
ABSTRACT
Macroautophagy/autophagy is essential for maintaining cellular homeostasis through the degradation of organelles and proteins. It also has a prominent role in modulating aging. However, the role of autophagy in the neuronal response to axon injury and axon regeneration, particularly in the context of aging, remains largely unknown. Our candidate genetic screen for axon regeneration regulators has identified genes in the autophagy pathway. Using a reporter that monitors autophagosomes and autolysosomes, we were able to monitor the dynamics of autophagy during axon regeneration. In response to axon injury, there was a significant increase in the number of autophagic vesicles. Injury-triggered autophagy activation and axon regeneration capacity undergo an age-dependent decline, and autophagy-activating agents partially rescued these declines. We found that DLK-1 was both required and sufficient for injury-induced autophagy activation. Autophagic vesicles co-localized with the NOTCH4 ortholog, LIN-12 receptor, a previously identified inhibitor of axon regeneration. Epistasis analyses indicate that LIN-12 might be a target of autophagy in axon regeneration. Together, our data suggest that DLK-mediated injury signaling can activate autophagy, which might limit the level of LIN-12 and NOTCH proteins to promote axon regeneration. Our findings reveal that autophagy activation can promote axon regeneration in neurons that lack maximal regrowth capacity, providing a promising therapeutic strategy for axon injury.
Abbreviations: 3-MA: 3-methyladenine; ALs: autolysosomes; APs: autophagosomes; ARF-6: ADP-Ribosylation Factor related 6; ATG-9: AuTophaGy (yeast Atg homolog) 9; ATG9A: autophagy related 9A; BA1: bafilomycin A1; BEC-1: BEClin (human autophagy) homolog; BECN1: beclin 1; C. elegans: Caenorhabditis elegans; CEBP-1: C/EBP (CCAAT/enhancer-binding protein) homolog; CNS: central nervous system; DLK-1: Dual-Leucine zipper Kinase; DMSO: dimethyl sulfoxide; DRG: dorsal root ganglion; FOS: Fos proto-oncogene, AP-1 transcription factor subunit; GABA: gamma-aminobutyric acid; GFP: green fluorescent protein; HDA-3: Histone DeAcetylase; IP3: inositol trisphosphate; ITR-1: Inositol Triphosphate Receptor; KLF-2: Kruppel-Like Factor (zinc finger protein) 2; LGG-1: LC3, GABARAP and GATE-16 family; MAK-2: MAP kinase Activated protein Kinase; MAP kinase: mitogen-activated protein kinase; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MKK-4: mitogen activated protein kinase kinase 4; MTOR: mechanistic target of rapamycin kinase; NGM: nematode growth medium; NICD: Notch intracellular domain; NOTCH: notch receptor; PLM: posterior lateral microtubule; PMK-3: P38 Map kinase family; PNS: peripheral nervous system; SCG10: superior cervical ganglion protein 10; SCI: spinal cord injury; UNC-51: UNCoordinated 51; ULK1: unc-51 like autophagy activating kinase 1; wnd: wallenda
Introduction
Characterization of the mechanisms underlying axon regeneration is of great interest to human health. Injured mature axons in the mammalian central nervous system (CNS) can regrow into sciatic nerve grafts transplanted into the lesion site [Citation1,Citation2], prompting the notion that successful axon regeneration requires a permissive environment that promotes regeneration. Research in recent years has also supported that the intrinsic growth ability of adult neurons is as important as the extrinsic inhibitory factors in determining axon regeneration capability. In contrast to the CNS, the mammalian peripheral nervous system (PNS) can regenerate axons robustly after injury, but this regenerative ability undergoes an age-dependent decline [Citation3,Citation4]. The regenerative ability of axons varies widely among different types of neurons and are regulated by both positive and negative signaling pathways [Citation5–8].
One of the positive signaling pathways that promote regeneration is the DLK pathway, which was initially shown in C. elegans to be essential for axon regeneration in motor and mechanosensory neurons [Citation9,Citation10]. DLK-1 is a conserved MAP kinase kinase kinase (MAPKKK) that acts upstream in a MAP kinase cascade including the MKK-4, PMK-3 and MAK-2 kinases. Loss of DLK-1 completely blocks axon regrowth, and overexpression of DLK-1 is sufficient to promote regenerative growth. The critical role of the DLK-1 pathway in axon regeneration has been confirmed in other species. The Drosophila ortholog of DLK-1, wnd (wallenda), acts upstream in a signaling cascade that involves MAPK/JNK and FOS homologs and is required for axon regrowth [Citation11]. In mice, MAP3K12/DLK functions upstream of JUN/c-Jun to promote axon regeneration in dorsal root ganglion (DRG) neurons [Citation12]. Remarkably, MAP3K12 is transported in injured axons and is required acutely for the generation of injury signals that are retrogradely transported to the cell body [Citation13].
The NOTCH signaling pathway regulates axon regeneration negatively [Citation14]. NOTCH signaling is highly conserved and regulates a wide variety of cellular processes, including cell proliferation, cell fate differentiation, and cell death [Citation15]. NOTCH is a cell-surface receptor that transduces short-range signals. The interaction of the NOTCH receptor with its transmembrane ligand triggers the cleavage of the NOTCH intracellular domain (NICD) by ADAM-family metalloproteases and γ-secretase. The NICD is then released into the cytosol and translocated into the nucleus to regulate transcription. Recently, NOTCH signaling was demonstrated to play an inhibitory role in axon regeneration in mature C. elegans neurons. Genetic disruption or pharmacological inhibition of NOTCH signaling improves axon regeneration in C. elegans motor neurons after injury [Citation14].
Axon regeneration is a highly regulated process in response to injury. Axonal injury is acute neuronal stress that triggers drastic responses in both the axon and cell body, including calcium influx, axonal membrane sealing, injury signal transduction, and transcriptional alterations [Citation16]. Autophagy is a tightly regulated process that is a central component of the cellular stress response [Citation17]. It plays a conserved role in maintaining cellular homeostasis by degrading dysfunctional proteins, lipids, and organelles through an autophagosome-lysosome pathway [Citation18]. Autophagy was first discovered in conditions of starvation, which activates autophagy [Citation19]. Autophagy initiation forms a double-membrane structure around cellular substances followed by autophagosome formation, which then fuses with a lysosome. The engulfed substances are then degraded in the lysosome and recycled back to the cell as amino acids and fatty acids to enable cell survival [Citation20]. Beyond its function in protecting cells in response to stress conditions, autophagy has a prominent role in modulating aging. Reduced autophagy is associated with accelerated aging, whereas activation of autophagy appears to have anti-aging effects [Citation21]. Genetic inhibition of autophagy causes neurodegeneration in various brain regions [Citation22,Citation23]. More recently, the role of autophagy in axon regeneration post-injury has been assessed. The expression of LC3, which is critical for autophagosome formation and a marker of autophagy, is upregulated in DRGs after acute spinal cord injury [Citation24]. Activating autophagy using an autophagy-inducing peptide can promote axon regeneration through limiting SCG10, a microtubule destabilizing protein [Citation25]. However, autophagy regulation after axon injury and how it promotes axon regeneration, particularly during aging, is not well understood.
In the present study, we investigated the regulation of autophagy in axon regeneration after injury using C. elegans as a model. We found that genetic loss-of-function mutants of genes involved in different steps of the autophagy process displayed impaired axon regeneration. Taking advantage of a tandem reporter, we were able to monitor the dynamics of autophagosomes (APs) and autolysosomes (ALs) in injured neurons. We found that axotomy induced an elevation of both APs and ALs in an age-dependent manner and that injury-induced autophagy requires DLK-1. Pharmacologically blocking autophagic flux inhibited axon regrowth, while activating autophagy with rapamycin promoted axon regeneration. However, the regrowth-promoting effect of rapamycin treatment was only seen in aged animals. Our data also demonstrate that LIN-12/NOTCH4 co-localizes with autophagic vesicles and that autophagy and LIN-12 might function through the same pathway to regulate axon regrowth. Together, our findings suggest that axon injury activates autophagy, which is required for effective axon regeneration by limiting NOTCH signaling.
Results
Proper autophagy function is required for normal axon regeneration
Despite a general agreement that autophagy alteration occurs following axon injury [Citation24,Citation26–28], the role of autophagy in the neuronal response to axon injury and axon regeneration is unclear. Autophagy is a multistep process, and many autophagy genes are involved. The initial step of autophagy is the formation of a crescent-shaped double-membrane vesicle called the phagophore, which is followed by elongation and engulfment of cytosolic cargos to form APs. APs then fuse with lysosomes to form ALs, where the degradation of cargo occurs [Citation29]. To determine whether a specific step of the autophagy cascade is critical for axon regeneration, we examined axon regeneration in loss-of-function mutants of genes involved in autophagy regulation. We assayed axon regrowth in vivo by using touch sensory PLM neurons, which are a well-established model for axon regeneration [Citation30,Citation31]. We used touch neuron-specific reporter alleles muIs32 (Pmec-7-GFP) and zdIs5 (Pmec-4-GFP) to label the touch sensory neurons for morphological analysis and single-neuron laser axotomy. We performed axotomy at the day 1 young adult stage and measured axon regrowth 24 h post-injury. All the autophagy mutants tested displayed impaired axon regeneration (–). It includes the genes regulating autophagy induction (unc-51/ULK1), vesicle nucleation (bec-1/BECN1 and atg-9/ATG9A), and vesicle elongation (lgg-1 and lgg-2/LC3), as well as transcription factors associated with autophagy (klf-2 and klf-3 [Kruppel-like factors]). These findings indicate that axon regeneration requires proper autophagic activity. The bec-1(ok700) mutation had the most severe defect in PLM axon regrowth, while atg-9(bp564) mutants showed a modest reduction in axon regeneration. The less severe defects in regrowth might be due to gene redundancy or partial loss-of-function mutations. Autophagy mutants displayed normal PLM neuron morphology when examined at the day 1 young adult stage, except for unc-51(e369), which had branched and wavy touch axons (Fig. S1).
Figure 1. Autophagy genes are required for effective axon regeneration. (A-D) Representative images and quantification of PLM axon regeneration 24 h post-laser axotomy in the genetic mutants of indicated autophagy-related genes; arrows indicate lesion sites. Axon is visualized by touch neuron-specific GFP expression driven by Pmec-4 or Pmec-7. (E-F) Time course of regrowth in lgg-1(tm3489) and wildtype control. PLM axon regrowth is impaired in lgg-1 mutant in all time periods measured. Total regrowth length measured at each time point (F) and regrowth rate (G) at each time window were plotted. (G-H) Representative images and quantification of total PLM axon regeneration length in worm strains with indicated genotypes. Scale bar: 20 µm. Statistics: one-way ANOVA; mean ± SEM; n ≥ 10 for each group (number in the bar); *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
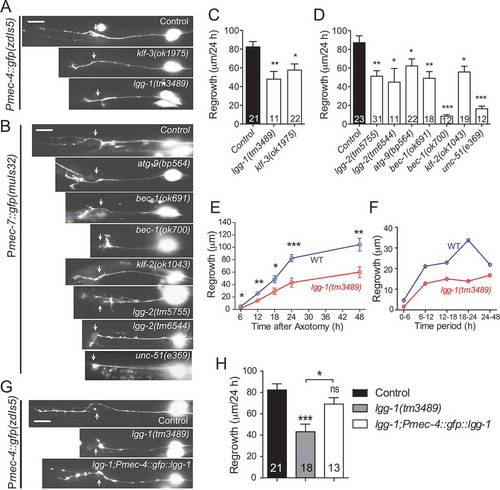
Altered regrowth examined at 24 h post-axotomy could reflect defects in regrowth initiation (i.e., regenerative growth cone formation) or the later processes of axon extension. We measured axon regrowth in lgg-1(tm3489) mutants at 6, 12, 18, 24, and 48 h post-injury and calculated the regrowth rate (µm/hour) between each time point. lgg-1 mutants showed reduced total regrowth length measured at different time points, and decreased axon regrowth rate at all times measured ( and ), suggesting that the axon regeneration process requires autophagy throughout. Expression of LGG-1 driven by a touch neuron-specific promoter rescued the axon regrowth defect ( and ), supporting that autophagy is required cell-autonomously for axon regeneration.
Axonal injury induces autophagy flux in C. elegans neurons
To understand how autophagy is regulated in response to axon injury and during axon regeneration, we constructed and expressed a dual-fluorescent mCherry::GFP::LGG-1 protein under the control of a touch neuron-specific promoter. This dual-fluorescent reporter has been used to monitor both APs and ALs and was confirmed functionally by rescuing an embryonic lethal lgg-1(tm3489) mutant [Citation32,Citation33]. In cells expressing this reporter, APs are positive for both GFP and mCherry, while ALs are only positive for mCherry because the low pH quenches GFP in ALs. We observed mCherry/GFP double-positive AP puncta and mCherry single-positive AL puncta in the intact PLM cell bodies. We found that the number of both AP and AL vesicles increased when examined at 24 h post-axon injury ( and ), indicating that axon injury induces autophagic vesicle formation and promotes autophagic flux. We were able to detect an increased number of puncta as early as 3 h post-injury, although the increase was not as dramatic as it was at 24 h. We attempted to examine autophagic vesicles in axons, particularly at the injury site. However, we were not able to detect vesicles in the majority of injured axons. Therefore, we have focused on studying autophagy regulation in response to injury within the cell bodies. To test whether axon injury-induced autophagy also occurs in other neuron types, we expressed the dual-fluorescent reporter in GABAergic neurons. Laser axotomy at the commissures of these motor neurons also induced autophagic puncta like those observed in PLM neurons (Fig. S2A).
Figure 2. Axon injury activates autophagy in PLM neurons. (A) Representative images of PLM neurons of transgenic WT animals expressing Pmec-4-mCherry::GFP::LGG-1. Axotomy was performed on day 1 of adulthood. Images were taken immediately before axotomy (before injury) and 24 h post-axotomy (injured). For the uninjured controls, animals underwent sham injury at day 1, and PLM neurons were imaged 24 h later. AP, autophagosome; AL, autolysosome. (B) Quantification of APs and ALs in PLM cell bodies for experiments in (A). (C) Representative images of APs and ALs in PLM neurons of transgenic WT animals expressing Pmec-4-mCherry::GFP::LGG-1 with indicated treatments. 50 µM of BA1 in 0.2% DMSO (or 0.2% DMSO control) was injected into the body cavity of the tail region at day 1 of adulthood 1 h before axotomy. Images were taken 24 h post axotomy (or sham injury). (D) Quantification of APs and ALs in PLM cell bodies for experiments in (C) to show that BA1 treatment blocked AL formation. Scale bar: 5 µm. Statistics: one-way ANOVA; mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
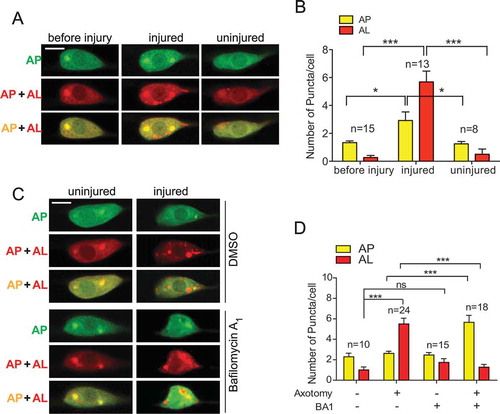
Bafilomycin A1 (BA1) is a well-known inhibitor of the late phase of autophagy. BA1 blocks vacuolar-type H+-ATPases and prevents maturation of autophagic vacuoles by inhibiting fusion between APs and lysosomes. Consistent with its role in blocking autophagic flux, BA1 treatment increased in mCherry/GFP double-positive puncta (an increase of APs) and a decrease in mCherry single-positive puncta (decrease of ALs) in cells expressing the dual-fluorescent reporter [Citation33]. When we treated the animals with BA1 by injecting the drug into the body cavity, we observed a significant increase in APs and decreased in ALs in injured PLM neurons expressing the dual-fluorescent reporter ( and ). However, BA1 treatment did not affect the numbers of APs or ALs in uninjured neurons, which is likely due to the low level of autophagic flux in intact neurons. Together, these data suggest that axon injury can induce autophagic vesicle formation and promote autophagic flux.
Autophagy-inducing drugs fail to enhance axon regrowth in young PLM neurons
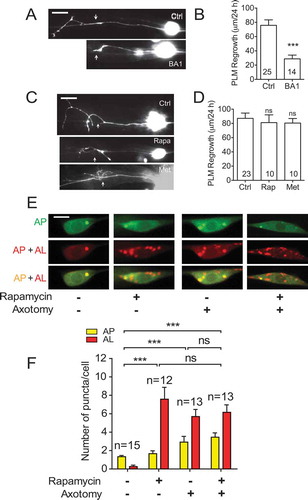
As BA1 treatment resulted in impaired autophagic flux in injured PLM neurons, we next examined the effect of BA1 on axon regeneration by injecting BA1 prior to axotomy. Compared to control animals injected with DMSO, animals injected with BA1 displayed significantly impaired axon regrowth ( and ), again indicating that axon regeneration requires proper autophagy function. Given that blocking autophagy genetically or pharmacologically resulted in impaired axon regeneration, we tested whether enhancing autophagy would promote axon regeneration. We treated the animals with rapamycin, a well-known autophagy inducer [Citation34]. Day 1 young adult wildtype animals were subjected to laser axotomy and then transferred to NGM plates containing 100 nM rapamycin for 24 h before axon regrowth was measured. However, rapamycin treatment did not result in enhanced axon regrowth ( and ). As an alternative approach to using rapamycin treatment to activate autophagy, we treated animals with metformin, another known autophagy inducer [Citation35]. Similar to rapamycin treatment, metformin treatment was not able to enhance PLM axon regrowth in day 1 animals ( and ). We next examined the effect of rapamycin on autophagic puncta in PLM neurons. Consistent with its role in promoting autophagy, rapamycin treatment significantly increased the numbers of both mCherry/GFP double-positive AP puncta and mCherry-positive AL puncta in uninjured neurons, to a level that was similar to untreated neurons subjected to laser axotomy ( and ). However, rapamycin failed to further increase autophagic puncta in injured neurons ( and ), possibly explaining why rapamycin treatment failed to enhance axon regeneration ( and ). We speculate that axon injury activates autophagy to the maximal level, and therefore, rapamycin is unable to promote autophagic activity further.
Figure 3. Rapamycin fails to enhance axon regrowth in young PLM neurons. (A-B) Representative images and quantification of PLM axon regeneration 24 h post-laser axotomy in animals injected with 50 µM of BA1 or 0.2% DMSO at day 1 of adulthood. Microinjection was performed 1 h before laser axotomy. Microinjection itself was detrimental to axon regrowth, as it decreased the average regrowth length from around 80 µm to less than 60 µm. Scale bar: 20 µm. (C-D) Representative images and quantification of PLM axon regeneration 24 h post-laser axotomy in animals with indicated treatments. L4 animals were treated with 100 nM rapamycin or metformin, and axotomy was performed 24 h later (day 1 adult). After axotomy, animals were allowed to recover for 24 h in the presence of rapamycin or metformin. Scale bar: 20 µm. (E) Representative images of APs and ALs in PLM cell bodies in day 1 adult animals expressing Pmec-4-mCherry::GFP::LGG-1 with indicated treatments. Rapamycin or axotomy alone was sufficient to enhance the numbers of APs and ALs, but the two treatments combined did not lead to further enhancement, suggesting that autophagic activity might have reached the maximal level with either rapamycin or axotomy treatment. Scale bar: 5 µm. (F) Quantification of APs and ALs in PLM neurons with indicated treatments. Statistics: Student’s t-test (A, B) and one-way ANOVA (C); mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
Autophagy induction by injury is defective in old neurons, and autophagy-activating agents can partially rescue the defect
Axon regeneration undergoes age-related decline, but the mechanisms linking aging to loss of axon regeneration capacity remain elusive. As injury can activate autophagy, which axon regeneration requires, we evaluated the effect of aging on injury-triggered autophagy induction. We first examined the dual-fluorescent mCherry::GFP::LGG-1 reporter in intact neurons from adult day 10 animals. We observed no obvious difference in the number of AP puncta in day 10 neurons compared to day 1. However, we observed slightly increased ALs ( and ), indicating that autophagy remains active in aged neurons. Remarkably, when we examined the autophagic vesicles in injured old neurons, we found that autophagy induction by injury was abolished completely in PLM neurons of adult day 10 animals ( and ), suggesting that PLM neurons lose their capacity to activate autophagy in response to injury with aging.
Figure 4. Age-related decline of autophagy induction can be partially rescued by autophagy-activating agents. (A) Representative images of AP and AL in PLM cell bodies in day 10 adult animals expressing Pmec-4-mCherry::GFP::LGG-1 with indicated treatments. Day 9 animals were picked to NGM plates containing rapamycin (or DMSO control) and cultured for 24 h before laser axotomy, followed by 24 h of culture on NGM plates with rapamycin (or DMSO control) immediately before imaging. (B) Quantification of APs and ALs in PLM cell bodies in day 1 and day 10 animals. Axon injury in aged neurons did not induce the formation of autophagic vesicles. (C) Quantification of APs and ALs in PLM cell bodies of day 10 animals with indicated treatments. Rapamycin treatment was able to enhance AL number in aged neurons. (D) Representative images of APs and ALs in PLM cell bodies in day 10 adult animals treated with metformin or control. Day 9 animals were treated with metformin for 24 h before laser axotomy, followed by 24 h of incubation with metformin immediately before imaging. (E) Quantification of APs and ALs in PLM cell bodies in day 10 animals with indicated treatments. Metformin treatment was able to enhance AL number in aged neurons. Scale bar: 5 µm. Statistics: One-way ANOVA (B, C) and Student’s t-test (E); mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
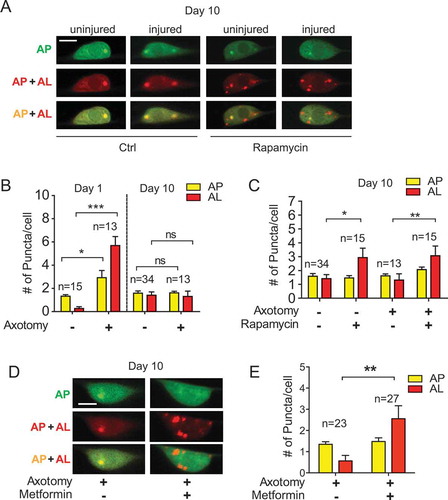
We next examined whether rapamycin was able to promote autophagy in injured day 10 PLM neurons, where the pool size of autophagic vesicles was much smaller than the pool size on day 1. We observed significantly increased numbers of both APs and ALs in injured day 10 neurons with rapamycin treatment (), supporting that rapamycin can enhance autophagic activity in cells with a low basal level of autophagic activity (such as intact young PLM neurons or injured old PLM neurons), but not in cells with a high or maximal autophagic activity level (e.g., injured young PLM neurons). Similar to rapamycin treatment, we observed enhanced autophagic vesicles in day 10 animals treated with metformin ( and ).
Enhancing autophagy in aged neurons can partly rescue the age-dependent decline in axon regrowth
The increased number of autophagic vesicles in rapamycin-treated day 10 neurons prompted us to evaluate the effect of rapamycin on the axon regrowth of aged neurons. We performed laser axotomy on PLM neurons from day 6 and day 10 animals and measured axon regrowth 24 h post-injury. Consistent with previously reported data [Citation36], PLM axon regeneration gradually declined with aging (, and ). This age-related reduction in axon regrowth appears to correlate with the decrease in injury-induced autophagic activity. As shown above, rapamycin treatment significantly enhanced axon regrowth in day 6 and day 10 neurons ( and ), but not in day 1 neurons (). Thus, the effect of rapamycin on promoting axon regeneration is associated with its effect on enhancing autophagic activity. Similar to rapamycin treatment, we observed enhanced axon regrowth in animals treated with metformin ( and ), further supporting that activating autophagy can promote axon regeneration in aged neurons.
Figure 5. Autophagy-activating agents promote axon regrowth in aged PLM neurons. (A-B) Representative images and quantification of axon regrowth in wildtype animals expressing muIs32 (Pmec-7::GFP) reporter. Day 5 or day 9 animals were treated with rapamycin or metformin for 24 h before laser axotomy. After axotomy at day 6 or day 10, animals were allowed to recover for 24 h in the presence of rapamycin or metformin. Scale bar: 20 µm. (C) Quantification of PLM axon regrowth in day 6 animals with indicated genotypes and treatments. Rapamycin treatment was able to enhance axon regrowth at day 6 in wildtype animals, but not in lgg-1 mutant animals. Transgenic expression of LGG-1 in touch neurons was able to restore the regrowth promoting effect of rapamycin in lgg-1 mutant animals. (D-E) Representative images and quantification of APs and ALs in PLM neurons of transgenic WT animals expressing Pmec-4-mCherry::GFP::LGG-1 with indicated treatments to show that Tat-ceBec peptide was able to enhance autophagic activity in injured PLM neurons at day 6. Tat-ceBec peptide (or Tat-scramble peptide) was injected into the body cavity of the tail region at day 6 of adulthood 1 h before axotomy. Images were taken 24 h post-axotomy. Scale bar: 5 µm. (F) Quantification of PLM axon regrowth in day 6 animals with indicated treatments. Tat-ceBec peptide was sufficient to promote axon regeneration on day 6. (G-H) Representative images and quantification of APs and ALs with indicated treatments at day 10. Scale bar: 5 µm. (I) Quantification of PLM axon regrowth in day 10 animals with indicated treatments. Statistics: One-way ANOVA; mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
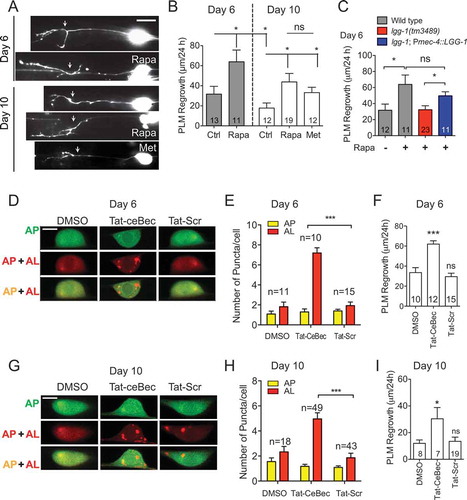
The MTOR (mechanistic target of rapamycin) signaling pathway is a master regulator of cell growth and metabolism. It integrates diverse upstream signals and controls various essential downstream cellular processes, including autophagy [Citation37]. We have shown that the effect of rapamycin on promoting axon regeneration is associated with its effect on enhancing autophagic activity. However, we were unable to rule out the possibility that the effect of rapamycin on axon regrowth was independent of autophagy. To test whether rapamycin promotes axon regrowth in aged neurons by activating autophagy, we compared PLM axon regrowth in wildtype and lgg-1(tm3489) mutants treated with rapamycin at day 6. Unlike in wildtype animals, rapamycin treatment failed to enhance axon regrowth in lgg-1(tm3489) mutants at day 6 (), suggesting that the regrowth-promoting effect of rapamycin requires proper autophagy function. Furthermore, the transgenic expression of LGG-1 in the touch neurons of the lgg-1(tm3489) mutant was sufficient to suppress the loss-of-function effect of lgg-1 on rapamycin-enhanced axon regrowth in aged animals, indicating that cell-autonomous autophagy function mediates the regrowth-promoting effect of rapamycin in old neurons.
As both rapamycin and metformin are inhibitors of MTOR, which regulates many other essential cellular processes in addition to autophagy, we used an autophagy-inducing peptide Tat-ceBec to activate autophagy specifically and examined its effects on axon regrowth. The Tat-Bec peptide induces autophagic vesicle-like structures in cultured mouse cortical neurons as well as increasing LC3-II expression. Local application of Tat-Bec peptide to the injured spinal cord can promote axon regeneration and improve motor function recovery after spinal cord injury [Citation25]. We synthesized the Tat-ceBec peptide based on the homologous BEC-1 sequence of the C. elegans BEC-1 protein (Fig. S3) and injected the peptide into the tail region, in the location of the PLM cell body. We found that Tat-ceBec treatment significantly increased the number of autophagic puncta and enhanced axon regeneration in both day 6 and day 10 animals to a level similar to those with rapamycin treatment (). Together, these data suggest that enhancing autophagic activity is sufficient to promote axon regeneration in PLM neurons and that the loss of injury-induced autophagy activation might contribute to the reduced axon regeneration capacity in older neurons.
Injury-induced autophagy activation is dependent on DLK-1 and Ca2+ signaling
As aged PLM neurons fail to activate autophagy in response to injury, we wondered how axon injury induces autophagy. Axon injury triggers a series of processes in the injured neuron, including a rapid and transient increase in axonal calcium. The level of Ca2+ influx correlates with subsequent regeneration ability [Citation38]. The effects of elevating Ca2+ in promoting axon regeneration requires DLK-1, a dual leucine zipper-bearing MAPKKK that was initially identified in C. elegans [Citation9,Citation10]. In mouse, DLK functions to promote axon regeneration in a c-Jun dependent manner [Citation12]. A previous study found that DLK is transported in injured axons and is required acutely for the generation of injury signals that are retrogradely transported to the cell body [Citation13]. To test whether DLK-1 regulates injury-induced autophagy, we examined the mCherry::GFP::LGG-1 reporter in dlk-1(tm4024) mutants. In intact neurons, dlk-1 mutants showed similar autophagic vesicle density as wildtype controls ( and ). However, unlike in young wildtype neurons, injury failed to activate autophagy in PLM neurons of dlk-1 mutants, indicating that injury-induced autophagy activation requires DLK-1.
Figure 6. Injury-induced autophagy activation is DLK-1-dependent. (A-B) Representative images and quantification of APs and ALs in PLM cell bodies in day 1 wildtype and dlk-1 mutant animals expressing Pmec-4-mCherry::GFP::LGG-1 with indicated treatments. Injury-induced autophagy activation was abolished in the absence of DLK-1. The autophagy induction defect in dlk-1 mutant was rescued by rapamycin treatment. Scale bar: 5 µm. (C-D) Representative images and quantification of axon regrowth in day 1 wildtype and dlk-1 mutant animals with muIs32 reporter. PLM axon regrowth was completely blocked in dlk-1 mutants and rapamycin treatment was able to rescue the regrowth defect in dlk-1 mutants partially. Scale bar: 20 µm. (E-F) Representative images and quantification of APs and ALs in PLM cell bodies in day 10 wildtype and transgenic animals with DLK-1 overexpression in touch neurons. DLK-1 overexpression was sufficient to significantly increase the number of ALs in day 10 injured neurons. Scale bar: 5 µm. (G) Quantification of PLM axon regrowth in day 10 animals with indicated treatments. Like rapamycin treatment, DLK-1 overexpression promoted axon regeneration in aged neurons. Statistics: One-way ANOVA; mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
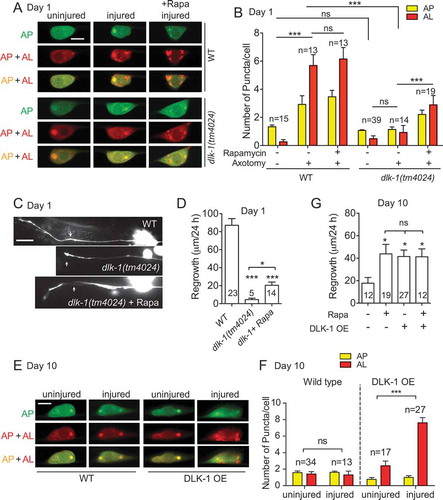
We next tested whether the autophagy-inducing drug rapamycin could augment autophagic activity in dlk-1 mutant neurons, which had lower overall autophagic activity. We observed a significantly increased number of autophagic puncta in neurons from dlk-1 mutant animals treated with rapamycin. However, rapamycin treatment was not able to boost the number of puncta to a wildtype level ( and ). Functionally, rapamycin treatment was able to significantly improve axon regeneration in dlk-1 mutants, which normally have completely blocked axon regrowth ( and ). Together, these data suggest that DLK-1 regulates injury-activated autophagy and that enhancing autophagy can partially bypass the requirement of DLK-1 in axon regeneration.
To further confirm the role of DLK-1 in regulating injury-activated autophagy, we overexpressed DLK-1 in touch neurons. Overexpression of the active form of DLK-1 results in constitutive activation of the DLK-1 pathway and enhanced axon regeneration [Citation9,Citation10]. We found that DLK-1 overexpression significantly enhances the number of APs and ALs in day 10 neurons ( and ). Therefore, DLK-1 is both required and sufficient to induce autophagy activation after injury. Given the effect of DLK-1 overexpression, we wondered to what degree DLK-1 was overexpressed compared to endogenous DLK-1 expression. To measure the endogenous level of DLK-1 expression, we generated MosSCI transgenic animals that express a single copy of GFP-tagged DLK-1 under its endogenous promoter to reflect the endogenous expression level. We then compared the fluorescence intensity in these overexpression transgenic animals to those in animals with a single copy of the transgene. We found that GFP::DLK-1 intensity in the overexpression transgenic animals were only about 4-fold of that in the single-copy transgenic line (Fig. S4). The low degree of overexpression suggests a need for tight control of the DLK-1 level. Similar to rapamycin treatment, DLK-1 overexpression was able to enhance axon regrowth in day 10 animals (), suggesting that enhancing autophagic activity in injured aged neurons can promote axon regeneration and that the age-dependent decline in autophagy induction might contribute to the decline in axon regrowth capacity. Treating animals with DLK-1 overexpression with rapamycin did not further enhance axon regrowth at day 10 (), suggesting that DLK-1 and rapamycin might function through the same pathway to promote axon regrowth.
As DLK-1 is a downstream effector of elevated Ca2+ in response to injury [Citation38], we examined the effect of Ca2+ inhibition on injury-induced autophagy induction. Calcium (Ca2+) plays a role in autophagic signaling pathways encompassing both MTOR and AMPK [Citation39]. The inositol trisphosphate (IP3) receptor is critical for injury-induced calcium release from internal stores [Citation40]. Overexpression of the N-terminal IP3-binding domain of ITR-1 can repress the ITR-1 function in a cell-autonomous manner, likely by titrating endogenous IP3 as “IP3 sponges” [Citation41]. We overexpressed either the low-affinity (“control sponge”) or high-affinity (“super sponge”) ITR-1 IP3-binding domain mutant forms in touch neurons. The expression of the super sponge led to decreased PLM regrowth, while the control sponge did not affect regrowth [Citation38]. Consistent with their effect on axon regrowth, the control sponge did not affect the number or the pattern of autophagic vesicles. In contrast, the super sponge significantly abolished injury-induced autophagy activation (Fig. S5), while rapamycin treatment can rescue this effect. These data indicate the essential role of Ca2+ in autophagy induction in response to injury.
DLK-1 signaling regulates autophagy induction
Previous studies have shown that mutations eliminate axon regeneration in the DLK-1/MKK-4/PMK-3/MAK-2/CEBP-1 signaling cascade [Citation9,Citation10]. To test whether the downstream components in this cascade are involved in injury-induced autophagy activation, we examined the flux reporter in young adult animals with the following genetic mutations: mkk-4(ju91), pmk-3(ok169), mak-2(gk1110), and cebp-1(tm2807). We found that injury did not increase autophagic vesicle numbers in any of these mutants, except for pmk-3(ok169) (). This result is likely because pmk-3(ok169) is only a partial loss of function allele with a small deletion at the C-terminus of the PMK-3 protein. These data suggest that the downstream components in the DLK-1 cascade are all required for injury-triggered autophagy induction.
Figure 7. DLK-1 signaling regulates injury-induced autophagy activation. (A) Schematic illustration of DLK-1 signaling transduction cascade in axon regeneration. (B) Quantification of APs and ALs in PLM cell bodies in day 1 transgenic animals expressing Pmec-4-mCherry::GFP::LGG-1 in the background of indicated genetic mutants. Mutations in all the downstream genes of the DLK-1 cascade, except for pmk-3(ok169), abolished injury-induced autophagy activation. (C) Quantification of APs and ALs in PLM cell bodies in day 10 transgenic animals expressing the tandem reporter Pmec-4-mCherry::GFP::LGG-1 and overexpressing DLK-1 in the background of indicated genetic mutants. DLK-1 overexpression failed to enhance autophagy induction after injury in the genetic mutants of the downstream genes of the DLK-1 cascade. (D) Quantification of APs and ALs in PLM cell bodies transgenic animals expressing the tandem reporter Pmec-4-mCherry::GFP::LGG-1 with or without overexpressing DLK-1 in the background of indicated genetic mutants and at the indicated stages. We examined the unc-51;DLK-1 OE animals on day 6 because they become very weak on day 10. Statistics: One-way ANOVA; mean ± SEM; ***p < 0.001. ns, not significant
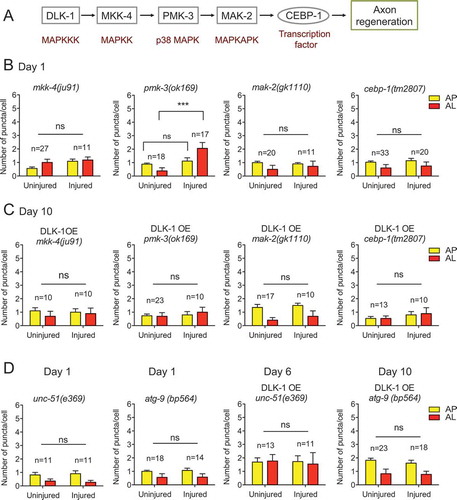
We have shown above that overexpressing DLK-1 in aged animals was sufficient to rescue the age-dependent decline of injury-induced autophagy activation partially. We next tested whether the downstream effectors are required to mediate the effect of DLK-1 overexpression. We expressed the tandem reporter and DLK-1 in these mutants and examined injury-triggered autophagy induction at day 10. We found that DLK-1 overexpression was no longer able to enhance autophagy induction in aged neurons in the loss-of-function mutants of the downstream genes (). These data further suggest that autophagy induction requires downstream factors in the DLK-1 signaling cascade in response to injury. Notably, although the partial loss-of-function mutation pmk-3(ok169) did not completely block injury-induced autophagy activation in young animals, it was sufficient to abolish the effect of DLK-1 overexpression at day 10, suggesting that aged neurons might be more sensitive to the changes in DLK-1 signaling.
To further delineate the genetic pathway of how DLK-1 controls autophagy, we tested whether the effect of DLK-1 overexpression on promoting injury-induced autophagy activation was dependent on UNC-51 or ATG-9, two regulators that occupy the upstream positions in the autophagy pathway. Consistent with their key roles in the autophagy pathway, loss of UNC-51 or ATG-9 completely abolished injury-induced autophagy activation on day 1. DLK-1 overexpression was no longer able to enhance autophagy induction in aged neurons when unc-51 or atg-9 were mutated (), supporting that the DLK-1 pathway acts upstream of the autophagy pathway.
Autophagy regulates LIN-12/NOTCH in axon regeneration
Autophagy is a process for degrading and recycling cellular constituents. Since autophagy functions to promote axon regeneration, it might do so by degrading inhibitors of axon regeneration. To test this hypothesis, we first examined the protein localization pattern of previously identified inhibitors of axon regrowth [Citation14,Citation30] to check whether any of these inhibitors co-localize with autophagic vesicles. We co-expressed a GFP-tagged inhibitor and mCherry::LGG-1 in touch neurons. If the protein were a substrate of autophagy, we would expect to see GFP signals co-localize with mCherry-labeled autophagic vesicles. Similar to the tandem reporter, mCherry::LGG-1 displayed a punctate pattern in PLM neurons, and the number of mCherry puncta increased in injured neurons (Fig. S6A and 8A). GFP-tagged regrowth inhibitors displayed various patterns. As previously reported, EFA-6 protein localized on the cell membrane. GFP::EFA-6 did not co-localize with mCherry::LGG-1 in either uninjured or injured neurons. ARF-6, like EFA-6, is also known to localize to the cell membrane. GFP::ARF-6 evenly distributed on the cell membrane with a distinct pattern compared to mCherry::LGG-1 in both intact and injured neurons. GFP::HDA-3 localized predominantly to the nucleus, and we observed a cytoplasm-to-nucleus shift of HDA-3 in injured neurons (Fig. S6A). LIN-12 is the worm homolog of the NOTCH transmembrane receptor. GFP::LIN-12 showed a membrane localization pattern similar to GFP::ARF-6. However, we also detected some GFP puncta that co-localized with mCherry puncta (). Therefore, among the inhibitors we examined, only LIN-12 showed co-localization with autophagic vesicles. Since low pH environment of the AL lumen quench GFP, these GFP and mCherry double-positive puncta might be APs that contain LIN-12 protein.
Figure 8. Autophagy regulates LIN-12 in axon regeneration. (A) Representative images of intact and injured PLM neurons from day 1 and day 10 animals co-expressing mCherry::LGG-1 and GFP::LIN-12 in touch neurons with indicated treatments. For rapamycin treatment, L4 and day 9 animals were treated for 24 h prior to laser axotomy. After axotomy, animals were cultured for 24 h in the presence of rapamycin before imaging. For BA1 treatment, BA1 or 0.2% DMSO was injected to day 1 and day 10 animals 1 h prior to laser axotomy and animals were then recovered for 24 h before imaging. For the uninjured controls, animals underwent sham injury and imaged 24 h later. Scale bar: 5 µm. (B) Quantification of GFP::LIN-12+, mCherry::LGG-1+, and double-positive puncta in PLM neurons from day 1 and day 10 animals co-expressing mCherry::LGG-1 and GFP::LIN-12 in touch neurons with indicated treatments. (C) Quantification of PLM axon regrowth in day 1 and day 10 animals of indicated genotypes and with indicated treatments following the same protocol as (A). (D) Schematic illustration of the summary/working model. Axon injury induces autophagy in a Ca2+ and DLK-1-dependent manner, and the injury-induced autophagy activation declines with aging. Autophagy limits the level of LIN-12/NOTCH to promote axon regeneration. Statistics: One-way ANOVA; mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant
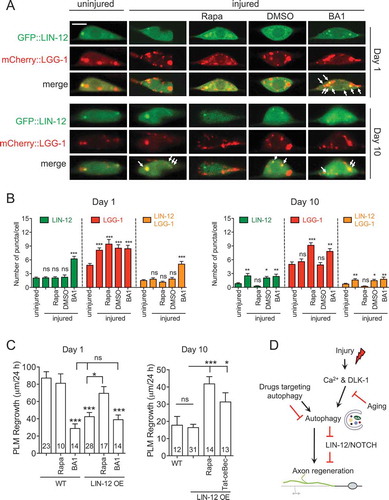
LIN-12 inhibits axon regeneration in a cell-autonomous manner [Citation14]. To test whether LIN-12 is a target of autophagy in axon regeneration, we examined the GFP::LIN-12 and mCherry::LGG-1 pattern in PLM neurons treated with autophagy drugs. Treatment with rapamycin or axotomy significantly increased LGG-1 puncta, but not LIN-12 puncta. In animals treated with BA1, we found significantly increased GFP puncta that co-localized with mCherry::LGG-1 ( and ), suggesting that when the autophagic flux is blocked, there might be more LIN-12 protein trapped in APs. In intact day 10 neurons, we observed a similar level of GFP::LIN-12 puncta compared to day 1, but enhanced its levels after axotomy. Rapamycin treatment on day 10 animals was sufficient to rescue the injury-triggered elevation of the GFP::LIN-12 puncta number ( and ), suggesting that injury might elevate the uptake of NOTCH to autophagic vesicles and that activating autophagy with rapamycin could promote NOTCH degradation. Although axon injury or rapamycin treatment did not show an impact on LIN-12 puncta number on day 1, this could reflect homeostasis between increased NOTCH uptake and increased autophagic activity (NOTCH degradation) in young neurons.
To confirm that the co-localization between LIN-12 and LGG-1 is specific, we examined a mutant form of LGG-1 for its co-localization with LIN-12. A point mutation (G116A) in LGG-1 protein has been shown to affect its post-translational lipidation and subsequently affect its autophagosome membrane targeting [Citation42]. We generated transgenic strains co-expressing mCherry::LGG-1G116A and GFP::LIN-12 in touch neurons. Unexpectedly, lipidation-deficient mCherry::LGG-1G116A resulted in mCherry-positive puncta (Fig. S6B). But unlike the wildtype LGG-1 puncta, the density of mutant LGG-1 puncta were not affected by injury or autophagy drugs (Fig. S6C). We detected very few GFP::LIN-12 puncta in these neurons, except for when we treated animals with BA1 (Fig. S6B and S6C). We also found that GFP::LIN-12 puncta did not co-localize with mCherry::LGG-1G116A. These GFP puncta might be endogenous APs and GFP::LIN-12 might be trapped in these vesicles in the presence of BA1. Therefore, LIN-12 appears to specifically co-localize with wildtype LGG-1.
We next examined axon regeneration in animals that overexpressed LIN-12. Consistent with a previous study, LIN-12 overexpression led to impaired axon regrowth in day 1 animals (). We also checked the level of LIN-12 overexpression by comparing it to the expression of a single-copy transgene under its endogenous promoter. The expression level of GFP::LIN-12 in the overexpression line was around 4-fold greater than the expression in the single-copy transgenic line (Fig. S7). Rapamycin treatment on LIN-12 transgenic animals was able to partially rescue the defect in axon regrowth, although rapamycin did not promote axon regrowth in wildtype animals. In contrast, BA1 treatment inhibited axon regrowth in wildtype animals but did not further reduce axon regrowth in animals overexpressing LIN-12. These epistasis analyses suggest that autophagy agents and LIN-12 overexpression might function through the same pathway to affect axon regeneration. Unexpectedly, we found that LIN-12 overexpression did not affect axon regrowth in day 10 neurons. This result might be due to an increased level of endogenous LIN-12 in aged neurons caused by impaired autophagy induction. Rapamycin or Tat-ceBec peptide treatment was able to promote axon regrowth in the LIN-12 transgenic animals (). Overall, these data are consistent with our working model that autophagy might limit LIN-12 to promote axon regeneration ().
Discussion
Autophagy is previously known to be associated with axonal injury. For instance, after axotomy, autophagosome-like vesicles accumulate in axon terminals [Citation43,Citation44]. There are speculations that genes in the autophagy pathway could serve as therapeutic targets to promote axon regrowth in diseases and conditions involving axonal injuries. However, the role of autophagy in axon regeneration remains unclear. In this study, we have examined loss-of-function mutants of genes in the autophagy pathway and used a dual-color LGG-1 reporter to comprehensively study the role of autophagy in axon regeneration and the neuronal autophagic response to axon injury in vivo at single-cell resolution. We found that all autophagy mutants tested impaired axon regeneration. Axon injury resulted in increased autophagic vesicles within the cell body, and this injury-induced autophagy activation was diminished in aged neurons and required DLK-1. Elevating autophagy with autophagy-inducing agents was sufficient to promote axon regrowth, but only in aged neurons. Finally, a previously identified axon regrowth inhibitor, LIN-12/NOTCH, co-localized with autophagic vesicles, and its levels negatively correlated with autophagic activity. Together, these data suggest that DLK-1-mediated injury signaling activates autophagy that promotes axon regeneration by limiting the level of NOTCH protein.
Injury-induced autophagy activation correlates with axon regeneration capacity
The potential role of autophagy in axon regrowth has been elusive, with controversial data on whether its effects are positive or negative. For instance, inhibiting autophagy with 3-MA has been shown to enhance neurite outgrowth in cultured cortical neurons [Citation45], and autophagy activation can prevent retinal ganglion cell death after optic nerve transection [Citation46]. However, 3-MA treatment also inhibits DRG neuron survival and axon growth [Citation47]. The lack of consensus might reflect the difference between CNS and PNS, but it might also be due to the nonspecific effects of autophagy-modulating agents. We tested genetic mutants of autophagy genes and found that loss of autophagy resulted in reduced axon regrowth. The growth rate in autophagy mutants was lower at different stages of axon regrowth, suggesting that axon regeneration requires autophagy throughout the process.
Autophagy induction after axon injury has been previously investigated [Citation24,Citation26–28]. Visualizing the accumulation of LC3-positive puncta using histological analysis or measuring the relative levels of LC3-II by western blot analysis is commonly used to measure autophagy activation. However, it’s well-characterized that interpretation of these analyses can be misleading because LC3 accumulation also results from autophagy blockage. SQSTM1/p62 is a receptor protein that mediates the delivery of ubiquitinated cargo to autophagosomes and is degraded by autophagy alongside its cargo. Accumulation of SQSTM1 indicates a disrupted autophagic degradation. By examining the level of SQSTM1, previous studies have revealed that increased autophagosome numbers after spinal cord injury (SCI) are due to impaired autophagy flux [Citation25,Citation27]. In this study, we used the mCherry::GFP::LGG-1 reporter to quantify AP and AL numbers in live C. elegans at different time points post-axotomy. As in vertebrate neurons, we detected increased numbers of autophagic vesicles in injured C. elegans neurons. After examining the reporter at 3, 6, 12, and 24 h post-axotomy, we decided to quantify AP and AL numbers at 24 h because of the convenience of the experiments and the low variability in AP and AL numbers at this time point. Compared to intact PLM neurons that typically have 1–2 AP puncta and no AL puncta, the neurons subjected to laser axotomy showed significantly increased numbers of both AP and AL (3 and 5 on average, respectively). This autophagy induction is gradually diminished with aging and correlates to the gradually reduced axon regeneration capacity in PLM neurons [Citation36]. In the dlk-1 mutant, in which there is complete inhibition of PLM axon regrowth, axotomy fails to induce autophagy; although, loss of DLK-1 does not affect the basal level of autophagy in uninjured neurons. Similarly, blocking internal calcium release is sufficient to inhibit axon regrowth [Citation38] and block autophagy induction after injury (Fig. S4). Therefore, injury-induced autophagy activation, but not the basal autophagy level in intact neurons, appears to correlate with axon regeneration capacity.
Treating young adult animals with autophagy-inducing agents, including rapamycin, metformin, and Tat-ceBec peptide, can elevate AP and AL numbers in uninjured neurons but not in neurons post-axotomy. In contrast, in the dlk-1 mutant or the aged wildtype animals, these autophagy-activating agents are sufficient to boost the number of autophagic vesicles in injured neurons, which normally show defective injury-induced autophagy. This result suggests that axon injury might have activated autophagy to its maximal level in young wildtype neurons, and thus, autophagy-inducing agents fail to augment autophagy further. Whereas in neurons with a defective injury response, such as those in the dlk-1 mutants or aged animals, autophagic activity is not at the maximal level and, therefore, can be elevated by autophagy-activating agents. Consistent with its effect on elevating injury-induced autophagy, treating animals with rapamycin or Tat-ceBec peptide is sufficient to enhance axon regrowth of aged neurons or young neurons in the dlk-1 mutant, but not those of young wildtype neurons. These results further support that the level of autophagy activation after injury, rather than the basal level before injury, might predict the regeneration capacity of PLM neurons.
DLK-1 signaling pathway mediates injury signaling to activate autophagy
DLK-1 is a conserved MAPKKK that acts upstream in a MAP kinase cascade including the MKK-4 and MAK-2 kinases. The DLK-1 pathway was one of the first signaling pathways identified in C. elegans as essential for axon regeneration in motor and mechanosensory neurons [Citation9,Citation10]. The critical role of DLK in axon regeneration is evolutionarily conserved [Citation11,Citation12]. DLK is an essential axon injury sensor. DLK is transported in injured axons and required acutely for the generation of injury signals that are retrogradely transported to the cell body [Citation13]. Also, axon injury triggers a rapid and transient increase in axonal calcium, and the effect of elevated Ca2+ in promoting axon regeneration is dependent on DLK-1 [Citation38]. We monitored autophagic activity in neurons expressing the Ca2+ “super sponge” that results in defective internal calcium release, in wildtype and dlk-1 mutant neurons. Although the basal level of autophagy in intact neurons was not affected, it greatly abolished injury-induced autophagy activation, suggesting that autophagy activation in response to axon injury requires the Ca2+-DLK-1 pathway-mediated injury signaling. Decreased DLK-1 expression in adult neurons accounts for the reduced injury response and regeneration observed in aged neurons [Citation48]. Consistent with this notion, we found that diminished autophagy activation post-injury in aged neurons could be rescued by overexpressing DLK-1 ( and ). Therefore, the DLK pathway might respond to axon injury and subsequently activate autophagy in the injured neurons to promote axon regeneration. Furthermore, we found that loss-of-function mutations eliminated injury-induced autophagy activation in the DLK-1/MKK-4/PMK-3/MAK-2/CEBP-1 signaling cascade. The effect of DLK-1 overexpression on autophagy induction was also abolished in these mutants, indicating that the DLK cascade is required to transduce injury signals that activate autophagy. Interestingly, in Drosophila neurons, the autophagy pathway has been shown to degrade the E3 ubiquitin ligase hiw (highwire) [Citation49,Citation50], which is a negative regulator of DLK. Thus, the DLK pathway and autophagy pathway might inter-regulate each other, but future studies will be required to understand how the DLK pathway regulates autophagy.
NOTCH as a target of autophagy in axon regeneration
As the autophagy pathway plays a positive role in regulating axon regeneration, we reasoned that it might promote axon regrowth by degrading inhibitory factors of axon regeneration. We, therefore, tested previously known inhibitors of axon regeneration for co-localization with autophagic vesicles by co-expressing GFP-tagged inhibitor proteins and mCherry::LGG-1. Among the 4 inhibitors we tested (EFA-6, ARF-6, HDA-3, and LIN-12), only GFP::LIN-12 showed co-localization with mCherry::LGG-1, suggesting that LIN-12 might be a substrate of autophagy. LIN-12/NOTCH inhibits axon regeneration via a canonical activation pathway, involving LIN-12, the metalloprotease SUP-17/ADAM10, and the gamma-secretase complex [Citation14]. As NOTCH function in axon regeneration does not require individual ligands, it is suspected that nerve injury and consequent relaxation of plasma membrane tension might alter the conformation of NOTCH relative to the membrane and cause the cleavage of NOTCH without ligand activation [Citation14]. Notch signaling activates autophagy in various biological contexts, including the survival of natural T regulatory cells [Citation51].
Conversely, autophagy can regulate NOTCH signaling by directly targeting NOTCH for degradation. For instance, the NOTCH receptor is uptaken into ATG16L1-positive autophagosome-precursor vesicles and subsequently degraded to modulate stem cell differentiation and neurogenesis [Citation52]. We found that in PLM neurons co-expressing GFP::LIN-12 and mCherry::LGG-1, GFP was evenly distributed on the cell membrane, with some GFP puncta co-localized with mCherry-positive puncta. These double-positive puncta might be APs containing LIN-12 protein, as GFP is quenched in ALs. We did not detect an obvious difference in the number of GFP::LIN-12 puncta between controls and animals treated with rapamycin in young neurons. This could reflect the homeostasis of increased APs (NOTCH uptake to vesicles) and ALs (NOTCH degradation) resulting from rapamycin treatment. Blocking autophagy flux with BA1 significantly increased the number of double-positive puncta in both young and old neurons. This observation is consistent with the notion that double-positive puncta are APs containing GFP::LIN-12 and that GFP::LIN-12 is trapped in APs upon BA1 treatment. Our data showing that rapamycin was able to rescue the regrowth defect caused by LIN-12 overexpression, but that BA1 could not further inhibit axon regrowth on top of LIN-12 overexpression () also support that autophagy might limit the level of NOTCH to promote axon regeneration. Notably, it has been suggested that NOTCH functions at the time of injury to inhibit axon regrowth [Citation14], plus our data suggest that autophagy is required throughout the regeneration process (), indicating that there might be additional autophagy targets. Indeed, a previous study has revealed that autophagy can downregulate SCG10 to stabilize microtubules and promote axon regeneration [Citation25]. Therefore, LIN-12/NOTCH might be one of the many substrates of autophagy in the axon regeneration process.
In summary, our results indicate that axon injury increases autophagic vesicles in injured neurons, and that injury-induced autophagy activation is critical for axon regeneration, possibly by limiting inhibitory factors. This injury-triggered autophagy response requires the DLK-1 pathway and is age-dependent. Our findings suggest that autophagy activation can promote axon regeneration in neurons that lack maximal regrowth capacity, for instance, in adult mammalian CNS and aged PNS neurons.
Materials & Methods
C. elegans genetics
C. elegans worms were maintained on nematode growth medium (NGM) agar plates seeded with E. coli strain OP50 at 20°C using standard procedures (http://www.wormbook.org) unless otherwise stated. Mutant strains carrying each of the following alleles, atg-9(bp564), bec-1(ok691), bec-1(ok700), klf-2(ok1043), klf-3(ok1975), lgg-1(tm3489), lgg-2(tm5755), lgg-2(tm6544), dlk-1(tm4024), and egl-19(ad695), were obtained from the C. elegans gene knockout consortium (CGC, funded by NIH Office of Research Infrastructure Programs P40 OD010440) or the Japan National Bioresource Project. Genotyping was done using standard PCR. For experiments using day 6 and day 10 adult animals, L4 animals were picked at day 0 and transferred to fresh NGM plates every other day. Transgenic animals were generated using microinjection (See Supplemental Experimental Procedures for more information).
Quantification of autophagic vesicles and LIN-12 puncta
For quantitative analysis of vesicles and puncta, live animals were immobilized with either 0.7% 1-phenoxy-2-propanol (MilliporeSigma,484,423,) or 30 mM muscimol (Sigma, M1523) on a 5% agarose pad. We collected fluorescence images on an Olympus IX83 fluorescence microscope or a Zeiss LSM780 confocal microscope, using 100x or 63x oil objectives, respectively. Z-stack confocal images were acquired at 0.5 µm slice intervals. Autophagic vesicles with puncta labeled by GFP::LGG-1, mCherry::LGG-1 or mKate2::LGG-1 in the cell body of PLM neurons were quantified with ImageJ (http://imagej.nih.gov) or manually. For neurons expressing the tandem sensor, the total number of mCherry-positive puncta, GFP-positive puncta, and double-positive puncta (autophagosome) were counted. The number of mCherry-positive puncta (autolysosome) were counted after merging the images from red and green channels or calculated by subtracting the number of GFP-positive puncta from the total number of mCherry-positive puncta.
Laser axotomy
We performed laser axotomy, as described previously (Wu et al., 2007) with some slight modifications. All experiments were performed in parallel with matched control animals. Adult day 1 animals (or animals of other indicated stages) were immobilized with 0.7% 1-phenoxy-2-propanol in batches of 12 animals per slide. GFP-labeled axons were visualized with an Olympus IX83 microscope using a 100x objective. PLM axons were axotomized 50 µm from the cell body of the PLM using a Micropoint UV laser (Andor Technology, Oxford Instrument). Axotomized animals were recovered, transferred to agar plates, and remounted 24 h later (unless indicated otherwise) for scoring. At least 20 animals were axotomized for each genotype, and 10 axons were measured using ImageJ for each genotype.
Rapamycin and bafilomycin A1 treatment
Rapamycin (AdipoGen, AG-CN2-0025) was dissolved in DMSO and added to NGM agar plates at a final concentration of 100 nM. Before imaging, animals were cultured on rapamycin plates for 24 h unless otherwise indicated. Bafilomycin A1 (BA1; BioViotica, BVT-0252-C100,) is not cuticle-permeable and was administrated via injection to the body cavity at the tail position. BA1 (50 µM) in 0.2% DMSO or just 0.2% DMSO control were co-injected with phenol red (0.1%; MilliporeSigma, P3532) 24 h before imaging. For axotomy experiments, animals were cultured on NGM plates to recover for 2 h post-BA1-injection, and before undergoing axotomy.
Synthesis and administration of tat-cebec and control peptides
The small peptides were synthesized by Thermo Fisher Scientific (Pierce Biotechnology). The Tat-CeBec small peptide (YGRKKRRQRRRGGTNVLDLCFHIWVDGIVGE) consisted of a TAT protein transduction domain (TAT/PTD) with 11 amino acids at the N terminus, a GG linker to increase flexibility, and 18 amino acids derived from the C. elegans BEC-1 protein (aa196-213, homologous to human BECN1 aa267-284) at the C terminus. The control peptide, Tat-scrambled (YGRKKRRQRRRGGHDLLCIDTVIGFVGEVWN), consisted of the Tat protein transduction domain, a GG linker, and a scrambled version of the 18 amino acids from Tat-CeBec 1 C terminus (YGRKKRRQRRRGGTNVLDLCFHIWVDGIVGE). Tat-CeBec and Tat-scrambled peptides were dissolved in 10% DMSO (50 μM of peptides) and stored at −80°C. For peptide injection, the stock was diluted using H2O to make a final concentration of 10 μM. The peptides were injected into the body cavity past the bend of the posterior arm of the gonad. Two hours after injection, the PLM axon was subjected to laser axotomy. Axotomized or intact neurons were imaged 24 h later.
Statistical analysis
We used the GraphPad Prism program for all statistical analyses. For two-way comparisons, we used the Student’s t-test. For comparisons involving more than one group in the design, we used a one-way ANOVA test.
Supplemental Material
Download Zip (13.5 MB)Acknowledgments
We thank Dr. Exing Wang from the UTHSCSA Imaging Core Facility for assistance in microscopy, and members in Chen and Liu Labs, particularly Karen Hernandez, for comments and discussion.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- AJ A, David S, Bray GM. Influences of the glial environment on the elongation of axons after injury: transplantation studies in adult rodents. J Exp Biol. 1981;95:231–240.
- David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214(4523):931–933.
- Pestronk A, Drachman DB, Griffin JW. Effects of aging on nerve sprouting and regeneration. Exp Neurol. 1980;70(1):65–82.
- Verdu E, Navarro X. [Degeneration and regeneration of the peripheral nervous system with aging]. Rev Neurol. 1995;23(121):648–655.
- Filbin MT. PirB, a second receptor for the myelin inhibitors of axonal regeneration Nogo66, MAG, and OMgp: implications for regeneration in vivo. Neuron. 2008;60(5):740–742.
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26(4):193–198.
- Rossi F, Gianola S, Corvetti L. Regulation of intrinsic neuronal properties for axon growth and regeneration. Prog Neurobiol. 2007;81(1):1–28.
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7(8):617–627.
- Hammarlund M, Nix P, Hauth L, et al. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323(5915):802–806.
- Yan D, Wu Z, Chisholm AD, et al. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138(5):1005–1018.
- Xiong X, Wang X, Ewanek R, et al. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191(1):211–223.
- Itoh A, Horiuchi M, Bannerman P, et al. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383(2):258–262.
- Shin JE, Cho Y, Beirowski B, et al. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74(6):1015–1022.
- El Bejjani R, Hammarlund M. Notch signaling inhibits axon regeneration. Neuron. 2012;73(2):268–278.
- Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138(17):3593–3612.
- He Z, Jin Y. Intrinsic control of axon regeneration. Neuron. 2016;90(3):437–451.
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293.
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477.
- Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53–67.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326.
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12(9):842–846.
- Zhao YG, Sun L, Miao G, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11(6):881–890.
- Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104(36):14489–14494.
- Hou H, Zhang L, Zhang L, et al. Acute spinal cord injury could cause activation of autophagy in dorsal root ganglia. Spinal Cord. 2013;51(9):679–682.
- He M, Ding Y, Chu C, et al. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc Natl Acad Sci U S A. 2016;113(40):11324–11329.
- Kanno H, Ozawa H, Sekiguchi A, et al. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine (Phila Pa 1976). 2011;36(22):E1427–1434.
- Liu S, Sarkar C, Dinizo M, et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6:e1582.
- Kanno H, Ozawa H, Sekiguchi A, et al. The role of autophagy in spinal cord injury. Autophagy. 2009;5(3):390–392.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873.
- Chen L, Wang Z, Ghosh-Roy A, et al. Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron. 2011;71(6):1043–1057.
- Wu Z, Ghosh-Roy A, Yanik MF, et al. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104(38):15132–15137.
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460.
- Chang JT, Kumsta C, Hellman AB, et al. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife. 2017;6.
- Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295.
- Guo Y, Wang F, Li H, et al. Metformin protects against spinal cord injury by regulating autophagy via the mTOR signaling pathway. Neurochem Res. 2018;43(5):1111–1117.
- Hubert T, Wu Z, Chisholm AD, et al. S6 kinase inhibits intrinsic axon regeneration capacity via AMP kinase in Caenorhabditis elegans. J Neurosci. 2014;34(3):758–763.
- Li J, SG K, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379.
- Ghosh-Roy A, Wu Z, Goncharov A, et al. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30(9):3175–3183.
- Bootman MD, Chehab T, Bultynck G. Parys JB and Rietdorf K. The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium. 2018;70:32–46.
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–325.
- Walker DS, Gower NJ, Ly S, et al. Regulated disruption of inositol 1,4,5-trisphosphate signaling in Caenorhabditis elegans reveals new functions in feeding and embryogenesis. Mol Biol Cell. 2002;13(4):1329–1337.
- Manil-Segalen M, Lefebvre C, Jenzer C, et al. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev Cell. 2014;28(1):43–55.
- Dixon JS. “Phagocytic” lysosomes in chromatolytic neurones. Nature. 1967;215(5101):657–658.
- Matthews MR, Raisman G. A light and electron microscopic study of the cellular response to axonal injury in the superior cervical ganglion of the rat. Proc R Soc Lond B Biol Sci. 1972;181(1062):43–79.
- Ban BK, Jun MH, Ryu HH, et al. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33(19):3907–3919.
- Rodriguez-Muela N, Germain F, Marino G, et al. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19(1):162–169.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
- Belin S, Norsworthy M, He Z. Independent control of aging and axon regeneration. Cell Metab. 2014;19(3):354–356.
- Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187(1):71–79.
- Clark SG, Graybeal LL, Bhattacharjee S, et al. Basal autophagy is required for promoting dendritic terminal branching in Drosophila sensory neurons. PloS One. 2018;13(11):e0206743.
- Marcel N, Sarin A. Notch1 regulated autophagy controls survival and suppressor activity of activated murine T-regulatory cells. eLife. 2016;5.
- Wu X, Fleming A, Ricketts T, et al. Autophagy regulates notch degradation and modulates stem cell development and neurogenesis. Nat Commun. 2016;7:10533.