS (BD, 556,433; 1:10,000), anti-FLAG (Sigma-Aldrich, F1804; 1:2000; Proteintech, 20,543–1-AP; 1:100), anti-GAPDH (CWBIO, 0100A; 1:5000), anti-GFP (produced in-house, 1:10,000) [Citation47], anti-HA (Sigma-Aldrich, H9658; 1:10,000), anti-LAMP1 (Developmental Studies Hybridoma Ban [DSBH], H4A3; 1:100 for immunofluorescence and 1:1000 for western blotting), anti-LC3 (MBL, M186-3; 1:10,000; MBL, M152-3; 1:100 for immunofluorescence), anti-MT-CO2 (Proteintech, 55,070–1-AP; 1:5000), anti-MFN2 (Proteintech, 12,186–1-AP; 1:1000), anti-PARP1 (Cell Signaling Technology, 9542; 1:1000), anti-SQSTM1 (MBL, PM045; 1:10,000), anti-REEP1 (Proteintech, 17,988; 1:3000), anti-REEP5 (Proteintech, 14,643; 1:1000), anti-RTN4 (Proteintech, 10,740; 1:50 for immunofluorescence), anti-STX17 (MBL, M212-3; 1:1000; Sigma-Aldrich, HPA001204; 1:2000; Proteintech, 17,815–1-AP; 10 μL for immunoprecipitation), anti-TOMM20 (Abcam, ab56783; 1:5000), anti-VAMP7 (Abcam, ab36195; 1:1000; Santa Cruz Biotechnology, sc-166,394; 1:20–1:100), and anti-TUBA4A (Sigma-Aldrich, T6199; 1:5000). Secondary antibodies for western blotting were Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch, 111–035-003; 1:5000), Peroxidase-AffiniPure Goat anti-Mouse IgG (H + L) (Jackson ImmunoResearch, 115–035-003; 1:5000), and IPKine™ HRP, Goat Anti-Mouse IgG HCS (Abbkine, a25112; 1:5000). Secondary antibodies for immunofluorescence were Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Invitrogen, A-11,034; 1:200), Alexa Fluor 568 (Invitrogen, A-11,031; 1:200) and Alexa Fluor 647 (Invitrogen, A-21,245; 1:200). The antibody for DIPK2A was raised against the C terminal peptide (ELREYLAQLSNNVR) of human DIPK2A in rabbits as previously reported [Citation24].
Cell culture and transfection
HeLa, HEK293T and U2OS cells were cultured in DMEM (GIBCO, 12,100–046) supplemented with 10% FBS (CellMax, SA212.02) at 37°C with 5% CO2. For cell transfection, PEI (Polysciences, Inc., 23,966) was used.
Vector construction
The sequences encoding human DIPK2A, MFN2, VAMP7A, VAMP7B and STX17 were amplified from HEK293T cell cDNA and cloned into the pcDNA3.1 (Invitrogen, V79020), p3× FLAG-CMV-7.1 (Sigma-Aldrich, E7533), pEGFP-N3 (Clontech, 6080–1) or pmCherry-N1 (Clontech, 632,523) vector. The ER marker ER-GFP was cloned into the pcDNA3.1 vector with the signal peptide of CALU (calumenin) at the N terminus and the ‘KDEL’ ER retrieval signal at the C terminus of GFP [Citation23]. Mitochondrial marker Mito-BFP was obtained by ligating BFP with the C terminus mitochondria localization sequence of human COX8A (cytochrome c oxidase subunit 8A) [Citation48], followed by cloning into the pcDNA3.1 vector. Truncation mutants for STX17 and VAMP7 were constructed by overlap PCR. The shRNA sequence for STX17 [Citation3] was obtained from Invitrogen and cloned into the pLKO.1 vector (Addgene, 10,878; deposited by David Root) after annealing.
Generation of knockout cells and knock-in cells
To generate DIPK2A, STX17 and VAMP7 knockout cell lines, two target oligos (5′-GGCGGGCGGTATGTGGCGCC-3′ and 5′-GCTTCAGCGAGCGGGACAGG-3′ for DIPK2A, 5′-GATAGTAATCCCAACAGACC-3′ and 5′-TCTTGAACCAGCTATCCAGA-3′ for STX17, 5′-AACAGCAAAAAGAATCGCCA-3′ and 5′-CTATCCTTGCCAAACATGCT-3′ for VAMP7) ligated into the gRNA vectors were transfected into HeLa or U2OS cells with a plasmid expressing the Cas9 protein [Citation49]. To generate DIPK2A-mCherry knock-in cell lines, one target oligo (5′-TGAAGTTCACAGGAATAAAC-3′) ligated into the gRNA vector, a plasmid expressing the Cas9 protein and an arm sequence (mCherry flanked by 1 kb homologous sequence of DIPK2A genome) constructed into the pcDNA3.1 vector were co-transfected into HEK293T cells. Twenty-four h after transfection, the cells were monocloned by flow cytometry (BeckMan Coulter, MoFlo XDP) and seeded into 96-well plates. Successful knockout clones were confirmed by western blotting and sequencing analysis.
Sucrose gradient separation
For sucrose gradient separation, cells were homogenized in solution buffer (20 mM HEPES, 1 mM EDTA, 250 mM sucrose [amresco, M1117], pH 7.4; protease inhibitor cocktail [bimake.com, B14001] was added before use) on ice with a homogenizer and centrifuged at low speed (1000 × g, 10 min) to remove unbroken cells. The supernatant was adjusted to 25% sucrose and placed on top of a preformed sucrose gradient consisting of 0.96 mL 45% sucrose, 2.08 mL 35% sucrose and 1.56 mL 25% sucrose. The sample was centrifuged at 150,000 × g for 3 h at 4°C. Twelve fractions were collected from the top of the gradient and subjected to western blotting analysis.
Western blotting and immunoprecipitation
Western blotting assays were performed as previously described [Citation23]. In brief, protein samples were transferred to PVDF membranes (Millipore, IPVH00010) after SDS-PAGE separation. The membranes were successively incubated with primary and HRP-conjugated secondary antibodies. Protein bands were detected by a chemiluminescence reagent with either a film exposure machine (Kodak) or Tanon 5200 chemiluminescence imager. Band intensities were analyzed by ImageJ software (NIH).
For the immunoprecipitation assays, HEK293T cells were transfected and lysed in immunoprecipitation buffer (25 mM HEPES, 150 mM KAc, 2 mM Mg[Ac]2, 0.5% Triton X-100 [amresco, 0694-1L]; protease inhibitor cocktail was added before use) on ice. The cell lysates were incubated with the appropriate antibodies overnight, followed by combination with Protein A Sepharose beads (GE Healthcare Life Sciences, 17–5280-02). The immunoprecipitates were subjected to western blotting analysis.
Fluorescence microscopy
For the fluorescence assays, cells were plated on acid-washed glass coverslips in advance. Twenty-four h after transfection, cells were fixed in 4% PFA (dissolved in PBS [137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4, 10 mM Na2HPO4]) at 37°C for 15 min followed by permeabilization in 0.15% Triton X-100 (in PBS) and washing with PBS. After blocking in 3% BSA (in PBS), the samples were incubated with the indicated primary antibodies, after which they were incubated with Alexa Fluor-conjugated secondary antibodies. Superfluous antibodies were washed away using PBS. The samples were sealed with nail polish and observed under an LSM-710 NLO (Zeiss) confocal fluorescent microscope equipped with a 100×/1.42 numerical aperture (NA) objective lens. The images were analyzed by ZEN 2009 software (Zeiss). 3D-SIM was performed at room temperature with an N-SIM system (Nikon) equipped with a 100×/1.49 NA objective lens (Nikon). The images were reconstructed and analyzed with NIS-Elements AR software (Nikon) and ImageJ (NIH).
Tandem mRFP-GFP-LC3 assay
HeLa cells were transfected with mRFP-GFP-LC3 or co-transfected with the indicated plasmids for 24 h. Next, cells were fixed with 4% PFA (dissolved in PBS) at 37°C for 15 min, sealed with nail polish and observed under an LSM-710 NLO (Zeiss) confocal fluorescent microscope equipped with a 100×/1.42 numerical aperture (NA) objective lens. Cells were randomly selected, and the numbers of GFP puncta and mRFP puncta were counted manually in a two-blinded manner. The percentage of GFP+ mRFP+ puncta, number of GFP+ mRFP+ puncta and number of GFP– mRFP+ puncta were calculated based on the numbers of GFP puncta and mRFP puncta. The statistical significance (p-value) was analyzed with one-way ANOVA or two-way ANOVA using Graphpad.
Proximity ligation assay (PLA)
For analyzing the interaction between VAMP7B and STX17, cells were transfected with the corresponding plasmids. PLA assay (Sigma-Aldrich, DUO9008, DUO92002 and DUO92004) was performed according to the manufacturer’s instructions by simultaneously probing tagged VAMP7B and STX17. Images were captured by N-SIM system (Nikon) equipped with a 100×/1.49 NA objective lens, confocal microscope (Zeiss, LSM-710 NLO) equipped with a 100×/1.4 NA objective lens, or fluorescence microscope (Olympus, TH4-200) equipped with a 60×/1.42 NA objective lens.
MTT assay
MTT assays were performed with the CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega, G3581) as previously reported [Citation50]. In brief, cells were seeded in 96-well plates, transfected and treated as indicated. At each time point, the relative viability of the cells was determined by measuring the optical density at an absorbance wavelength of 492 nm. Each experiment was repeated six times.
Statistical analysis
The results are shown as mean ± S.D. The statistical significance of the results was analyzed using unpaired two-tailed Student’s t-tests, one-way ANOVA or two-way ANOVA. The p-values are shown in the figures. ns, not significant.
Supplemental Material
Download MS Word (14.6 MB)Acknowledgments
We thank Dr. Rui Jia at National Institute of Child Health and Human Development for providing advices for the project. We thank Dr. Jingwei Xiong at the Institute of Molecular Medicine of Peking University for kindly providing Cas9 and gRNA plasmids. We thank Liying Du, Xiaochen Li, Chunyan Shan and Hongxia Lv at the Core Facilities of Life Sciences of Peking University for flow cytometry, super-resolution microscopic and statistical analysis. This work was supported by the National Natural Science Foundation of China (NSFC) (31871353 and 31671392) and the National Key Research and Development Program of China, Stem Cell and Translational Research (2016YFA0100501).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PubMed PMID: 21801009.
- Takats S, Nagy P, Varga A, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013 May 13;201(4):531–539. PubMed PMID: 23671310; PubMed Central PMCID: PMC3653357.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 7;151(6):1256–1269. PubMed PMID: 23217709.
- Matsui T, Jiang P, Nakano S, et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol. 2018 Aug 6;217(8):2633–2645. PubMed PMID: 29789439; PubMed Central PMCID: PMCPMC6080929.
- Daste F, Galli T, Tareste D. Structure and function of longin SNAREs. J Cell Sci. 2015 Dec 1;128(23):4263–4272. PubMed PMID: 26567219.
- Rossi V, Banfield DK, Vacca M, et al. Longins and their longin domains: regulated SNAREs and multifunctional SNARE regulators. Trends Biochem Sci. 2004 Dec;29(12):682–688. PubMed PMID: 15544955.
- Takats S, Glatz G, Szenci G, et al. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet. 2018 Apr;14(4):e1007359. PubMed PMID: 29694367; PubMed Central PMCID: PMCPMC5937789.
- Vacca M, Albania L, Della Ragione F, et al. Alternative splicing of the human gene SYBL1 modulates protein domain architecture of Longin VAMP7/TI-VAMP, showing both non-SNARE and synaptobrevin-like isoforms. BMC Mol Biol. 2011 May 24;12:26. PubMed PMID: 21609427; PubMed Central PMCID: PMC3123573.
- Martinez-Arca S, Rudge R, Vacca M, et al. A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc Natl Acad Sci U S A. 2003 Jul 22 100(15):9011–9016. PubMed PMID: 12853575; PubMed Central PMCID: PMCPMC166429.
- McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016 Aug 1;214(3):275–291. PubMed PMID: 27458136; PubMed Central PMCID: PMC4970327.
- Yamashita SI, Jin X, Furukawa K, et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol. 2016 Dec 5;215(5):649–665. PubMed PMID: 27903607; PubMed Central PMCID: PMC5147001.
- Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016 Dec 19;215(6):857–874. PubMed PMID: 27864321; PubMed Central PMCID: PMC5166504.
- Sugo M, Kimura H, Arasaki K, et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. Embo J. 2018 Nov 2;37(21). PubMed PMID: 30237312; PubMed Central PMCID: PMCPMC6213275.
- Morrow EM, Yoo SY, Flavell SW, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008 Jul 11;321(5886):218–223. PubMed PMID: 18621663; PubMed Central PMCID: PMC2586171.
- Aziz A, Harrop SP, Bishop NE. Characterization of the deleted in autism 1 protein family: implications for studying cognitive disorders. PloS One. 2011 Jan 19;6(1):e14547. PubMed PMID: 21283809; PubMed Central PMCID: PMC3023760.
- Takatalo MS, Tummers M, Thesleff I, et al. Novel Golgi protein, GoPro49, is a specific dental follicle marker. J Dent Res. 2009 Jun;88(6):534–538. PubMed PMID: 19587158.
- Hareza A, Bakun M, Swiderska B, et al. Phosphoproteomic insights into processes influenced by the kinase-like protein DIA1/C3orf58. PeerJ. 2018;6:e4599. PubMed PMID: 29666759; PubMed Central PMCID: PMC5896498.
- Dudkiewicz M, Lenart A, Pawlowski K. A novel predicted calcium-regulated kinase family implicated in neurological disorders. PloS One. 2013;86:e66427. PubMed PMID: 23840464; PubMed Central PMCID: PMC3696010.
- Beigi F, Schmeckpeper J, Pow-Anpongkul P, et al. C3orf58, a novel paracrine protein, stimulates cardiomyocyte cell-cycle progression through the PI3K-AKT-CDK7 pathway. Circ Res. 2013 Aug 02;113(4):372–380. PubMed PMID: 23784961; PubMed Central PMCID: PMC3870268.
- Bareja A, Patel S, Hodgkinson CP, et al. Understanding the mechanism of bias signaling of the insulin-like growth factor 1 receptor: effects of LL37 and HASF. Cell Signal. 2018 Jun;46:113–119. PubMed PMID: 29499305.
- Bareja A, Hodgkinson CP, Payne AJ, et al. HASF (C3orf58) is a novel ligand of the insulin-like growth factor 1 receptor. Biochem J. 2017 Feb 20;474(5):771–780. PubMed PMID: 28096202.
- Huang J, Guo J, Beigi F, et al. HASF is a stem cell paracrine factor that activates PKC epsilon mediated cytoprotection. J Mol Cell Cardiol. 2014 Jan;66:157–164. PubMed PMID: 24269490; PubMed Central PMCID: PMC3897274.
- Zheng P, Chen Q, Tian X, et al. DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling. Cell Res. 2018 Aug;28(8):833–854. PubMed PMID: 30030520; PubMed Central PMCID: PMCPMC6063967.
- Takatalo M, Jarvinen E, Laitinen S, et al. Expression of the novel Golgi protein GoPro49 is developmentally regulated during mesenchymal differentiation. Dev Dyn. 2008 Aug;237(8):2243–2255. PubMed PMID: 18651652.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013 Feb 15;339(6121):819–823. PubMed PMID: 23287718; PubMed Central PMCID: PMC3795411.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010 Feb 5;140(3):313–326. PubMed PMID: 20144757; PubMed Central PMCID: PMC2852113.
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007 Sep-Oct;3(5):452–460. PubMed PMID: 17534139.
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006 Sep;7(9):631–643. PubMed PMID: 16912714.
- Vivona S, Liu CW, Strop P, et al. The longin SNARE VAMP7/TI-VAMP adopts a closed conformation. J Biol Chem. 2010 Jun 4;285(23):17965–17973. PubMed PMID: 20378544; PubMed Central PMCID: PMC2878558.
- Abada A, Levin-Zaidman S, Porat Z, et al. SNARE priming is essential for maturation of autophagosomes but not for their formation. Proc Natl Acad Sci U S A. 2017 Nov 28 114(48):12749–12754. PubMed PMID: 29138318; PubMed Central PMCID: PMCPMC5715740.
- Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014 Feb;15(2):81–94. PubMed PMID: 24401948; PubMed Central PMCID: PMC3970201.
- Chandra D, Liu JW, Tang DG. Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J Biol Chem. 2002 Dec 27;277(52):50842–50854. PubMed PMID: 12407106.
- Wang W, Cheng X, Lu J, et al. Mitofusin-2 is a novel direct target of p53. Biochem Biophys Res Commun. 2010 Oct 01;400(4):587–592. PubMed PMID: 20804729.
- Guo X, Chen KH, Guo Y, et al. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. 2007 Nov 26;101(11):1113–1122. PubMed PMID: 17901359.
- Muzerelle A, Alberts P, Martinez-Arca S, et al. Tetanus neurotoxin-insensitive vesicle-associated membrane protein localizes to a presynaptic membrane compartment in selected terminal subsets of the rat brain. Neuroscience. 2003;122(1):59–75. PubMed PMID: 14596849.
- Hua Z, Leal-Ortiz S, Foss SM, et al. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 2011 Aug 11;71(3):474–487. PubMed PMID: 21835344; PubMed Central PMCID: PMC3155686.
- Martinez-Arca S, Alberts P, Zahraoui A, et al. Role of tetanus neurotoxin insensitive vesicle-associated membrane protein (TI-VAMP) in vesicular transport mediating neurite outgrowth. J Cell Biol. 2000 May 15;149(4):889–900. PubMed PMID: 10811829; PubMed Central PMCID: PMC2174569.
- Proux-Gillardeaux V, Raposo G, Irinopoulou T, et al. Expression of the Longin domain of TI-VAMP impairs lysosomal secretion and epithelial cell migration. Biol Cell. 2007 May;99(5):261–271. PubMed PMID: 17288539.
- Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. 2015 Apr;28(2):91–102. PubMed PMID: 25695134.
- Giulivi C, Zhang YF, Omanska-Klusek A, et al. Mitochondrial dysfunction in autism. JAMA. 2010 Dec 1;304(21):2389–2396. PubMed PMID: 21119085; PubMed Central PMCID: PMCPMC3915058.
- Tang G, Gudsnuk K, Kuo SH, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014 Sep 3;83(5):1131–1143. PubMed PMID: 25155956; PubMed Central PMCID: PMCPMC4159743.
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016 Oct 18;17(4):1053–1070. PubMed PMID: 27760312; PubMed Central PMCID: PMCPMC5078873.
- Yan J, Porch MW, Court-Vazquez B, et al. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc Natl Acad Sci U S A. 2018 Oct 9 115(41):E9707–E9716. PubMed PMID: 30242133; PubMed Central PMCID: PMCPMC6187122.
- Napoli E, Song G, Panoutsopoulos A, et al. Beyond autophagy: a novel role for autism-linked Wdfy3 in brain mitophagy. Sci Rep. 2018 Jul 27;8(1):11348. PubMed PMID: 30054502; PubMed Central PMCID: PMCPMC6063930.
- Poultney CS, Goldberg AP, Drapeau E, et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am J Hum Genet. 2013 Oct 3;93(4):607–619. PubMed PMID: 24094742; PubMed Central PMCID: PMCPMC3791269.
- Zhang J, Zhang JX, Zhang QL. PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res Bull. 2016 Jul;125:152–158. PubMed PMID: 27320472.
- Wang Q, Feng H, Zheng P, et al. The intracellular transport and secretion of calumenin-1/2 in living cells. PloS One. 2012;7(4):e35344. PubMed PMID: 22514732; PubMed Central PMCID: PMCPMC3325945.
- Rizzuto R, Nakase H, Darras B, et al. A gene specifying subunit VIII of human cytochrome c oxidase is localized to chromosome 11 and is expressed in both muscle and non-muscle tissues. J Biol Chem. 1989 Jun 25;264(18):10595–10600. PubMed PMID: 2543673.
- Chang N, Sun C, Gao L, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013 Apr;23(4):465–472. PubMed PMID: 23528705; PubMed Central PMCID: PMCPMC3616424.
- Chen L, Xu S, Xu Y, et al. Cab45S promotes cell proliferation through SERCA2b inhibition and Ca2+ signaling. Oncogene. 2016 Jan 7;35(1):35–46. PubMed PMID: 25772237.