
ABSTRACT
The etiology of preeclampsia (PE), a serious pregnancy complication, remains an enigma. We have demonstrated that proteinopathy, a pathologic feature of neurodegenerative diseases, is a key observation in the placenta and serum from PE patients. We hypothesize that the macroautophagy/autophagy machinery that mediates degradation of aggregated proteins and damaged organelles is impaired in PE. Here, we show that TFEB (transcription factor EB), a master transcriptional regulator of lysosomal biogenesis, and its regulated proteins, LAMP1, LAMP2, and CTSD (cathepsin D), were dysregulated in the placenta from early and late onset PE deliveries. Primary human trophoblasts and immortalized extravillous trophoblasts (EVTs) showed reduced TFEB expression and nuclear translocation as well as lysosomal protein content in response to hypoxia. Hypoxia-exposed trophoblasts also showed decreased PPP3/calcineurin phosphatase activity and increased XPO1/CRM1 (exportin 1), events that inhibit TFEB nuclear translocation. These proteins were also dysregulated in the PE placenta. These results are supported by observed lysosomal ultrastructural defects with decreased number of autolysosomes in hypoxia-treated primary human trophoblasts. Autophagy-deficient human EVTs exhibited poor TFEB nuclear translocation, reduced lysosomal protein expression and function, and increased MTORC1 activity. Sera from PE patients induced these features and protein aggregation in EVTs. Importantly, trophoblast-specific conditional atg7 knockout mice exhibited reduced TFEB expression with increased deposition of protein aggregates in the placenta. These results provide compelling evidence for a regulatory link between accumulation of protein aggregates and TFEB-mediated impaired lysosomal biogenesis and autophagy in the placenta of PE patients.
Abbreviation: atg7: autophagy related 7; CTSD: cathepsin D; ER: endoplasmic reticulum; EVTs: extravillous trophoblasts; KRT7: keratin 7; LAMP1: lysosomal associated membrane protein 1; LAMP2: lysosomal associated membrane protein 2; mSt: mStrawberry; MTORC1: mechanistic target of rapamycin complex 1; NP: normal pregnancy; NPS: normal pregnancy serum; PE: preeclampsia; PES: preeclampsia serum; p-RPS6KB: phosphorylated ribosomal protein S6 kinase B1; SQSTM1/p62: sequestosome 1; TEM: transmission electron microscopy; TFEB: transcription factor EB; XPO1/CRM1: exportin 1
Introduction
Preeclampsia (PE) is a pregnancy-specific, multifactorial and multiorgan disorder [Citation1,Citation2]. It is a major cause of maternal and neonatal morbidity and mortality [Citation3]. PE affects 5–8% of pregnant women and the incidence is higher in low resource settings [Citation4,Citation5]. PE is characterized by the new onset of hypertension and proteinuria in pregnant women after 20 weeks of gestation and with subtypes of early (delivered before 34 weeks) and late onset (delivered after 34 weeks) syndrome [Citation2,Citation4]. Despite decades of investigation and improved clinical care, the pathogenesis of PE remains enigmatic, and no effective treatment(s) besides delivery is available. Furthermore, this pregnancy complication is a significant risk factor for chronic diseases later in life, including cardiovascular disease, diabetes mellitus, kidney disease, hypothyroidism, and chronic hypertension [Citation5 Citation6 Citation7 Citation8–Citation9]. It is thus critical that the etiology of PE be better explained.
It is widely accepted that the placenta is one of the primary sites of dysregulation that contributes to the pathophysiology of PE. This occurs as a result of detrimental events at the maternal-fetal interface, including oxidative and endoplasmic reticulum (ER) stress, anti-angiogenesis, and inflammation [Citation10 Citation11 Citation12–Citation14]. We and others have recently suggested that PE is a syndrome of protein misfolding and aggregation [Citation15 Citation10–Citation19]. It is well documented that neurodegenerative diseases, such as Alzheimer disease, are caused by deposition of aggregated proteins [Citation20,Citation21]. Aggregated proteins can also be detected in the serum and urine of PE patients [Citation15 Citation16 Citation17–Citation18] and have been shown to induce PE-like features in pregnant mice [Citation15]. Thus, understanding the molecular events that lead to accumulation of aggregated proteins and their deposition in the placenta is a critical issue in the pathophysiology of PE.
Autophagy is a cellular homeostasis pathway targeting aggregated proteins and damaged organelles for lysosomal degradation [Citation22–Citation26]. Mechanistically, autophagy-lysosomal biogenesis is tightly regulated by TFEB (transcription factor EB), which controls the expression of lysosomal proteins, LAMP1 (lysosomal associated membrane protein 1), LAMP2 and CTSD (cathepsin D) [Citation27,Citation28]. When activated, intracellular inhibitors of TFEB nuclear translocation such as MTORC1 (mechanistic target of rapamycin kinase complex 1) and XPO1/CRM1 (exportin 1) dysregulate lysosomal biogenesis and impair autophagy [Citation29,Citation30]. Studies on the human placenta reveal that autophagy protects the placenta against pathogens and stress [Citation31 Citation32 Citation33 Citation34–Citation35]. Placental autophagy can also be exploited by pathogens to evade surveillance from host defenses [Citation36]. Accumulation of protein aggregates in the PE placenta poses an important question whether autophagy is activated or inhibited in the PE placenta. In this regard, several reports have suggested that autophagy is hyperactivated in PE [Citation37 Citation39 Citation40 Citation41 Citation44 Citation45–Citation46]. However, these observations of excessive autophagy as demonstrated by activation of early stage markers do not explain the presence of aggregated proteins in serum and the placenta from PE patients. We propose that the lysosomal degradation pathway, a key regulatory step for aggregated protein clearance and cellular homeostasis, is impaired as a result of poor lysosomal biogenesis in PE.
To demonstrate the importance of protein aggregation and impaired autophagy in PE, we examined lysosomal biogenesis proteins and accumulation of protein aggregates in placental tissue, primary human trophoblasts, autophagy-deficient human extravillous trophoblasts (EVTs), and trophoblast-specific conditional atg7 (autophagy related 7) knockout (atg7 cKO) mice. Taken together, our data strongly suggest that lysosomal biogenesis and autophagy are impaired in PE patients, implying that toxic protein aggregates accumulate in the PE placenta as a result of these abnormalities.
Results
Placental tissue from PE exhibits significantly reduced presence of TFEB and increased content of protein aggregates
Schematic details of the architecture of the human placenta and the sites of expression and action of TFEB and its regulated proteins are shown in . To investigate TFEB expression, we performed immunostaining using antibody to TFEB and compared villous regions () and EVTs () in placental tissue. The PE placental tissue and serum samples were collected from women with early (n = 6) and late onset PE (n = 10) and gestational age-matched preterm birth (n = 6) or normal pregnancy (NP) (n = 10) deliveries (see clinical details in Table S1). Significantly reduced presence of TFEB was consistently observed in PE placental tissue (PE) in the syncytiotrophoblast layer () and in KRT7 (keratin 7)-positive EVTs () when compared to tissue from normal pregnancy (NP) deliveries. It is important to note that besides the trophoblast layer in NP placental tissue, variable TFEB staining was also observed in other cell types in the stroma including endothelial cells around blood vessels. Colocalization of TFEB and nuclear DAPI staining in a subpopulation of EVTs from normal pregnancies () suggested nuclear translocation of TFEB. This was not observed in EVTs from PE placental tissue (). Nuclear translocation of TFEB in EVTs (see boxed EVTs in ) from NP not PE is further demonstrated at a higher magnification (). To provide further evidence for reduced presence of TFEB in the PE placenta, we performed western blotting using protein extracts from placental tissues from early onset PE (e-PE) vs. gestational age-matched preterm birth deliveries () as well as late onset PE (l-PE) vs. normal term delivery (Fig. S1A). These quantitative results () support the immunohistochemical data presented in . We next attempted to correlate reduced TFEB expression with the presence of protein aggregates in the PE placenta. We have developed a novel assay to detect protein aggregates by employing a rotor dye, ProteoStatTM [Citation47], which binds selectively to aggregated protein structures [Citation48]. The ProteoStat-specific signal was prominently detected in the trophoblast layer of the villi and EVTs in placental tissue from PE deliveries (Fig. S1B and C). These results suggest that reduced TFEB expression in the trophoblast layer and in EVTs in the PE placenta possibly result in poor autophagy-mediated degradation of protein aggregates thus leading to their accumulation.
Figure 1. Impairment of TFEB expression in the trophoblast layer and anchoring EVTs in placental tissue from PE patients. (A) Diagram of human placental architecture. Symbols: Villi, villous structures; ST, syncytiotrophoblast; CT, cytotrophoblast; AV, anchoring villi; EVT, extravillous trophoblast. Boxes a and b represent villous structures and anchoring villi containing EVTs, respectively. (B) TFEB staining of placental tissue from normal pregnancy (NP) and from preeclampsia (PE) deliveries. TFEB was predominantly detected in the trophoblast layer including cytotrophoblasts (arrowheads) and syncytiotrophoblasts (arrows) in the NP, not PE, placenta. It was also detected at moderate levels in endothelial cells of blood vessels (gray arrows) of the villi in control placenta, but significantly reduced in the PE placenta. Quantification shows significantly difference between NP and PE samples (p = 0.005). Bar: 50 µm. (C) Extravillous trophoblast domains in the placenta were co-stained for KRT7 (keratin 7), TFEB and nuclei with DAPI. TFEB was mainly observed in NP EVTs that co-localized with KRT7 staining. Arrows show TFEB nuclear localization in NP EVTs. Quantification of the data is presented C. Bar: 25 µm. (D) Amplified view of nuclear TFEB in EVTs of NP and PE shown in boxes in Panel C. (E) Western blotting of TFEB in NP and PE placental protein extracts. Decreased levels of TFEB expression were observed in the placentas from early onset PE vs. gestational age-matched preterm birth deliveries (p < 0.01, n = 5). Quantification of TFEB signal is presented here as normalized to ACTB. Data are presented as mean ± SEM and analyzed by a Student t-test.
LAMP1, LAMP2, and CTSD are poorly expressed in the PE placenta with increased accumulation of SQSTM1/p62
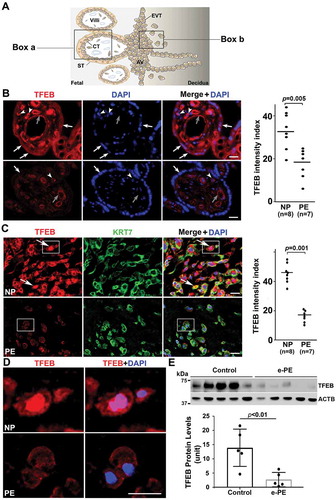
The lysosomal-associated membrane proteins, LAMP1 and LAMP2, constitute a major part of the lysosomal proteome and are regulated by TFEB [Citation49,Citation50]. Immunohistochemical analysis was performed and the mean fluorescence intensity index was calculated for both LAMP1 and LAMP2 in placental tissue from NP and PE. As shown in , LAMP1 and LAMP2 were expressed predominantly in the trophoblast layer in the NP placenta. In contrast, little or no staining was observed in the PE placenta. The mean fluorescent intensity index of LAMPs was significantly lower in placental tissue (n = 7) from PE than the NP placenta (n = 8) (), p = 0.037 and 0.021, respectively).
Figure 2. Autophagy protein expression in placental tissue from normal pregnancy (NP) and PE deliveries. Tissue was stained for LAMP1, LAMP2, CTSD (cathepsin D), and SQSTM1 (p62). Nuclei were stained with DAPI. (A) The trophoblast layer in NP, not PE, tissue, was significantly stained for LAMP1. Analysis of LAMP1 staining in NP and PE tissue samples suggested statistically significant differences. (B) LAMP2 staining in NP and PE tissues and its intensity index analysis support the data presented in Panel A. (C) A representative experiment for CTSD and quantitative analysis are shown in this panel with statistically significant differences between NP and PE placenta. It demonstrates that the trophoblast layer in the villi was predominantly positive for CTSD in NP as compared to PE. (D) A representative experiment is shown for SQSTM1 staining. Sporadic SQSTM1 staining was mainly detected in PE placental tissue, not NP tissue. Data are presented as mean ± SEM and analyzed by a Student t-test. Bar: 20 µm.
We next examined the presence of CTSD and SQSTM1/p62 proteins in placental tissue from PE deliveries. CTSD was poorly expressed in the trophoblast layer of the PE placenta as compared to the NP placenta (). In contrast, sporadic accumulation of SQSTM1/p62 was detected in the PE, not NP placenta (). Of note, SQSTM1/p62 clusters usually accumulate in the absence of functional autophagy [Citation22,Citation23,Citation24,Citation25]. The quantification of immunostaining in NP (n = 8) and PE (n = 7) placental tissue further supports the visual detection of these proteins (). To demonstrate that these observations were typical of all PE placentas analyzed, we show data from additional placentas from PE and gestational age-matched deliveries (Fig. S2). These results demonstrate that lysosomal biogenesis proteins are poorly expressed in the PE placenta and suggest that impaired lysosomal degradation may be a key feature of PE.
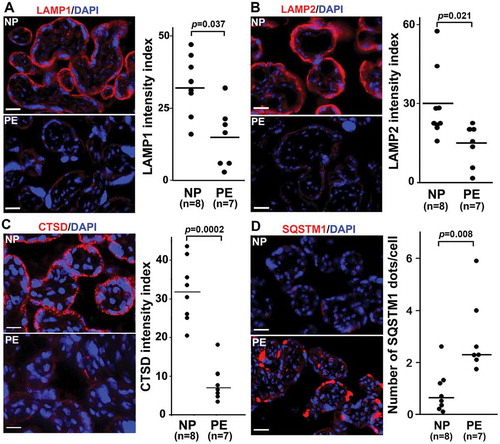
Hypoxia inhibits TFEB, lamps and CTSD expression as well as TFEB nuclear translocation in primary human trophoblasts
Hypoxia-induced ER stress has been associated with the pathogenesis of PE [Citation13,Citation47]. To determine if lysosomal biogenesis is perturbed by hypoxic environment in human trophoblasts, freshly-isolated third trimester trophoblast preparations from three different deliveries were cultured under normoxic or hypoxic (1% O2) conditions for different time periods. Proteins were analyzed by western blotting to assess the effect on LAMPs, TFEB, and CTSD. Nuclear translocation of TFEB in hypoxia-exposed trophoblasts was visualized by immunofluorescence staining. Our results show that exposure to hypoxia (results shown for 72 h treatment) significantly reduced the protein content and mRNA level of TFEB (). In addition, attenuated LAMP1, LAMP2 and CTSD were also observed in hypoxia-exposed cells (). Intriguingly, hypoxia treatment blocked TFEB nuclear translocation as shown by lack of colocalization of DAPI and TFEB (). We also examined expression of these proteins in hypoxia-treated TCL-1 cell, a third trimester EVT cell line (Fig. S3A-D). The data from western blots and quantification of the signal intensity in TCL-1 cells support our observations in primary human trophoblasts. The results in Fig. S4 show hypoxia-induced accumulation of ProteoStat-positive clusters of protein aggregates in primary trophoblast cells, which is consistent with the data observed in placental tissue (Fig. S1B and C).
Figure 3. Effect of low oxygen tension (1% O2) on the expression of TFEB protein and mRNA, lysosomal proteome, and TFEB nuclear translocation in primary human trophoblasts. Primary trophoblasts from three different preparations were cultured under normoxic or hypoxic (1% O2) conditions. (A–C) TFEB protein abundance and mRNA levels were significantly decreased in hypoxia-treated cells. (D, E) LAMP1, LAMP2, and CTSD were significantly reduced in primary trophoblasts in response to hypoxia. (F) TFEB immunofluorescent signals were predominantly concentrated in the nuclei (arrows) of the cells under normoxic, not hypoxic, condition. Cells were cultured in media containing 2.5% FBS. Images are representatives of 3 independent experiments. Bar: 10 μm. Data are presented as mean ± SEM and analyzed by a Student t-test (n = 3).
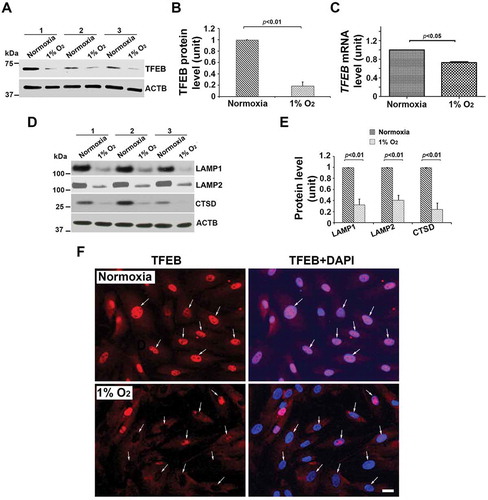
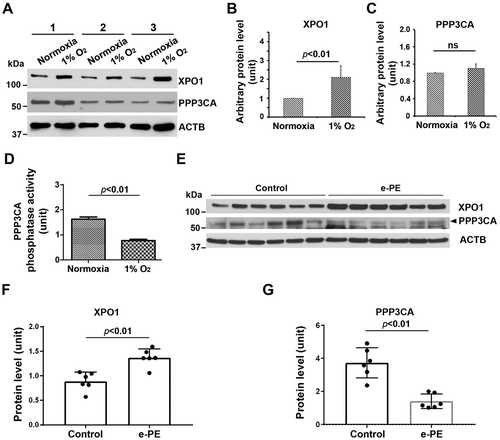
Decreased PPP3/calcineurin activity and increased XPO1 abundance in hypoxia-exposed primary human trophoblasts and early onset PE (e-PE) placenta
Since TFEB nuclear translocation is regulated by serine/threonine protein phosphatase, PPP3/calcineurin and XPO1, a regulator of protein nuclear export [Citation29,Citation30], we next examined the expression of these two proteins in normoxia/hypoxia-exposed primary trophoblasts and the placenta from PE deliveries and gestational age-matched controls. As shown in , hypoxia upregulated XPO1 expression (p < 0.01) did not significantly alter PPP3/calcineurin abundance (p = 0.31). However, PPP3/calcineurin phosphatase activity was significantly inhibited in hypoxia-exposed trophoblasts (). Similar assessment was also performed in placental tissue. As shown in , placental tissue from e-PE exhibited higher levels of XPO1 (p < 0.01) and somewhat lower levels of PPP3/calcineurin compared to gestational age-matched controls (p < 0.01). These data suggest that inhibition of PPP3/calcineurin phosphatase activity and increase in XPO1 expression in hypoxia-exposed trophoblasts and the PE placenta may be responsible for decreased TFEB nuclear translocation.
Figure 4. Analysis of XPO1 and PPP3/calcineurin in normoxia/hypoxia-exposed primary human trophoblasts and the placenta from e-PE vs. controls. (A–C) The expression of XPO1 and PPP3 in cells with indicated treatment was evaluated using western blotting in A and statistically analyzed in B and C. (D) The phosphatase activity of PPP3/calcineurin was assessed and compared between hypoxia- and normoxia-treated cells. (E–G) Protein abundance of XPO1 and PPP3 was detected using western blotting (E) and statistically analyzed (F and G) in the placenta from e-PE and gestational age-matched deliveries.
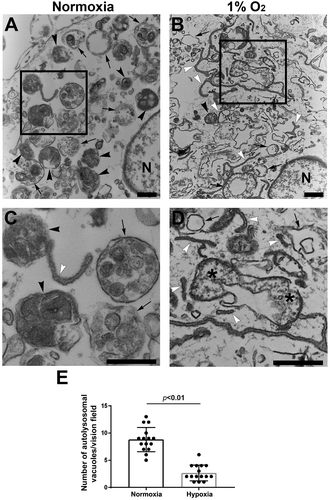
Ultrastructural evidence for autophagy impairment in primary human trophoblasts in response to hypoxia
Our results discussed above suggest that hypoxia dysregulated TFEB and inhibited lysosomal biogenesis, implying impairment of autophagic flux. To verify this, we assessed autophagic vacuoles using transmission electron microscopy (TEM). Under normal conditions, autophagosome is characterized by double-membrane structure containing the cytoplasmic contents and/or damaged organelles, and autolysosome appears as single-membrane vacuole containing electron-dense lysosomal materials and cytoplasmic contents and/organelles at various state of degradation (). Semi-quantitative analysis revealed a significantly lower number of autophagosome and autolysosome in hypoxia-exposed trophoblasts vs. control cells (, p < 0.01). Moreover, hypoxia-treated cells displayed a large number of phagophores (). These data indicate that hypoxia inhibits the formation of autophagosome and autolysosome, thereby disrupting autophagic flux.
Figure 5. Ultrastructural analysis of autophagic vacuoles in primary human trophoblasts treated with hypoxia or normoxia. (A, B) Micrographs show autophagosomes (arrows), autolysosomes (black arrowheads) and isolation membranes or phagophores (white arrowheads) in normoxia- (A) and hypoxia- (B) treated cells. (C, D) Boxed areas in A and B were magnified in C and D, respectively, to show details of autophagic vacuoles. (E) Semi quantification was performed to evaluate the number of autolysosomes in normoxia- and hypoxia-treated trophoblasts. Bar: 600 nm.
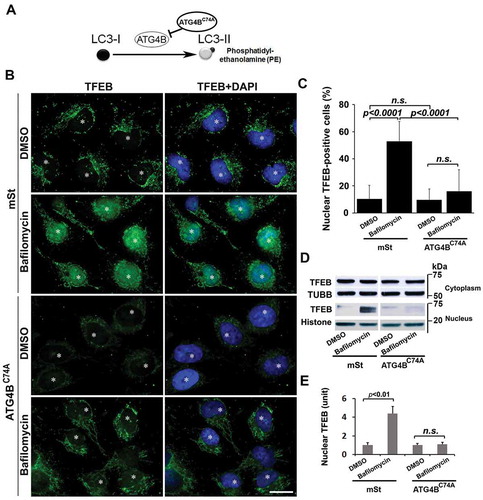
Autophagy-deficient EVTs exhibit poor TFEB nuclear translocation and lysosomal function
To demonstrate that poor lysosomal function and dysregulated TFEB expression/nuclear translocation are linked with impaired autophagy in stressed human trophoblasts, we employed human first trimester EVTs engineered to exhibit autophagy deficiency [Citation51]. An autophagy-deficient human EVT trophoblast cell line (HchEpC1b-ATG4BC74A) was established by stable transfection of EVT trophoblasts with ATG4BC74A, an inactive mutant of ATG4B (see Materials and methods for details). The mutant ATG4BC74A inhibits conversion of LC3-I to LC3-II () [Citation52]. Stably transfected, autophagy-deficient HchEpC1b-ATG4BC74A and autophagy-proficient cells derived by stable transfection with control HchEpC1b-mStrawberry (mSt) were analyzed for dysregulated expression of TFEB and LAMPs, impaired lysosome function, and TFEB nuclear translocation. Both western blotting and immunohistochemical analyses showed that LAMP1 and LAMP2 protein levels were significantly reduced in autophagy-deficient HchEpC1b-ATG4BC74A cells, compared to the autophagy-proficient mSt cells (Fig. S5A-C). We next examined whether reduced expression of LAMP1 and LAMP2 was associated with impaired lysosomal function in autophagy-deficient HchEpC1b-ATG4BC74A cells (Fig. S5A-C). A hallmark of lysosomal function is the translocation of lysosomal membrane markers to the plasma membrane [Citation49,Citation50,Citation53]. LysoTracker is an acidotropic dye that stains cellular acidic compartments such as lysosomes and autolysosomes [Citation54]. This dye has been used to detect autophagy-associated lysosomal activity. LysoTracker red staining, which normally accumulates in intraluminal vesicles with low pH, showed poor staining intensity in HchEpC1b-ATG4BC74A cells compared to control mSt cells (Fig. S5D). Taken together, these data suggest that lysosomal function was significantly reduced in the autophagy-deficient HchEpC1b-ATG4BC74A trophoblast cells. To examine the TFEB content and nuclear translocation in HchEpC1b-ATG4BC74A cells, we cultured control mSt and autophagy-deficient EVTs in the presence or absence of bafilomycin A1, a drug that induces TFEB nuclear translocation [Citation55]. We assessed TFEB expression by immunofluorescence in fixed cells and western blotting of fractionated cytoplasmic and nuclear protein contents. The data presented in show that bafilomycin A1 failed to induce TFEB nuclear translocation in the autophagy-deficient EVTs. This is further supported by the absence of TFEB in the nuclear fraction of autophagy-deficient EVTs (). The observations presented in and Fig. S5 support a mechanistic link between autophagy deficiency and abnormal TFEB localization and impaired trophoblast lysosomal function.
Figure 6. Lysosomal defects in autophagy-deficient extravillous trophoblast (EVT) cells stably transfected with vehicle plasmid mStrawberry (mSt) lacking or harboring mutant ATG4BC74A coding region. (A) A schematic model is shown that depicts the inhibition of autophagy by an inactive mutant ATG4BC74A, which inhibits the conjugation of phosphatidylethanolamine (PE) to LC3 for conversion of LC3-I to LC3-II. Deficiency in lysosome function was depicted in ATG4BC74A cells by LysoTracker staining (see Figure S5). (B) Assessment of TFEB nuclear translocation in bafilomycin A1-treated or untreated mSt and ATG4BC74A cells for 24 h. mSt cells exhibited significant nuclear translocation in response to bafilomycin A1. In contrast, ATG4BC74A showed poor overall expression of TFEB. Importantly, no nuclear TFEB translocation was observed in these cells in response to bafilomycin A1. (C) Quantification of at least three experiments supported the data presented in B. (D) Nuclear and cytoplasmic fractions were isolated from bafilomycin A1-treated and untreated cells and probed for the TFEB content in a western blot analysis. TUBB/β-tubulin and Histone were used as protein loading controls for cytoplasmic and nuclear fractions, respectively. Data are presented as mean ± SEM with at least n = 3 per group, and analyzed by one-way ANOVA with Bonferroni post hoc test. Bar: 20 µm.
Sera from PE patients augment RPS6KB phosphorylation, block nuclear translocation of TFEB, and induce protein aggregation in EVTs
Sera from patients with PE have been shown to induce PE-like features in pregnant mice [Citation56] and to inhibit activation of autophagy in peripheral blood mononuclear cells [Citation57]. The data in suggest that TFEB nuclear translocation was blocked in autophagy-deficient EVTs which also show increased accumulation of protein aggregates. Activated MTORC1 has been shown to block TFEB nuclear translocation [Citation29,Citation30] which can be evaluated by elevated phosphorylation of its substrate RPS6KB/S6 kinase (p-RPS6KB). To investigate a possible mechanism for lack of TFEB nuclear translocation in HchEpC1b-ATG4BC74A cells, we compared the content of p-RPS6KB in control mSt and HchEpC1b-ATG4BC74A cells. Our results clearly demonstrated that autophagy-deficient cells exhibited significantly higher (p = 0.021) content of p-RPS6KB (), suggesting constitutive activation of MTORC1 in these cells. This may contribute to a mechanism for poor TFEB nuclear translocation in autophagy-deficient cells.
Figure 7. MTORC1 activation in autophagy-deficient ATG4C74A cells and lack of TFEB nuclear translocation in mSt cells in response to sera from PE patients. (A) Increased MTORC1 activity was observed in ATG4BC74A cells that show higher ratio of p-RPS6KB:RPS6KB. (B) PE sera (PES), not NP sera (NPS), increase phosphorylation of MTORC1 target, RPS6KB. (C) A representative western blot shows evidence for PES-mediated block of TFEB nuclear translocation. mSt cells were treated with NPS or PES samples for 24 h in the presence of bafilomycin A1, and nuclear protein fraction was then isolated. (D) PES, not NPS, downregulated the expression of LAMP1, LAMP2 and CTSD in mSt cells. Data are presented as mean ± SEM with at least n = 3 per group, and analyzed by a Student t-test.
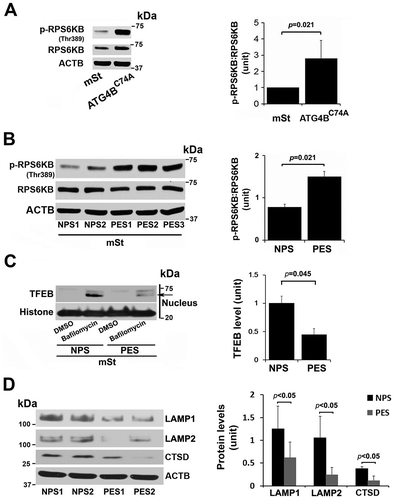
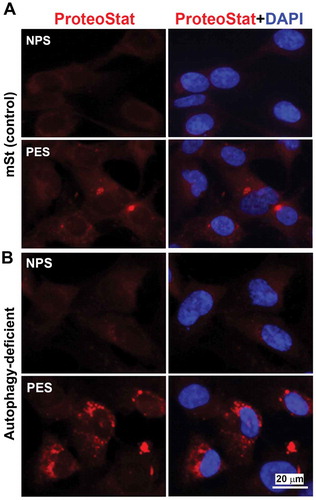
Next, using serum samples from women with normal pregnancy (NPS) and PE (PES), we demonstrated that sera from women with PE induced significantly higher (p = 0.021) phosphorylation of RPS6KB in mSt EVTs (), indicative of enhanced MTORC1 activity. We then examined whether PES blocked nuclear translocation of TFEB induced by bafilomycin A1. As shown in , PES significantly blocked TFEB nuclear translocation. Thus, it appears that sera from women with PE contain factors that can induce MTORC1 activity and disrupt TFEB nuclear translocation. To further provide evidence that PES dysregulated TFEB and impaired autophagy-lysosomal degradation machinery, we evaluated expression of LAMP1/2 and CTSD as well as accumulation of protein aggregates in mSt EVTs and autophagy-deficient EVTs in response to NPS and PES. For western blot analysis, mSt cells were used because the expression of LAMPs and CTSD is intrinsically very poor in autophagy-deficient EVTs. EVTs were cultured with NPS or PES for 24 h, subjected to western blot analysis to evaluate the lysosomal proteome. As shown in , PES, not NPS, downregulated the expression of LAMP1, LAMP2 and CTSD in autophagy normal mSt EVTs. We then evaluated accumulation of protein aggregates in autophagy-deficient and autophagy-proficient cells in response to NPS or PES. The results presented in demonstrated that PES, not NPS, caused accumulation of protein aggregates in autophagy-deficient EVTs. Unlike autophagy-deficient cells, PES did not elicit significant protein aggregation in control mSt cells.
Figure 8. Evidence for accumulation of ProteoStat-positive protein aggregates in autophagy deficient EVTs in response to gestational age matched sera from NP and PE women. Autophagy deficient ATG4BC74A cells and autophagy proficient control cells (mSt) were grown in FBS-free media supplemented with 10% NPS or PES for 24 h and then stained with ProteoStat. Images are representative of independent experiments using six serum samples from each. Bar: 20 µm.
Trophoblast-specific conditional atg7 knockout (atg7 cKO) mice exhibit accumulation of protein aggregates in the placental junctional zone
atg7 cKO mice were generated by the unique lentiviral (LV) vector-mediated trophoblast -specific gene knockout system involving atg7 gene [Citation58]. This LV vector only infects the trophoectoderm and not the inner cell mass in the zona pellucida-less blastocyst. As described by Aoki et al [Citation58], atg7 knockout allele was only detected in the placenta, suggesting that the placenta-specific effects of autophagy deficiency could be studied using this mouse model. atg7 cKO mice experience pregnancy-specific hypertension as compared to control EGFP mice [Citation58] and provide a model for PE-associated abnormalities. We took advantage of this mouse model and investigated whether placental accumulation of protein aggregates was associated with lower autophagy activity in atg7 cKO mice. depicted representative histological analyses of TFEB expression and protein aggregation in the placenta of atg7 cKO mice. Immunohistochemical staining showed lower levels of TFEB expression in the labyrinth and junctional zone or spongiotrophoblast layer of placenta-specific autophagy deficient mice vs. control mice (). This is consistent with a prior finding showing that syncytiotrophoblast-specific atg7 knockout decreases lysosomal biogenesis in mouse placenta using western blotting [Citation59]. The ProteoStat dye detection assay revealed that the junctional zone of the placenta of atg7 cKO mice exhibited intense ProteoStat signal compared to the decidua and the labyrinth zones. The increased presence of protein aggregates in the junctional zone was statistically significant as analyzed by ImageJ software for the ProteoStat intensity (). These results support our observations in the PE placenta and in hypoxia-treated human trophoblasts (Fig. S4 and S8) that autophagy deficiency leads to accumulation of protein aggregates.
Figure 9. Evidence for reduced TFEB expression and enhanced intrinsic protein aggregation in trophoblast-specific atg7 cKO placentas. (A, B) TFEB staining was performed in placenta-specific atg7 knockout mice (cKO) and control mice (EGFP) (A). TFEB-positive cell rate in the placentas from cKO and EGFP were calculated and statistically analyzed (B). La: labyrinth layer, Sp: spongiotrophoblast layer, De: decidua. Arrows indicated TFEB-positive trophoblasts. (C, D) Protein aggregates in the labyrinth layer (La) and spongiotrophoblast layer (Sp) in the placentas from atg7 cKO and EGFP control mice were identified by ProteoStat staining (C). H&E staining showed the structure of mouse placental tissue. The intensity of ProteoStat signal in the placentas was compared between EGFP control and atg7 cKO as shown in D. Data are expressed as mean ± SEM and analyzed by a Student t-test (n = 4). Bar: 50 µm.
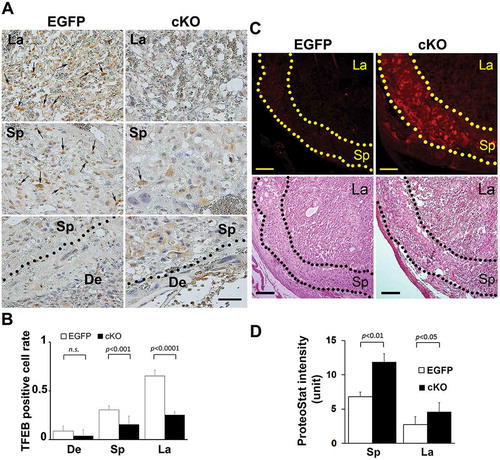
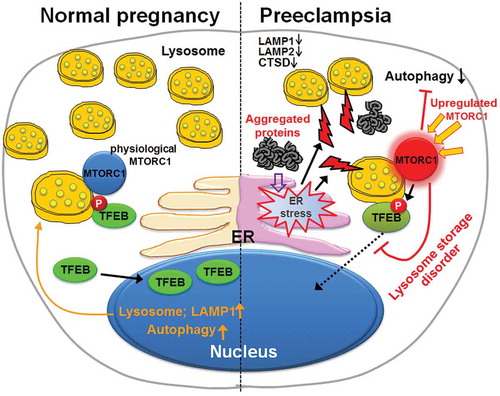
Figure 10. Schematic representation of a model depicting lysosomal biogenesis defects, impaired autophagy and aggregated proteins in PE. The model focuses on ER stress, poor TFEB regulation and function, upregulated MTORC1, impaired autophagy, and lysosome storage-like condition in PE.
Discussion
We systematically evaluated the autophagy lysosomal machinery in the PE placenta by focusing on the components of lysosomal biogenesis, including TFEB, LAMP1, LAMP2, and CTSD, and explored whether impaired autophagy is associated with accumulation of protein aggregates observed in the PE placenta. TFEB, a master regulator of lysosomal biogenesis, plays a pivotal role in mediating autophagy-mediated degradation of toxic aggregated proteins and has been associated with the etiology of lysosomal storage diseases [Citation60 Citation61–Citation62]. Here, we show for the first time that TFEB protein levels and its nuclear translocation were significantly reduced in the trophoblast layer and the anchoring EVTs in the placenta from PE patients ( and S1A). PPP3/calcineurin, a serine/threonine protein phosphatase, can dephosphorylate TFEB, which induces TFEB translocation to the nucleus [Citation29]. Additionally, Napolitano et al. recently showed that TFEB nuclear translocation is negatively regulated by nuclear export protein, XPO1 [Citation30]. As shown in , reduced PPP3/calcineurin phosphatase activity and increased XPO1 abundance may contribute to poor TFEB nuclear translocation in hypoxia-treated trophoblasts and the PE placenta. Moreover, the lysosome proteome members, LAMP1, LAMP2, and CTSD, were also significantly reduced, suggesting impaired lysosomal biogenesis ultrastructural changes. Indeed, TEM analysis revealed significantly reduced numbers of autophagic vacuoles in hypoxia-exposed trophoblasts ().
Our observations on inhibition of TFEB nuclear translocation and accumulation of protein aggregates in human trophoblasts in response to sera from PE patients provide support for the link of impaired autophagy with buildup of protein aggregates in the PE placenta (–). Furthermore, autophagy-deficient human trophoblasts cells exhibited significantly reduced TFEB nuclear translocation and protein content of LAMPs, poor lysosomal function, and hypoxia-induced accumulation of protein aggregates (, , S4 and S5). Importantly, these data are supported by in vivo observations in trophoblast-specific conditional atg7−/- mice wherein no or very little immunostaining of placental TFEB was associated with accumulation of protein aggregates in the placental junctional zone. These results provide compelling evidence for an involvement of impaired autophagy in the PE pathophysiology.
The ER stress can serve as a double-edged sword for the autophagy machinery in the placenta. Physiological low oxygen tension-mediated autophagy activation enhances trophoblast invasion, a requirement for placental-maternal crosstalk and endometrial spiral artery remodeling [Citation51]. On the other hand, excessive hypoxic stress has been shown to inhibit invasive properties of trophoblast cells [Citation35,Citation51,Citation63]. Our results describing the detrimental effects of excessive hypoxia and sera from PE patients on the lysosomal biogenesis proteome and TFEB nuclear translocation are noteworthy (, S3 and ). It is quite possible that these effects may be even more prominent in the scenario of hypoxia-reoxygenation as suggested in the case of cardiac cells [Citation64]. Importantly, accumulation of protein aggregates as a result of dysregulation of lysosomal biogenesis in the PE placenta and stressed trophoblasts could be a critical event contributing to the onset of PE. Although it is difficult to pinpoint the source of excessive ER stress, these events resulting in impaired autophagy may cooperatively disrupt the growth, differentiation and invasion of trophoblasts via lysosomal dysregulation. Future studies should focus on therapeutic options for prevention and treatment of PE that can restore lysosomal biogenesis and autophagy as well as inhibit protein aggregation.
Although the exact etiology of PE is still unclear, the observations on accumulation of toxic protein aggregates in the PE placenta and serum provide a new line of investigation for better understanding the events that program this multifactorial pregnancy complication. Protein misfolding and aggregation have been shown to be the key pathologic features in a number of neurodegenerative diseases [Citation20,Citation21,Citation47]. Thus, predictive detection of the protein cargo of these aggregates in serum will be of immense importance. Our data on the detection assay for protein aggregates in serum using autophagy-deficient trophoblast cells and in the placenta is a cutting-edge approach. This approach is highly reproducible and is likely to be of great value for setting up a point of care assay for detection of serum-based protein aggregates not only in the case of PE but also other pregnancy complications and Alzheimer disease-related dementias.
There have been controversies regarding whether autophagy activity is increased or impaired in the placenta form women with PE [Citation37–Citation46]. Most studies focused on detection of initial stage markers of autophagy activation, such as LC3-II, BECN1/Beclin-1, and SQSTM1/p62, and suggested that PE was associated with excessive autophagy. For instance, Oh and colleagues demonstrated an increased content of LC3-II and unchanged expression of BECN1 in the placenta from PE vs. controls [Citation37]. Gao et al. showed elevated expression of LC3 and BECN1 in the placenta from early onset PE [Citation38]. Akaishi and coworkers demonstrated upregulated LC3-II and downregulated SQSTM1/p62 in the PE placenta [Citation43]. Most recently, Akcora-Yildiz et al. reported increased autophagy activity based on the findings showing BECN1 upregulation and cyclin E downregulation in the PE placenta, although higher levels of SQSTM1/p62 were observed in the placenta from women with PE vs. normal pregnancy [Citation42]. In contrast, our results strongly suggest that PE is associated with impaired autophagy due to defective lysosomal biogenesis machinery. To reconcile both sets of observations, we propose that the number of autophagosomes significantly increases in response to ER stress as represented by increased LC3-II and BECN1, whereas the number of autolysosomes is significantly reduced as reflected by our results on poor lysosomal biogenesis. Such a scenario will result in the impairment of autophagosome-lysosome fusion by excessive ER stress leading to inhibited autophagy. Increased numbers of autophagosomes as shown by appearance of LC3-II dots [Citation37,Citation38,Citation43] do not necessarily suggest enhanced autophagy activity because this may be caused by failure of LC3 degradation in dysregulated autolysosome at the final stage of autophagic flux. In the present study, the accumulation of aggregated proteins in the PE placenta, PE serum-treated autophagy-deficient EVTs and atg7 cKO mouse placenta strongly supports the concept of defective autophagy in PE. Notably, placental autophagy deficiency induces PE-like features in atg7 cKO mice [Citation58], which points to a direct causative link between autophagy impairment and the PE pathology.
Collectively, we provide evidence for TFEB dysregulation, impaired lysosomal biogenesis, and defective autophagy in placental tissue from PE and in cellular models of the syndrome. Suppression of autophagy conversely dysregulates TFEB expression, exacerbates lysosomal biogenesis defect and supports accumulation of protein aggregates. Toxic protein aggregates accumulated in the placenta may lead to poor placentation and production of pregnancy-incompatible milieu, including anti-angiogenic factors, extracellular vesicles and alarmins (). Thus, pharmacological options that can restore lysosomal biogenesis and autophagy hold promise as attractive therapeutic avenues for PE.
Materials and methods
Ethics statement and human subjects
Serum and placental samples were obtained from pregnant women with severe PE and normotensive gestational age-matched pregnant women with informed consents under the approved protocols by the Institutional Review Board of Women & Infants Hospital of Rhode Island, Providence, USA. The clinical information on pregnant women recruited for this study is detailed in Table S1. Serum was separated and frozen as aliquots at −80ºC until further use. Placental tissue was collected at the time of delivery and processed within 2 h of collection. Exclusion criteria included women who were smokers, had a multifetal pregnancy, were < 18 and > 45-years old, were known HIV positive, and had pre-pregnancy hypertension, gestational or preexisting diabetes, fetal demise, fetal anomalies and prior administration of intramuscular steroids.
Reagents
The following mouse monoclonal antibodies (Ab) were used: anti-ACTB/β-actin (Abcam, ab6276), anti-LAMP1 (Abcam, ab25630), anti-LAMP2 (Abcam, ab25631), and anti- SQSTM1/p62 (MBL, M162-3). The following rabbit polyclonal Ab were used: anti-TUBB/β-tubulin (Cell Signaling Technology, 2146), anti-KRT7 (Novus Biological, NB120-17069), anti-MAP1LC3B (MBL, PM036), anti-TFEB (Millipore, ABF234), anti-PPP3CA/calcineurin α subunit (Sigma-Aldrich C1956) and anti-XPO1/CRM1 (Cell Signaling Technology). The following horseradish peroxidases (HRP)-conjugated secondary Abs were used: anti-mouse IgG-HRP conjugate (Alpha Diagnostic, 40320–200), anti-rabbit IgG-HRP conjugate (Cell Signaling Technology, 7074). Bafiloymcin A1 (Cayman Chemical, 11038) was used as an inducer of TEFB nuclear translocation and inhibitor of autophagy. ProteoStat Aggresome Detection Kit (ENZO, ENZ-51023-KP002) was used for detecting aggregated proteins.
Placenta procurement and trophoblast culture
Tissue pieces of placentas from normal singleton pregnancy and PE deliveries were taken from the middle of the placenta. After extensive wash, samples were either stored in 10% formalin or immediately frozen at −80ºC. For trophoblast isolation and culture, placentas were procured on a protocol approved at the University of Pittsburgh. Primary human trophoblasts were dispersed using the trypsin (Sigma, T5266) -deoxyribonuclease I (Sigma, D5025) -dispase (Collaborative Research, 40235) -Percoll (Sigma, GE17-0891-01) as described previously [Citation65,Citation66]. Cells were plated at a density of 300,000 cells/cm2 and cultured for 4 h in standard culture atmosphere that supports cell differentiation into syncytiotrophoblast [Citation67]. After 4 h, we exposed primary trophoblast cells to varying concentrations of atmospheric oxygen for 48–72 h. A lower FiO2 (≈1%) was maintained using a hermetically-enclosed incubator (Thermo Electron, Marietta, OH, USA) that provided the desired level of oxygen, (O2 = 0%, 5% CO2, 10% H2, and 85% N2), with continuous digital recording of atmospheric oxygen using a sensor connected to a data acquisition module (Scope; Data Translation, Marlboro, MA, USA). All media (DMEM with “high glucose” of 4.5 g/L D-glucose (ThermoFisher, 11965084) were pre-equilibrated to the gas mixture before addition to the culture plate, and refreshed every 24 h.
Cell lines and stable transfection
EVT cell lines, the third trimester TCL-1 and the first trimester HchEpC1b, were generated as described previously [Citation51,Citation68]. HchEpC1b is an HPV E6 and hTERT-transfected immortalized EVT cell line [Citation51]. An autophagy-deficient cell line, HchEpC1b-ATG4BC74A cells (ATG4BC74A), was constructed by stable transfection with pMRX-IRES-puro-mStrawberry-ATG4BC74A (a gift from Fujita et al [Citation52]), an ATG4BC74A mutant expression vector that inhibits MAP1LC3B-II formation [Citation51]. HchEpC1b-mSt cells, a control cell line, were stably transfected with pMRX-IRES-puro-mStrawberry (a gift from Fujita et al [Citation52]), a control vector encoding monomeric red fluorescent protein [Citation51]. Cells were cultured in RPMI1640 medium (GIBCO, 11875) supplemented with 10% FBS (Fisher Scientific, 10-082-147) 100 U/ml penicillin and 100 µg/ml streptomycin (GIBCO, 15,140) at 37°C in a 5% CO2 atmosphere. For starved cultures, the culture medium was exchanged for Hanks’ balanced salt solution (GIBCO, 14025092).
Atg7 cKO mice
Conditional atg7 knockout (atg7 cKO) mice were generated as described previously [Citation58]. In brief, C57BL/6 Atg7flox/flox mice (Japan SLC Inc. Shizuoka, Japan) were crossbred at least 3 times with B6D2F1 mice (Japan SLC) and female Atg7flox/flox mice were obtained. The Atg7flox/flox blastocysts were infected with lentivirus expressing the Cre recombinase. For the control placentas, the blastocysts were infected with EGFP-lentivirus in place of the Cre recombinase. Blastocysts were implanted into a pseudo-pregnant mouse, which has a normal autophagy phenotype. Generation of atg7 cKO was confirmed by the absence of the ATG7 protein in the placenta. Western blotting showed reduced ATG7 expression coupled with accumulation of SQSTM1 [Citation58]. Mouse placental tissues were fixed and processed for detecting TFEB expression using immunohistochemistry as described previously [Citation58]. The TFEB-positive signal was quantified by the positive cell number divided with the number of nuclei in labyrinth and spongiotrophoblast layers.
Immunofluorescence
Immunofluorescent staining was carried out as described previously [Citation15,Citation51]. Paraffin-embedded placental sections from term-matched normal pregnant and preeclamptic women were de-paraffinized, treated with 0.1% Sudan Black B (Sigma, 199,664) for 20 min at room temperature and incubated overnight at 4°C with the primary antibodies diluted with Pierce immunostaining enhancer (ThermoFisher Scientific, 46,644). After extensive wash in phosphate buffered saline (PBS)-Tween 20 solution (PBS: VWR Life Science, 3467C459; Tween-20: Fisher Scientific, BP337-100), the sections were incubated for 1 h with the secondary antibodies. For cell culture, human primary trophoblasts (purchased from ScienCell Research Laboratories, 7120) or HchEpC1b cells were fixed in 4% paraformaldehyde in PBS (VWR Life Science, 3467C459) for 15 min, blocked with 0.1% Triton X-100 (BIO-RAD, 1610407), 5% BSA (Fisher Scientific, BP9706100) in PBS and then labeled with the primary antibodies. Labeled proteins were visualized with Alexa Fluor 488 donkey anti-rabbit IgG (ThermoFisher, A21206) or Alexa Fluor 594 donkey anti-rabbit IgG (ThermoFisher, A32754). Lysosomal activity was examined by staining with LysoTracker DND-99 (1 μM; Molecular Probes, L7528) for 40 min at 37°C prior to fixation. Negative controls were performed by replacing the primary antibody with purified rabbit IgG or mouse IgG. Fluorescence images were captured using a Nikon Eclipse TE2000 (Nikon, Tokyo, Japan) fluorescent microscope and analyzed using MetaVue Imaging software (Molecular Devices, CA, USA). Mean fluorescent intensity in each section was measured from at least 50 villi from ten randomly selected fields at 40X magnification using ImageJ software (http://imagej.nih.gov/).
PPP3/calcineurin phosphatase activity assay
The phosphatase activity of PPP3/calcineurin was measured using a Calcineurin Phosphatase Activity Assay Kit (colorimetric) (Abcam, ab139461), according to the manufacturer’s instructions.
Transmission electron microscopy (TEM)
Ultrathin sections were cut, double-stained with uranyl acetate and lead citrate, and examined under a Philips 410 Transmission Electron Microscope. For quantification of autolysosomes, photomicrographs showing the perinuclear area from hypoxia-treated (n = 15) or control cells (n = 15) (x21000 magnification) were randomly taken and blindly analyzed.
Detection of aggregated proteins in the placenta and trophoblasts cells
Aggregated proteins were detected using ProteoStat Aggresome Detection Kit (ENZO, ENZ-51,035-K100) according to a modified protocol described previously [Citation48]. Briefly, human and mouse placental sections were de-paraffinized and then treated with 0.1% Sudan Black B for 20 min at room temperature. Sections were then fixed with 4% formaldehyde in PBS for 15 min at 37°C. After washes in deionized water, the sections were stained with ProteoStat dye for 15 min at room temperature. Nuclei were stained with DAPI (VECTOR LABORATORIES, H-1200). For detection of protein aggregates in trophoblast cells, primary trophoblasts or immortalized trophoblasts were plated on glass cover slips and treated with sera from normal pregnancy or preeclamptic deliveries for 24 h or exposed to low oxygen tension (1% O2) for 72 h. Cells were fixed, permeabilized, and then incubated with ProteoStat dye for 15 min.
The intensity of ProteoStat specific signal in each section was measured from at least 50 villi from ten randomly selected fields at x40 magnification using ImageJ software (http://imagej.nih.gov/). The mean fluorescent intensity was obtained by the signal intensity of the regions in the trophoblast layers divided by measured area.
Western blotting and nuclear and cytoplasmic protein extraction
Cells were washed with cold PBS and lysed in a lysis buffer containing 25 mM Tris-HCl, 150 mM NaCl, 1% sodium deoxycholate, 1% NP40, and 0.1% SDS (RIPA Buffer [10X], Cell Signaling Technology, 9806S) mixed with protease inhibitor cocktail (Roche, 0469316001), and 1% phosphatase inhibitor (Sigma-Aldrich, P5726). To isolate the nuclear and cytosolic fractions, cell pellet was treated with NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific, 78833). Equal amounts of protein were then subjected to western blotting. The polyvinylidene difluoride membranes (BIO-RAD Laboratories, 162–0177) were incubated for 1 h at room temperature in a 5% nonfat dry milk blocking buffer (BIO-RAD Laboratories, 170–6404), overnight with primary antibodies at 4ºC, and then with secondary antibodies for 1 h. The blots were visualized with an enhanced chemiluminescence detection system (ECL detection kit; PIERCE, Rockford, IL, USA). Only blots with exposure time within the linear range of detection were used for quantification.
Statistical analysis
Results were presented as the mean ± SEM. Comparisons between experimental groups were performed using a Student t test or one-way ANOVA followed by Bonferroni post hoc test, Fisher’s exact test or Wilcoxon rank-sum test. Values of p < 0.05 were considered statistically significant. P > 0.05 was considered not significant (ns).
Supplemental Material
Download Zip (6.1 MB)Acknowledgments
We thank Phil Gruppuso and Michael Soares for critical reading of the manuscript and Paula Krueger for technical assistance. We also thank the Department of Pediatrics, Women & Infants’ Hospital of Rhode Island, Warren Alpert Medical School of Brown University, for continued support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365:785–799.
- Staff AC, Benton SJ, von Dadelszen P, et al. Redefining preeclampsia using placenta-derived biomarkers. Hypertension. 2013;61:932–942.
- Wildman K, Bouvier-Colle MH. Maternal mortality as an indicator of obstetric care in Europe. BJOG. 2004;111:164–169.
- Steegers EA, von Dadelszen P, Duvekot JJ, et al. Pre-clampsia. Lancet. 2010;376:631–644.
- S-B C, Sharma S. Preeclampsia and health risks later in life: an immunological link. Semin Immunopathol. 2016;38:699–708.
- Vikse BE, Irgens LM, Leivestad T, et al. Preeclampsia and the risk of end-stage renal disease. N Engl J Med. 2008;359:800–809.
- Saade GR. Pregnancy as a window to future health. Obstet Gynecol. 2009;114:958–960.
- Bellamy L, Casas JP, Hingorani AD, et al. Preeclampsia and risk of cardiovascular disease and cancer in later life: a systemic review and meta-analysis. BMJ. 2007;335:974–977.
- Smith GC, Pell JP, Walsh D. Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129,290 births. Lancet. 2001;357:2002–2006.
- Zhou Y, Gormley MJ, Hunkapiller NM, et al. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J Clin Invest. 2013;123:2862–2872.
- Roberts JM, Hubel CA. Is oxidative stress the link in the two stage model of preeclampsia? Lancet. 1999;354:788–789.
- Redman CW, Sacks GP, Sargent IL. Preeclampsia:anexcessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180:499–506.
- Burton GJ, Yung HW. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens. 2011;1:72–78.
- Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683.
- Kalkunte SS, Neubeck S, Norris WE, et al. Transthyretin is dysregulated in preeclampsia, and its native form prevents the onset of disease in a preclinical mouse model. Am J Pathol. 2013;183:1425–1436.
- Buhimschi IA, Nayeri UA, Zhao G, et al. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci Transl Med. 2014;6:245ra292.
- McCarthy FP, Adetoba A, Gill C, et al. Urinary congophilia in women with hypertensive disorders of pregnancy and preexisting proteinuria or hypertensin. Am J Obstet Gynecol. 2016;215:464.e1–7.
- Tong M, Cheng SB, Chen Q, et al. Aggregated transthyretin is specifically packaged into placental nano-vesicles in preeclampsia. Sci Rep. 2017;7:6694.
- Cater JH, Kumita JR, Zeineddine Abdallah R, et al. Human pregnancy zone protein stabilizes misfolded proteins including preeclampsia- and Alzheimer’s-associated amyloid beta peptide. Proc Natl Acad Sci U S A. 2019;116:6101–6110.
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11:301–307.
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative diseases. Nat Med. 2004;10:S10–7.
- Yoshimori T. Autophagy: paying Charon’s toll. Cell. 2007;128:833–836.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075.
- Cuervo AM, Bergamini E, Brunk UT, et al. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1(3):131–140.
- Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–1117.
- Shimada Y, Klionsky DJ. Autophagy contributes to lysosomal storage disorders. Autophagy. 2012;8:715–716.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. 2018;20(12):1338–1348.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–299.
- Napolitano G, Esposito A, Choi H, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9:3312.
- Delorme-Axford E, Bayer A, Sadovsky Y, et al. Autophagy as a mechanism of antiviral defense at the maternal-fetal interface. Autophagy. 2013;9:2173–2174.
- Delorme-Axford E, Donker RB, Mouillet JF, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci USA. 2013;110:12048–12053.
- Cao B, Macones M, Mysorekar IU. ATG16L1 governs placental infection risk and preterm birth in mice and women. JCI Insight. 2016;1:e86654.
- Hung TH, Hsieh TT, Chen SF, et al. Autophagy in the human placenta throughout gestation. PLoS One. 2013;8:e83475.
- Nakashima A, Aoki A, Kusabiraki T, et al. Autophagy regulation in preeclampsia: pros and cons. J Reprod Immunol. 2017;123:17–23.
- Cao B, Parnell LA, Diamond MS, et al. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med. 2017;214:2303–2313.
- Oh SY, Choi SJ, Kim KH, et al. Autophagy-related proteins, LC3 and Beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod Sci. 2008;15:912–920.
- Gao L, Qui HB, Kamana KC, et al. Excessive autophagy induces the failure of trophoblast invasion and vasculature: possible relevance to the pathogenesis of preeclampsia. J Hypertens. 2015;33:106–117.
- Melland-Smith M, Ermini L, Chauvin S, et al. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy. 2015;11:653–669.
- Saito S, Nakashima A. A review of the mechanism for poor placentation in early onset preeclampsia: the role of autophagy in trophoblast invasion and vascular invasion. J Reprod Immunol. 2014;101–102:80–88.
- Hutabarat M, Wibowo N, Huppertz B. The trophoblast survival capacity in preeclampsia. PLoS ONE. 2017;12:e0186909.
- Akcora Yildiz D, Irtegun Kandemir S, Agacayak E, et al. Evaluation of protein levels of autophagy markers (Beclin 1 and SQSTM1/p62) and phosphorylation of cyclin E in the placenta of women with preeclampsia. Cell Mol Biol (Noisy-le-grand). 2017;63(12):51–55.
- Akaishi R, Yamada T, Nakabayashi K, et al. Autophagy in the placenta of women with hypertensive disorders in pregnancy. Placenta. 2014;35(12):974–980.
- Ozsoy AZ, Cayli S, Sahin C, et al. Altered expression of p97/Valosin containing protein and impaired autophagy in preeclamptic human placenta. Placenta. 2018;67:45–53.
- Chen B, Longtine MS, Nelson DM. Hypoxia induces autophagy in primary human trophoblasts. Endocrinology. 2012;153(10):4946–4954.
- Zhang Y, Hu X, Gao G, et al. Autophagy protects against oxidized low density lipoprotein-mediated inflammation associated with preeclampsia. Placenta. 2016;48:136–143.
- Cheng SB, Nakashima A, Sharma S. Protein misfolding and aggregation: a novel mechanism for preeclampsia. Am J Reprod Immunol. 2014;71:40.
- Guo Z-J, Tao L-X, Dong X-Y, et al. Characterization of aggregate/aggresome structures formed by polyhedron of Bombyx mori nucleoployhedrovirus. Sci Rep. 2015;5:14601.
- Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control center for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296.
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632.
- Nakashima A, Yamanaka-Tatematsu M, Fujita N, et al. Impaired autophagy by soluble endoglin, under physiological hypoxia in early pregnant period, is involved in poor placentation in preeclampsia. Autophagy. 2013;9:303–316.
- Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659.
- Saftig P, Klumperiman J. Lysosome biogenesis and lysosome membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635.
- DeVorkin L, Gorski SM. LysoTracker staining to aid in monitoring autophagy in Drosophilla. Cold Spring Harb Protoc. 2014;2014:951–958.
- Mauvezin M, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438.
- Kalkunte S, Boij R, Norris W, et al. Sera from preeclampsia patients elicit symptoms of human disease in mice and provide a basis for an in vitro predictive assay. Am J Pathol. 2010;177:2387–2398.
- Kanninen TT, Jayaram A, Jaffe-Lifshitz S, et al. Altered autophagy induction by sera from pregnant women with preeclampsia. A case-control study. BJOG. 2014;121:958–964.
- Aoki A, Nakashima A, Kusabiraki T, et al. Trophoblast-specific conditional ATG7 knockout mice develop gestational hypertension. Am J Path. 2018;188:2474–2486.
- Muralimanoharan S, Gao X, Weintraub S, et al. Sexual dimorphism in activation of placental autophagy in obese women with evidence for fetal programming from a placenta-specific mouse model. Autophagy. 2016;12:752–769.
- Spampanato C, Feeney E, Li L, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 2013;5:691–696.
- Parenti G, Andria G, Ballabio A. Lysosome storage diseases: from pathophysiology to therapy. Ann Rev Med. 2015;66:471–486.
- Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–430.
- Yamanaka-Tatematsu M, Nakashima A, Fujita N, et al. Autophagy induced by HIF1α overexpression supports trophoblast invasion by supplying cellular energy. PLoS One. 2013;8(10):e76605.
- Matsui Y, Kyoi S, Takagi H, et al. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy. 2008;4(4):409–415.
- Kliman HJ, Nestler JE, Sermasi E, et al. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582.
- Schaiff WT, Bildirici I, Cheong M, et al. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor signaling regulate fatty acid uptake by primary human placental trophoblasts. J Clin Endocrinol Metab. 2005;90:4267–4275.
- Nelson DM, Johnson RD, Smith SD, et al. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am J Obstet Gynecol. 1999;180:896–902.
- Kalkunte S, Lai Z, Tewari N, et al. In vitro and in vivo evidence for lack of endovascular remodeling by third trimester trophoblasts. Placenta. 2008;29:871–878.