
ABSTRACT
Removal of damaged mitochondria is vital for cellular homeostasis especially in non-dividing cells, like neurons. Damaged mitochondria that cannot be repaired by the ubiquitin-proteasomal system are cleared by a form of selective autophagy known as mitophagy. Following damage, mitochondria become labelled with ‘eat-me’ signals that selectively determine their degradation. Recently, we identified the mitochondrial matrix proteins, NIPSNAP1 (nipsnap homolog 1) and NIPSNAP2 as ‘eat-me’ signals for damaged mitochondria. NIPSNAP1 and NIPSNAP2 accumulate on the mitochondrial outer membrane following mitochondrial depolarization, recruiting autophagy receptors and adaptors, as well as human Atg8 (autophagy-related 8)-family proteins to facilitate mitophagy. The NIPSNAPs allow a sustained recruitment of SQSTM1-like receptors (SLRs) to ensure efficient mitophagy. Zebrafish lacking Nipsnap1 show decreased mitophagy in the brain coupled with increased ROS production, loss of dopaminergic neurons and strongly reduced locomotion.
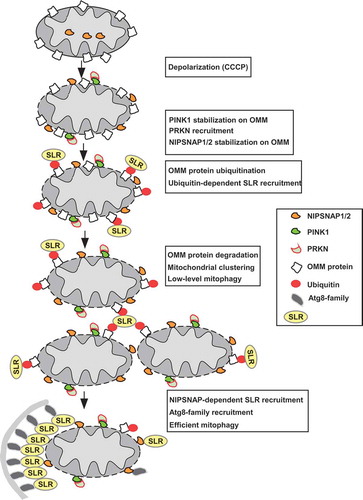
Clearance of damaged or dysfunctional mitochondria and mitochondrial proteins is an important aspect of cellular quality control required for maintenance of homeostasis, which is especially important in slow-dividing and non-dividing cells, such as neurons. While outer mitochondria membrane (OMM) proteins become ubiquitinated and degraded in the proteasome upon mitochondrial damage, entire mitochondria can be eliminated by mitophagy in the event of serious damage to the organelle. Upon induction of mitophagy, damaged mitochondria are labelled with ‘eat me’ signals that are specifically recognized by specialized autophagy receptors and degraded in the lysosome. In a recent study, we identified 2 mitochondrial matrix proteins, NIPSNAP1 and NIPSNAP2 that function as ‘eat me’ signals to facilitate mitophagy [Citation1]. PINK1 (PTEN induced kinase 1) and the E3 ligase PRKN/Parkin, whose mutations are associated with Parkinson disease, have been identified as key players in mitophagy. PINK1 is normally imported into the mitochondria and degraded by several mitochondrial proteases. Upon mitochondrial depolarization and/or damage, protein import into the mitochondria is blocked resulting in accumulation of PINK1 on the OMM (). PINK1 phosphorylates both ubiquitin and PRKN, activating and recruiting the latter to the mitochondria. PRKN then ubiquitinates several OMM proteins leading to their proteasomal degradation. Ubiquitination of OMM proteins also facilitates recruitment of ubiquitin-interacting autophagy receptors, so called SQSTM1-like receptors (SLRs, including SQSTM1 [sequestosome 1], CALCOCO2/NDP52 [calcium binding and coiled-coil domain 2], OPTN [optineurin], NBR1 [NBR1 autophagy cargo receptor] and TAX1BP1 [Tax1 binding protein 1]), causing degradation of the entire mitochondria by mitophagy. It is, however, not entirely clear how ubiquitination and proteasomal degradation of OMM proteins versus mitophagy is regulated. We discovered that mitochondrial depolarization and/or damage induces OMM accumulation of the matrix proteins NIPSNAP1 and NIPSNAP2, where they act as signals to sustain recruitment of the SLRs and Atg8-family proteins that drive efficient mitophagy ().
Figure 1. Working model of PINK1-PRKN-dependent mitophagy illustrating the role of NIPSNAP1 and NIPSNAP2 as ‘eat me’ signals for recruiting autophagy receptors. Mitochondrial damage results in stabilization of PINK1 and recruitment of PRKN. PRKN-dependent ubiquitination of OMM protein leads to proteasome-dependent degradation of OMM proteins and ubiquitin-dependent recruitment of the SLRs. This is accompanied by localization of NIPSNAP1 and NIPSNAP2 to the OMM. The result is the sustained recruitment of the SLRs and Atg8-family proteins to mediate robust mitophagy. The PINK1-PRKN pathway contains important loops; ubiquitination of OMM proteins leads to both the recruitment of the SLRs and proteasomal degradation of the ubiquitinated OMM proteins. The ubiquitin-dependent recruitment of SLRs leads to mitochondrial clustering and low-level mitophagy. Continuous damage and increased degradation of the OMM proteins prime the mitochondria, allowing the OMM-localized NIPSNAP1 and NIPSNAP2 to act as ‘eat-me’ signals, recruiting SLRs and Atg8-family proteins to mediate robust mitophagy.
NIPSNAP1 and NIPSNAP2 are members of an evolutionarily conserved protein family that also includes NIPSNAP3A and NIPSNAP3B. NIPSNAP1 and NIPSNAP2, like other members of the NIPSNAP protein family, have an N-terminal mitochondrial targeting sequence (MTS) that is sufficient for their import into the mitochondria. In addition, NIPSNAP1 and NIPSNAP2 also possess an internal mitochondrial localization sequence, which is absent in the other members of the NIPSNAP protein family, and is vital for the accumulation of NIPSNAP1 and NIPSNAP2 on the OMM following mitochondrial damage. Using a combination of split-fluorescent and enzymatic complementation assays, we discovered that NIPSNAP1 and NIPSNAP2 accumulate on the OMM following treatment with the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP). The interactions between the SLRs and the NIPSNAPs increase following mitochondrial depolarization.
Single and combined clustered regularly interspaced short palindromic repeats (CRISPR)-mediated knockout (KO) of NIPSNAP1 and NIPSNAP2 in HeLa cells revealed that both proteins are required and act redundantly in PINK1-PRKN-mediated mitophagy. While single KO does not affect PINK1-PRKN-mediated mitophagy, double knockout (DKO) of both NIPSNAP1 and NIPSNAP2 almost completely blocks PRKN-mediated mitophagy. Reconstitution of the DKO with either NIPSNAP1 or NIPSNAP2 restores mitophagy, suggesting that they act redundantly. Confocal fluorescence microscopy analysis shows mitochondrial DNA nucleoid clumping in wild-type (WT) cells, but not in the DKO cells. PRKN-dependent mitochondrial aggregation has been reported to depend on SQSTM1/p62. We therefore hypothesized that the NIPSNAPs may play a role in recruiting the SLRs to damaged mitochondria. Indeed, while PRKN recruitment is unaffected, recruitment of SLRs to mitochondria is strongly reduced in DKO cells treated with CCCP compared to control cells. Thus, our results clearly showed that NIPSNAP1/2 are required for OMM recruitment of the SLRs during PINK1-PRKN-mediated mitophagy. To understand how this may occur, we mapped the interaction between CALCOCO2 and the NIPSNAPs using an in vitro GST-affinity-isolation assay. We discovered that NIPSNAPs bind to the C-terminal zinc finger II (ZF-2) domain of CALCOCO2, the same region as required for binding to ubiquitin, but found that several mutations that abolish binding to ubiquitin do not affect binding to NIPSNAP1 or NIPSNAP2. Consequently, we tested the recruitment of EGFP-tagged WT, NIPSNAP- and/or ubiquitin-binding deficient mutants of CALCOCO2 to the mitochondria in CALCOCO2 KO cells expressing mCherry-PRKN. Analysis of these cells by confocal fluorescence microscopy following 6 h of CCCP treatment showed recruitment of both WT CALCOCO2 and CALCOCO2L446A (which binds the NIPSNAPs, but not ubiquitin) to the mitochondria, but not CALCOCO2[ΔZF2] (that neither binds NIPSNAPs nor ubiquitin). Moreover, NIPSNAPs lacking a functional MTS, but retaining the ability to bind to the OMM, can rescue mitophagy in the NIPSNAP1 and NIPSNAP2 DKO cells.
Together, these results demonstrate that while PRKN-dependent ubiquitination of OMM proteins is necessary for mitophagy, it probably acts as a priming event that allows the OMM localized NIPSNAP1 and NIPSNAP2 to recruit the autophagy receptors. This is in line with studies showing that PRKN-mediated ubiquitination of several OMM proteins results in their proteasomal degradation and that this occurs early and prior to mitophagy. However, PRKN-mediated ubiquitination may also participate in early recruitment of the SLRs, resulting in mitochondrial aggregation and low level mitophagy. The OMM-localized NIPSNAP1 and NIPSNAP2 then act as ‘eat me’ signals to sustain recruitment of the SLRs and the human Atg8-family proteins to mediate effective clearance of damaged mitochondria. The SLRs may further recruit autophagy adaptor proteins and more Atg8-family proteins in a positive feedback loop allowing efficient expansion of phagophores on the damaged mitochondria. Our results and current knowledge can be integrated into the following working model (): Mitochondrial damage induces stabilization of PINK1 and subsequent recruitment of PRKN to damaged mitochondria accompanied with OMM-localization of NIPSNAP1 and NIPSNAP2. PRKN-mediated ubiquitination coupled with ubiquitin-dependent recruitment of the SLRs induce mitochondrial clustering and proteasomal degradation of the ubiquitinated OMM proteins. These events prime the mitochondria, allowing the OMM-localized NIPSNAP1 and NIPSNAP2 to sustain the recruitment of SLRs and Atg8-family proteins to ensure efficient and robust clearance of damaged mitochondria ().
Furthermore, we discovered that Zebrafish depleted of Nipsnap1 have reduced brain mitophagy coupled with increased reactive oxygen species (ROS) production, locomotive defects and neuronal death. These phenotypes are similar to other reports on depletion of proteins associated with Parkinson disease in Zebrafish including Pink1, Prkn and Lrrk2 (leucine-rich repeat kinase 2), demonstrating the importance of the Nipsnap1 protein in vivo and confirming results obtained from cells.
In conclusion, our results have led to the identification of novel ‘eat me’ signals for PRKN-mediated mitophagy that is required for ubiquitin-independent recruitment of both the SLRs and Atg8-family proteins and describe new proteins that may be relevant in the concept of Parkinson disease and perhaps also other neurodegenerative diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Princely Abudu Y, Pankiv S, Mathai BJ, et al. NIPSNAP1 and NIPSNAP2 act as “eat me” signals for mitophagy. Dev Cell. 2019;49(4):509–525 e12.