
ABSTRACT
Opportunistic bacterial infections amongst HIV–infected individuals contribute significantly to HIV-associated mortality. The role of HIV-mediated modulation of innate mechanisms like autophagy in promoting opportunistic infections, however, remains obscure. Here we show, HIV reactivation in or infection of macrophages inhibits autophagy and helps the survival of pathogenic Mycobacterium tuberculosis (Mtb) and nonpathogenic non-tuberculous mycobacterial strains (NTMs). The HIV-mediated impairment of xenophagy flux facilitated bacterial survival. Activation of autophagy by trehalose could induce xenophagy flux and kill intracellular Mtb or NTMs either during single or co-infections. Trehalose, we delineate, activates PIKFYVE leading to TFEB nuclear translocation in MCOLN1-dependent manner to induce autophagy. Remarkably, trehalose significantly reduced HIV-p24 levels in ex–vivo-infected PBMCs or PBMCs from treatment-naive HIV patients and also controlled mycobacterial survival within Mtb-infected animals. To conclude, we report leveraging of HIV-mediated perturbed host innate-immunity by opportunistic bacterial pathogens and show an attractive therapeutic strategy for HIV and associated co-morbidities.
Abbreviations: AIDS: acquired immune deficiency syndrome; AMPK: AMP-activated protein kinase; ATG5: autophagy related 5; BafA1: bafilomycin A1; CFU: colony forming unit; CTSD: cathepsin D; CD63: CD63 molecule; EGFP: enhanced green fluorescent protein; FRET: Förster resonance energy transfer; GABARAP: gamma-aminobutyric acid receptor-associated protein; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; GLUT: glucose transporter; HIV: human immunodeficiency virus; hMDMs: human monocyte derived macrophages; IL2: interleukin 2; LAMP1: lysosomal-associated membrane protein 1; LC3B-II: lipidated microtubule-associated proteins 1A/1B light chain 3B; Mtb: Mycobacterium tuberculosis; MTOR: mechanistic target of rapamycin; mRFP: monomeric red fluorescent protein; M6PR: mannose-6-phosphate receptor; NAC: N- acetyl- L –cysteine; NTM’s: non-tuberculous mycobacteria; PBMC: Peripheral Blood Mononuclear cells; PIKFYVE: phosphoinositide kinase; FYVE-Type Zinc Finger; PHA: phytohemagglutinin; PMA: phorbol 12-myristate 13-acetate; PtdIns(3,5)P2: Phosphatidylinositol 3,5-bisphosphate; ptfLC3: pEGFP-mRFP-LC3; ROS: reactive oxygen species; SQSTM1: sequestosome1; TFEB: transcription factor EB; MCOLN1/TRPML1: mucolipin 1; PIP4P1/TMEM55B: Human trans-membrane Protein 55B; UVRAG: UV Radiation Resistance Associate; VPS35: vacuolar protein sorting associated protein 35; WDR45: WD repeat domain 45; YCAM: Yellow Chameleon.
Introduction
Opportunistic infections in human immunodeficiency virus-1 (HIV-1)-infected individuals are one of the significant causes leading to acquired immunodeficiency syndrome (AIDS)-associated mortality. The CD4+ T lymphocytes are major targets for HIV-1 infection. The decline in CD4+ T cells is directly correlated to the increased viral load and is used as a clinical benchmark to establish the onset of AIDS [Citation1]. However, several opportunistic bacterial infections, which aggravate the clinical presentation of AIDS patients, do not require a strong T cell-response initially for their effective containment by the host immune system. These bacterial infections instead require a strong innate defense response to get controlled, in the absence of which, they could establish a successful infection. It is, therefore, critical to understand whether and how HIV-1 infection also perturbs innate defense mechanisms. Cells of myeloid lineage like monocytes, macrophages, and dendritic cells infected with HIV-1 serve as reservoirs, underscoring the importance of innate defense mechanisms in the clinical presentation and management of AIDS [Citation2,Citation3]. Several studies suggest HIV-1-mediated perturbation of host innate immune system, including those involving surfactants and effector cell functions like phagocytosis, efferocytosis, and response to inflammatory stimuli like TNFs [Citation4]. However, there are also studies, which did not report any defect in alveolar macrophage function among HIV-1-infected individuals unless they were also tobacco smokers [Citation5].
Tuberculosis (TB), which infects a large section of the population, especially in the endemic countries, remains an asymptomatic infection in more than 90% of cases. However, among HIV-AIDS patients, nearly 40% of them may show active TB [Citation6]. Similarly, certain non-tuberculous mycobacterial strains (NTMs) like Mycobacterium avium, Mycobacterium fortuitum, Mycobacterium kansasi, Mycobacterium terrai and Mycobacterium intracellulare, most of which are mainly nonpathogenic in humans, may cause active disease among HIV-1-infected individuals [Citation7]. HIV-1-TB co-infection, as co-morbidity has been studied extensively, and features like increased HIV-1 replication at the site of co-infection, HIV-1 induced killing of CD4+-T cells in the granulomas to prevent Mtb killing by manipulating macrophage function and functional changes in Mtb-specific T cells has been reported previously [Citation8].
Among various anti-bacterial innate defense mechanisms, macroautophagy/autophagy has emerged as a central player in recent times. Several pathogenic bacterial species, including Mtb, Salmonella typhimurium, Legionella sp., Listeria, etc. are known to perturb the host autophagy machinery in order to survive within the host cells [Citation9]. Interestingly, HIV-1 infection is also known to inhibit the final stage of autophagy, i.e., the fusion of autophagosomes with the lysosomes for maturation into autolysosomes. The rate of autophagic cargo degradation through autophagosome maturation is described by autophagy flux, which compares cargo accumulation in the presence of a lysosomal function inhibitor relative to control [Citation10]. Importantly HIV-1 infection triggers the early stages of autophagy, leading to the formation and accumulation of more autophagosomes [Citation11–13] and more virion biogenesis. Despite both HIV-1 and other bacterial infections perturbing the host autophagy pathway, very little is known about whether within the same host, autophagy perturbation by a viral infection could inadvertently help other bacterial infections. There are minimal systematic studies, which connect the high opportunistic bacterial infections with HIV-1-induced perturbation of innate defense mechanisms, especially autophagy, in the host. Few studies, which somewhat explored this exciting crosstalk, reported a role of vitamin D3-mediated induction of autophagy to control both HIV-1 replication and Mtb survival within macrophages, either singly or during co-infections [Citation14–16]. At least one study suggests an increased bacterial burden of Mtb or NTMs in ex vivo models during co-infection with HIV [Citation17]. However, there is no such study yet that explores whether HIV-mediated impairment of autophagy could promote opportunistic infection of Mtb or other NTMs during HIV co-infection.
Modulation of autophagy can influence xenophagy, as has been shown for several small-molecule autophagy modulators like rapamycin [Citation18,Citation19]. A potent, safe autophagy inducer is the naturally occurring disaccharide trehalose. Trehalose induces autophagy independently of the MTOR (mechanistic target of rapamycin) pathway and first reported to facilitate aggrephagy (clearance of aggregation-prone proteins) in mammalian and neuronal cells [Citation20]. The exact mechanism of trehalose-mediated induction of autophagy, however, remains debatable, despite some recent developments like its role as a competitive inhibitor of glucose transporters (GLUT) and the identification of SLC2A8/GLUT8 as mammalian trehalose transporter [Citation21–23].
In this study, we show that active HIV-1 replication, either through reactivation or active infection, results in inhibition of autophagy flux and the accumulation of autophagosomes in macrophages. More importantly, HIV-1 mediated block in autophagy flux, and we show for the first time that autophagy helps enhance the survival of Mtb, as well as, NTMs in HIV-1-infected macrophages. Using trehalose, we showed that autophagy induction not only controlled Mtb survival alone or when co-infected with HIV-1 but could also control p24 levels in peripheral blood mononuclear cells (PBMC) cultures and CD4+ T cells. Moreover, we report here a previously unrecognized mechanism of action of trehalose for inducing autophagy in macrophages.
Materials and methods
Ethics statement
All the experiments with PBMCs, either from healthy volunteers or HIV–infected donors, were approved by the institutional ethics committee of AIIMS, New Delhi (IEC/NP-295/2011). The blood samples were obtained from HIV-1-infected pediatric donors after obtaining written informed consent from the parent/guardian of the children. IEC approval from ICGEB-IEC was also obtained for obtaining PBMCs from healthy volunteers (ICGEB/IEC/2017/06).
Animal ethics statement
Animal experiments were performed as approved by the Institutional Animal Ethics Committee (ICGEB/IAEC/08/2016/CI-1) following the guidelines of the Committee for the purpose of control and supervision of experiments on animals (CPSCEA).
Cell culture, media, and reagents
Human pro-monocytic cell lines U937 and U1.1 (NIH reagents program) were maintained in RPMI 1640 (Cell Clone, CC3014) with 10% FCS (Gibco Life Technologies, 10,270) at 37°C in 5% CO2, humidified incubator. Adherent cells HEK293T (Clonetech) and TZM-bl (NIH reagents program) cells were maintained in DMEM (Cell Clone, CC3004) and 10% FCS, as described above. Monocytic cells were differentiated into macrophages by treating them with phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich Co, P8139) at 10 ng/ml for 24 h. Adherent cells were seeded in tissue culture-treated Petri-plates or six-well plates in complete DMEM media overnight and transfection and titration experiments were carried out as explained below. Reagents like HEPES (N-2-hydroxyethylpiperazine-N9-2-ethane sulfonic acid; Sigma- Aldrich Co, H3784) sodium bicarbonate (Sigma- Aldrich Co, S5761), glutamine (Sigma- Aldrich Co, G3126) etc. were used for media preparations. Transfection reagents Jet Prime and Jet Prime buffer, (PolyplusTm, 114–07), Retro-concentrin (Clonetech Laboratories Inc., RV100A-1), MCSF (R&D systems, 216-MC-025) and used at a concentration at 50 ng/ml for activating MDM’s. IL2 (eBiosciences, 14-8029-63) was used at a concentration of 20 units/ml. PHA (Sigma Aldrich Co, L8902-5mg) was used at 1 μg/ml.
Inhibitors, antibodies, plasmids, constructs, and other reagents
Bafilomycin A1 (Sigma Aldrich Co, B1793-10ug), rapamycin, (Sigma Aldrich Co, R8781), 3-methyladenine, (Sigma Aldrich Co, 900525) amikacin sulfate, (Sigma Aldrich Co, A2324), PKH67, (Sigma Aldrich Co, PKH67GL) MTT (1-[4,5- dimethylthiazol-2-yl]-3,5-diphenylformazan; Sigma Aldrich Co, M5655), beta-galactosidase, (Sigma Aldrich Co, G3153), DMSO, (Sigma Aldrich Co, D2650) polybrene (1,5-dimethyl-1,5-diazaundecamethylene polymethobromide, hexadimethrine bromide; Sigma Aldrich Co, TR-1003), BSA (Bovine Serum Albumin; Sigma Aldrich Co, 05470) and paraformaldehyde, (Sigma Aldrich Co., 158127) Primary antibodies MAP1LC3B, (Novus Biologicals, NB 100–2220). GAPDH, (Santa Cruz Biotechnology, PA-1988). TFEB, (Bethyl Laboratories, Inc., A303-673A-T), Anti-HIV-1 p24 (Abcam, ab9071). Anti-HIV-1 p24 FITC (Beckman Coulter, 6604665), p-RPS6KB1/p-p70S6K, (Novus Biologicals, NB600-1049). Anti-MCOLN1/TRPML1, (Novus Biologicals, NB100-82375SS), anti-ATG5, (Novus Biologicals, NB100-53818SS). Anti-PIKFYVE (R&D Systems, H00200576-BO2P) and RSP6KB1/p70S6K, (R&D systems, AF8962). IR-conjugated secondary antibodies anti-mouse IR780 (LI-COR, 926–32210) and anti-rabbit IR780 (LI-COR, 925–68071) were obtained from LI-COR Biosciences. Alexa Flour dye conjugated secondary antibodies used were anti-mouse Alexa Fluor 488 (Invitrogen, A28175), anti-mouse Alexa Fluor 405 (Invitrogen, A31553), anti-mouse Alexa Fluor 568 (Invitrogen, A11031), anti-mouse Alexa Fluor 647 (Invitrogen, A21235), anti-rabbit Alexa Fluor 488 (Invitrogen, A11034), anti-rabbit Alexa Fluor 405 (Invitrogen, A31556), anti-rabbit Alexa Fluor 568 (Invitrogen, A11011), anti-rabbit Alexa Fluor 647 (Invitrogen, A21245). JC-1 was procured from Thermo Fischer SCIENTIFIC, (Thermo, T3168). Plasmids used in the study are: pEGFP-mRFP-LC3/ptfLC3 (Addgene, 21074, Tamotsu Yoshimori), YCAM-3.6 (Addgene, 58182, Michael Davidson), LAMP-1-YCAM (a kind gift from Peter Haynes) and mEGFP-PI(3,5)P2 (Addgene, 92419, Geert van den Bogaart). 1X PBS (1 L- 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4) and to make 1X PBST 0.1% Tween-20 (Sigma Aldrich Co., P1379) was added to 1X PBS. D-(+)-Trehalose, (MP Biomedicals, 103097).
Mycobacterial cultures and HIV −1 clones
HIV-1 clones (pNL[AD8] and pNL-AD8Δnef) were from NIH AIDS Reagent program and obtained from Dr. Shahid Jameel, pSG3Δenv strain was from NIH AIDS Reagent program. Mycobacterial strain H37Rv was obtained from the Colorado State University. Clinical strains of Mycobacterium aviuum Complex and Mycobacterium fortuitum were obtained from All India Institute of Medical Sciences, New Delhi. All mycobacterial cultures were grown in Middlebrook 7H9 broth, (Becton Dickinson, 271310)
Transfection experiments and virus preparation
For the preparation of HIV-1 viruses, molecular clones of HIV-1 (pNL[AD8]) and VSVG pseudotyped pSG3Δenv were transfected in HEK293T cells using jet PRIME. In brief, 10 ug of plasmid DNA was taken and diluted with jet prime buffer according to the manufacturer’s protocol. Jet prime reagent was mixed with DNA containing buffer, vortexed briefly and incubated for 10 min. The mixture was added onto the cells and kept for 4–8 h after which fresh media was added to the culture and kept for an additional 36–48 h. Viruses were harvested after 48 h of transfection by collecting the supernatant and filtered with a 0.45 μm filter. The harvested supernatant was concentrated by mixing it with Retro-concentrin following the manufacturer’s protocol. The concentrated virus was resuspended in filtered 1X PBS, and aliquots were stored in −80°C for further use.
Titration of viruses
For titration experiments, TZM-bl cells were seeded in 6-well plates at a density of .3 million cells/well. Cells were treated with polybrene (8 ug/ml) for 10 min and viruses at different dilutions were added onto the cells. Cells were incubated for 4 h with the virus for adsorption and internalization and were subsequently washed with PBS. For supernatant assay from PBMC’s and CD4 T cells, stored supernatants were added on TZM-bl cells for 24 h. Then cells were washed twice with 1X PBS and fresh complete DMEM was added and kept in an incubator for an additional 36 h. Beta-galactosidase assay was performed on these cells for titration of viruses. In brief, the media was removed, cells were fixed in 0.05% glutaraldehyde, (Amresco, 0875) for 10 min and washed twice with 1X PBS. The working solution of beta-galactosidase was added (2/3 ml per well) and kept for 2–16 h (according to the signal) in the CO2 incubator. Blue cells were counted in different areas in each well and titer was calculated according to the following formula: Number of virions present in viral stock/ml = Number of blue cells*conversion factor of the plate*dilution of virus used.
Bacterial cultures and infection experiments
All bacterial cultures were grown in 7H9 media supplemented with 10% ADC, (Becton Dickinson, 211887), until log phase. For CFU counts, the plating was performed on 7H11 agar (Becton Dickinson, 8801671) supplemented with 10% OADC, (Becton Dickinson, 211886). For infection, single-cell suspension of bacteria was used obtained by passaging the log phase culture of bacteria through needles as follows: 23 gauge (7 times), 26 gauge (5 times) and 30 gauge (3 times). For bacterial quantification, OD600 was considered equivalent to 100 million bacteria. The required number of bacteria was added onto the seeded cells in complete media at MOI of 1:10. For confocal experiments, bacteria were stained with PKH67 dye according to the manufacturer’s protocol and resuspended into RPMI for infection.
For all bacterial infection, 4 h after adding bacteria, amikacin sulfate (Sigma Aldrich Co., A2324) treatment was given at a final concentration of 200 μg/ml for 2 h and washed, and subsequently kept for desired time points for different assays.
For CFU experiments, culture media was removed and cells were lysed in 7H9 containing 0.06% SDS and plated on 7H11 plates supplemented with OADC and incubated at 37°C for three weeks. In parallel wells, cell viability was monitored by MTT assay using MTT (1 mg/ml) and colorimetric reading was taken at 645 nm.
HIV-1 infection and co-infection experiments
In all HIV-1 infection experiments, U937-derived macrophages were infected with 1:0.1 MOI of pNL-AD8 strain of HIV-1 by incubating the cells with the virus through spinoculation (centrifuged for 1 h at 450 x g). Then, cells were kept for 4 h in the CO2 incubator for virus internalization and washed subsequently to remove any unbound virus and kept for further experiments. For co-infection, U937 macrophages were first infected with HIV-1, as described above. After 24 h of HIV-1 infection, bacteria were added to the cells as described above.
Flow cytometry
For flow cytometry experiments, cells were seeded in a 6-well plate at a density of 1 × 106 cells/well. At different time points, cells were scraped off and pelleted down at 1000 RPM. For LC3B staining cells were permeabilized with 0.1% saponin (Sigma Aldrich Co., 84510) for 10 min and subsequently blocked in 3% BSA in 1X PBS (1 h), incubated with primary antibody for 1 h in blocking buffer followed by incubation in Alexa-flour 488-conjugated secondary antibody for 1 h. Cells were washed and re-suspended in 1X PBS and acquired in BD FACS Canto II using FACSDiva acquisition software. The data were analyzed using FlowJo software (FlowJo LLC, FlowJo V 10.2). For JC1 staining, HIV-1-infected U937 cells (1:0.1 MOI, as described above) were treated with JC1 stain (2 μM, 20 min) and kept in 37oC CO2 incubator and analyzed with BD FACS Canto II. Analyses were done using Flow Jo V. Surface receptor staining for ITGAM/CD11B (Becton Dickinson, 347557) and intracellular staining for HIV-p24 was performed according to the protocols from BD biosciences.
siRNA transfections
U937-derived macrophages or hMDMs were either treated with control siRNA or with specific siRNA using Dharmafect 2 reagent (Dharmacon, T-2002-02) against MCOLN1/TRPML1 (Dharmacon, M-006281-00-0005), PIKFYVE (Dharmacon, M-005058-05-0005), ATG5 (Dharmacon, M-004374-04-0005) or TFEB (Dharmacon, M-009798-02-0005) (100 nM) each and kept for 36 h. In experiments involving infections, U937-derived macrophages were infected with H37Rv or HIV-1 and H37Rv as described earlier and siRNA against MCOLN1, PIKFYVE, or ATG5 were added after infection. Trehalose was also added after infection and continued until the desired time point.
Nucleofection
U1.1 cells were nucleofected according to the manufacturer’s protocol by using Amaxa 2D-Nucleofector Tm (Lonza, AAB-1001). In brief, the cells were nucleofected by suspending the required amount of DNA (ptfLC3) in the provided suspension buffer and nucleofection reagent was added subsequently according to the kit protocol. The mixture was then taken for nucleofection and cells were kept for 12 h post nucleofection. The cells were either left untreated or treated with 10 ng/ml of PMA for 24 h. BafA1 was added for 3 h before the time point. At the desired time points, cells were fixed and mounted for confocal microscopy.
Real-time PCR
Total RNA was isolated at the required time point/treatments from U937-derived macrophages or hMDMs using total RNA preparation kit (mdi membrane technologies). RNA was reverse transcribed using iscriptTM cDNA synthesis kit (BIO-RAD, 170–8891) to get cDNAs using random hexamer primers. cDNA was used for setting up individual qPCR reactions. For internal control, 18s rRNA was used in U937 experiments. In hMDMs, 18s rRNA and ACTB were used as internal controls for siMCOLN1 experiment and siTFEB experiments, respectively. Primer sequences were as follows (5ʹ-3ʹ), MCOLN1, F-GGAAAGCAGCTCCAGTTACA, R- GATGAGGCTCTGGAGGTTAATG, CTSD, F- GACCAGAACATCTTCTCCTTCTAC, R- GGACAGAGAACCCTTGTAATACTT, CD-63, F- GGGCTGCTAACTACACAGATT, R- CTTATGGATCGCCTTCTCGTT, LAMP1, F- CGTCAGCAGCAATGTTTATGG, R- CATGTTCTTAGGGCCACTCTT, PIP4P1, F- CTGTACAGAACCACAGGAACTG, R- AAGGTCGGGAGGAGGAATTA, M6PR, F- GCCTCCATGAGAATCTGGTATT, R- CATCTCCAGGCTCAGACTAAAC, BECN1, F- CCCGTGGAATGGAATGAGATTA, R- CCGTAAGGAACAAGTCGGTATC, GABARAP, F- CCCTTCCTTTCACTACCTTCTTT, R- GCTTGGTGGTTTCCTCTTTATTG, SQSTM1, F- CTCTGGACACCATCCAGTATTC, R- TGCAATTCTACGCAAGCTTAAC, UVRAG, F- AAGAGGCTAGACGAGACATAGA, R- GAAATCTGAATGCGGGAATGAC, VPS35, F- TGGCTGCCTGTAGTGAAATAG, R- GCAGACACACAAAGCACAAA, WDR45, F- CTCTGCGCTTCCAGTGATAA, R- CTCTGCGCTTCCAGTGATAA which were obtained from Sigma-Aldrich Co (St Louis, MO, USA).
Immunofluorescence (IFA) staining and confocal microscopy
U937-derived macrophages were fixed (4% PFA) and permeabilized with 0.25% TX-100 (Sigma Aldrich Co, X100PC) for 15 min and subsequently blocked in 3% BSA (1 h). Samples were incubated with primary antibody (anti-LC3B or anti-TFEB), at the dilution of 1:200 (1 h), and washed twice with PBST followed by incubation with secondary antibody tagged with Alexa Flour 565 nm (1:200, 45 min). DAPI (Sigma Aldrich Co, D9542) (300 nM) was used to stain the nucleus. For HIV p24 intracellular staining, cells were fixed and permeabilized with 0.25% TX-100 followed by blocking with 5% BSA. Cells were stained with HIV-p24 primary antibody followed by secondary antibody (Alexa Anti-mouse 405), as described above, washed and mounted for confocal microscopy. For LysoTracker (Thermo Fisher Scientific, L7528) staining, samples were treated with 100 nM of LysoTracker for 30 min prior to respective time points and fixed before the acquisition.
Confocal imaging was done on a Nikon Ti-E microscope equipped with 60X/1.4 NA plan-apochromat DIC objective lens. For FRET experiments, U937 cells were first nucleofected (Neon transfection system, Thermo Fisher Scientific), and subsequently treated with PMA for 24 h followed by control siRNA or siRNA for PIKFYVE or MCOLN1 and kept for 36 h, after which trehalose (100 mM) was added for 12 h. At 48 h post-transfection cells were fixed and analyzed using the FRET module to calculate % FRET efficiency as follows: (1-[IB – IA]/IB)*100 where IB and IA denote mean fluorescence intensity before and after photobleaching.
Autophagy and xenophagy flux measurement
Autophagy flux was calculated, ratios of GAPDH normalized densitometric values for LC3-II in BafA1-treated set to that of BafA1-untreated sets i.e., LC3-II(BafA1+)/LC3-II(BafA1−). For xenophagy flux, total bacteria and bacteria in the autophagosomes were counted manually using Imaris software (Bitplane, Imaris 7.6.4), represented as percent LC3B co-localization. This analysis was performed in both BafA1-untreated or treated sets. An increase in the LC3B co-localization upon BafA1 treatment with respect to that of the untreated set was considered indicative of xenophagy flux. Counting of LC3B and PtdIns(3,5)P2 puncta was performed using the spot-detection tool in Imaris software.
PBMC, CD4 + T cells and hMDM experiments and HIV-p24 ELISA
PBMCs from healthy donors were stimulated with phytohemagglutinin (PHA, 1 µg/ml) (Sigma Aldrich Co, L8902-5 mg) for 3 d at 5x106/ml cell density. Then cells were washed thrice and seeded at a cell density of 5x106/ml each in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES buffer, and IL2 (20 U/ml). The activated PBMCs were then infected with 200x and 1000x of TCID50 of HIV-1 (corresponding to 1:0.08 and 1:0.4 MOIs, respectively). After the establishment of infection, trehalose (100 mM) was added to the cells and a parallel set was left untreated. On day 3 and day 7, supernatant from infected PBMC’s was used to perform ELISA for HIV-p24 following manufacturer’s protocol (SKU, XB-1000). The HIV-1 inoculum used to infect the cells were kept in a cell-free well with complete medium and used as the baseline control for day 3 and day 7 ELISA.
Similarly, CD4+T cells were first purified from PBMC’s by using CD4+T cells negative isolation kit, (Miltenyi Biotech, 130-091-153), followed by culturing as described above. Cells were then infected with HIV-1. After the establishment of infection, trehalose (100 mM) was added to the cells and a parallel set was left untreated. For hMDMs, PBMCs were isolated from healthy donors followed by magnetic separation of human monocytes by Monocyte Isolation Kit (II) Human (Milteny Biotech, 130-091-153) following the manufacturer’s protocol. Monocytes were then differentiated using MCSF (50 ng/ml) for further experiments.
For HIV-p24 ELISA with PBMCs from the seropositive HIV-1-infected ART, naïve donors were stimulated with phytohemagglutinin (PHA) for 3 d at 5x106/ml (5 million) cell density. Then cells were washed thrice and cultured at a cell density of 5x106/ml each in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, antibiotic cocktail including penicillin (250 U/ml) and streptomycin (250 mg/ml) (Thermo Scientific, SV30079.01), 10 mM HEPES buffer, and IL2 (20 U/ml). Cells were cultured for 7 d in the absence or presence of trehalose (100 mM). Supernatants were collected from each group and ELISA was performed as described above.
Mice experiments
C57BL/6 mice (6 weeks old) were infected with H37Rv by the aerosol challenge (~200 bacilli/per lung). After 4 weeks of infection, animals were grouped into control and treated groups of 4 mice each and trehalose was fed to the animals through oral gavage (3 mg/kg). Trehalose dose was decided following IPCS (International Program on Chemical Safety) guidelines. Control animals received an equal volume of the solvent. Four weeks after the start of the treatment, animals were sacrificed and lung homogenates were plated on 7H11 plates for CFU counting.
Statistical analysis
All multiple/complex treatment experiments were analyzed by ANOVA using GraphPad Prism (indicated in Figure legends). Pair-wise analyses were performed using two-tailed t-test and p-values were calculated using Microsoft Excel.
Results
Activation of HIV-1 replication in macrophages results in impaired autophagy flux
To understand the effect of HIV-1 replication on macrophage autophagy response, we used U937 and U1.1 monocytic cell systems. U1.1 cell line is a sub-clone of U937 cells, which is chronically infected with HIV-1 and contains an integrated copy of HIV-1 in its genome. This cell line shows a minimal constitutive viral expression. The viral expression can be induced upon treatment with specific cytokines or PMA [Citation24]. In both U937 and U1.1 monocytes, bafilomycin A1 (BafA1) treatment resulted in the accumulation of MAP1LC3B-II/LC3-II, indicating basal autophagy flux in these cells (). We calculated the ratio of GAPDH normalized LC3-II level of BafA1+ to that of BafA1− to quantify the autophagy flux [Citation25]. Both U937 and U1.1 monocytes showed comparable levels of basal autophagy flux (). Treatment with PMA, known to activate these monocytes into macrophages, led to a decline in autophagy flux in both U937, as well as, U1.1 cells (). Hereinafter, we refer to PMA-treated U937 and U1.1 cells as U937-derived and U1.1-derived macrophages, respectively, in this manuscript. However, while U937-derived macrophages still showed a certain level of autophagy flux (), U1.1-derived macrophages showed increased LC3-II protein levels even in the absence of BafA1, which did not increase further upon BafA1 treatment, suggesting a block in the autophagy flux (). Immunoblot analysis of p24 in the lysates of PMA-treated U1.1 cells confirmed activation of HIV-1 replication at 24 and 48 h of PMA treatment (). In U1.1-derived macrophages, p24 staining revealed that nearly 99% of cells were p24-positive (Fig. S1A). We also confirmed the decrease in autophagy flux upon HIV-1 activation in U1.1-derived macrophages by flow cytometry, where we stained cells for detergent-insoluble LC3-II proteins in untreated, PMA-treated, or PMA followed by chloroquine-treated cells (). In agreement with the immunoblot data, U937 cells showed autophagic flux in both untreated and PMA-stimulated conditions (). In contrast, U1.1 cells did not show flux when we stimulated them with PMA (). Finally, we analyzed autophagosome maturation via the pH-sensitive tandem-fluorescent-tagged autophagy reporter, mRFP-EGFP-LC3, which can distinguish between the autophagosomes (mRFP+/EGFP+) and the autolysosomes (mRFP+/EGFP), since EGFP gets quenched in the acidic environment of the latter compartment [Citation10,Citation26]. In U1.1 monocytes, BafA1 prevented autophagosome maturation, as evidenced by increased autophagosome/autolysosome ratio (ratio of mRFP+/EGFP+ to mRFP+/EGFP− puncta), which remained unchanged in U1.1-derived macrophages in the absence or presence of BafA1 (). Together, these data suggest that activation of HIV-1 replication in macrophages results in the impairment of autophagic flux.
Figure 1. Replication of HIV-1 in macrophages results in autophagy block. (A) Cell lysates of U937 and U1.1 monocytes without PMA treatment or with 10 ng/ml PMA treatment for 24 h were prepared from cells, in the absence or presence of bafilomycin A1 (BafA1, 100 nM) and immunoblotted for LC3B. BafA1 was added 3 h before the time point as described in materials and methods. (B) Fold autophagy flux for U937 and U1.1 monocytes (with and without PMA) corresponding to the blots at 24 and 48 h is shown. (C) U1.1 cells were treated with PMA and HIV-1 activation was monitored using p24 immunoblotting at indicated time points post-PMA treatment (D) U937 and U1.1 monocytes (without PMA treatment) or macrophages (with PMA treatment) were stained for endogenous LC3B upon mild permeabilization using saponin (0.1%) and flow cytometry was performed to monitor autophagy flux in the absence or presence of autophagy inhibitor chloroquine (100 nM, 3 h) at 48 h. (E) U1.1 cells were nucleofected with ptfLC3 construct and cells were kept with or without PMA (10 ng/ml) after transfection for 24 h. BafA1 was added 3 h before the time point. (F) Ratio of autophagosomes (yellow puncta) to autolysosomes (red puncta of LC3B) for images in Figure 1E was quantified for autophagy flux using Imaris software (see Materials and Methods). Scale bar: 10 µm (G) U937-derived macrophages were infected with HIV-1 (pNL[AD8]) at 1:0.1 MOI and p24 levels were probed by immunoblotting in the total cell lysates generated at 48 h post-infection. (H) In the above experiment, HIV-1-infected U937 cells were either treated with BafA1 or left untreated during the final three h of the assay time (48 h). LC3 levels were probed in control and HIV-1-infected U937 cells in BafA1-treated or untreated sets by immunoblotting. (I) Fold autophagy flux in U937-derived macrophages in the absence and presence of HIV is shown. Student’s t-test was performed to calculate p-values (*p value<.05, ** p value <.01; ***p-value<0.001; **** p value <0.0001)
![Figure 1. Replication of HIV-1 in macrophages results in autophagy block. (A) Cell lysates of U937 and U1.1 monocytes without PMA treatment or with 10 ng/ml PMA treatment for 24 h were prepared from cells, in the absence or presence of bafilomycin A1 (BafA1, 100 nM) and immunoblotted for LC3B. BafA1 was added 3 h before the time point as described in materials and methods. (B) Fold autophagy flux for U937 and U1.1 monocytes (with and without PMA) corresponding to the blots at 24 and 48 h is shown. (C) U1.1 cells were treated with PMA and HIV-1 activation was monitored using p24 immunoblotting at indicated time points post-PMA treatment (D) U937 and U1.1 monocytes (without PMA treatment) or macrophages (with PMA treatment) were stained for endogenous LC3B upon mild permeabilization using saponin (0.1%) and flow cytometry was performed to monitor autophagy flux in the absence or presence of autophagy inhibitor chloroquine (100 nM, 3 h) at 48 h. (E) U1.1 cells were nucleofected with ptfLC3 construct and cells were kept with or without PMA (10 ng/ml) after transfection for 24 h. BafA1 was added 3 h before the time point. (F) Ratio of autophagosomes (yellow puncta) to autolysosomes (red puncta of LC3B) for images in Figure 1E was quantified for autophagy flux using Imaris software (see Materials and Methods). Scale bar: 10 µm (G) U937-derived macrophages were infected with HIV-1 (pNL[AD8]) at 1:0.1 MOI and p24 levels were probed by immunoblotting in the total cell lysates generated at 48 h post-infection. (H) In the above experiment, HIV-1-infected U937 cells were either treated with BafA1 or left untreated during the final three h of the assay time (48 h). LC3 levels were probed in control and HIV-1-infected U937 cells in BafA1-treated or untreated sets by immunoblotting. (I) Fold autophagy flux in U937-derived macrophages in the absence and presence of HIV is shown. Student’s t-test was performed to calculate p-values (*p value<.05, ** p value <.01; ***p-value<0.001; **** p value <0.0001)](/cms/asset/f61c793d-3226-416c-a662-2b7b5f37ac63/kaup_a_1725374_f0001_c.jpg)
Infection of U937-derived macrophages with HIV-1 results in impairment of autophagic flux
While results in the previous section show impairment of autophagic flux upon reactivation of latent HIV-1 infection, we next assessed how host autophagy response became regulated during infection with HIV-1. We infected U937-derived macrophages with HIV-1 ([pNL{AD8}]; CXCR5 tropic) at 1:0.1 MOI using spinoculation and at 48 h post-infection (see methods). We treated cells with BafA1 or left untreated for 3 h before lysing the cells and probed for LC3 by immunoblotting. We confirmed the infection by p24 immunoblotting in the cell lysates () and by p24 staining, which revealed that nearly 40-50% of cells were HIV-p24-positive (Fig. S1A). Similar to our data in the activated U1.1-derived macrophages, autophagic flux became blocked in U937-derived macrophages upon infection with HIV-1, as evidenced by HIV-1-induced increase in LC3-II levels, which modestly increased only upon BafA1 treatment (). Densitometric analysis of BafA1-treated versus untreated sets, as described above, showed more than a 2-fold decline in autophagy flux in HIV-1-infected U937 macrophages (). Thus, our data point to an impairment of autophagic flux both during reactivation of HIV-1 and during HIV-1 infection in macrophages.
Inhibition of autophagic flux in activated U1.1-derived macrophages or HIV-1 infection in U937-derived macrophages helps better survival of Mtb and NTMs strains
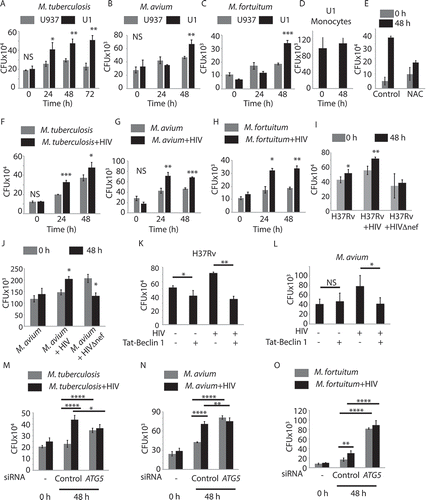
Autophagic flux depicts the crucial step of autophagosome maturation that governs the autophagic cargo degradation in the autolysosomes. Several bacterial pathogens are known to inhibit this late step of autophagy (or xenophagy) in order to survive within the macrophages, which is particularly true in the context of Mtb [Citation18,Citation27]. We, therefore, investigated whether the block in autophagic flux, caused by active HIV-1 infection, also facilitated Mtb survival within the macrophages. Since the virulent Mtb strain H37Rv is known to inhibit xenophagy flux in the infected macrophages to help its survival [Citation18], we analyzed some NTMs like M. avium and M. fortuitum, which are known to have relatively poor survival within human macrophages but can establish opportunistic infection in HIV–infected individuals. We infected U937-derived macrophages (used as a control without HIV) or U1.1 (chronically infected with HIV)-derived macrophages with H37Rv, M.avium, or M. fortuitum at an MOI of 1:10, and we monitored their survival at 48 h post-infection. Evidently, the activation of HIV-1 replication in U1.1-derived macrophages resulted in better survival/growth of each of the three mycobacterial strains, with respect to the U937-derived macrophage controls (). To confirm that the better survival was due to HIV activation and not due to intrinsic differences between U1.1 and U937-derived macrophages, we infected U1.1 monocytes with H37Rv, where we observed no such increase in H37Rv CFU (). We thus confirmed that H37Rv infection alone did not induce HIV reactivation in U1.1 monocytes (Fig. S1B). In U937 monocytes, a similar infection led to a marginal decline in H37Rv CFU (Fig. S1C). HIV reactivation in U1.1 cells is dependent on NFKB-mediated transcriptional activation [Citation24]. We used the generic NFKB inhibitor NAC to study its role in HIV-p24 activation upon PMA activation. In parallel, we also checked ITGAM/CD11B levels, whose surface expression is known to increase upon activation of monocytes [Citation28]. At 15 mM, NAC completely blocked PMA-mediated activation of U1.1 into macrophages, while also observing no HIV-p24 expression (Fig. S1D). At 7.5 mM, we observed a partial reduction in macrophage activation and a significant decline in HIV-p24 levels (Fig. S1D). In U1.1 cells, when treated with 7.5 mM NAC along with PMA, H37Rv did not grow as efficiently as in cells, which did not receive NAC treatment (). In U937 cells, NAC treatment, along with PMA, did not have any significant effect on H37Rv CFU (Fig. S1E). These results confirmed that HIV reactivation in these cells resulted in better H37Rv survival in U1.1-derived macrophages (Fig. S1E). Similarly, infection of U937-derived macrophages with HIV-1 also led to much better survival of H37Rv, M. avium, and M. fortuitum, with respect to the single-infected control conditions (). Interestingly, in U937 macrophages infected with a variant of HIV-1 lacking the accessory protein nef (HIVΔnef), there was no increase in survival of either H37Rv or M. avium, with respect to single infections (). Since HIV-nef is known for its role in executing HIV-mediated autophagy block [Citation13], we verified in HIVΔnef-infected U937-derived macrophages that autophagy flux became significantly higher with respect to wild type HIV-1 infection (Fig. S1F). To further confirm that HIV-nef-mediated autophagy block indeed occurred, which allowed better bacterial survival, we used Tat-Beclin-1 peptide, a peptide derived from HIV-tat, which prevents HIV-nef-mediated autophagy inhibition by blocking the interaction between HIV-nef and BECN1/Beclin 1 [Citation29]. We confirmed, in U937 macrophages infected with HIV-1, that Tat-Beclin-1 peptide indeed induced autophagy flux (Fig. S1G). Survival of both H37Rv and M. avium was significantly curtailed when we added Tat-Beclin-1 peptide during co-infection (, respectively). While the peptide was able to control H37Rv survival even in the absence of HIV-1 co-infection, no such effect was seen on M. avium during a single infection (). Finally, to conclusively show the role of autophagy in enhanced mycobacterial survival during HIV co-infection, we knocked down the ATG5 transcript using siRNA. In ATG5 knockdown cells, HIV co-infection did not result in an increase in the bacterial survival for either of the three mycobacterial strains with respect to the corresponding singly-infected bacterial sets (). ATG5 knockdown alone, however, led to an increase in bacterial survival ().
Figure 2. Better survival of Mtb and NTMs in activated U1.1 macrophages or in HIV-1-infected U937 macrophages. U937 and U1.1-derived macrophages were infected with either H37Rv (A), M. avium (B) or M.fortuitum (C) at 1:10 MOI and total surviving bacteria were counted at 24 and 48 h post-infection as CFU/ml. U1.1 monocytes were infected with H37Rv and CFUs were enumerated at 0 and 48 h post-infection (D). In (E) U1.1 cells were treated with PMA either alone or along with NAC (7.5 mM) for 24 h followed by infection with H37Rv at 1:10 MOI and bacterial CFU were enumerated at 0 and 48 h post-infection. In (F), (G) and (H) U937-derived macrophages were infected first with HIV-1 at an MOI of 1:0.1 for 24 h followed by infection with either H37Rv, M. avium or M. fortuitum respectively at MOI of 1:10. Total surviving bacteria were counted at 24 and 48 h post-infection as CFU/ml. U937-derived macrophages were infected with HIV-1 or HIVΔnef strain followed by H37Rv (I) or M. avium (J) infection. Intracellular bacteria at 0 h and 48 h post-infection were enumerated by CFU analysis. (K, L) U937-derived macrophages were infected with HIV-1 followed by H37Rv (K) or M. avium (L) infection. Cells were then treated with Tat-Beclin-1 peptide (100 nM) and bacterial CFU was enumerated at 48 h. U937 macrophages were infected with 0.1 MOI of HIV-1 as described above followed by (M) M.tuberculosis, (N) M.avium (O) M.fortuitum infection, respectively, at 1:10 MOI. In all the above experiment siATG5 was added after bacterial infection and CFU plating was done at 48 h post-bacterial infection. Student’s t test was performed for calculating p values. For (M), (N), (O) ANOVA followed by multiple comparison test was performed to calculate p values. *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001. Data are representative of more than three independent experiments with more than five replicates each
Thus, from the above results, we concluded that HIV helps opportunistic bacterial survival within macrophages during co-infection by impairing the host autophagy response.
Xenophagy flux is inhibited in HIV-1 infected U937-derived macrophages
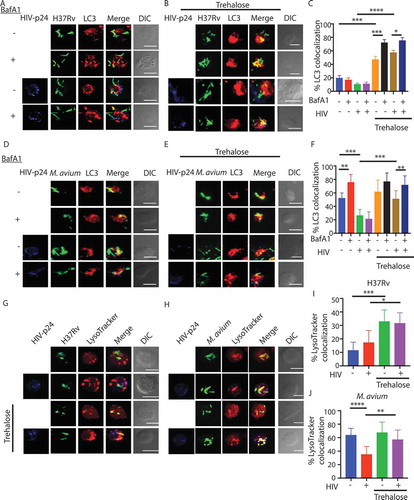
We previously demonstrated in H37Rv–infected macrophages, that xenophagy (anti-bacterial autophagy) became selectively impaired without compromising the basal autophagy flux [Citation18]. This phenomenon enabled better survival of Mtb within macrophages [Citation18]. Since, HIV-1 infection, too, inhibited autophagy flux, which we verified using LC3 puncta count upon HIV-1 infection and BafA1 treatment (Fig. S1H), we examined whether the increased bacterial survival in HIV-1-infected cells was, at least in part, due to an additional block in the xenophagy flux. Xenophagy flux is measured by comparing the localization of bacteria to the LC3+ compartment in the absence or presence of BafA1 treatment [Citation10]. As reported earlier, H37Rv–infected cells did not show xenophagic flux either alone or when co-infected with HIV-1 (). In the context of M.avium, there was significant xenophagic flux in the non-HIV-1 condition, which was mostly abolished in the HIV-1 co-infected cells (). Interestingly, when co-infected with HIVΔnef, M. avium showed a significant xenophagy flux (Fig. S1I).
Figure 3. Modulation of xenophagy flux in single and co-infected U937 macrophages in the absence and presence of autophagy modulator trehalose. U937-derived macrophages either uninfected or infected with HIV-1 (1:0.1 MOI), as described in methods followed by infection with PKH67-labeled (green) H37Rv (A) or M. avium (D) at an MOI of 1:10. Three hours before the experimental time point (48 h), one set of the cells were treated with BafA1 (100 nM). At 48 h, cells were fixed and stained with anti-LC3 and anti-HIV p24 antibodies followed by anti-rabbit-Alexa568 (red) and anti-mouse Alexa 405, respectively (blue), and analyzed under the confocal microscope. Co-localization between LC3 compartment and H37Rv (C) or M. avium (F) in the presence or absence of BafA1 treatment was calculated by analyzing the confocal images in Imaris (see methods, values are mean±SD). For (B) and (E), U937-derived macrophages either uninfected or infected with HIV-1 (MOI 1:0.1) for 48 h were subsequently infected with PKH67-labeled H37Rv (B) or M. avium (E) (green), respectively, at 1:10 MOI. Trehalose (100 mM) was added immediately after bacterial infection. Finally, before the sample harvest at 48 h, cells from each of the groups were either treated with BafA1 (100 nM, 3 h) or left untreated. At 48 h, cells were fixed and stained with anti-LC3 and anti-HIV-p24 antibody followed by anti-rabbit-Alexa568 (red) and anti-mouse Alexa 405, respectively (blue), and analyzed under the confocal microscope. Scale bar: 10 µm in A, B, C and D, respectively. Co-localization between LC3 compartment and H37Rv (C) or M. avium (F) in the trehalose-treated sets was calculated by analyzing the confocal images in Imaris (see methods, values are mean±SD). U937-derived macrophages with or without HIV infection were infected with H37Rv (G) or M. avium (H) followed by treatment with trehalose. Lysotracker staining was performed 30 min prior to harvesting and fixing the cells at 48 h post-bacterial infection followed by antibody staining for anti-HIV-p24 as described above. Co-localization between H37Rv (I) or M. avium (J) with the LysoTracker compartment was calculated by analyzing the confocal images in Imaris. (see methods, values are mean±SD; ANOVA analysis followed by multiple comparison analysis was performed to get the p values. *p-value<0.05; **p-value<0.005; ***p-value<0.001; ****p- value<0.0001). Data represents more than 150 phagosomes, from more than three different coverslips, generated across three independent experiments
The known autophagy inducer trehalose can induce xenophagy flux
To further establish that the effect of HIV-1 infection on mycobacterial survival was primarily due to the inhibition of autophagic/xenophagic flux in the host macrophages, we assessed whether stimulation of autophagy could reverse this phenomenon and facilitate bacterial killing. For inducing autophagy, we used known autophagy enhancers, such as rapamycin, 1, 25-dihydroxy cholecalciferol (vitamin D3), and trehalose. Treatment with either rapamycin or vitamin D3 led to a significant reduction in the bacterial CFU in H37Rv–infected macrophages alone, consistent with existing literature [Citation16,Citation19,Citation30] (Fig. S2A). Trehalose was included in our study because it is considered a safer, MTOR-independent autophagy inducer, and has an established role in rescuing the disease phenotypes in mouse models of neurodegenerative diseases [Citation31–33]; therefore, clinically more acceptable for treatment. Consistent with its known pro-autophagy function, trehalose treatment resulted in a dramatically reduced bacterial load in H37Rv–infected U937-derived macrophages (Fig. S2A). During in vitro cultures, the presence of excess trehalose did not interfere or increase bacterial growth (Fig. S2B and S2C). We confirmed that trehalose indeed induced high autophagy flux in U937-derived macrophages (Fig. S2D), which was even higher than starvation-induced autophagy flux in these macrophages (Fig. S2D). Also, there was no effect on RPS6KB1 phosphorylation, corroborating with the fact that trehalose acts in an MTOR-independent manner [Citation20] (Fig. S2E). We next measured xenophagy flux in H37Rv–infected cells or H37Rv-HIV-1 co-infected cells upon trehalose treatment. Trehalose treatment caused a substantial increase in H37Rv-specific xenophagy flux, as evidenced by the increased bacterial co-localization to LC3+ compartment in the presence of BafA1 (). Trehalose-induced H37Rv-specific xenophagy flux was equally significant in the absence or presence of HIV-1 co-infection (). We saw similar effects of trehalose with M.avium (). Thus, we concluded that trehalose facilitated xenophagic flux of infecting mycobacteria in the host macrophages. Moreover, increased xenophagy flux upon trehalose treatment led to increased targeting of Mtb () or M.avium () to the lysosome compartment either during single or HIV-1 co-infection.
Trehalose-induced xenophagy flux enables the killing of mycobacterial species
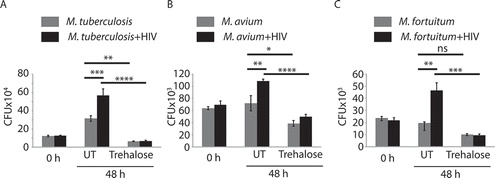
Since trehalose treatment could induce both autophagic and xenophagic flux in macrophages and higher lysosomal targeting of infecting bacteria, irrespective of the presence or absence of HIV-1 co-infection, we next assessed intracellular survival of Mtb, M.avium, and M. fortuitum under these conditions. Trehalose treatment led to a significant decline in the intracellular survival of H37Rv, M.avium, and M. fortuitum in U937-derived macrophages, as evidenced by the reduced intracellular bacterial load (). Likewise, trehalose exerted similar effects in enhancing bacterial killing in HIV-1-infected U937-derived macrophages, where bacterial survival is otherwise more favorable (). The effect of trehalose treatment on bacterial survival was robust since both pre-activation and treatment with trehalose post-infection was equally effective in reducing the bacterial survival within U937 macrophages (Fig. S2F).
Figure 4. Trehalose-induced xenophagy flux helps kill mycobacterial species. U937-derived macrophages uninfected or infected with HIV-1 (MOI 1:0.1) for 48 h were subsequently infected with H37Rv (A), M. avium (B), or M.fortuitum (C) at the MOI of 1:10. For each group of infections, samples were subsequently either treated with trehalose (100 mM) or left untreated for 24 or 48 h post-bacterial infection. Cells were lysed and plated for CFU counting. Values represent mean±SD, from nearly three independent experiments; ANOVA analysis followed by multiple comparison was performed to calculate p values, *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001. Data are representative of more than three independent experiments with more than five replicates each
Trehalose treatment results in nuclear translocation of TFEB
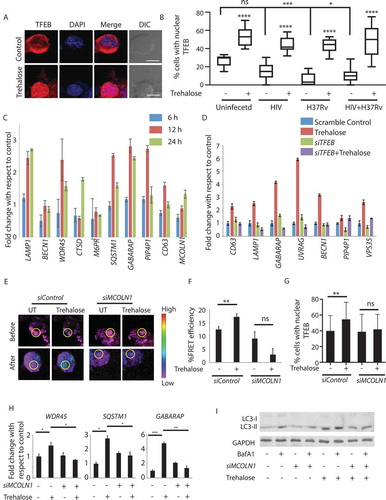
Trehalose is known to induce autophagy in an MTOR-independent manner [Citation20]. Recently trehalose was shown to inhibit glucose transporters at the cell surface, specifically SLC2A3/GLUT3 and SLC2A8, thereby causing a pseudo-starvation-like response to induce autophagy [Citation21,Citation22]. However, more recently, it was also reported that trehalose could enter the host cells through SLC2A8 transporter and may have intracellular targets for autophagy induction [Citation23]. Since trehalose treatment in our experiments lasted for 12 h or more, we hypothesized that an early increase in autophagy might be followed by a transcriptional increase in the expression of autophagy- and lysosome-related genes. The increased expression of genes related to autophagy and lysosome function is dependent on the activation of TFEB, a key transcription factor upstream to the autophagy pathway and lysosomal biogenesis genes [Citation32–34]. Using immunostaining, we confirmed that trehalose treatment in U937 macrophages led to a substantial increase in the translocation of TFEB to the nucleus (). In HIV–infected, H37Rv–infected, or HIV-H37Rv-co-infected U937 macrophages, there was a substantial decline in TFEB nuclear translocation (). Treatment with trehalose induced TFEB nuclear translocation efficiently across all the three conditions (). We tested a panel of known TFEB target genes for their expression upon trehalose treatment of U937 cells. Trehalose treatment resulted in substantial upregulation in the expression of several such genes including LAMP1, WDR45, CTSD, SQSTM1, GABARAP and PIP4P1/TMEM55B (), which was dependent on TFEB since in cells where TFEB was knocked down using specific siRNAs (Fig. S3A), trehalose-mediated increase in the expression of several of these genes was prevented ().
Figure 5. Trehalose treatment results in Lysosomal Ca2+ release-dependent activation of TFEB. (A) U937-derived macrophages either untreated or treated with trehalose (100 mm, 12 h) were fixed and stained with anti-TFEB antibody followed by anti-rabbit Alexa 568 (Red) and DAPI (Blue) and images were acquired under a confocal microscope. Scale bar corresponds to 10 µm. Similar experiments were done for uninfected, H37Rv–infected, HIV–infected or HIV-H37Rv-co-infected cells and percent cells showing nuclear translocation of TFEB were calculated by manually counting the cells in Imaris software (B). For (C), a set of known TFEB target genes were probed for expression upon trehalose treatment of U937-derived macrophages and shown here as fold change with respect to the untreated control set. For (D) U937-derived macrophages were first treated with siTFEB or control siRNA for 24 h, followed by treatment with trehalose (100 mm). Cells were collected at 24 h post-trehalose treatment and total RNA was isolated. The expression of known TFEB target genes was monitored by RT-PCR and shown here as fold-change with respect to scramble siRNA set. (E) U937 cells were nucleofected with LAMP1 YCAM plasmid for 24 h, PMA-treated (24 h), and subsequently transfected with control siRNA or siMCOLN1. At 36 h post-nucleofection trehalose (100 mM) was added for 12 h, cells were fixed, and images were acquired under a confocal microscope using the FRET module. Scale bar corresponds to 10 µm. FRET efficiency was calculated after obtaining MFI’s before and after photobleaching (F, see methods). For (G), U937-derived macrophages were treated either control siRNA or siMCOLN1 (100 nM for 36 h). Subsequently, cells were treated with trehalose (100 mM, 12 h), fixed and stained with anti-TFEB antibody followed by anti-rabbit Alexa 568 (Red) and DAPI (Blue) and images were acquired under confocal microscope. Percent cells showing nuclear translocation of TFEB was calculated by manually counting the cells in Imaris software. (H) U937-derived macrophages were first treated with siMCOLN1 or control siRNA for 24 h, followed by treatment with trehalose (100 mM). Cells were collected at 24 h post-trehalose treatment and total RNA was isolated. Expression of genes showing significant regulation in panel D was probed under MCOLN1 knockdown cells. (I) U937-derived macrophages were treated with siMCOLN1 or control siRNA. At 36 h of siRNA transfection, cells were treated with trehalose (100 mM, 12 h). Cell lysates were generated keeping a BafA1-treated set and untreated set for each of the siRNA groups and were probed for LC3 using immunoblotting; ANOVA analysis followed by multiple comparisons was performed to calculate p-value. *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001
Trehalose induces lysosomal Ca2+ release to induce TFEB nuclear translocation
We next wanted to understand how trehalose activated the TFEB pathway. Activation of TFEB requires its dephosphorylation by calcineurin, a serine-threonine phosphatase that is activated by intracellular Ca2+ [Citation35]. The release of Ca2+ from lysosomal lumen is an important signal for the activation of calcineurin [Citation35]. While the elevation in intracellular Ca2+ under any condition is easy to monitor using intracellular Ca2+ dyes, we wanted to ascertain whether trehalose causes lysosomal Ca2+ release. To that end, we used the Ca2+-dependent FRET reporter yellow-chameleon fused with the LAMP1-lysosomal localization signal (LAMP1-YCAM) [Citation36]. We nucleofected U937 cells with LAMP1-YCAM, followed by PMA treatment, and at 36 h post-transfection, we treated these cells with trehalose for another 12 h. In the instance of increased Ca2+ release from the lysosome, the LAMP1-YCAM traps the Ca2+ and increases FRET signals. In trehalose-treated cells, we observed enhanced FRET signal from the LAMP1-YCAM reporter, suggesting increased Ca2+ release from the lysosome (see methods, ). The lysosome localized channel protein MCOLN1/TRPML1 regulates the release of Ca2+ from the lysosome [Citation35,Citation37]. We, therefore, performed a siRNA-mediated knockdown of MCOLN1 and monitored the LAMP1-YCAM FRET signals in trehalose-treated cells. We achieved more than 60% knockdown of MCOLN1 in U937-derived macrophages (Fig. S3B). In MCOLN1 knockdown cells, there was no increase in LAMP1-YCAM FRET signal upon treatment with trehalose (). Consistent with this data, trehalose failed to induce nuclear translocation of TFEB in MCOLN1 knockdown cells (), which also abolished trehalose-induced expression of genes like WDR45, SQSTM1, and GABARAP (). The knockdown of MCOLN1 also led to the decline in trehalose-mediated autophagy flux in these cells (,S3C). These results revealed a new mechanism of autophagy induction by trehalose in macrophages via lysosomal Ca2+ release and TFEB activation.
Trehalose induces Ptdins(3,5)p2 levels to activate MCOLN1
How trehalose triggers lysosomal Ca2+ release via MCOLN1, as shown above, is not clear. Activation of MCOLN1 depends on PIKFYVE, a class I PI3Kinase (PtdIns3K), which enzymatically converts PtdIns5P into PtdIns(3,5)P2 [Citation38]. Generated PtdIns(3,5)P2, thus, serves as MCOLN1 agonist and activates the channel for Ca2+ release from the lysosomal lumen [Citation39]. We found that the siRNA-mediated knockdown of PIKFYVE in U937-derived macrophages resulted in a loss in trehalose-induced lysosomal Ca2+ release and trehalose-induced TFEB localization to the nucleus (Fig. S4A–S4C). To further ascertain the role of PtdIns(3,5)P2 in trehalose-induced autophagy, we transiently expressed an EGFP construct fused to PtdIns(3,5)P2-interacting domain of MCOLN1 in HEK293T cells [Citation40]. We observed a significant increase in PtdIns(3,5)P2 puncta in cells treated with trehalose compared to the untreated control (). In cells treated with PIKFYVE inhibitor YM201636, PtdIns(3,5)P2 levels reduced dramatically with respect to the control cells (). Treatment with YM201636 completely abolished the increase in PtdIns(3,5)P2 puncta upon trehalose treatment (). Knocking down PIKFYVE using specific siRNA led to a moderate decline in PtdIns(3,5)P2 levels in control cells with respect to scramble siRNA control. Upon trehalose treatment, we observed a significant increase in scramble control cells but only a marginal insignificant increase in PIKFYVE siRNA-treated cells ().
Figure 6. Trehalose increases PtdIns(3,5)P2 levels to activate MCOLN1. (A) HEK293T cells were transfected with mEGFP-(PtdIns[3,5]P2) and after 36 h were treated with trehalose (100 mM) for an additional 12 h. In parallel, cells were either transfected with PIKFYVE siRNA (36 h) or treated with YM201636 (100 nM for 12 h) before trehalose treatment. Cells were fixed and images were acquired using confocal microscopy. Scale bar: 10 µm. (B) PtdIns(3,5)P2 puncta/cell were counted across the samples by Imaris software for the barplot (see methods, *p < 0.05). (C) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by siRNA treatment against MCOLN1 at 6 h post-addition of bacteria. Trehalose (100 mM) was added after bacterial infection. Cells were harvested at 48 h post-infection, lysed and plated to get total CFU count. A gray bar represents the initial bacterial load. (D) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by treatment with trehalose. YM201636 was added 4 h before the time point for harvesting the samples. Cells were harvested at 48 h post-infection, lysed and plated to get total CFU count. A gray bar represents the initial bacterial load. (E) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by siRNA treatment against ATG5 at 6 h post bacterial infection. Trehalose (100 mM) was added after bacterial infection. Cells were harvested at 24 and 48 h post-infection, lysed and plated to get total CFU count. (F) U937-derived macrophages were infected with HIV-1 followed by infection with H37Rv at 1:10 MOI. Cells were treated with control siRNA or siRNA against PIKFYVE, MCOLN1, or ATG5 after bacterial infection. Parallel groups also received trehalose (100 mM) post-bacterial infection. Cells were harvested at 48 h post H37Rv infection, lysed and plated to get total CFU count. Values represent mean±SD; *p- ANOVA analysis was performed to calculate p values. *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001. Data are representative of more than two different experiments with five replicates each. Data are representative of more than two different experiments with five replicates each
![Figure 6. Trehalose increases PtdIns(3,5)P2 levels to activate MCOLN1. (A) HEK293T cells were transfected with mEGFP-(PtdIns[3,5]P2) and after 36 h were treated with trehalose (100 mM) for an additional 12 h. In parallel, cells were either transfected with PIKFYVE siRNA (36 h) or treated with YM201636 (100 nM for 12 h) before trehalose treatment. Cells were fixed and images were acquired using confocal microscopy. Scale bar: 10 µm. (B) PtdIns(3,5)P2 puncta/cell were counted across the samples by Imaris software for the barplot (see methods, *p < 0.05). (C) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by siRNA treatment against MCOLN1 at 6 h post-addition of bacteria. Trehalose (100 mM) was added after bacterial infection. Cells were harvested at 48 h post-infection, lysed and plated to get total CFU count. A gray bar represents the initial bacterial load. (D) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by treatment with trehalose. YM201636 was added 4 h before the time point for harvesting the samples. Cells were harvested at 48 h post-infection, lysed and plated to get total CFU count. A gray bar represents the initial bacterial load. (E) U937-derived macrophages were infected with H37Rv at 1:10 MOI followed by siRNA treatment against ATG5 at 6 h post bacterial infection. Trehalose (100 mM) was added after bacterial infection. Cells were harvested at 24 and 48 h post-infection, lysed and plated to get total CFU count. (F) U937-derived macrophages were infected with HIV-1 followed by infection with H37Rv at 1:10 MOI. Cells were treated with control siRNA or siRNA against PIKFYVE, MCOLN1, or ATG5 after bacterial infection. Parallel groups also received trehalose (100 mM) post-bacterial infection. Cells were harvested at 48 h post H37Rv infection, lysed and plated to get total CFU count. Values represent mean±SD; *p- ANOVA analysis was performed to calculate p values. *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001. Data are representative of more than two different experiments with five replicates each. Data are representative of more than two different experiments with five replicates each](/cms/asset/c797f79c-48a4-40bb-9f30-f55cc87162bf/kaup_a_1725374_f0006_c.jpg)
The trehalose-mediated killing of Mtb is dependent on the PIKFYVE-MCOLN1 pathway and requires ATG5
Having established the lysosomal Ca2+ release via MCOLN1 as the critical pathway for trehalose-mediated autophagy induction, we next wanted to see whether PIKFYVE and MCOLN1-dependent autophagy induced by trehalose was indeed essential for the trehalose-mediated killing of mycobacteria in U937-derived macrophages, as previously demonstrated (). We found that while treatment with trehalose led to a significant decline in H37Rv CFU in U937-derived macrophages transfected with scrambled siRNA, siRNA-mediated MCOLN1 knockdown significantly prevented the effect of trehalose on mycobacterial killing (). Consistent with this result, inhibition of PIKFYVE activity by YM201636 also rescued H37Rv from trehalose-mediated killing in U937-derived macrophages, albeit, partial but significant (). Finally, in U937-derived macrophages, where we knocked down ATG5 using siRNA (Fig. S4D), the trehalose-mediated killing of H37Rv became partially but significantly rescued (). Interestingly, ATG5 knockdown alone led to a significant increase in the H37Rv CFU, suggesting some level of autophagy-dependent killing of Mtb (). Knocking down either PIKFYVE, MCOLN1, or ATG5 also rescued trehalose-mediated killing of H37Rv during HIV-TB co-infection experiments ().
Trehalose controls H37Rv infection during HIV-1 co-infection in human monocyte-derived macrophages (hMDMs)
While all the results obtained so far revealed the bacterial survival during HIV-1 co-infection in cell lines, we wanted to establish whether a similar mechanism existed even in the human primary cells. We confirmed that HIV-1 infection of hMDMs results in a block in autophagy flux (). Moreover, while a VSVG-pseudotyped strain of HIV-1 (pSG3Δenv) was still able to block autophagy flux, which is consistent with the previous reports [Citation13], a nef-deleted strain failed to inhibit autophagy flux (). Subsequently, we showed that trehalose treatment reversed the HIV-1-mediated block in autophagy flux in MDMs (). Next, we verified that the trehalose treatment of hMDMs resulted in an increased expression of genes involved in autophagy and lysosomal pathways in TFEB and MCOLN1-dependent manner (). Next, we confirmed that trehalose could efficiently kill Mtb within hMDMs, either during single or HIV-1 co-infection (). Further, using ATG5 siRNA, we also confirmed that the trehalose-mediated killing of Mtb in hMDMs was ATG5-dependent (). Finally, in hMDMs, we could also see a significant reduction in the production of infectious HIV-1 particles upon trehalose treatment (). Trehalose-mediated control of HIV-1 in macrophages was dependent on autophagy, as knocking down ATG5 using specific siRNAs in U937-derived macrophages resulted in a decrease in HIV-p24 by ELISA (Fig. S4E), which also corroborates with earlier reports [Citation16]. Furthermore, the strain pSG3Δenv, which inhibits autophagy flux despite being replication-deficient, helps better survival of M. avium (Fig. S4F).
Figure 7. Human monocyte-derived macrophages (hMDMs) show similar autophagy function during HIV-TB co-infection and trehalose-mediated autophagy induction. (A) Monocytes were isolated from healthy donors PBMCs and cultured in the presence of MCSF (50 ng/ml) to get hMDMs. Cells were then infected with pNL(AD8), pSG3Δenv and pNL(AD8ΔNef) strain of HIV as described in methods. Three days post-infection cell lysates were prepared for immunoblotting with or without BafA1 treatment (3 h) and probed for LC3B protein by western blot. Fold autophagy flux as described earlier, is shown in the plot at the right. (B) hMDMs were infected with HIV-1 for 4 h followed by treatment with trehalose (100 mM). Three days post-infection cell lysates were prepared with or without BafA1 treatment and immunoblotted with anti-LC3 antibody. Fold autophagy flux as described earlier, is shown in the plot at the right. hMDMs were transfected either with control siRNA or siMCOLN1 (C) or siTFEB (D) for 24 h followed by trehalose (100 mM) treatment for 24 h. At 48 h post-transfection total RNA was isolated and expression of indicated genes monitored by RT-PCR. (E) hMDMs were infected with HIV-1(1:0.1) followed by H37Rv infection at an MOI of 1:10 and transfected either with control siRNA or siATG5. Further cells were treated with trehalose for 48 h and CFU plating was performed. (F) Supernatant saved from the hMDMs experiment shown in 7G was used to perform TZM-bl assay to calculate infectious viral particles in the presence or absence of trehalose as described in the methods. Western blot (data) and CFUs are from three separate donors. RT-PCRs were done in triplicates from single but distinct donors for MCOLN1 and TFEB siRNAs. ANOVA was performed to calculate p values. *p-value<0.05; **p-value<0.01; ***p-value<0.001; ****p- value<0.0001
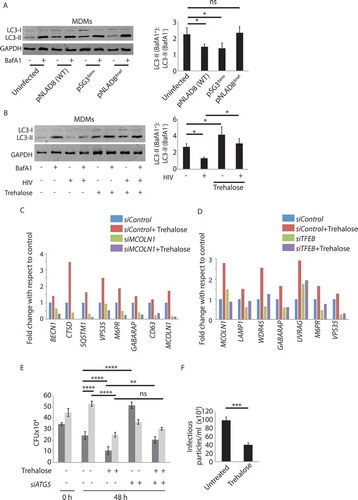
Trehalose treatment results in reduced HIV load in pbmcs from HIV patients
The reduced production of infectious HIV-1 particles upon trehalose treatment in MDMs led us to examine whether trehalose treatment could also directly impact on the HIV-1 replication or survival in primary T cells. We activated PBMCs isolated from healthy volunteers with PHA and IL2, thereby enriching it with T cells, followed by infection with clinical strains of HIV-1 subtype C at 200x (equivalent of MOI 1:0.08) and 1000x (MOI 1:0.4) of TCID50. We cultured the HIV-1-infected activated-PBMCs in the presence or absence of trehalose for 7 d, and we measured p24 in the supernatant on 3rd and 7th d. At both the doses of HIV-1 and the time-points studied, treatment with trehalose led to a massive decline in the p24 levels in the supernatant (). In a separate experiment, we confirmed that trehalose treatment also resulted in a decline in the release of infectious virus particles from activated-PBMCs (,S5A). This observation was also true in PHA and IL2 activated-purified-CD4+ T cells isolated from the PBMCs (,S5B). Western blot analysis revealed that unlike in the macrophages, HIV-1 infection did not inhibit autophagy flux in PBMCs or activated CD4+ T cells (Fig. S5C,S5D). While trehalose could increase autophagy flux either in uninfected or HIV–infected CD4+ T cells, in PBMCs, trehalose could increase autophagy flux only during HIV-1 infection (Fig. S5C,S5D). Having observed the effect of trehalose on the ex vivo infection model, we subsequently investigated whether trehalose could also impact the survival/replication of HIV-1 in PBMCs from HIV-1-infected donors. We recruited 4 treatment-naïve seropositive HIV-1-infected donors, and we activated isolated PBMCs with IL2 and PHA and cultured for 7 d in the presence or absence of trehalose. Strikingly, trehalose treatment was also able to inhibit HIV-1 replication/survival in these clinically relevant cellular platforms ().
Figure 8. Effective control of HIV-1 and Mtb by trehalose in clinically relevant settings. PBMC from seronegative healthy donors were isolated and infected with a clinical isolate of HIV-1 subtype C at doses of TCID200 and TCID1000 for 24 h. Trehalose (100 mM) was added post-infection and supernatants were collected on Day 3 (A) and Day 7 (B) post-treatment for p24 ELISA. Values are mean±SD from duplicates. The first data point in both (A) and (B), marked ‘C1’ and ‘C2’against HIV is the control sample, where the initial viral inoculum corresponding to TCID200 and TCID1000, respectively, used to infect hMDMs were kept in the media without cells for 3 and 7 d, respectively, and used as a control for ELISA. (C) In a separate experiment, supernatants from stimulated (IL2/PHA) PBMCs infected with HIV-1 were used for TZM-bl assay for infectious virus particle count in control or trehalose-treated (3 d) groups. (D) TZM-bl assay performed using supernatants from activated or HIV-1-infected CD4+ T cells which were either control or trehalose-treated for 3 d post-infection (E) Infected PBMCs isolated from four ART-naïve HIV–infected donors were treated with trehalose and p24 ELISA was performed on the supernatant at day 7. Values are mean±SD from triplicates; *p-value<0.05; **p-value<0.005; ***p-value<0.001. (F) C57BL/6 mice were infected with H37Rv by the aerosol challenge (~200 bacilli/per lung). After 4 weeks of infection, trehalose was fed to the animals through oral gavage (3 mg/kg). Control animals received an equal volume of the solvent. Four weeks after the start of the treatment, animals were sacrificed, and lung homogenates were plated on 7H11 plates for CFU counting. ***p-value<0.001
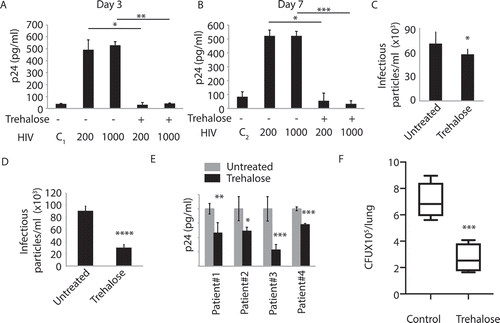
Trehalose treatment controls Mtb survival in the animal model of tuberculosis
So far, results in this study established that trehalose could control opportunistic mycobacterial infections, as well as HIV infection either singly or as co-infection in U937 macrophages and hMDMs. We finally wanted to test whether trehalose retained its anti-mycobacterial activity in the animal models. We infected C57/BL6 mice with H37Rv using aerosol and confirmed the infectivity to be around 150–200 bacilli per lung on day 1. Subsequently, we allowed the infection to establish for the next 4 weeks and started with trehalose treatment at a dose of 3 mg/kg body weight orally for the next 4 weeks. Control animals were fed with an equal volume of solvent orally. After 4 weeks of treatment, we sacrificed animals, and we plated lung homogenates on 7H11 plates for CFU. As shown in (), animals treated with trehalose showed remarkably controlled bacterial count in their lungs with respect to the control animals.
Discussion
We designed this study to address whether the host innate defense mechanisms perturbed upon HIV-1 infection inadvertently ends up facilitating opportunistic bacterial pathogens like Mtb and other NTMs. The association between these two pathogens is well studied in the context of disease manifestation, exacerbation, and immune response [Citation41]. The innate defense pathway of interest in this study is autophagy, which has been extensively characterized during pathogenic bacterial infections, including Mtb infections [Citation19,Citation30,Citation42]. Autophagy regulation has also been explored in the context of several viral infections, including HIV-1, dengue, and influenza, among others [Citation43]. In case of HIV-1, it was initially reported that while its infection results in a surge in the number of autophagosomes formed, it, however, impairs the maturation of autophagosomes and blocks its fusion with the lysosomes, which was subsequently reported to be dependent on the viral accessory protein Nef [Citation13,Citation29]. Therefore, our findings that either upon activation of latently integrated HIV-1 in U1.1 cells or by infecting U937 macrophages with HIV-1 caused a block in autophagy flux are consistent with the literature. In different cells, including activated macrophages, a block in autophagy flux could result in the accumulation of damaged and depolarized mitochondria, leading to increased cellular ROS and induction of apoptosis [Citation25]. However, upon HIV-1 infection, while autophagic flux was suppressed, we observed no mitochondrial depolarization in these infected cells (Fig. S6). Curiously, it is known that HIV-1 Tat, another accessory protein of HIV-1, can localize to the mitochondria and ensure its hyperpolarized state by modulating the Ca2+ channel activities [Citation44].
Interestingly, recent studies show that a slight increase in cellular glutathione redox state may activate latent HIV-1 infections into active replicative forms [Citation24]. As our data show, activation of HIV-1 replication in U1.1 cells inhibited autophagic flux, thus, ensuring mitochondria in a hyperpolarized state would certainly help prolong host cell survival. The role of autophagy in targeting intracellular pathogens to the lysosomes for degradation is well-established [Citation9,Citation18,Citation27]. We reported that in cells with active HIV-1 replication, Mtb, M. avium, and M. fortuitum survived better than the control cells, consistent with previous reports [Citation17]. We also showed an HIV-mediated block in autophagy flux increased bacterial survival during co-infections.
Using autophagy modulators to target both HIV-1 and Mtb has been explored earlier, where it was shown that vitamin D3 could kill both Mtb and HIV either alone or under co-infection conditions [Citation14–16]. However, so far, there is no study directly establishing HIV-1-mediated block in autophagy as the causative mechanism behind opportunistic bacterial infections. Using NTMs in this study, we demonstrated that the usually nonpathogenic strains could also benefit from the HIV-1-induced autophagy block, and therefore, could cause opportunistic infections. In fact, virulent Mtb itself can inhibit xenophagy flux without perturbing the homeostatic autophagy flux in the cells [Citation18,Citation27]. This aspect is evident in our results, where H37Rv localization to LC3+ compartments did not increase in the presence of BafA1 or the presence of HIV-1 infection. In the presence of HIV-1, H37Rv localization to LC3+ compartments rather declined marginally in the BafA1-treated set. Nonetheless, it raises a question on whether the increased bacterial survival observed in our experiments, at least in the case of H37Rv infection, was indeed due to impairment of autophagy flux by HIV-1. It is plausible that any residual xenophagy flux occurring during H37Rv infection gets stronger or entirely blocked in the presence of HIV-1, which possibly is beyond the detection limits of our experimentation. Alternatively, in the context of HIV-1-induced increase in H37Rv survival, improved mitochondrial health possibly plays a more defining role than HIV-1-induced autophagy block. However, since ATG5 knockdown alone could cause a better H37Rv survival in U937 cells, the ATG5-mediated basal killing mechanism seems operational in these cells. Moreover, the fact that MTOR-dependent or MTOR-independent autophagy inducers like vitamin D3, rapamycin, and trehalose could kill H37Rv as efficiently as M. avium and M. fortuitum in HIV-1 co-infection conditions, affirms that autophagy, indeed, is a major player in intracellular bacterial survival even during co-infection with HIV-1.
We focused on trehalose for inducing autophagy because of multiple reasons: a) its ability to induce autophagy is being explored currently for future clinical trials against neurodegenerative diseases associated with accumulation of cytosolic protein aggregates due to dysfunctional autophagy [Citation45,Citation46]; b) it is a natural disaccharide and nontoxic; c) it was more efficient in reducing bacterial load compared to standard inducers like rapamycin and vitamin D3. Interestingly, trehalose is an important metabolite for Mtb, which is secreted out of the cell during mycolic acid biosynthesis by antigen 85B as the byproduct [Citation47]. The secreted trehalose must be taken back by the bacteria to ensure the continued synthesis of cell-wall components. The sugar transport operon of Mtb consisting of genes like lpqY, sugA, sugB, and sugC regulates the uptake of extracellular trehalose [Citation47]. The same study also suggested that the inability to recycle trehalose due to loss of sugA operon genes resulted in compromised virulence and intracellular survival; however, there was no effect on in vitro growth of the mutants [Citation47]. It is possible that due to the loss of trehalose transporters from Mtb, trehalose, thus, excluded out in the infected macrophages, may induce autophagy in the host cells, thereby killing the bacterium more efficiently. Experimentally proving this hypothesis, however, will be technically challenging for reasons like quantifying the amount of trehalose secreted by mutant strains lacking trehalose transporter.
Moreover, there is no apparent intracellular target known for trehalose, making it even difficult to establish the role of trehalose released from intracellular Mtb for inducing autophagy. Initial reports posit trehalose as an inducer of autophagy through a mechanism that was independent of MTOR regulation [Citation20]. Although the mechanism of action was unclear, recent reports presume that trehalose causes pseudo-starvation like phenotype by competitively inhibiting GLUT transporters at the cell surface, resulting in activation of autophagy [Citation21]. Starvation-induced autophagy, however, is known to rely on inhibition of MTOR signaling, in addition to activation of the AMPK pathway [Citation48,Citation49]. A subsequent report, however, suggests that at least in certain cell types, SLC2A8 may serve as trehalose transporter, and trehalose may have some intracellular targets for inducing autophagy [Citation23]. Given our new findings in this study, trehalose may influence autophagy through multiple cellular pathways.
Since the duration of treatment with trehalose in our study ranged between 4 to 48 h, we opined that trehalose must also increase the autophagy response through transcriptional activation of autophagy-and lysosome-related genes. This assumption led us to study the nuclear translocation of TFEB and the activation of the MCOLN1 channel on the lysosomes. Incidentally, cytosolic sequestration of TFEB by both HIV-1 and Mtb has been reported earlier [Citation50,Citation51], which we also verified here. While single or co-infection with HIV and/or Mtb caused TFEB retention in the cytosol, trehalose was able to reverse it across these conditions and induced substantial nuclear translocation of TFEB. Trehalose-mediated nuclear translocation of TFEB and autophagy activation in macrophages was also recently implicated as a potential therapy for atherosclerosis [Citation52]. Activation of the MCOLN1 channel is dependent on its interaction with PtdIns(3,5)P2, a product of the enzymatic action of PIKFYVE [Citation39,Citation53]. The role of PIKFYVE in regulating autophagy in itself is debatable, with literature references supporting both pro- and anti-autophagy properties. Thus, while PIKFYVE through PtdIns(3,5)P2 can activate MCOLN1 [Citation39,Citation53], it is also reported that both PtdIns(3,5)P2, as well as PndIns5P, can activate MTOR pathway, which should negatively regulate autophagy [Citation54,Citation55]. In fact, PtdIns(3,5)P2 serves as the precursor for entire cellular PtdIns5P content, most likely through some phosphatase activity [Citation38]. However, there is no contradiction in the requirement of PtdIns(3,5)P2 in the activation of the MCOLN1 channel. We, therefore, used PtdIns(3,5)P2 binding domain of MCOLN1 fused with EGFP to conclusively show that trehalose induces PtdIns(3,5)P2 levels, which explains the autophagy-inducing property of this molecule. Alternatively, MCOLN1 activation could also regulate bacterial survival through phagosome maturation, as reported previously [Citation56]. The results with hMDMs confirmed that the entire mechanism of trehalose-induced autophagy and its ability to control HIV-TB co-infection identified in the U937-derived macrophages is also operational in the primary cells.
The most fascinating and clinically relevant finding in this study, however, was the effect of trehalose treatment on HIV-1 replication during the ex vivo infections, as well as in the PBMC cultures derived from treatment naïve HIV-1-infected donors. The substantial decline in HIV-p24 that was noted in the above conditions suggests the viral killing ability of trehalose. Since the majority of cells are lymphocytes in activated PMBC cultures, it is evident that trehalose is equally effective to clear HIV-1 infection in monocyte/macrophages, as well as also in CD4+ T cells. Furthermore, HIV-1-induced neuropathy is known to get manifested through inappropriate autophagy response [Citation57]. Since trehalose has been shown to be beneficial in the transgenic models of neurodegenerative disorders, there is an immense potential of this disaccharide in the management of HIV-1 infection and disease progression, which must be further explored in combination with ART. Finally, the efficacy of trehalose in controlling mycobacterial survival in the animal models provided a strong lead to pursue this disaccharide for potentially managing HIV-TB co-infected patients. One limitation of this study is that we could not check the efficacy of trehalose using an in vivo HIV-TB model. However, given the highly encouraging results shown here, it may be possible to test it in a clinical setting in the future.
In conclusion, we demonstrated that HIV-1-mediated inhibition of autophagy is an important regulator of opportunistic bacterial infections. Furthermore, we showed that trehalose, a naturally occurring disaccharide, can induce autophagy and kill Mtb and NTMs alone or during co-infection with HIV-1. Interestingly, we also delineated the mechanism of trehalose-mediated autophagy induction and showed that it acts via PtdIns(3,5)P2. Finally, the ability of trehalose to kill HIV-1 in PBMCs from healthy volunteers infected in vitro or from treatment naïve HIV-1-infected donors or to kill Mtb in mice models show the tremendous potential of this molecule in combating HIV-1 and associated pathogenesis.
Supplemental Material
Download Zip (7.9 MB)Acknowledgments
We thank Lee Haynes for LAMP1-Yellow-Cameleon construct, Shahid Jameel for infectious clones and cell lines, N. Siddappa for providing cell lines, Amit Kumar Singh for animal experiments and Murali K Kaja for his support to CNIHR grant. All works involving mycobacteria were performed in the Department of Biotechnology, Govt. of India supported Tuberculosis Aerosol Challenge Facility (TACF), the BSL3 facility at ICGEB.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Crowe SM, Carlin JB, Stewart KI, et al. Predictive value of CD4 lymphocyte numbers for the development of opportunistic infections and malignancies in HIV-infected persons. J Acquir Immune Defic Syndr. 1991;4:770–776.
- Lebargy F, Branellec A, Deforges L, et al. HIV-1 in human alveolar macrophages from infected patients is latent in vivo but replicates after in vitro stimulation. Am J Respir Cell Mol Biol. 1994;10:72–78.
- Orenstein JM, Fox C, Wahl SM. Macrophages as a source of HIV during opportunistic infections. Science. 1997;276:1857–1861.
- Staitieh BS, Egea EE, Guidot DM. Pulmonary innate immune dysfunction in human immunodeficiency virus. Am J Respir Cell Mol Biol. 2017;56:563–567.
- Elssner A, Carter JE, Yunger TM, et al. HIV-1 infection does not impair human alveolar macrophage phagocytic function unless combined with cigarette smoking. Chest. 2004;125:1071–1076.
- Zumla A, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745–755.
- Singh S, Gopinath K, Shahdad S, et al. Nontuberculous mycobacterial infections in Indian AIDS patients detected by a novel set of ESAT-6 polymerase chain reaction primers. Jpn J Infect Dis. 2007;60:14–18.
- Diedrich CR, Flynn JL, Maurelli AT. HIV-1/mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect Immun. 2011;79:1407–1417.
- Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial subversion of host innate immune pathways. Science. 2013;340:697–701.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Killian MS. Dual role of autophagy in HIV-1 replication and pathogenesis. AIDS Res Ther. 2012;9:16.
- Dinkins C, Arko-Mensah J. Deretic V and Autophagy HIV. Seminars in cell & developmental biology; 2010; 21. p. 712–718.
- Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268.
- Campbell GR, Spector SA. Hormonally active vitamin D3 (1alpha,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286:18890–18902.
- Campbell GR, Spector SA. Autophagy induction by vitamin D inhibits both Mycobacterium tuberculosis and human immunodeficiency virus type 1. Autophagy. 2012;8:1523–1525.
- Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8:e1002689.
- Pathak S, Wentzel-Larsen T, Asjo B. Effects of in vitro HIV-1 infection on mycobacterial growth in peripheral blood monocyte-derived macrophages. Infect Immun. 2010;78:4022–4032.
- Chandra P, Ghanwat S, Matta SK, et al. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep. 2015;5:16320.
- Kumar D, Nath L, Kamal MA, et al. Genome-wide analysis of the host intracellular network that regulates survival of mycobacterium tuberculosis. Cell. 2010;140:731–743.
- Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652.
- DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9:ra21–ra.
- Mardones P, Rubinsztein DC, Hetz C. Mystery solved: trehalose kickstarts autophagy by blocking glucose transport. Sci Signal. 2016;9:fs2.
- Mayer AL, Higgins CB, Heitmeier MR, et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep. 2016;6:38586.
- Bhaskar A, Munshi M, Khan SZ, et al. Measuring glutathione redox potential of HIV-1-infected macrophages. J Biol Chem. 2015;290:1020–1038.
- Matta SK, Kumar D. AKT mediated glycolytic shift regulates autophagy in classically activated macrophages. Int J Biochem Cell Biol. 2015;66:121–133.
- Sarkar S, Carroll B, Buganim Y, et al. Impaired autophagy in the lipid storage disorder Niemann–Pick type C1 disease. Cell Rep. 2013;5:1302–1315.
- Chandra P, Kumar D. Selective autophagy gets more selective: uncoupling of autophagy flux and xenophagy flux in mycobacterium tuberculosis-infected macrophages. Autophagy. 2016;12:608–609.
- Lundahl J, Hallden G, Skold CM. Human blood monocytes, but not alveolar macrophages, reveal increased CD11b/CD18 expression and adhesion properties upon receptor-dependent activation. Eur Respir J. 1996;9:1188–1194.
- Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206.
- Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766.
- Kaplon RE, Hill SD, Bispham NZ, et al. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging (Albany NY). 2016;8:1167–1183.
- Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Tong Y, Song F. Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy. 2015;11:1192–1195.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signaling regulates autophagy via calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299.
- McCue Hannah V, Wardyn Joanna D, Burgoyne Robert D, et al. Generation and characterization of a lysosomally targeted, genetically encoded Ca(2+)-sensor. Biochem J. 2013;449:449–457.
- Wang W, Gao Q, Yang M, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America; 2015; 112. p. E1373–E81.
- Zolov SN, Bridges D, Zhang Y, et al. In vivo, Pikfyve generates PI(3,5)P(2), which serves as both a signaling lipid and the major precursor for PI5P. Proceedings of the National Academy of Sciences of the United States of America; 2012; 109. p. 17472–17477.
- Dong X-P, Shen D, Wang X, et al. PI(3,5)P(2) controls membrane traffic by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38.
- Baranov MV, Revelo NH, Verboogen DRJ, et al. SWAP70 is a universal GEF-like adapter for tethering actin to phagosomes. Small GTPases. 2017;10:311–323.
- Bell LCK, Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat Rev Microbiol. 2018;16:80–90.
- Kumar D, Rao KV. Regulation between survival, persistence, and elimination of intracellular mycobacteria: a nested equilibrium of delicate balances. Microbes Infect. 2011;13:121–133.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335.
- Perry SW, Norman JP, Barbieri J, et al. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50:98–115.
- Emanuele E. Can trehalose prevent neurodegeneration? Insights from experimental studies. Curr Drug Targets. 2014;15:551–557.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997.
- Kalscheuer R, Weinrick B, Veeraraghavan U, et al. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America; 2010; 107. p. 21761–21766.
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075.
- Jayachandran R, Sundaramurthy V, Combaluzier B, et al. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 2007;130:37–50.
- Campbell GR, Rawat P, Bruckman RS, et al. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015;11:e1005018.
- Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750.
- Waller-Evans H, Lloyd-Evans E. Regulation of TRPML1 function. Biochem Soc Trans. 2015;43:442–446.
- Bridges D, Ma JT, Park S, et al. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012;23:2955–2962.
- Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584:1302–1312.
- Dayam RM, Saric A, Shilliday RE, et al. The phosphoinositide-gated lysosomal Ca(2+) channel, TRPML1, is required for phagosome maturation. Traffic. 2015;16:1010–1026.
- Spector SA, Zhou D. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy. 2008;4:704–706.