
ABSTRACT
The mechanism by which ATG2 (ATG2A and ATG2B in mammals) regulates autophagosome biogenesis remains largely unknown. In our recent study, we showed that ATG2A translocates to the mitochondria-associated ER membranes (MAM) to promote phagophore growth during nutrient starvation. Mechanistically, the mitochondrial translocase TOMM40 binds to a C-terminal domain of ATG2A, termed the MAM localization domain (MLD), and mediates its MAM translocation in a manner dependent on the TOMM receptor TOMM70. Moreover, ATG2A associates with ATG9A through its N-terminal domain and this interaction is required for phagophore expansion and efficient autophagic flux. These observations suggest that ATG2 operates a mechanism for phagophore expansion at the MAM through the TOMM40-TOMM70 complex and ATG9 during autophagy.
Macroautophagy (hereafter referred as autophagy) is an evolutionarily conserved intracellular degradation process that plays a vital role in the maintenance of cellular homeostasis. During this catabolic process, a membranous cistern known as the phagophore elongates and sequesters cytoplasmic components to form the double-membrane autophagosome. A series of autophagy-related (ATG) genes have been identified to orchestrate the process of autophagosome biogenesis. ATG2 is the largest ATG protein whose function in autophagosome formation remains unclear. In mammalian cells, there are two functionally redundant ATG2 homologs, namely ATG2A and ATG2B. Using the newly developed HaloTag-LC3 assay together with transmission electron microscopy and correlative light and electron microscopy, our recent study provided mechanistic insights into the role of ATG2A/B in phagophore expansion [Citation1]. We found that ATG2A accumulates on the mitochondria-associated ER membranes (MAM) in response to nutrient starvation. The MAM localization of ATG2A is mediated through the C-terminal 45-amino acid domain (amino acids 1776–1820) that we have termed the MAM localization domain (MLD), comprising amphipathic α-helices that are located within the previously characterized autophagosome and lipid droplet localization region. Through a proteomic analysis, we identified the translocase of outer mitochondrial membrane (TOMM) components TOMM40 and TOMM70 as ATG2A-interacting proteins that bind to the MLD domain and are responsible for the MAM localization of ATG2A. Disruption of the MLD abolishes the ATG2A-TOMM40 interaction and impairs phagophore growth, whereas artificial targeting of an MLD-lacking ATG2A mutant to the MAM via rapamycin-induced dimerization with TOMM70, results in enlarged autophagic membranes in ATG2A/B-deficient cells. These results highlight the importance of the MAM localization of ATG2 in phagophore expansion and support the notion that autophagosomes grow at the ER-mitochondria contact sites.
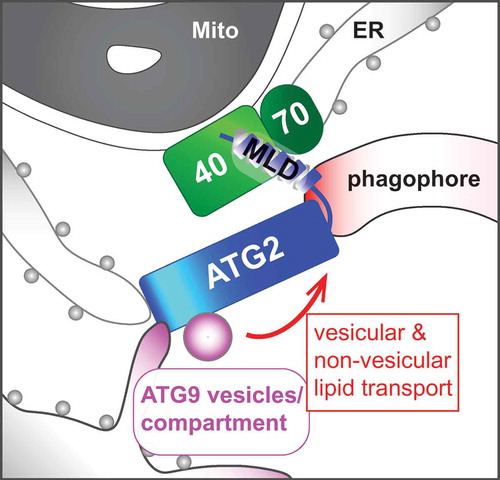
ATG9 is the sole mammalian transmembrane ATG protein required for phagophore elongation. Upon the induction of autophagy, vesicles containing ATG9 bud off from the ATG9 reservoir at the trans-Golgi network or recycling endosomes, and traffic to the autophagosome formation sites where they coalesce with the ER to form tubulovesicular ATG9 compartments near mitochondria. Atg2 interacts with Atg9 to regulate the retrieval of Atg9 from the phagophore assembly site in yeast. Consistently, we found that ATG2A interacts with ATG9A through the N-terminal region (amino acids 237–431). Moreover, whereas the deletion of the N-terminal region impairs the ATG2A-dependent phagophore expansion without affecting the ATG2A-TOMM40 interaction, ATG2A/B loss results in the accumulation of ATG9-positive vesicular structures around autophagic structures at the MAM. Intriguingly, other groups have recently reported an in vitro lipid-transfer activity of the N-terminal region of ATG2. As ATG2 forms a rod-shaped structure capable of bridging highly curved membranes, we propose that ATG2 at the MAM tethers the phagophore to the ATG9 tubulovesicular compartments to mediate non-vesicular and/or vesicular lipid transport for phagophore growth ().
Figure 1. A hypothetical model of ATG2-mediated phagophore expansion. The C-terminal MAM localization domain (MLD) of ATG2 interacts with the TOMM40-TOMM70 complex to facilitate its N-terminal region binding to ATG9-positive donor membranes and ER for non-vesicular and/or vesicular lipid transport into the phagophore. Mito, mitochondrion; 40, TOMM40; 70, TOMM70.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Tang Z, Takahashi Y, He H, et al. TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 2019;28(7):1744–1757 e1745.