
ABSTRACT
Many diseases are caused by aberrant accumulation of certain proteins that are misfolded and cytotoxic, and lowering the level of these proteins provides promising treatment strategies for these diseases. We hypothesized that compounds that interact with both the disease-causing protein and the phagophore (autophagosome precursor) protein LC3 may tether the former to phagophores for subsequent autophagic degradation. If true, this autophagosome-tethering compound (ATTEC) concept could be applied to many disease-causing proteins to treat diseases. We tested this hypothesis in the scenario of Huntington disease (HD), a neurodegenerative disorder that is caused by the mutant HTT (mHTT) protein with an expanded polyglutamine (polyQ) stretch. In our recent study, we designed a small-molecule microarray-based screening and identified four mHTT-lowering compounds that interact with both mHTT and LC3, but not wild-type (WT) HTT. These compounds target mHTT to phagophores for autophagic degradation without influencing the WT HTT level, and rescue HD-relevant phenotypes in HD cells and in vivo in the fly and mouse HD models. Interestingly, these compounds interact with the expanded polyQ stretch directly and are able to reduce other disease-causing proteins with expanded polyQ. In summary, our study provides the initial validation of lowering mHTT by ATTEC, providing entry points to new treatment strategies of HD and similar diseases.
In classical targeted drug discovery, drugs are identified as “inhibitors” or “activators” that alter activities of disease-relevant proteins. This strategy is inapplicable to most neurodegenerative disorders and many other proteinopathies, which are caused by proteins with unknown “activities”. A largely unexplored area is manipulating the levels of disease proteins by low-molecular-weight compounds (compounds for short) to treat these diseases. An emerging approach called “PROTAC” may achieve this goal by enhancing the ubiquitination of disease proteins and targeting them to proteasomal degradation, but the proteasomes alone may be inefficient in degrading certain large disease proteins or aggregates that cause neurodegenerative diseases. In addition, acquired resistance may occur due to genomic alterations of targeted E3 ligase complexes. Another important protein degradation pathway is macroautophagy (referred to as autophagy hereafter), which is a bulk degradation system that engulfs proteins into phagophores for subsequent autophagic degradation. Autophagy is ubiquitously present in eukaryotic cells but its selectivity is limited. Thus, harnessing the power of autophagy to degrade certain target proteins may open new windows for drug discovery. In a recent study, we investigated a potential strategy to discover such compounds in the context of lowering mutant HTT protein, which causes Huntington disease [Citation1].
HD is an incurable monogenetic neurodegenerative disorder caused by the cytotoxicity of mHTT with an expanded polyglutamine (polyQ) stretch (≥36Q). Lowering mHTT levels by genetic or RNA-targeting approaches using large biomolecules or viruses ameliorates pathogenic effects, and has gained interest as a promising treatment strategy for HD. The search for small-molecule compounds to lower levels of mHTT has been less active even though the need for such compounds is acute for the therapeutic treatment of HD. Compounds that enhance autophagy or act through other targets to degrade total HTT may lead to undesired global effects due to increase of autophagy flux and/or reduction of WT HTT, which has critical biological functions. In addition, most HTT-lowering compounds lack in vivo validation and/or have low efficacy (effective at ~10 μM), and act indirectly by modulating other targets to regulate the mHTT level. Thus, mHTT-lowering compounds that target mHTT directly and lower mHTT in an allele-selective manner (i.e., without influencing WT HTT) would be ideal for HD treatment.
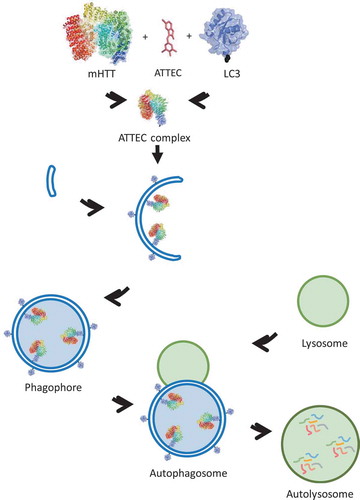
To achieve this goal, we proposed the concept of autophagosome-tethering compounds. We hypothesized that compounds capable of interacting with both mHTT and LC3 may target the former to phagophores for autophagic degradation, based on the fact that the autophagic protein substrates are engulfed within double-membrane phagophores where lipidated LC3 proteins are associated (). In addition, we can then perform counter-screens to exclude WT HTT-interacting compounds, and identify those compounds that only interact with mHTT and LC3, offering allele-selective degradation of mHTT. We thus carried out small-molecule microarray-based screening for desired compounds, and utilized WT HTT for the counter-screen to identify allele-selective ones.
Figure 1. A schematic picture illustrating the mechanism of action of ATTEC. The identified ATTEC compounds interact with mHTT and LC3, tethering these 2 together and targeting mHTT to phagophores for degradation. Similar principles could be applied to other diseases.
We stamped 3375 compounds in duplicates into a microarray chip, and then measured the compound-protein interaction using an optical biosensor, the scanning oblique-incidence reflectivity difference (OI-RD) microscope. Using this system, we measured the compounds’ interaction with the purified human LC3B protein, a pathogenic mHTT exon 1 fragment with expanded polyQ (mHTTexon1-Q72), and a control WT HTT exon 1 fragment (HTTexon1-Q25). We identified 2 hits from the screen, and 2 additional active compounds by searching for similar structures and validation experiments.
We confirmed the compound-protein interaction for these 4 compounds, and validated their efficacy in lowering the mHTT level without influencing the WT in various cellular and animal models, including primary cultured cortical neurons, HD patient fibroblasts, patient induced pluripotent cell-derived neurons, HD Drosophila models expressing human mHTT, and a well-characterized HD knockin mouse model (HdhQ140/Q7). Importantly, 2 of these compounds are able to pass the blood-brain-barrier and are effective in vivo in the HD mice using intraperitoneal injections. We also confirmed the specific mHTT lowering by western blots and proteomics analysis using both cultured neurons and in vivo brain tissues.
We further tested the therapeutic potential of these compounds and validated that these compounds were able to significantly rescue various HD-relevant phenotypes including neuronal shrinkage and apoptosis, HD Drosophila lifespan and climbing abilities, as well as HD mouse motor-function deficits.
To verify the mechanism of action of these compounds, we tested their mHTT-lowering effects with or without the inhibition of autophagy by treatment with autophagy inhibitors or knockout of the key autophagy gene Atg5. We confirmed that the mHTT-lowering effects were mediated through autophagy. To further test whether these compounds function as ATTECs to tether mHTT with the phagophores, we performed in vitro affinity isolation experiments and in vivo colocalization experiments to confirm that the compounds tethered mHTT and LC3 together, targeting mHTT to phagophores. Thus, these compounds truly functioned as ATTECs to target mHTT for autophagic degradation.
While these compounds were able to interact with the key autophagosome protein LC3, we confirmed that they did not influence global autophagy function and autophagosome size/numbers. This property is fundamentally different from other compounds that are autophagy activators/enhancers.
Finally, we investigated why the discovered compounds were able to distinguish between mHTT and WT HTT. We revealed that these compounds interact with mHTT’s expanded polyQ stretch, which is absent in WT HTT. We further confirmed that these compounds are able to drastically reduce mutant ATXN3, another polyQ expansion protein that causes spinocerebellar ataxia 3 (SCA3).
Taken together, we have provided initial evidence to demonstrate the concept of using ATTEC to tether a target protein (mHTT or other polyQ expansion proteins) and LC3 for autophagy-mediated degradation. We think this approach may open a new window for drug discovery, different from the PROTAC strategy and the autophagy-enhancing strategy. The critical next step is to resolve the core chemical compound structure that interacts with LC3 without influencing its function, and we may then utilize it to develop a general degradation tool by conjugating the core compound structure with other compounds interacting with specific targets.
Disclosure statement
Some of the authors (B.L., Y.F. and Y.D.) have applied for patents related to the study commented in this paper.
Additional information
Funding
Reference
- Li Z, Wang C, Wang Z, et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature. 2019. DOI:10.1038/s41586-019-1722-1.