
ABSTRACT
More evidence is emerging of the roles long non-coding RNAs (lncRNAs) play as regulatory factors in a variety of biological processes, but the mechanisms underlying the function of lncRNAs in acute myocardial infarction (AMI) have not been explicitly delineated. The present study identified the lncRNA 2810403D21Rik/AK007586/Mirf (myocardial infarction-regulatory factor), that inhibited macroautophagy/autophagy by modulating Mir26a (microRNA 26a). Inhibition of Mir26a led to cardiac injury both in vitro and in vivo, whereas overexpression of Mir26a attenuated ischemic stress-induced cell death by activating autophagy through targeting Usp15 (ubiquitin specific peptidase 15). More importantly, 2810403D21Rik/Mirf acted as a competitive endogenous RNA (ceRNA) of Mir26a; forced expression of 2810403D21Rik/Mirf downregulated Mir26a to inhibit autophagy. In contrast, loss of 2810403D21Rik/Mirf resulted in upregulation of Mir26a to promote autophagy and alleviate cardiac injury, which in turn improved cardiac function in MI mice. This study identified a lncRNA 2810403D21Rik/Mirf that functions as an anti-autophagic molecule via ceRNA activity toward Mir26a. Our findings suggest that knockdown of 2810403D21Rik/Mirf might be a novel therapeutic approach for cardiac diseases associated with autophagy.
Abbreviations
3-MA: 3-methyladenine; AAV-9: adenovirus associated virus-9; agoMir26a: cholesterol-conjugated Mir26a mimic; AMI: acute myocardial infarction; AMO-26a: Mir26a inhibitor; ATG: autophagy related; BECN1: beclin 1; ceRNA: competitive endogenous RNAs; EF: ejection fraction; f-2810403D21Rik/Mirf: fragment encompassing the Mir26a binding site; FS: fraction shortening; GFP-mRFP: a plasmid expressing green fluorescent protein-monomeric red fluorescent protein; lncRNA: long non-coding RNA; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; Mirf: myocardial infarction-regulatory factor; miRNAs: microRNAs; NC: negative control; NMCMs: neonatal mice cardiomyocytes; shRNA: short hairpin RNA; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; TEM: transmission electron microscopy; Usp15: ubiquitin specific peptidase 15.
Introduction
Acute myocardial infarction (AMI), which is induced by acute coronary artery occlusion, is one of the leading causes of death worldwide [Citation1]. Cardiac cell death, along with subsequent excessive inflammation, is the main cause of cardiac injury in AMI, and can eventually lead to heart failure and sudden death [Citation2]. Because cardiomyocytes are terminally differentiated and have little potentiality for division, strategies that prevent the irreversible loss of cardiomyocytes in AMI have great therapeutic value.
Numerous studies have found that autophagy, which is generally thought to be a protective factor against ischemic injury in cardiomyocytes [Citation3,Citation4], is involved in cardiac cell death associated with AMI, though damaged cardiomyocytes exhibit characteristics of macroautophagy/autophagy during heart failure [Citation5]. Our previous study reveals that PPP3/calcineurin (protein phosphatase 3) suppresses cytoprotective autophagy in cardiomyocytes under oxidative stress. Correspondingly, loss of PPP3 restores autophagy and decreases the extent of apoptosis induced by oxidative stress [Citation6]. However, the role of autophagy in the progression of ischemic heart disease is not well understood, and the upstream factors that control autophagy require elucidation.
Non-coding RNAs are a class of RNAs that do not encode proteins [Citation7]. Among non-coding RNAs, microRNAs (miRNAs) are known to participate widely in numerous biological and pathophysiological coronary processes, such as arrhythmia, myocardial infarction, and cardiac fibrosis [Citation8,Citation9]. Our previous study finds that Mir1 is a master regulator for arrhythmia, and inhibition of Mir1 in infarcted rat hearts relieves arrhythmogenesis [Citation9]. Mir26a is a highly conserved miRNA and is dysregulated in a number of cardiovascular diseases. For example, overexpression of Mir26a offers protection against atrial fibrillation [Citation10], cardiac hypertrophy [Citation11] and endothelial cell apoptosis [Citation12]. However, it remains to be determined whether Mir26a is involved in AMI.
Besides miRNAs, it has recently been demonstrated that long non-coding RNAs (lncRNAs) play a crucial role in cardiovascular diseases [Citation13–Citation15]. More interestingly, the cross-regulatory network between miRNAs and lncRNAs has recently been identified, providing insights into the comprehensive functionality of these non-coding transcripts and their potential as therapeutics [Citation16]. However, it remains unclear whether lncRNAs participate in the regulation of cardiomyocyte autophagy.
In the current study we found that Mir26a directly targeted Usp15 (ubiquitin specific peptidase 15), activating autophagy and, thereby, alleviating ischemic stress-induced cardiac injury. Furthermore, we identified lncRNA 2810403D21Rik/AK007586/Mirf (myocardial infarction-regulatory factor), and found that upregulation of this lncRNA induced cardiac injury by inhibiting autophagy through its interactions with Mir26a. More importantly, knockdown of 2810403D21Rik/Mirf mitigated cardiac injury and improved heart function in MI mice. Our results suggest that 2810403D21Rik/Mirf and Mir26a could be novel therapeutic targets for ischemic heart disease.
Results
Cardioprotective effects of Mir26a in MI: role of autophagy
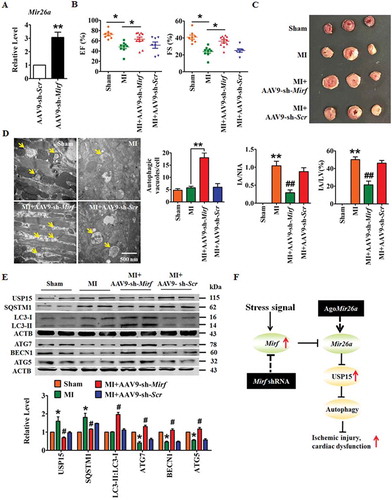
To determine whether Mir26a participates in progression of cardiac injury, we developed a MI model in mice. Expression of Mir26a was decreased in the MI mice after 24 h (), and was also decreased in normal cardiomyocytes treated with H2O2 ().
Figure 1. Knockdown of Mir26a decreases autophagy to impair heart function. (A) Downregulation of Mir26a in the peri-infarct area determined by qRT-PCR. n = 6. *p < 0.05 vs. Sham. (B) Downregulation of Mir26a in cultured neonatal mouse cardiomyocytes (NMCMs) treated with 200 μmol/L H2O2 for 12 h. n = 5. **p < 0.01 vs. Control. (C) Knockdown of Mir26a inhibited autophagic flux in NMCMs. NMCMs were transfected with AMO-26a for 24 h, followed by transfection of tandem-LC3 construct (GFP-mRFP-LC3) for another 24 h. The images were obtained using a confocal microscope. The yellow and red puncta represent autophagosomes and autolysosomes, respectively. The data were obtained from 3 independent experiments and 10 cells were scored in each experiment. *p < 0.05. NC, negative control of AMO-26a. (D) Knockdown of Mir26a increased the level of SQSTM1/p62 protein and reduced the expression of LC3-II, as determined by western blot. n = 5. *p < 0.05 vs. Control. NC, negative control of AMO-26a. (E) Downregulation of Mir26a in Mir26a KD mice. n = 6. **p < 0.01 vs. WT. (F) Knockdown of Mir26a impaired heart function in mice. n = 15. **p < 0.01 vs. WT. (G) Knockdown of Mir26a resulted in the deregulation of autophagy-related proteins in mouse hearts. n = 6. *p < 0.05 vs. WT.
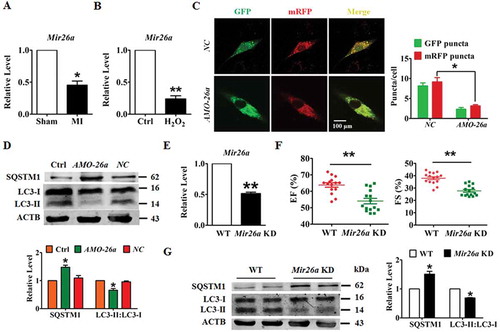
To examine the role of Mir26a in the pathogenesis of ischemic injury in neonatal mice cardiomyocytes (NMCMs), we applied an Mir26a inhibitor (AMO-26a) to inhibit expression of endogenous Mir26a. Meanwhile, a plasmid expressing green fluorescent protein/monomeric red fluorescent protein (GFP-mRFP) in tandem tagged MAP1LC3/LC3 protein were transfected into NMCMs to assess the influence of Mir26a inhibition on the final step of autophagic breakdown (autophagosome formation and the autophagosome-lysosome fusion process). Silencing Mir26a reduced the number of both autophagosomes (yellow puncta) and autolysosomes (red puncta) in NMCMs (). Consistent with this result, knockdown of Mir26a retarded expression of LC3-II and increased expression of SQSTM1/p62 ().
To further investigate the function of Mir26a in progression of myocardial infarction, we generated a Mir26a sponge transgenic mouse (Mir26a KD mice) to knockdown expression of endogenous Mir26a. As shown in , the level of Mir26a was markedly decreased in the Mir26a KD mice. Moreover, silencing of endogenous Mir26a reduced the ejection fraction and fraction shortening (EF and FS, respectively; ), indicating that Mir26a inhibition impaired the left ventricular function. Furthermore, inhibition of Mir26a blocked the formation of autophagosomes and caused downregulation of autophagy in the hearts of Mir26a KD mice ().
These results indicated that knockdown of Mir26a resulted in heart defects in mice. To examine whether overexpression of Mir26a has a beneficial effect on the pathogenesis of ischemic injury in cardiomyocytes, we transfected Mir26a mimics into NMCMs. Overexpression of Mir26a alleviated H2O2-induced inhibition of cell viability in NMCMs, inhibition which was abolished by the autophagy antagonist 3-methyladenine (3-MA). No significant changes were elicited in the negative control (). Meanwhile, enhanced expression of Mir26a promoted formation of autophagosomes and autophagosome-lysosome fusion. Notably, this effect was attenuated if autophagy had been inhibited by 3-MA ( and Figure S1A). To further examine whether autophagy is necessary for the protective effect of Mir26a, autophagy flux in NMCMs was blocked by application of siRNA against Atg5 or treated with either chloroquine or pepstatin A-E64d. Blocking autophagy diminished the pro-survival and pro-autophagic effects of Mir26a in H2O2-treated cardiomyocytes (Figure S1B,C).
Figure 2. Forced expression of Mir26a protects cardiomyocytes from injury by activating autophagy. (A) Overexpression of Mir26a alleviated H2O2-induced inhibition of cell viability in NMCMs. This effect was reversed by application of 3-MA, an autophagic antagonist. n = 8. **p < 0.01 vs. Control; ##p < 0.01 vs. H2O2; &&p < 0.01 vs. H2O2+ Mir26a. MirNC, negative control of Mir26a. (B) Forced expression of Mir26a restored the inhibition of autophagy induced by H2O2. Data were obtained from 3 independent experiments and 10 cells were scored in each experiment. **p < 0.01. MirNC, negative control of Mir26a. (C) Overexpression of Mir26a improved heart function (n = 9. *p < 0.05 vs. Sham; #p < 0.05 vs. MI) and reduced the infarct size (both IA/NIA and IA/LV) in MI mice (D) (n = 9. **p < 0.01 vs. MI+ago MirNC). (E) Overexpression of Mir26a enhanced autophagic activity in MI mice. n = 5. **p < 0.01 vs. MI. AgomiR-NC, negative control of agoMir26a. (F) Overexpression of Mir26a normalized the dysregulated expression of autophagy-related proteins in MI mice. n = 6. *p < 0.05 vs. Sham; #p < 0.05 vs. MI. Ago MirNC, negative control of agoMir26a.
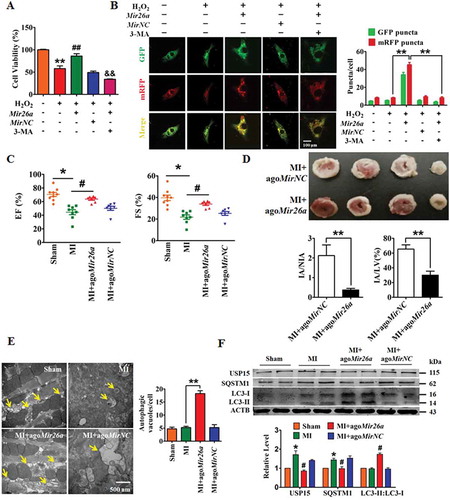
To assess whether Mir26a activates autophagy to mitigate ischemic myocardial injury we injected a cholesterol-conjugated Mir26a mimic (agoMir26a) through the tail-vein into mice for overexpression, and then created MI conditions in the mice. As expected, overexpression of Mir26a improved heart function and reduced infarct size in the model MI mouse (,). Promotion of autophagy by Mir26a in MI mice was observed directly by transmission electron microscopy (). In accordance with these results, western blot showed that forced expression of Mir26a promoted expression of proteins related to autophagy in MI mice ().
Signaling mechanisms for the cardioprotective effects Mir26a: repression of Usp15
To delineate the downstream signaling mechanisms responsible for the beneficial effects of Mir26a, the TargetScan miRNA database (http://www.targetscan.org/vert_71/) was used for identification of autophagy-related genes that could be targeted by Mir26a. Computational -analyzes identified that the gene encoding Usp15, which is broadly conserved among human, mouse and rat species, contains the seed sequence for Mir26a (). Usp15 has previously been reported to antagonize mitophagy in fibroblasts of patients [Citation17], and, intriguingly, was upregulated in the border zone of MI hearts and in the cardiomyocytes exposed to H2O2 (,).
Figure 3. Usp15 is a direct target of Mir26a. (A) Sequence alignment showing complementarity between mouse, rat, and human Mir26a and Usp15 genes. The matched base pairs in the seed region are outlined by red and green. (B) Increased expression of USP15 in infarct border area in MI mice. n = 6. **p < 0.01 vs. Sham. (C) Increased expression of USP15 in NMCMs after treatment with 200 μmol/L H2O2 for 12 h. n = 6. (D) Luciferase reporter activity of chimeric vectors carrying a luciferase gene and a fragment of the Usp15 3′-UTR containing the wild-type or mutant binding sites for Mir26a were detected in HEK293 cell line. n = 6. *p < 0.05. MirNC, negative control of Mir26a. (E) Overexpression of Mir26a depressed protein levels of USP15 (n = 6. *p < 0.05 vs. Control. MirNC, negative control of Mir26a; NC, negative control of AMO-26a), (F) but did not affect its mRNA levels (n = 6). (G) Upregulation of USP15 in Mir26a KD mice, the three lanes of WT and KD correspond to three different mice. n = 6. **p < 0.01 vs. Sham. (H) Forced expression of Mir26a inhibited expression of SQSTM1 and promoted the transition of LC3-I into LC3-II. n = 6. *p < 0.05 vs. Control. MirNC, negative control of Mir26a.
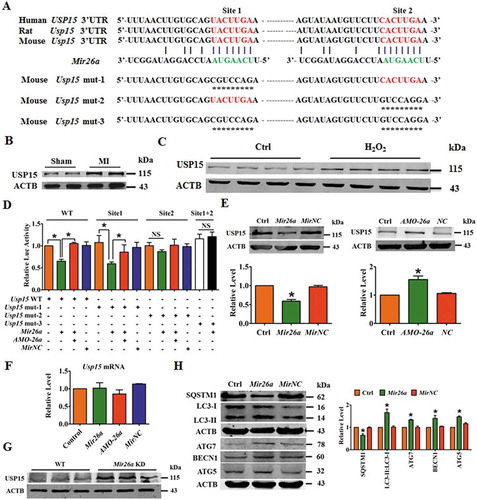
Subsequent experiments validated the direct regulatory effect of Mir26a on Usp15. Luciferase expression vectors carrying the wild-type (WT) Mir26a binding sites or one of the mutated versions of the Mir26a binding sites () were constructed. Overexpression of Mir26a by co-transfection inhibited luciferase activity by the luciferase vectors carrying the WT Usp15 binding sites or the mutation in binding site 1 (mut-1), but not by the luciferase vectors carrying mutations in site 2 (mut-2) or both sites 1 and 2 (mut-3) (). This suggests that the binding site 2 of Usp15 might play the major role in Mir26a regulation of Usp15. Moreover, the level of USP15 proteins was reduced by Mir26a but increased by AMO-26a, although neither affected transcription of Usp15 (,). Consistent with the in vitro study, expression of USP15 was also increased in Mir26a KD mice (). More importantly, overexpression of Mir26a in NMCMs inhibited expression of SQSTM1/p62 and promoted expression of LC3-II, ATG7, BECN1/Beclin1, and ATG5 ().
To determine whether Mir26a promotes cardiac autophagy through Usp15, we constructed siRNA against Usp15 to inhibit endogenous Usp15 expression. Inhibition of Mir26a caused a decrease in cell viability that could be rescued by Usp15 knockdown (Figure S2A). Silencing Usp15 also attenuated the inhibitory effect of Mir26a inhibition on autophagosomes and autolysosomes formation (Figure S2B). Furthermore, western blot analysis showed that knockdown of Mir26a resulted in higher expression of USP15, SQSTM1 and downregulation of LC3-II:LC3-I, ATG7, BECN1, and ATG5. Changes in protein expression by Mir26a knockdown were all relieved by Usp15/USP15 inhibition (Figure S2C). These results show that Mir26a regulated autophagy in cardiomyocytes by targeting Usp15.
LncRNA 2810403D21Rik/Mirf is able to bind to and regulate activity of Mir26a
Evidence is mounting that lncRNAs act as ceRNA (competitive endogenous RNA) that competitively bind to and restrain the activity of miRNAs [Citation18]. To determine whether lncRNAs inhibit expression of Mir26a in the murine MI model, we used the miRanda database (http://www.microrna.org/microrna/getMirnaForm.do) to identify lncRNA in mice with Mir26a biding sites. The candidate lncRNAs were filtered based on their expression profiles in the MI mouse (GSE46395) and their expression profiles were tested by qRT-PCR. The lncRNA AK007586 (Ensembl ID: ENSMUSG00000086629), which was significantly upregulated in the MI mice (), was selected for further analysis and designated 2810403D21Rik/Mirf. Expression of 2810403D21Rik/Mirf was significantly increased in NMCMs after treatment with H2O2 (). In addition, we found that 2810403D21Rik/Mirf was mainly located in the cytoplasm of NMCMs (). Over-expression of 2810403D21Rik/Mirf inhibited expression of Mir26a in NMCMs (), whereas silencing of 2810403D21Rik/Mirf by specific short hairpin RNA (shRNA) resulted in Mir26a upregulation ().
Figure 4. LncRNA 2810403D21Rik/Mirf regulates the expression and activity of Mir26a. (A) Upregulation of 2810403D21Rik/Mirf in MI mice. n = 6. **p < 0.01 vs. Sham. (B) Upregulation of 2810403D21Rik/Mirf in NMCMs after treatment with 200 μmol/L H2O2 for 12 h. n = 6. **p < 0.01 vs. Control. (C) In situ hybridization showed the location of 2810403D21Rik/Mirf in NMCMs. (D) Overexpression of 2810403D21Rik/Mirf downregulated Mir26a. n = 6. *p < 0.05 vs. Control. pcDNA3.1, negative control. (E) Silencing of 2810403D21Rik/Mirf by the specific shRNA upregulated Mir26a. The black bar indicates three different shRNA sequences against 2810403D21Rik/Mirf. n = 6. **p < 0.01 vs. sh-Scramble. sh-Scramble, negative control of sh-2810403D21Rik/Mirf. (F) 2810403D21Rik/Mirf contained a sequence domain complementary to the seed motif of Mir26a. (G and H) 2810403D21Rik/Mirf binds to Mir26a and regulates its activity in NMCMs. n = 6. *p < 0.05, **p < 0.01. (I) Luciferase reporter activities of chimeric vectors carrying luciferase gene and a fragment of 2810403D21Rik/Mirf RNA containing WT binding site or mutant binding site for Mir26a were detected in HEK293 cell line. n = 6. *p < 0.05. pcDNA3.1, negative control of 2810403D21Rik/Mirf. sh-Scramble, negative control of sh-2810403D21Rik/Mirf. MirNC, negative control of Mir26a. (J) RNA affinity isolation in NMCMs using biotin-labeled 2810403D21Rik/Mirf probes. Western blot was used to detected enrichments of AGO2. n = 3. (K) Mir26a was pulled down by 2810403D21Rik/Mirf probe, and the expression of Mir26a was analyzed by qRT-PCR. Cardiomyocytes were transfected with biotinylated Mir26a and then harvested for biotin-based affinity-isolation assay. n = 6. **p < 0.01. Scramble, negative control of 2810403D21Rik/Mirf. (L) 2810403D21Rik/Mirf was pulled down by Mir26a as analyzed by real-time RT-PCR. n = 6. **p < 0.01. MirNC, negative control of Mir26a. (M) The product of real-time RT-PCR was identified by agarose gel electrophoresis. MirNC, negative control of Mir26a.
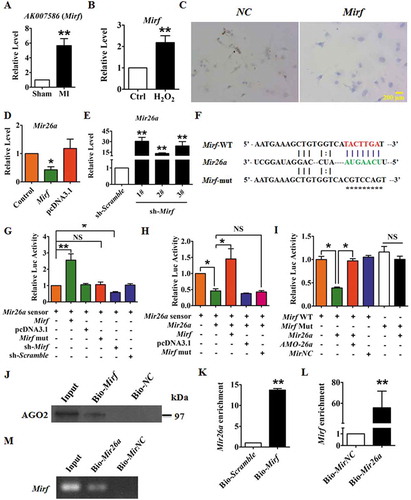
To further characterize the relationship between 2810403D21Rik/Mirf and Mir26a, a Mir26a sensor luciferase vector containing a fragment of the 2810403D21Rik/Mirf sequence that included the Mir26a binding site incorporated into the 3′-UTR of the luciferase gene was constructed (). Overexpression of 2810403D21Rik/Mirf increased luciferase activity and silencing of 2810403D21Rik/Mirf had the opposite effect (), indicating that 2810403D21Rik/Mirf could bind to Mir26a and reduce the availability of functional Mir26a. Furthermore, Mir26a significantly inhibited the luciferase activity of the sensor vector, inhibition which was alleviated by co-expression of WT 2810403D21Rik/Mirf. The mutated 2810403D21Rik/Mirf (mut-2810403D21Rik/Mirf), which lacked the Mir26a binding site, failed to alleviate inhibition of luciferase activity by Mir26a (). More importantly, overexpression of Mir26a suppressed luciferase activity of the WT 2810403D21Rik/Mirf luciferase vector, but not of the mut-2810403D21Rik/Mirf luciferase vector ().
Next, we constructed a biotin-labeled 2810403D21Rik/Mirf-specific probe and performed an RNA affinity-isolation assay to test whether 2810403D21Rik/Mirf could pull down Mir26a. It has been found that miRNA, miRNA target RNA, and AGO2 form the RNA-induced silencing complex (RISC) to mediate miRNA-induced gene silencing [Citation19]. The RNA affinity-isolation assay showed that 2810403D21Rik/Mirf interacted with AGO2 (). Furthermore, qRT-PCR revealed binding of 2810403D21Rik/Mirf to Mir26a (), indicating that Mir26a and 2810403D21Rik/Mirf interacted with AGO2. We also applied a biotin-avidin affinity-isolation system to test whether Mir26a could pull down 2810403D21Rik/Mirf. Cardiomyocytes were transfected with biotinylated Mir26a and then harvested for a biotin-based affinity-isolation assay in which 2810403D21Rik/Mirf was pulled down by Mir26a, as determined by qRT-PCR () and agarose gel electrophoresis (), indicating that the recognition of Mir26a to 2810403D21Rik/Mirf is sequence specific. Taken together these results suggest that 2810403D21Rik/Mirf interacts directly with Mir26a and AGO2 to form RISC and regulate the expression and activity of Mir26a.
LncRNA 2810403D21Rik/Mirf as a regulator of autophagy through the Mir26a-Usp15 axis
NMCMs were transfected with 2810403D21Rik/Mirf to verify that 2810403D21Rik/Mirf contributed to the process of myocardial injury and determine whether this effect was mediated by Mir26a. Overexpression of 2810403D21Rik/Mirf significantly reduced the viability of NMCMs and aggravated H2O2-provoked inhibition of cell viability, while this effect was partly alleviated by Mir26a or si-Usp15 (,). As expected, forced expression of mut-2810403D21Rik/Mirf did not affect cell viability (). Furthermore, forced expression of 2810403D21Rik/Mirf reduced the number of autophagosomes and autolysosomes in NMCMs (), whereas mut-2810403D21Rik/Mirf failed to do so. Overexpression of 2810403D21Rik/Mirf increased expression of SQSTM1 and USP15 and blocked expression of LC3-II, ATG7, BECN1 and ATG5 ( and Figure S3), indicating that 2810403D21Rik/Mirf suppresses autophagy in cardiomyocytes. Moreover, forced expression of Mir26a or silencing Usp15 mitigated the inhibition of autophagy by 2810403D21Rik/Mirf (, and Figure S3). These results suggest that 2810403D21Rik/Mirf limits the functionality of the Mir26a/Usp15 axis, thereby depressing autophagy and causing myocardial injury via mitigation of intrinsic cardioprotective activity.
Figure 5. 2810403D21Rik/Mirf modulated autophagy by regulating Mir26a and Usp15, promoting cardiac injury in NMCMs. (A) Overexpression of 2810403D21Rik/Mirf decreased viability of NMCMs; this effect was attenuated by Mir26a or si-Usp15. n = 5. *p < 0.05. pcDNA3.1, negative control of 2810403D21Rik/Mirf. MirNC, negative control of Mir26a. si-Scramble, negative control of si-Usp15. (B) 2810403D21Rik/Mirf aggravated H2O2-induced cardiac injury in NMCMs. n = 6. *p < 0.05. pcDNA3.1, negative control of 2810403D21Rik/Mirf. MirNC, negative control of Mir26a. (C) Overexpression of 2810403D21Rik/Mirf blocked the autophagic activity of NMCMs. n = 10. *p < 0.05, **p < 0.01. pcDNA3.1, negative control of 2810403D21Rik/Mirf. MirNC, negative control of Mir26a. si-Scramble, negative control of si-Usp15. (D) Overexpression of 2810403D21Rik/Mirf produced anti-autophagic action through regulation of various relevant proteins, which was abolished by Mir26a. n = 5. *p < 0.05 vs. pcDNA3.1; #p < 0.05 vs. 2810403D21Rik/Mirf. pcDNA3.1, negative control of 2810403D21Rik/Mirf. MirNC, negative control of Mir26a.
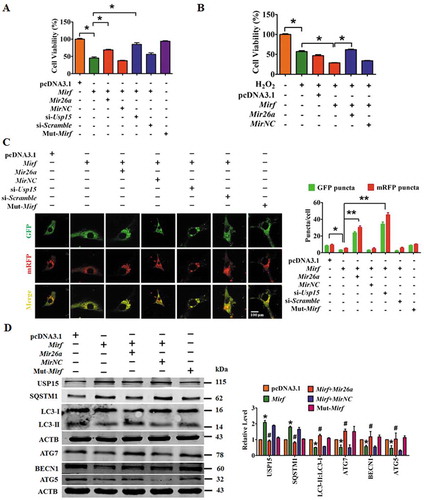
We then examined the potential for silencing of 2810403D21Rik/Mirf to exert a protective effect against cardiac injury in NMCMs exposed to H2O2. H2O2 attenuated cell viability, an effect that was alleviated by silencing 2810403D21Rik/Mirf, whereas knockdown of Mir26a or inhibition of autophagy using 3-MA abolished the beneficial action of 2810403D21Rik/Mirf knockdown (). Consistently, blocking autophagy using si-Atg5, chloroquine or pepstatin A/E64d consistently restored the regulatory action of 2810403D21Rik/Mirf knockdown on cell viability in H2O2-treated NMCMs (). Moreover, a western blot showed that knockdown of 2810403D21Rik/Mirf restored the H2O2-induced upregulation of autophagy-related proteins SQSTM1 and USP15, and promoted expression of LC3-II, ATG7, BECN1 and ATG5. These regulatory changes were abated by addition of AMO-26a or 3-MA (). Blocking autophagy using si-Atg5, chloroquine or pepstatin A/E64d consistently restored the regulatory action of 2810403D21Rik/Mirf on autophagy-related proteins in H2O2-treated NMCMs (Figure S4). In agreement with these results, silencing of 2810403D21Rik/Mirf mitigated the H2O2-induced inhibition of autophagy in NMCMs, inhibition which was effectively restored by addition of the Mir26a inhibitor or 3-MA (). These data show that silencing of 2810403D21Rik/Mirf protected against H2O2-induced cardiomyocytes injury by activating Mir26a and autophagy.
Figure 6. Inhibition of 2810403D21Rik/Mirf antagonizes H2O2-induced injury in NMCMs through modulation of autophagy by increasing Mir26a. (A) Silencing of 2810403D21Rik/Mirf alleviated H2O2-induced cardiac injury, which was reversed by knockdown of Mir26a by AMO-26a or inhibition of autophagy by 3-MA in NMCMs. n = 6. *p < 0.05. sh-Scramble, negative control of sh-2810403D21Rik/Mirf. NC, negative control of AMO-26a. (B) Inhibition of autophagy by si-Atg5, chloroquine or pepstatin A/E64d mitigated the pro-survival effects of 2810403D21Rik/Mirf silencing in H2O2-treated NMCMs. n = 6. *p < 0.05. (C) Knockdown of 2810403D21Rik/Mirf restored abnormal expression of autophagy-related proteins induced by H2O2. n = 5. *p < 0.05 vs. or Control; #p < 0.05 vs. H2O2; &p < 0.05 vs. H2O2+ sh-2810403D21Rik/Mirf. sh-Scramble, negative control of sh-2810403D21Rik/Mirf. NC, negative control of AMO-26a.(D) Silencing of 2810403D21Rik/Mirf promoted autophagy through increasing expression of Mir26a. The data were obtained from 3 independent experiments and 10 cells were scored in each experiment. **p < 0.01. sh-Scramble, negative control of sh-2810403D21Rik/Mirf. NC, negative control of AMO-26a.
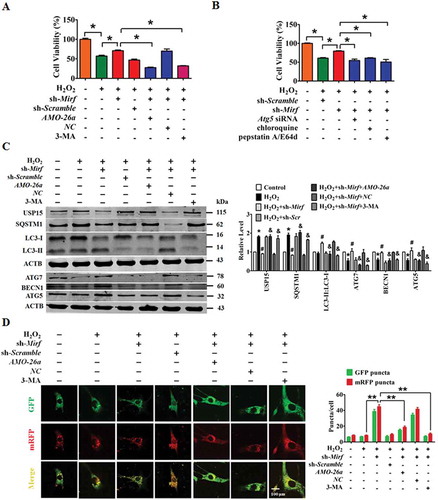
To identify the sequence domain of 2810403D21Rik/Mirf responsible for the observed ceRNA action toward Mir26a, we synthesized a 25 nt 2810403D21Rik/Mirf fragment (f-2810403D21Rik/Mirf) encompassing the Mir26a binding site (see the 2810403D21Rik/Mirf-WT in ). Similar to the full-length 2810403D21Rik/Mirf, f-2810403D21Rik/Mirf retained the ability to inhibit the viability of cardiac cells and the effect was abrogated by Mir26a (). Meanwhile, overexpression of f-2810403D21Rik/Mirf significantly reduced punctate accumulations of GFP-mRFP-LC3, whereas co-transfection of Mir26a alleviated the effects of f-2810403D21Rik/Mirf and promoted accumulation of autophagosomes and autolysosomes in NMCMs (). Moreover, forced expression of f-2810403D21Rik/Mirf increased expression of SQSTM1 and USP15, and inhibited expression of LC3-II, ATG7, BECN1 and ATG5; effects which were almost entirely reversed by Mir26a (). These results suggest that f-2810403D21Rik/Mirf could mimic the actions of 2810403D21Rik/Mirf on autophagy and cell injury, indicating that the Mir26a binding site is the functional domain of 2810403D21Rik/Mirf.
Figure 7. Overexpression of f-2810403D21Rik/Mirf provokes myocardial injury in cultured cardiomyocytes by inhibiting endogenous Mir26a. (A) Introduction of f-2810403D21Rik/Mirf, a 25 nt fragment of the 2810403D21Rik/Mirf sequence which contained the binding sites of Mir26a, inhibits the viability of NMCMs. n = 6. *p < 0.05 vs. Scramble; #p < 0.05 vs. f-2810403D21Rik/Mirf. Scramble, negative control of f-2810403D21Rik/Mirf. MirNC, negative control of Mir26a. (B) A confocal assay showed the anti-autophagic effects of f-2810403D21Rik/Mirf in NMCMs and could be abrogated by introduction of Mir26a. The data were obtained from 3 independent experiments and 10 cells were scored in each experiment. *p < 0.05, **p < 0.01. Scramble, negative control of f-2810403D21Rik/Mirf. MirNC, negative control of Mir26a. (C) Forced expression of f-2810403D21Rik/Mirf disrupts the expression of autophagy-related proteins by regulation of Mir26a. n = 6. *p < 0.05 vs. Scramble; #p < 0.05 vs. f-2810403D21Rik/Mirf. Scramble, negative control of f-2810403D21Rik/Mirf. MirNC, negative control of Mir26a.
Silencing of 2810403D21Rik/Mirf mitigates myocardial injury and protects heart function in mice subjected to myocardial infarction
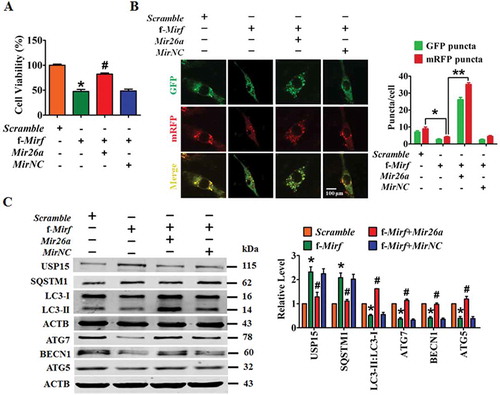
We injected adenovirus associated virus (AAV)-9 carrying the 2810403D21Rik/Mirf-specific shRNA into mice and evaluated the effects of 2810403D21Rik/Mirf inhibition on cardiac damage in a mouse model of MI. Knockdown of 2810403D21Rik/Mirf through tail-vein injection of AAV9-sh-2810403D21Rik/Mirf induced upregulation of Mir26a in WT mice (), consistent with data from in vitro experiments. Moreover, loss of 2810403D21Rik/Mirf improved the heart function of and diminished the infarct size in mice after MI (,). Meanwhile, knockdown of 2810403D21Rik/Mirf increased the number autophagic vesicles (arrows) and promoted cardiac autophagy in MI hearts (). In addition, a western blot showed that inhibition of 2810403D21Rik/Mirf in MI mice restored the upregulation of SQSTM1 and USP15, rescued the downregulating of ATG7, BECN1 and ATG5, and promoted the transition of LC3-I into LC3-II (). These results suggest that silencing of 2810403D21Rik/Mirf might regulate autophagy and afford protective effects against ischemic damage of cardiac function.
Figure 8. Silencing of 2810403D21Rik/Mirf alleviates myocardial injury and protects heart function in a mouse model of MI. (A) Upregulation of Mir26a in the hearts of WT mice after inhibition of 2810403D21Rik/Mirf. n = 6. **p < 0.01 vs. AAV9-sh-Scr. AAV9-sh-Scr, negative control of AAV9-sh-2810403D21Rik/Mirf. Silencing of 2810403D21Rik/Mirf by AAV9-sh-2810403D21Rik/Mirf improved heart function (B) (n = 11. *p < 0.05), reduced infarct size (C) (n = 5. **p < 0.01 vs. Sham; ##p < 0.01 vs. MI) in MI mice. Transmission electron microscopy (D) (n = 5. **p < 0.01) and western blot assay (E) (n = 6. *p < 0.05 vs. Sham; #p < 0.05 vs. MI) show the pro-autophagic effects of 2810403D21Rik/Mirf inhibition. The yellow arrows point to autophagic vesicles. AAV9-sh-Scr, negative control of AAV9-sh-2810403D21Rik/Mirf. (F) Proposed autophagic signaling mechanism for 2810403D21Rik/Mirf and Mir26a during MI. Cardiac stress inhibits expression of Mir26a by increasing the expression of 2810403D21Rik/Mirf and blocks autophagy, resulting in post-transcriptional de-repression of USP15 and leading to ischemic injury and cardiac dysfunction, respectively.
Discussion
The present study established that the lncRNA 2810403D21Rik/Mirf was a damaging factor in MI, whereas Mir26a was a protective miRNA in MI. That is, 2810403D21Rik/Mirf exaggerated and Mir26a attenuated ischemic cardiac injury through modulation of autophagy. Moreover, reciprocal alterations in the expression of these two non-coding RNAs (upregulation of 2810403D21Rik/Mirf and downregulation of Mir26a) in the ischemic zones of MI hearts were observed. The opposite functionalities and reciprocal expression of 2810403D21Rik/Mirf and Mir26a could be well explained by ceRNA regulation, in which the former acts as a sponge to limit the availability of the latter. In addition, we found that Mir26a plays a protective role in ischemic injury in cardiomyocytes at least in part by directly targeting regulation of USP15. Notably, both silencing 2810403D21Rik/Mirf and overexpressing Mir26a reduced the infarcted area and improved cardiac function. These findings lead to the conclusion that 2810403D21Rik/Mirf is a novel anti-autophagic lncRNA that absorbs the pro-autophagic miRNA Mir26a, which can exaggerate ischemic cardiac injury by relieving repression of anti-autophagic protein USP15 (). Both 2810403D21Rik/Mirf and Mir26a might be considered as potential non-coding RNA targets for the treatment of cardiac diseases associated with autophagy.
Cardiovascular disease, especially AMI, is one of the major causes of death worldwide and its morbidity is increasing year by year [Citation20]. Though our understanding of the pathogenesis of AMI has improved, there are still no effective methods for prevention and treatment. Thus, explorations of new therapeutic approaches at the molecular level are necessary. For the past decade, miRNAs have received worldwide attention for their active participation in various physiological and pathological cardiac processes [Citation21]. Specifically, Mir26a has been shown to act against atrial fibrillation and cardiac fibrosis [Citation10,Citation22], hence we investigated whether Mir26a has a similarly protective role in AMI. In this work we found that expression of Mir26a was decreased in mouse models of ischemic injury and in cardiomyocytes injured by H2O2. Overexpressing Mir26a improved the cardiac function and cardiomyocytes viability both in vivo and in vitro, respectively. Thus, our study expands the range of known protective effects of Mir26a against ischemic heart injuries.
Autophagy is an important process in aging, inflammation, tumor metabolism and cardiovascular diseases [Citation23]. In cardiomyocytes autophagy maintains the renewal of mitochondria, helping to meet the energy demands of the heart [Citation24]. Because of their non-dividing nature, cardiomyocytes are highly dependent on autophagy for the renewal of non-functional mitochondria, proteins and other organelles. However, the roles of autophagy in MI are still a matter of debate. Autophagy can be either protective or maladaptive in MI, depending on the context of stress. Some studies have found that autophagy is detrimental and that abnormal autophagy is associated with myriad diseases, including cardiovascular disease [Citation25,Citation26]. However, an increasing amount of evidence has demonstrated that activation of autophagy in cardiomyocytes is an adaptive response to stimuli and that inhibiting autophagy accelerates heart failure [Citation27]. Zhang et al. found that BAG3 promotes cell proliferation and inhibits apoptosis by activating autophagy in cardiomyocytes injury induced by hypoxia [Citation28]. Also, cardiac-specific over-expression of Mir221, which inhibits autophagy by modulating the CDKN1B-CDK2-MTOR axis, in mice promotes heart failure [Citation29]. Our present work focused on identifying the role of Mir26a in regulating autophagy, finding that Mir26a enhanced cell viability and suppressed cardiac injury by autophagy at least in part by targeting regulation of Usp15.
It has been well demonstrated that lncRNAs participate in multiple human diseases by acting as sponges to reduce the expression levels of miRNAs and correspondingly increase the levels of target genes [Citation30]. The lncRNA Chast (cardiac hypertrophy-associated transcript) is reported to impede cardiomyocyte autophagy and drive cardiac hypertrophy [Citation31] and another, Chaer1 (cardiac hypertrophy associated epigenetic regulator 1), negatively regulates AEBP2/PRC2 function and induces cardiac hypertrophy by defining an epigenetic checkpoint [Citation32]. LOC100129973 has been found to suppress apoptosis by sponging Mir4707-5p and Mir4767 in vascular endothelial cells [Citation33]. These reports demonstrate that lncRNAs can interact with miRNAs to exert biological functions. In the present study, we found that lncRNA 2810403D21Rik/Mirf is a distinct ceRNA for Mir26a. Expression of 2810403D21Rik/Mirf was significantly increased in MI hearts and H2O2 treated cardiomyocytes, suggesting meaningful participation of 2810403D21Rik/Mirf in MI. Theoretically, ceRNAs should not affect the level of miRNA expression, but our results, as well as other studies investigating interactions between lncRNAs and miRNAs, suggest that lncRNAs not only affect the activity of target miRNAs but also regulate their expression [Citation34,Citation35]. It is not likely that ceRNA regulation is the only mechanism by which lncRNAs affect levels of miRNAs. We concluded that lncRNA 2810403D21Rik/Mirf regulated Mir26a only partially through ceRNA, but the other mechanisms of Mir26a inhibition by 2810403D21Rik/Mirf need to be studied further.
USP15 is a member of a family of deubiquitinating enzymes, which are shown to inhibit mitochondrial autophagy by the activation of PINK1/PRKN pathway [Citation36]. PINK1 can be rapidly degraded by proteolytic enzymes in healthy mitochondria but the proteolytic enzyme responsible for PINK1 degradation is inhibited in injured mitochondria, leading PINK1 to gather on damaged mitochondria and recruit PRKN (parkin RBR E3 ubiquitin protein ligase) from the cytoplasm into the mitochondria. The resulting increased E3 ubiquitin ligase activity induces the ubiquitination of mitochondrial matrix proteins, which leads to accumulation of SQSTM1 in the mitochondrial matrix. SQSTM1 combines with LC3 to initiate autophagy. It has been confirmed that mammalian MFN (mitochondrial fusion proteins) 1 and 2, and the VDAC1 (voltage-dependent anion channel protein 1) are substrates for ubiquitination by PRKN. PRKN degrades MFN1 and MFN2 by ubiquitination and further inhibits the fusion of damaged and healthy mitochondria, activating mitochondrial autophagy to remove damaged mitochondria. Some deubiquitinating enzymes, such as USP8, USP15, and USP30, can remove the ubiquitin chain from PRKN or its substrate, thereby inhibiting the occurrence of mitochondrial autophagy [Citation17,Citation37,Citation38]. USP15 has been reported to antagonize mitophagy, a subtype of autophagy, in fibroblasts in patients [Citation17]. The present study found that Usp15 was a potential target of Mir26a and mediated the pro-autophagic and cardio-protective effects of Mir26a against ischemic injury.
Generally, the expression level lncRNAs is much lower in tissues [Citation39], but the biological functions of lncRNAs are not limited by their low expression. We found that 2810403D21Rik/Mirf regulated expression of Mir26a and overexpression of 2810403D21Rik/Mirf increased the level of USP15, likely due to repression of Mir26a. 2810403D21Rik/Mirf suppressed the autophagy in cardiomyocytes and knockdown of 2810403D21Rik/Mirf protected the heart and cardiomyocytes against cardiac injury by enhancing autophagy under both in vivo MI and in vitro H2O2 conditions. The present study focused mainly upstream, on Mir26a modulation of cardiac autophagy and the interactions between 2810403D21Rik/Mirf and Mir26a. Downstream, we have shown that Mir26a may activate autophagy by targeting Usp15, since USP15 can inhibit mitophagy. This mechanism will be studied in future work.
In short, the present work identified a novel anti-autophagic lncRNA, 2810403D21Rik/Mirf, which exaggerated ischemic cardiac injury by modulating Mir26a levels via ceRNA regulation. Thus, adjusting expression of 2810403D21Rik/Mirf may provide a novel approach for prevention and treatment of AMI.
Materials and methods
Animal treatment
Male C57BL/6 mice weighing 20–30 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Cholesterol-conjugated Mir26a mimic (agoMir26a) (RiboBio Company, Ltd., Guangzhou, China) was injected through tail-vein at a dosage of 80 mg/kg/d in 0.2 ml saline once a day for three consecutive days, followed by creation of a mouse MI model on day 4. To evaluate the role of 2810403D21Rik/Mirf in MI, adeno-associated virus 9 carrying the 2810403D21Rik/Mirf shRNA (AAV9-sh-2810403D21Rik/Mirf) (Biowit Technology, Shenzhen, China) or its scramble control construct (AAV9-sh-Scramble) was injected once through tail-vein at a dosage of 1x10^11 vg per mice followed by creation of a mouse MI model after two weeks.
To establish an MI model, mice were anesthetized with sodium pentobarbitone (40 mg/kg, i.p.) and xylazine (12.5 mg/kg, i.p.). And placed in the supine position with the upper limbs taped to the table. A 1–1.5 cm incision was made along the left side of the sternum. The muscle layers of the chest wall were bluntly dissected to avoid bleeding. The thorax was opened at the point of the most pronounced cardiac pulsation and the right side of the chest was pressed to push the heart out of the thoracic cavity. The left anterior descending coronary artery was occluded and then the chest was closed. All surgical procedures were performed under sterile conditions. After 24 h, mice were anesthetized with sodium pentobarbitone (40 mg/kg, i.p.) and xylazine (12.5 mg/kg, i.p.) and sacrificed with their hearts quickly isolated. The animals in the sham control group underwent open chest procedures without coronary artery occlusion. The procedures were in accordance with the regulations of the Ethics Committee of Harbin Medical University and the Guide for the Care and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, 2011).
Generation of Mir26a knockdown mice
The fragment of Spong-Mir26a (gtcgacgaattcagcctatccttcgtacttgaagcacagcctatccttcgtacttgaagcacagcctatccttcgtacttgaagcacagcctatccttcgtacttgaagcacagcctatccttcgtacttgaagcacagcctatccttcgtacttgaagcacaagcttgaattc) was synthesized by Shanghai Biological Engineering Inc. The fragment was subcloned into the SalI (R4054) and HindIII (R4044) sites of Bluescript vector (Huayueyang Biotechnology, Beijing, VECT4840) carrying the cardiac-specific Myh6/αMHC promoter and human GH (growth hormone) poly(A)+ signal, and then injected into fertilized eggs of BL6/SJL hybrid mice using the pronuclear injection method. Founder mice were then back-crossed with C57/BL6 mice many times to obtain a clean background.
TTC staining
We collected the heart at 24 h after myocardial infarction, and the hearts were washed by cold PBS (Beyotime, C0221A) to drain residual blood. The hearts were cut into 5–6 sections with the same thickness, then soaked in 1% TTC dyeing buffer (Solarbio, G3005), and incubated for 20 min at 37°C. The sections were fixed with 4% paraformaldehyde for 30 min after staining uniformly and take photos for the sections placing in order. And the infarcted area was calculated by ImageJ (NIH).
Echocardiograph
Mice were anesthetized using 4–5% isoflurane (Sigma, T48402)-inhalation anesthesia. Cardiac hemodynamics was analyzed using ultrasound with a Vevo2100 system (VisualSonics, Toronto, Canada) 24 h after MI. Mice were anesthetized and left ventricular function was assessed two-dimensional M-mode recording using a 30 MHZ transducer with continuous oxygen supply. Functional parameters were evaluated including fractional shortening (FS) and ejection fraction (EF).
Cell culture and treatment
Neonatal mice cardiomyocytes (NMCMs) were prepared by the following procedures: 1- to 3-d-old C57BL/6 mouse were anesthetized using 4–5% isoflurane-inhalation anesthesia. Mouse hearts were finely minced and placed together in 0.25% trypsin (Thermofisher, 25200056); 5-bromo-2-deoxyuridine (BrdU; Solarbio, B8010) was added at 0.1 mM to eliminate the cardiac fibroblasts. Dispersed NMCMs were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Biological Industries, 01–052-1-A) containing 10% fetal bovine serum (Biological Industries, 04–001-1C) in a 5% CO2 and 37°C humidified atmosphere. The cells were seeded in 6-well plates, cultured for 48 h and transfected with 100 nM Mir26a or AMO-26a, with X-treme GENE siRNA transfection reagent (Roche, 06366244 001). Forty-eight h after transfection, cells were used for total protein purification. 3-methyladenine (3-MA; 5 mM), an antagonist of autophagy, purchased from Sigma-Aldrich (M9281) was dissolved in the culture medium.
MTT assay
MTT assay was used to determine cell viability. NMCMs were plated in 96-well culture plates. After 48 h of transfection, the cells were incubated with 20 μl MTT (Sigma, M2128) (0.5 mg/ml) and 180 μl DMEM for 4 h. The culture medium was carefully removed and 150 μl DMSO was added to each well to dissolve the formazan. After rocking for 15 mins at room temperature, the absorbance values were read at 490 nm using an Infinite®200PRO microplate spectrophotometer (Tecan, Salzburg, Austria).
Luciferase assay
Usp15 3′-UTR containing the conserved Mir26a binding sites and mutated 3′-UTRs were synthesized by Invitrogen (Carlsbad, CA, USA). The fragment was subcloned into the SacI and HindIII sites downstream of the luciferase gene in the pMIR Reporter (Addgene, MLCC13738). HEK293 cells (American Type Culture Collection, ATCC#1531) were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. HEK293 cells were co-transfected in 24-well plates with WT Usp15 or mutant constructs and Mir26a, negative control or AMO-26a. The Mir26a sensor reporter was constructed according to the method previously described [Citation35]. Briefly, the mouse genomic sequence (200 bp) flanking pre-Mir26a was inserted into the pGL3 vector (Promega, E1761) in reverse orientation downstream of the luciferase gene coding region. NMCMs were co-transfected with Mir26a sensor and si-Mirf/Mirf/Mir26a or their negative control using the Lipofectamine 2000 reagent (Invitrogen,11668019). After 48 h of transfection, the cells were harvested and lysed. Luciferase activity was assayed using the Dual Luciferase Reporter Assay System (Promega, E1910) according to the manufacturer’s instructions. Firefly luciferase values were normalized to Renilla, and relative ratios of Firefly to Renilla activity was reported.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from heart tissues or cultured NMCMs using a Trizol standard protocol. The integrity, quantity, and purity of RNA were examined using Nano-Drop 8000 Spectrophotometer (Thermo Scientific, Wilmington, USA). For each sample, 500 ng of total RNA was converted to cDNA using High Capacity cDNA Reverse Transcription Kit (Transgene, AT341-02). The relative expression levels of mRNAs and miRNAs were quantified by qRT-PCR with SYBR Green I (Roche, 4913914001). Threshold cycle (Ct) was determined and relative mRNA and miRNA levels were calculated based on the Ct values and normalized to Actb or Rnu6 in each sample. And the primers are shown in .
Table 1. The primers in RT-qPCR assays.
Western blot
For western blot analysis, total protein samples were extracted from tissues or cells. Briefly, tissues or cells were lysed with RIPA lysis buffer (Beyotime, P0013K) containing a complete protease inhibitor cocktail (Roche, 04963159001). Protein samples (60 μg) were fractionated on a 10% or 12% SDS-PAGE. After electrophoretic transfer to a Pure Nitrocellulose Blotting membrane (Pall Life Science, 66485), the blots were probed with primary antibodies and ACTB/β-actin (Proteintech, 60008–1-Ig) as an internal control. Primary antibodies against USP15 was purchased from Proteintech (14354–1-AP), antibodies against ATG5 (2630s) and ATG7 (8558s) were obtained from Cell Signaling Technology, and antibodies against BECN1 (057k4822), LC3B (L7543) and SQSTM1 (103M4785V) were obtained from Sigma-Aldrich. The immunoreactivity was detected using Odyssey Infrared Imaging System (Gene Company Limited, Hongkong, China). The bands were quantified by measuring the band density.
Confocal imaging
As previously described [Citation6], NMCMs transfected with tandem GFP-mRFP-LC3 were examined by laser microscopy. The number of GFP and mRFP puncta per cell was manually quantified. At least 10 cells in each of three independent experiments were randomly analyzed.
Transmission electron microscopy
Left ventricular tissues were sliced into ultrathin sections. These sections were fixed with 4% paraformaldehyde and 1% glutaraldehyde in PBS at 4°C overnight. The preparations were washed and dehydrated with increasing concentrations of ethanol, followed by embedding and sectioning. The cardiac slices were examined by a microscopist under an H7650 transmission electron microscope (Hitachi, Japan). For each treatment, the micrographs of at least 15 unique cells were recorded.
Affinity-isolation assay with biotinylated miRNA
Cardiomyocytes were transfected with biotinylated miRNA (50 nM), harvested 48 h after transfection. The cells were washed with PBS and briefly vortexed then incubated in a lysis buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 2.5 mM MgCl2, 0.05% Igepal (Sigma, I8896), 60 U/ml Superase-In (Ambion, CA, USA), 1 mM DTT, protease inhibitors (Roche, 04963159001) on ice for 10 min. The lysates were pre-cleared by centrifugation and 50 ml of the samples were aliquoted for input. The remaining lysates were incubated with M-280 streptavidin magnetic beads (Invitrogen, 14203). To prevent nonspecific binding of RNA and protein complexes, the beads were coated with RNase-free bovine serum albumin (Sigma-Aldrich, C3023) and yeast tRNA (Invitrogen, 15401029). The beads were incubated at 4°C for 3 h, washed twice with ice-cold lysis buffer, then three times with the low salt buffer (0.1% SDS, 1% Triton X-100 (Thermofisher, HFH10), 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl), and once with the high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 500 mM NaCl). The bound RNAs were purified for qRT-PCR using Trizol (Invitrogen, 15596–026). Mir26a affinity-isolation probe, 5ʹ- UUCAAGUAAUCCAGGAUAGGCU-3ʹ, and NC affinity-isolation probe, 5ʹ- UUUGUACUACACAAAAGUACUG- 3ʹ.
RNA affinity-isolation assay with biotinylated DNA probe
The biotinylated DNA probe complementary to 2810403D21Rik/Mirf was synthesized and dissolved in 500 ml of wash/binding buffer (0.5 M NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM EDTA). The probes were incubated with streptavidin-coated magnetic beads (Pierce, 88816) at 25°C for 2 h to generate probe-coated magnetic beads. Cardiomyocyte lysates were incubated with probe-coated beads, and, after washing with the wash/binding buffer, the RNA complexes bound to the beads were eluted and extracted for qRT-PCR analysis. 2810403D21Rik/Mirf affinity-isolation probe,
5ʹ-TTCCTGTAAGGGCAATCGGACCCTTTCCGGTGAGGGAACCTCTCTCCAGACATGATCAAGAGAAACTGGCGAGGGACTGACATGATCTAGCTTACCTTGGGTCACCGAACGGATTTGGCATGTGACATAATTATCCCAGCGATCACTAAAACTGTAAATTTGGATGCAATTAAGAGAGAAAACTCCACACATTTATGGAAATTTAAGATTTGCAGCTTGCTGGCTTGCTGAGTGGAAAATGATTCTTTCCCAGTTCTCCTTGGTTTCTTCCTTTCATGAATTGATTAACACTGAGAGATTATGTTTTAATTATGTCGACTAGAGGTTTTAAATGATTCTAGGTTTCTGTGTCCATTTGTTTTCCTAGGTTTGGGGAATTTGCTACTGCAATATCGTTGTATAGATATTCTTTGCTTTAGTGAGAAACTCAGCTCCTTCTACTCTGTGGTTCTTAGCTTTAGTCTATTGTACACATCACAGAGTTAATGAAAGCTGTGGTCATACTTGATTTTTTCTTCATTACTATTGAAATAAAGCATATCCTTG −3ʹ, and Scramble affinity-isolation probe, 5ʹ- CTTGGTACCGAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTCTGCAGATATCCAGCACAGTGGCGGCCG-3ʹ.
Statistical analysis
All the data were expressed as the mean±SEM. GraphPad Prism 5.0 was used for our statistical analysis. For two group comparisons, Student t-test was used. For multiple group comparisons, one-way ANOVA was used with Bonferroni post-test for comparisons between selected two groups as well as Dunnett post-test for comparisons among all other treatment groups to the corresponding control. A value p < 0.05 was considered statistically significant.
Supplemental Material
Download MS Word (64.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Qian L, Van Laake LW, Huang Y, et al. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560.
- Orogo AM, Gustafsson AB. Cell death in the myocardium: my heart won’t go on. IUBMB Life. 2013;65:651–656.
- Shirakabe A, Zhai P, Ikeda Y, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263.
- Qin Q, Qu C, Niu T, et al. Nrf2-mediated cardiac maladaptive remodeling and dysfunction in a setting of autophagy insufficiency. Hypertension. 2016;67:107–117.
- Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. 2008;103:343–351.
- He H, Liu X, Lv L, et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell Death Dis. 2014;5:e997.
- Cai B, Ma W, Bi C, et al. Long noncoding RNA H19 mediates melatonin inhibition of premature senescence of c-kit(+) cardiac progenitor cells by promoting miR-675. J Pineal Res. 2016;61:82–95.
- Liang H, Zhang C, Ban T, et al. A novel reciprocal loop between microRNA-21 and TGFbetaRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol. 2012;44:2152–2160.
- Yang B, Lin H, Xiao J, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491.
- Luo X, Pan Z, Shan H, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123:1939–1951.
- Liu Y, Wang Z, Xiao W. MicroRNA-26a protects against cardiac hypertrophy via inhibiting GATA4 in rat model and cultured cardiomyocytes. Mol Med Rep. 2016;14:2860–2866.
- Zhang Y, Qin W, Zhang L, et al. MicroRNA-26a prevents endothelial cell apoptosis by directly targeting TRPC6 in the setting of atherosclerosis. Sci Rep. 2015;5:9401.
- Huang ZP, Ding Y, Chen J, et al. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc Res. 2016;112(2):543–554.
- Wang K, Liu F, Liu CY, et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016;23:1394–1405.
- Zhang X, Sha M, Yao Y, et al. Increased B-type-natriuretic peptide promotes myocardial cell apoptosis via the B-type-natriuretic peptide/long non-coding RNA LSINCT5/caspase-1/interleukin 1beta signaling pathway. Mol Med Rep. 2015;12:6761–6767.
- Boon RA, Jae N, Holdt L, et al. Long noncoding RNAs: from clinical genetics to therapeutic targets? J Am Coll Cardiol. 2016;67:1214–1226.
- Cornelissen T, Haddad D, Wauters F, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23:5227–5242.
- Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358.
- Liu J, Li Y, Lin B, et al. HBL1 is a human long noncoding RNA that modulates cardiomyocyte development from pluripotent stem cells by counteracting MIR1. Dev Cell. 2017;42:333–48 e5.
- Ounzain S, Crippa S, Pedrazzini T. Small and long non-coding RNAs in cardiac homeostasis and regeneration. Biochim Biophys Acta. 2013;1833:923–933.
- Barwari T, Joshi A, Mayr M. MicroRNAs in cardiovascular disease. J Am Coll Cardiol. 2016;68:2577–2584.
- Harada M, Luo X, Qi XY, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064.
- Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med. 2013;368:1845.
- Taneike M, Yamaguchi O, Nakai A, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606.
- Huangfu L, Liang H, Wang G, et al. miR-183 regulates autophagy and apoptosis in colorectal cancer through targeting of UVRAG. Oncotarget. 2016;7:4735–4745.
- Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793.
- Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59.
- Zhang J, He Z, Xiao W, et al. Overexpression of BAG3 attenuates hypoxia-induced cardiomyocyte apoptosis by inducing autophagy. Cell Physiol Biochem. 2016;39:491–500.
- Su M, Wang J, Wang C, et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986–999.
- Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–762.
- Viereck J, Kumarswamy R, Foinquinos A, et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra22.
- Wang Z, Zhang XJ, Ji YX, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131–1139.
- Lu W, Huang SY, Su L, et al. Long noncoding RNA LOC100129973 suppresses apoptosis by targeting miR-4707-5p and miR-4767 in vascular endothelial cells. Sci Rep. 2016;6:21620.
- Zhao X, Sun J, Chen Y, et al. lncRNA PFAR promotes lung fibroblast activation and fibrosis by targeting miR-138 to regulate the YAP1-twist axis. Mol Ther. 2018;26:2206–2217.
- Liang H, Pan Z, Zhao X, et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics. 2018;8:1180–1194.
- Wang Y, Serricchio M, Jauregui M, et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy. 2015;11:595–606.
- Durcan TM, Tang MY, Perusse JR, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. Embo J. 2014;33:2473–2491.
- Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375.
- Wang K, Liu CY, Zhou LY, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6:6779.