
ABSTRACT
A wide variety of genetic, pharmacological and nutrient manipulations that extend lifespan in model organisms do so in a manner dependent upon increased autophagic flux. However, our recent findings suggest that when mitochondrial membrane integrity is compromised, macroautophagy/autophagy can be detrimental. In C. elegans lacking the serine/threonine kinase mechanistic target of rapamycin kinase complex 2 and its downstream effector SGK-1 (Serum- and Glucocorticoid-inducible Kinase homolog), lifespan is shortened in spite of increased levels of autophagy, whereas reducing autophagy restores normal lifespan. This is due to a concomitant defect in mitochondrial permeability in mutants defective in either SGK-1 or mechanistic target of rapamycin kinase complex 2, attributable to increased VDAC-1 (VDAC Voltage Dependent Anion Channel homolog) protein level. More generally, we find that induction of mitochondrial permeability reverses each and every tested paradigm of autophagy-dependent lifespan extension and, further, exacerbates ischemia-reperfusion injury. In this punctum, we discuss our finding that autophagy with increased mitochondrial permeability is a detrimental combination conserved from nematode to mammals.
A hallmark of aging is the accumulation of various forms of molecular damage, dysfunctional organelles, and defective enzymes. Deficient autophagy in aging organisms is thought to be at least partially responsible for these findings. In contrast, elegant studies have proven that elevated autophagy levels are mechanistically required for lifespan extension in a number of conserved longevity paradigms. Therefore, autophagy is generally thought to have a prominent role in promoting healthy aging.
The protein kinase MTORC2 controls cell proliferation and survival primarily by phosphorylating several members of the AGC (PRKA/PKA-RPKG/PKG-PRKC/PKC) family of protein kinases including SGK1. In worms and mammals alike, we have shown that SGK-1/SGK1 is a major effector of metabolic regulation downstream of mechanistic target of rapamycin kinase complex 2/MTORC2. RICT-1 is the worm ortholog of the essential MTORC2 component RICTOR. C. elegans rict-1 mutants have slow development, high body fat mass, and shortened lifespan, which is phenocopied by loss-of-function mutations in sgk-1. Both rict-1 and sgk-1 mutant C. elegans exhibit elevated autophagic flux, which sharply contrasts these short-lived mutants with many long-lived animals that have elevated levels of autophagy. Remarkably, genetic inhibition of autophagy restores normal lifespan in both rict-1 and sgk-1 mutant worms, suggesting that mechanistic target of rapamycin kinase complex 2 and SGK-1 activity is critical for autophagy to have beneficial effects on longevity [Citation1]. This observation provided an opportunity to dissect the cellular context in which autophagy has detrimental rather than beneficial effects.
In order to determine the mechanism by which autophagy shortens lifespan in sgk-1 mutants, we examined SGK-1-interacting proteins. This analysis revealed that SGK-1 interacts with multiple protein regulators of the mitochondrial permeability transition pore (mPTP). A direct interaction between SGK-1/SGK1 and the mPTP regulator VDAC-1/VDAC1 was found in both C. elegans and mammalian cells. Under normal conditions, SGK1 is responsible for phosphorylating VDAC1, leading to proteasomal degradation. VDAC1 protein accumulation is associated with increased mPTP opening.
The mPTP is a critical regulator of mitochondrial homeostasis. Opening of the mPTP is triggered under pathological conditions such as oxidative stress and high cellular calcium concentrations. In turn, opening of the mPTP permits diffusion of molecules of less than 1500 Da freely across the mitochondrial inner membrane. Transient mPTP opening regulates calcium and mitochondrial reactive oxygen species (ROS) signaling, whereas prolonged mPTP opening results in mitochondrial energetic dysfunction, organelle swelling, rupture, and apoptotic or necrotic cell death. Opening of the pore in mitochondria also marks dysfunctional organelles for autophagic clearance. Generally, mPTP opening is also associated with activation of macroautophagy.
In line with the published literature, elevated levels of VDAC1 protein in sgk1 knockout hepatocytes increases mitochondrial permeability. Increased mitochondrial permeability in this setting in the worm increases autophagy at least in part by increasing mRNA levels of hlh-30 (a homolog of mammalian TFEB, a critical transcriptional regulator of autophagy). In sgk-1 mutant C. elegans, genetic or pharmacological inhibition of the mPTP significantly extends lifespan, suggesting that mPTP opening is mechanistically linked to premature aging in sgk-1 mutants. Confirming this possibility, increased mitochondrial permeability by vdac-1 overexpression alone is sufficient to shorten the lifespan of wild-type C. elegans. Importantly, inhibiting autophagy by bec-1 RNAi restores normal lifespan in vdac-1-overexpressing C. elegans. The implication of this work is that autophagy exerts a progeric, rather than a beneficial effect, in the setting of elevated mitochondrial permeability.
In multiple long-lived C. elegans models, including deficiency in germline stem cells (glp-1 mutants), caloric restriction (eat-2 mutants) and electron-transport chain dysfunction (nuo-6 or frh-1 RNAi), direct stimulation of mPTP opening by vdac-1 overexpression completely suppresses the autophagy-dependent lifespan extension. The conclusion that autophagy is detrimental when mitochondrial permeability is increased is further supported in an ischemia-reperfusion (I/R) injury model in mammals. mPTP opening, which is induced when blood flow is restored to tissue to which it had been restricted during I/R, causes cell death and tissue damage. Liver-specific sgk knockout mice, which have both elevated hepatic autophagy levels and increases in mPTP opening, are more susceptible to hepatic I/R injury, an effect completely reversed by pre-treatment with the mPTP inhibitor cyclosporine A.
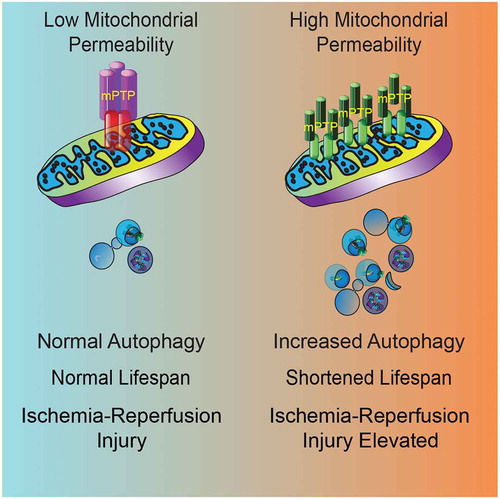
Overall, our work highlights the fact that mitochondrial membrane permeability is a critical determinant of the consequences of autophagy on organismal health and lifespan (). Recent emerging data indicate that mitochondrial permeability increases with aging. Mitoflashes, which are triggered by transient openings of the mPTP, increase as animals age, and are correlated with timing of death. Moreover, diverse manipulations that promote health and extend lifespan, including caloric restriction, induction of the mitochondrial unfolded protein response, reductions in MTOR activity and metformin treatment, all inhibit mPTP opening in various types of cells. These results, together with our findings, suggest that maintenance of low mitochondrial permeability may be an obligate step in order to extend lifespan. Future work will be needed to determine whether mitochondrial permeability can be favorably manipulated in preclinical and clinical studies in order to maximize the beneficial effects of autophagy, promoting healthy aging and reducing morbidity associated with diseases such as ischemic stroke, heart attack, ischemic renal and hepatic disease, and with organ transplantation.
Figure 1. Model of the effects of autophagy on lifespan and health with VDAC1 accumulation and increased mitochondrial permeability (right) versus normal mitochondrial permeability (left).
Acknowledgments
We thank the members of the Soukas Laboratory for helpful discussions, and our collaborators Malene Hansen, Wilhelm Haas, Johannes Kreuzer, Caroline Kumsta, Sudeshna Das, and Kimberli Kamer.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
Reference
- Zhou B, Kreuzer J, Kumsta C, et al. Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell. 2019 Apr 4;177(2):299–314.