
ABSTRACT
Influenza A virus (IAV) infection induces mitophagy, which is essential for the clearance of damaged mitochondria. Dysfunctional mitochondria can be selectively targeted by PINK1, which recruits PRKN/PARK2 and leads to subsequent mitochondrial sequestration within autophagosomes. The IAV PB1-F2 protein translocates to mitochondria, accelerates the mitochondrial fragmentation and impairs the innate immunity. However, whether PB1-F2 mediates IAV–induced mitophagy and the relation between mitophagy and PB1-F2-attenuated innate immunity remain obscure. Here, we showed that PB1-F2 translocated to mitochondria by interacting and colocalizing with TUFM (Tu translation elongation factor, mitochondrial). Further studies revealed that PB1-F2 induced complete mitophagy, which required the interactions of PB1-F2 with both TUFM and MAP1LC3B/LC3B that mediated the autophagosome formation. PB1-F2-induced mitophagy was critical for the MAVS (mitochondrial antiviral signaling protein) degradation and led to its suppression of the type I IFN production. Importantly, the C-terminal LIR motif of PB1-F2 protein was demonstrated to be essential for its mitophagy induction and attenuated innate immunity. In conclusion, PB1-F2-induced mitophagy strongly correlates with impaired cellular innate immunity, revealing it is a potential therapeutic target.
Abbreviations: BCL2L13: BCL2 like 13; BECN1: beclin 1; BNIP3L/Nix: BCL2 interacting protein 3 like; CQ: chloroquine; DDX58: DExD/H-box helicase 58; eGFP: enhanced green fluorescent protein; hpi: hours post infection; IAV: influenza A virus; IFN: interferon; IP: immunoprecipitation; LIR: LC3-interacting region; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAVS: mitochondrial antiviral signaling protein; MMP: mitochondrial membrane potential; MOI, multiplicity of infection; mRFP: monomeric red fluorescent protein; NBR1: NBR1 autophagy cargo receptor; NC: negative control; NLRP3: NLR family pyrin domain containing 3; PINK1: PTEN induced kinase 1; PRKN/PARK2: parkin RBR E3 ubiquitin protein ligase; RLR: RIG-I-like-receptor; ROS: reactive oxygen species; SEV: sendai virus; SQSTM1/p62: sequestosome 1; TAX1BP1: Tax1 binding protein 1; TM: transmembrane; TOMM20/40: translocase of outer mitochondrial membrane 20/40; TUFM: Tu translation elongation factor, mitochondrial.
Introduction
Mitophagy is a process of the selective engulfment of mitochondria by autophagosomes and their subsequent degradation by lysosomes, which functions as selective removal of damaged mitochondria [Citation1,Citation2]. So, the primary pathway in mitochondria quality control and cell survival is the clearance of damaged mitochondria via mitophagy [Citation3]. The mechanisms of mitophagy are classified broadly into 2 distinct groups: PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase)-dependent and PRKN-independent [Citation4]. PRKN-dependent mitophagy generally occurs after recognition of damaged or dysfunctional mitochondria with low membrane potential by some autophagic receptors [Citation4], such as SQSTM1/p62 (sequestosome 1), NBR1 (NBR1 autophagy cargo receptor), BCL2L13 (BCL2 like 13), TAX1BP1 (Tax1 binding protein 1), FUNDC1 (FUN14 domain containing 1) and BNIP3L/Nix (BCL2 interacting protein 3 like) [Citation5–9], which are recruited to the damaged mitochondria via ubiquitin binding and mediate autophagic engulfment through their association with LC3B [Citation10]. Apart from utilizing host mitophagy machinery to trigger PRKN-dependent or receptors-mediated mitophagy, some viruses can directly induce mitophagy via their viral factors [Citation10–12]. The mitochondrial dynamics (fusion and fission) are sensitive to virus infection. They can be either directly targeted by viral proteins or affected by the cellular physiological or metabolic changes during viral pathogenesis [Citation13]. Several viruses, such as HBV, HCV, HIV, CSFV, and PRRSV, have been reported to promote mitochondria fission and induce mitophagy for the maintenance of persistent infection and attenuation of the innate immune responses [Citation3,Citation14–19]. It also has been proposed that A/Puerto-Rico/8 (A/PR8/H1N1) influenza virus infection can induce mitophagy, which is independent of PRKN but a kinase-dependent manner by ULK1 (unc-51 like autophagy activating kinase 1) phosphorylation [Citation20]. However, whether other intermediates are involved in IAV–induced mitophagy and the mechanisms by which IAV protein(s) mediate(s) mitophagy occurrence remains unclear.
IAV is an enveloped virus with a segmented negative-sense RNA genome, causing significant morbidity and mortality in humans and animals worldwide, and threatening their populations with epidemics and pandemics [Citation21,Citation22]. IAV infection induces rapidly host innate immune response via a variety of regulatory mechanisms [Citation23], which is essential for pathogen clearance and host survival. However, IAV can evade the host innate immune response that is involved in several viral proteins (PB1, PB2, PA, NS1, and PB1-F2) with complicated mechanisms [Citation24–29]. Among them, the IAV PB1-F2 protein, a small protein encoded by a + 1 alternate open-reading frame of the RNA polymerase subunit (PB1 gene segment) of IAV [Citation30], has variable size with different polypeptide length of predominantly 87 or 90 amino acids [Citation31]. PB1-F2 proteins from highly pathogenic strains (for example, H5N1 subtype) encode a longer polypeptide containing 90 amino acids. In contrast, the low pathogenic strains (for example, H1N1, except the 1918 pandemic strain) tend to encode 57 amino acids known as the C-terminal truncated form [Citation32]. The C-terminal region of the PB1-F2 protein contains a mitochondrial targeting sequence, which is critical for its mitochondria translocation [Citation33] linked to its increased pathogenicity and secondary bacterial infection [Citation34–38]. The PB1-F2 protein derived from the A/PR8/H1N1 strain, which has 87 amino acids (longer polypeptide), was reported to translocate to mitochondrial inner membrane space via TOMM40 channel and lead to the reduction of mitochondrial membrane potential (MMP) [Citation30,Citation39–41]. In contrast, the PB1-F2 variant lacking C-terminal region does not affect mitochondrial function [Citation32]. PB1-F2-induced mitochondrial fission and fragmentation are involved in the attenuation of innate immune response and activation of NLRP3 (NLR family pyrin domain containing 3) inflammasome [Citation29,Citation32]. Furthermore, other studies also have documented that PB1-F2 affects IFN synthesis by binding to MAVS [Citation29,Citation42]. Viruses-induced mitophagy is closely associated with the attenuation of the innate immune response [Citation10,Citation43,Citation44]. However, the role that PB1-F2 plays in mediating the IAV–induced mitophagy process remains unclear. In addition, whether mitophagy is associated with PB1-F2-suppressed innate immunity is obscure.
TUFM/EFTu (Tu translation elongation factor, mitochondrial) is encoded by a nuclear gene, synthesized in the cytoplasm and imported into the mitochondria, along with a large number of other nuclear-encoded protein factors, constitutes the mitochondrial translation apparatus [Citation45–49]. Since TUFM is localized exclusively in mitochondria and among the most frequently identified components in the mitochondrial nucleoprotein complex [Citation50–54], it is known to participate in various biological processes such as mitochondrial protein translation elongation and biosynthesis [Citation50,Citation51,Citation55,Citation56], oncogenesis, oxidative phosphorylation, and protein quality control [Citation51,Citation57–59]. It possesses GTPase activity and RNA binding activity [Citation60,Citation61]. TUFM contains three domains including domain I, domain II, and domain III, among which, the N-terminal contains a mitochondria-targeting sequence and a GTP-binding domain I that was previously shown to be essential for optimal interaction with the ATG12–ATG5 conjugate, while domains II and III were not as critical [Citation62]. Previous studies have reported that TUFM acts as an intermediary partner between NLRX1 (NLR family member X1) and autophagic proteins ATG12–ATG5-ATG16L1 to form a molecular complex in mitochondria, which not only enhances VSV–induced autophagy but also inhibits MAVS-induced IFN-expression by acting against DDX58 (DExD/H-box helicase 58) [Citation62,Citation63]. Upon autophagy induction under cetuximab treatment, TUFM is able to gradually recruit BECN1/Beclin 1 to mitochondria and promotes its polyubiquitination, impeding BECN1 interaction with RUBCN (rubicon autophagy regulator) to intersect with autophagy machinery in HNSCC cells [Citation64]. A recent study demonstrated that human parainfluenza virus type 3 (HPIV3)-induced LC3B vacuole-like structures colocalize well with TUFM [Citation10]. Furthermore, both M protein of HPIV3 and Gn protein of Hantavirus (HTNV) translocate to mitochondria by interacting with TUFM, and then recruit LC3B to mitochondria, thus they function as autophagy receptors to mediate mitophagy, which results in the inhibition of the type I IFN response [Citation10,Citation12]. Importantly, it has been documented that TUFM promotes autophagy in avian-signature influenza virus PB2627E-infected human cells, and this TUFM-dependent autophagy may regulate avian influenza virus replication in human cells [Citation65]. However, whether TUFM can also involve in the modulation of IAV PB1-F2-induced mitophagy process and its suppression of innate immunity remains unknown.
In this study, we demonstrated that IAV PB1-F2 protein translocated to mitochondria via its interaction with TUFM, and subsequently mediated the engulfment of mitochondria into autophagosome by binding to LC3B, thereby promoting the clearance of MAVS protein, thus negatively modulated the MAVS-mediated antiviral innate immune signaling.
Results
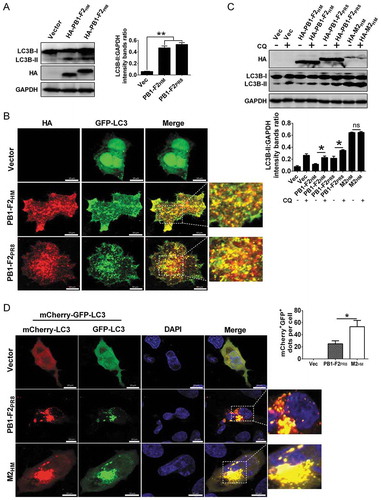
PB1-F2 protein overexpression induces mitophagy
A previous study has shown that A/PR8/H1N1 influenza virus infection induces incomplete autophagy by blocking autophagosome fusion with lysosome [Citation66] and mitophagy [Citation20], a form of autophagy that selectively cleared the damaged mitochondria with low membrane potential [Citation67]. The mitochondria-associating IAV protein PB1-F2 translocates into mitochondria and accelerates the mitochondrial fragmentation due to reduced ∆Ψm [Citation32]. Hence, we determined whether PB1-F2 is involved in the IAV–induced mitophagy process. To this end, we first assayed PB1-F2-induced autophagy level by western blot, and it demonstrated that PB1-F2 from both A/duck/Hubei/Hangmei01/2006(H5N1) (H5N1/HM) (PB1-F2HM) and A/PR8/H1N1 (PB1-F2PR8) overexpression resulted in a significant increase in the LC3B-II level (). Furthermore, results of immunofluorescence analysis indicated that numbers of GFP-LC3 puncta formed and were colocalized well with PB1-F2 in both PB1-F2HM- and PB1-F2PR8-transfected cells (). These results suggested that PB1-F2 indeed induced autophagosome formation. Our previous study showed that the H5N1/HM M2 (M2HM) protein could trigger autophagy initiation as well as block the autophagosome fusion with lysosome [Citation68]. To elucidate how PB1-F2 results in the accumulation of autophagosomes, LC3B lipidation was detected in the PB1-F2HM-, PB1-F2PR8- and M2HM (as a positive control)-expressing cells treated with or without chloroquine (CQ, an inhibitor blocking the fusion of the autophagosome with lysosome). Results in demonstrated that a higher level of LC3B-II accumulated in PB1-F2HM- and PB1-F2PR8-transfected cells upon CQ treatment than in PB1-F2-transfected cells without CQ treatment, indicating that PB1-F2 led to the autolysosome formation. Enhanced green fluorescent protein (eGFP) is sensitive to lysosomal proteolysis and may diminish quickly in acidic pH, while red fluorescent protein (RFP) retains fluorescence even at acidic pH [Citation69]; therefore, a tandem reporter construct, mCherry-GFP-LC3 (ptf-LC3), was also used to measure PB1-F2-induced autophagic flux. As shown in , many LC3-positive autophagic vacuoles were yellow in cells co-expressing M2HM and ptf-LC3, while cells-transfected with PB1-F2PR8 and ptf-LC3 resulted in a shift from yellow to partially red fluorescence, indicating that the increased level of autophagic flux by PB1-F2. Together, these results revealed that PB1-F2 induced complete autophagy.
Figure 1. IAV PB1-F2 induces autophagy. (A) HEK 293T cells were transfected with HA-PB1-F2PR8 and HA-PB1-F2HM from the A/PR8/H1N1 virus and H5N1/HM virus, respectively. Cell lyses were harvested and analyzed by western blot. (B) A549 cells were transfected with GFP-LC3 and vector, HA-PB1-F2PR8, or HA-PB1-F2HM, respectively. The LC3 puncta formation and the colocalization of LC3 puncta with PB1-F2 were analyzed. Scale bar: 10 μm. It was the representative of 20 cells. (C) HEK 293T cells were transfected with indicated plasmids for 12 h and then treated with or without chloroquine (CQ, 25 μM). 12 h later, cell lysates were analyzed by western blot. (D) A549 cells were transfected with ptf-LC3 (mCherry-GFP-LC3) and vector, HA-PB1-F2PR8, or HA-M2HM (as a control to induce incomplete autophagy), respectively. Cells were analyzed to detect the autophagosome formation. In the zoomed images, fluorescence signals indicated the expression of mRFP-LC3 and EGFP-LC3 (yellow color: incomplete autophagy, red color: complete autophagy). Scale bar: 10 μm. The graph shows the quantification of mRFP+GFP+ autophagosomes by taking the average number of dots in 20 cells (n = average number of dots in 20 cells). Mean ± SD (error bars) were determined for triplicates of three independent experiments (*p < 0.05; **p < 0.01; ns, nonsignificant)
Next, we determined whether PB1-F2 can induce mitophagy. The immunofluorescence analysis showed that the GFP-tagged LC3 puncta was partially colocalized with the RFP-tagged Mito, a mitochondrial marker protein, both in PB1-F2HM and PB1-F2PR8-transfected cells (), indicating that sequestrated mitochondria could be within PB1-F2-induced autophagosome. SQSTM1 binds to LC3B and accumulates with the inhibition of autophagy. The autophagy process after autophagosome fusion with lysosome degrades both [Citation70]. As complete mitophagy eliminates damaged mitochondria, we further observed significant decline in the levels of SQSTM1 (an indicator of autophagic degradation), the outer membrane mitochondrial protein TOMM20 (translocase of outer mitochondrial membrane 20) and the inner membrane mitochondrial protein MT-CO2 (mitochondrially encoded cytochrome c oxidase II, which indicates complete mitophagy [Citation71]) in PB1-F2PR8-expressing cells treated without CQ, but CQ reversed this effect (Fig. S1A and B).
Figure 2. PB1-F2 overexpression induces mitophagy. (A) A549 cells were co-transfected with indicated plasmids for 24 h and analyzed for mitophagosome formation. In the zoomed images, yellow color indicated the colocalization of GFP-LC3 and dsRed-mito (Mito). Scale bar: 10 μm. It was the representative of 20 cells. (B and C) HEK 293T cells were transfected with indicated plasmids. Lysates were evaluated by western blot. (D) pmRFP-GFP-Mito-transfected A549 cells were co-transfected with HA-PB1-F2PR8 or HA-PB1-F2PR8 and HA-M2HM for 24 h and then analyzed to detect mitophagosome formation. In the zoomed images, fluorescence signals indicated the expression of mRFP-EGFP-mito targeting the mitochondria (yellow color: incomplete mitophagy; red color: complete mitophagy). Scale bar: 10 μm. The graph shows the quantification of mRFP+GFP+ mitophagosomes by taking the average number of dots in 20 cells (n = average number of dots in 20 cells). Mean ± SD (error bars) were determined for triplicates of 3 independent experiments (*p < 0.05; **p < 0.01)
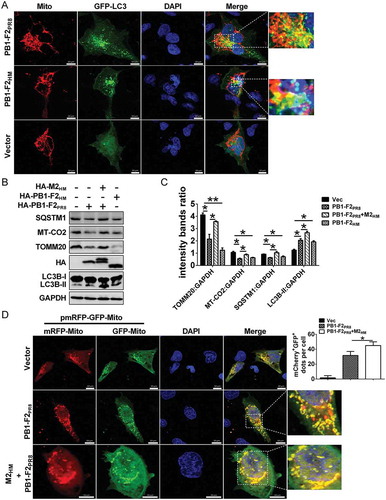
Similarly, the presence of M2HM blocked the degradation of LC3B-II, TOMM20, SQSTM1, and MT-CO2 caused by both PB1-F2PR8 and PB1-F2HM (). These results suggested that PB1-F2 possibly induced complete mitophagy. To confirm that, we further used a tandem reporter construct encoding a mitochondria-targeting signal sequence fused to RFP and EGFP genes, pmRFP-GFP-Mito. The GFP signal is attenuated in an acidic pH environment, while RFP signal remains stable in lower pH; therefore, the fusion of mitophagosome with lysosome results in the loss of yellow fluorescence and the appearance of only red fluorescence of RFP, indicating the completion of the mitophagy process [Citation18]. Results showed that the yellow-merged fluorescence, indicative of the presence of both EGFP and mRFP in mitochondria, are present in the control-transfected cells (, up); in contrast, the cells expressing PB1-F2PR8 displayed distinct red puncta and fewer green puncta (, middle); however, M2 blocked this effect (, down), suggesting that PB1-F2 expression promoted the fusion of mitophagosome with lysosome. Collectively, these results indicated that PB1-F2 induced complete mitophagy, and M2 inhibited the fusion of mitophagosome and lysosome, leading to the accumulation of mitophagosomes.
PB1-F2 interacts with TUFM
Next, we determined the mechanism of how PB1-F2 induced mitophagy. Recent studies showed that both M protein of HPIV3 and Gn protein of HTNV induces mitophagy, depending on TUFM [Citation10,Citation12]. TUFM also regulates influenza PB2627E virus-induced autophagy [Citation65]. We first performed reciprocal coIP assays to assess the interaction between ectopically expressed PB1-F2PR8 and TUFM to determine whether TUFM also regulates PB1-F2-induced mitophagy. Results showed that PB1-F2PR8 did physically interact with exogenous TUFM (). Notably, the endogenous TUFM also was coimmunoprecipitated with PB1-F2PR8 in HEK 293T cells (). Furthermore, we performed an in vitro glutathione S-transferase (GST) affinity-isolation assay with GST-fused TUFM. GST-TUFM, but not GST alone, was able to pull down the HA-tagged-PB1-F2PR8 in the mitochondrial fraction expressed in HEK293T cells (Fig. S2A and B). Coincidently, results of immunofluorescence analysis revealed that a significant portion of TUFM colocalized with PB1-F2PR8 at the mitochondria when cells were in the presence of PB1-F2PR8 ( and S2C). Together, our findings indicated that TUFM directly interacted with PB1-F2, and their interaction occurred at the mitochondria.
Figure 3. PB1-F2 interacts and colocalizes with TUFM. (A) HEK 293T cells were transfected with vector or Flag-TUFM and HA-PB1-F2PR8, respectively. Cell lysates were subjected to IP. (B) HEK 293T cells were transfected with vector or HA-PB1-F2PR8 and Flag-TUFM, respectively. Cell lysates were subjected to IP. An asterisk next to the blot indicated the protein. (C) Lysates of HA-PB1-F2PR8-transfected HEK 293T cells were prepared and immunoprecipitated with the anti-TUFM antibody or control IgG. (D) A549 cells were transfected with vector or HA-PB1-F2PR8. 24 h later, cells were analyzed for the distribution of PB1-F2 and endogenous TUFM. In the zoomed images, yellow color indicated the colocalization of PB1-F2 and TUFM. Scale bar: 10 μm. It was the representative of 20 cells. (E) HEK 293T cells were transfected with Flag-TUFM with PB1-F2PR8 truncation mutant HA-PB1-F2∆N37. Cell lysates were subjected to IP. (F) HEK 293T cells were transfected with Flag-TUFM and PB1-F2PR8 truncation mutant GFP-PB1-F2∆C50 for 24 h cell lysates were subjected to IP and analyzed by western blot. (G) Flag-tagged WT TUFM or domain truncation mutants (TUFM∆domain I, TUFM∆domain II and TUFM∆domain III) were co-transfected with HA-PB1-F2PR8 into HEK 293T cells. Cell lysates were immunoprecipitated with anti-FLAG and immunoblotted for HA-PB1-F2PR8.
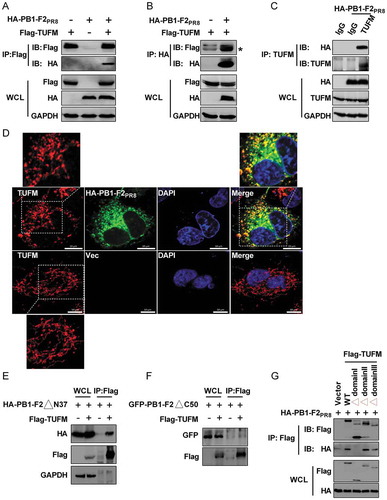
We constructed PB1-F2PR8 truncation mutants to map the region in PB1-F2 that is critical for its interaction with TUFM. We showed that deletion of the N-terminal of 37 residues of PB1-F2PR8 (PB1-F2∆N37) still was able to bind to TUFM (), while deletion of the C-terminal of 50 residues of PB1-F2PR8 (PB1-F2∆C50) impaired the interaction of PB1-F2PR8 with TUFM (), suggesting that the C terminal of 50 residues were essential for PB1-F2 to interact with TUFM. To further map the essential domain in TUFM necessary for its interaction with PB1-F2, we generated a series of mutant forms of TUFM, as described previously [Citation62]. CoIP results showed that mutant with N-terminal domain I of TUFM deleted scarcely coimmunoprecipitated with PB1-F2PR8, while mutants with domain II and domain III deleted still were able to bind to TUFM comparably to TUFM (), suggesting that the domain I of TUFM was required for the optimal interaction with PB1-F2. These results were in agreement with a previous study showing that optimal interaction with ATG12–ATG5 required the N-terminal domain I of TUFM, which contains a mitochondria-targeting sequence, while domains II and III are not as critical [Citation62].
PB1-F2 mediates TUFM-dependent mitophagy
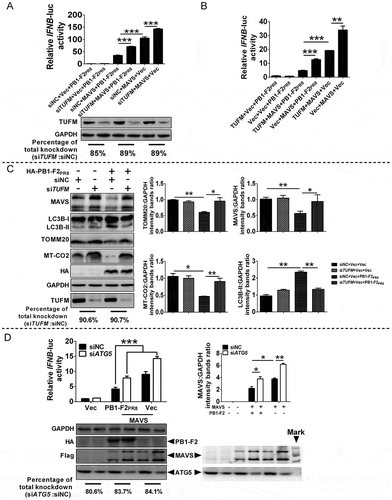
To explore whether TUFM regulated PB1-F2-induced mitophagy, we analyzed autophagy level both in cytosolic fractions and mitochondria-enriched fractions from NC (negative control, non-targeting siRNA) and TUFM-knockdown cells. As shown in and B, there was no obvious difference in the LC3B-II level between the NC and TUFM-knockdown cells in the cytosolic fractions with PB1-F2PR8 expression; however, cell lysates expressing PB1-F2PR8 with siTUFM exhibited a significant reduction in the level of LC3B-II in the mitochondrial fractions, in comparison with the siNC-transfected cells. Simultaneously, knockdown TUFM in PB1-F2PR8-expressing cells resulted in a significant increase in the levels of TOMM20 and MT-CO2 in comparison with the siNC-transfected cells ( and B), which were consistent with the above results that PB1-F2 induced complete mitophagy. Moreover, the translocation of PB1-F2PR8 from the cytoplasm to mitochondria significantly decreased in both TUFM-knockdown ( and B) and knockout (TUFM-KO) cells, compared to the control cells (Fig. S3A-C), which was reversed by WT TUFM (Fig. S3C). These results indicated that TUFM was necessary to recruit PB1-F2 to mitochondria and regulate PB1-F2-induced mitophagy.
Figure 4. PB1-F2 mediates TUFM-dependent mitophagy. (A and B) HEK 293T cells were transfected with siTUFM or siNC (negative control) for 12 h, and cells were further transfected with HA-PB1-F2PR8 for another 24 h. Cytoplasm and mitochondrial fractions were purified for western blot analysis (Fractions: Cyto, purified cytosolic; Mito, purified mitochondria. Organelle markers: TOMM20, mitochondria; HSPA5/GRP78, endoplasmic reticulum [ER]; LAMP1, lysosome; GAPDH, cytoplasm). (C) TUFM knockout (TUFM-KO) or wild type (WT) A549 cells were transfected with the indicated plasmids for 24 h and analyzed for mitophagosome formation. In the zoomed images, yellow color indicated the colocalization of GFP-LC3 and TOMM20 (as a mitochondrial marker), white color indicated the colocalization of GFP-LC3, mitochondria, and PB1-F2. Scale bar: 10 μm. It was the representative of 20 cells (**p < 0.01; ***p < 0.001)
![Figure 4. PB1-F2 mediates TUFM-dependent mitophagy. (A and B) HEK 293T cells were transfected with siTUFM or siNC (negative control) for 12 h, and cells were further transfected with HA-PB1-F2PR8 for another 24 h. Cytoplasm and mitochondrial fractions were purified for western blot analysis (Fractions: Cyto, purified cytosolic; Mito, purified mitochondria. Organelle markers: TOMM20, mitochondria; HSPA5/GRP78, endoplasmic reticulum [ER]; LAMP1, lysosome; GAPDH, cytoplasm). (C) TUFM knockout (TUFM-KO) or wild type (WT) A549 cells were transfected with the indicated plasmids for 24 h and analyzed for mitophagosome formation. In the zoomed images, yellow color indicated the colocalization of GFP-LC3 and TOMM20 (as a mitochondrial marker), white color indicated the colocalization of GFP-LC3, mitochondria, and PB1-F2. Scale bar: 10 μm. It was the representative of 20 cells (**p < 0.01; ***p < 0.001)](/cms/asset/0c50f01e-c9e7-45ef-b221-e354b85ffe10/kaup_a_1725375_f0004_c.jpg)
We performed rescue experiments in TUFM-KO cells with and without PB1-F2 transfection to confirm that TUFM is a special modulator of PB1-F2-induced mitophagy. Results showed that ectopically expressing WT TUFM, but not TUFM∆domain I, rescued the colocalization of LC3B puncta with mitochondria () and the conversion of LC3B-I to LC3B-II (Fig. S3D and E), and further reduced the expression of TOMM20 and MT-CO2 (Fig. S3D and E). Furthermore, WT TUFM but not TUFM∆domain I increased the translocation of PB1-F2 from the cytoplasm to mitochondria (Fig. S3A). These results suggested that the interaction and colocalization of TUFM with PB1-F2 on mitochondria were essential for recruiting PB1-F2 to mitochondria and PB1-F2-induced mitophagy. Next, we sought to determine whether PB1-F2 induced TUFM-dependent mitophagy under A/PR8/H1N1 virus infection. As shown in Fig. S4A and B, A/PR8/H1N1 virus infection induced autophagy and mitophagy in the WT cells, which were in agreement with the results of previous studies [Citation20,Citation66]. In contrast, the expression level of LC3B-II (Fig. S4A) and the colocalization of LC3B puncta with mitochondria (Fig. S4B) were significantly decreased in TUFM-KO cells, indicating that TUFM also regulated the IAV–induced autophagy and mitophagy processes. In addition, we demonstrated that WT TUFM but not TUFM∆domain I overexpression under IAV infection reversed the autophagy and mitophagy levels blocked by TUFM knockout (Fig. S4A and B). IAV infection also reduced the expression of MT-CO2, in comparison with the control-infected cells. We observed a further decrease in MT-CO2 expression by WT TUFM overexpression but not TUFM∆domain I overexpression. Finally, knockout of TUFM reversed the reduced MT-CO2 expression (Fig. S4A). Together, these results revealed that PB1-F2 mediated TUFM-dependent mitophagy.
PB1-F2 suppresses mitophagy-dependent type I IFN response
Both TUFM and IAV PB1-F2 attenuate the RIG-I-like-receptor (RLR)-mediated innate antiviral signaling [Citation29,Citation62]. Next, we determined whether PB1-F2-attenuated innate immune depends on TUFM. As shown in , knockdown TUFM by siTUFM reversed the inhibitory effect of PB1-F2PR8 on the IFNB promoter activity stimulated by MAVS. In contrast, overexpression of TUFM enhanced the depressing effect of PB1-F2PR8 on IFNB promoter activity triggered by MAVS (). Complete mitophagy can lead to selectively eliminate dysfunctional mitochondria, intracellular pathogens, and protein aggregates such as innate immune signaling protein MAVS [Citation19,Citation72]. Intriguingly, our studies further showed that PB1-F2PR8 overexpression markedly decreased the expression levels of TOMM20, MT-CO2, and MAVS compared with the control-transfected cells, while knockdown of TUFM reversed it ( and S5A), indicating that PB1-F2 inhibited the innate immune response by regulating MAVS expression and depending on TUFM. In addition, we also showed that the mutant PB1-F2PR8∆C50 failed to reduce the MAVS level (Fig. S5B), suggesting that the interaction of TUFM and PB1-F2 was critical for its inhibition of the type I IFN response. To confirm that PB1-F2 suppressed TUFM-dependent innate immune response in the context of viral infection, we further determined whether TUFM could benefit the IAV replication through inhibiting the IFNB production. Expectedly, results of TCID50 assays showed that the replication of A/PR8/H1N1 virus dramatically decreased in TUFM-KO cells (Fig. S5C), compared with the WT cells; however, overexpressing WT TUFM but not TUFM∆domain I resulted in a significant increase in viral yield (Fig. S5C). Moreover, IAV infection-induced IFNB production significantly increased in TUFM-KO cells, and WT TUFM but not TUFM∆domain I reversed this effect (Fig. S5D). These results suggested that TUFM was essential for PB1-F2-suppressed innate immune response.
Figure 5. PB1-F2 suppresses the innate immune response depending on the mitophagy. (A) HEK 293T cells were transfected with siTUFM or siNC for 12 h, the cells were further transfected with indicated plasmids for another 24 h and then luciferase assays were performed. The expression of TUFM was analyzed by western blot. (B) HEK 293T cells were transfected with the indicated plasmids. The luciferase assays were performed at 24 h post-transfection. (C) siNC- or siTUFM-transfected HEK 293T cells were transfected with indicated plasmids for 24 h. Cell lysates were harvested for the western blot analysis. (D) HEK 293T cells were transfected with siATG5 or siNC, and 24 h later, cells were further transfected with indicated plasmids. The luciferase assays were performed as (A). Expression levels of corresponding proteins were confirmed by western blot. Mean ± SD (error bars) were determined for triplicates of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001)
Having established that PB1-F2 could induce complete mitophagy, resulting in the removal of the damaged mitochondria, it was reasonable to speculate that the suppression of type I IFN response by PB1-F2 was dependent on the PB1-F2-induced mitophagy. As expected, knockdown ATG5 abolished the inhibitory effect of PB1-F2PR8 on the innate immune response (). In brief, these results indicated that PB1-F2-induced mitophagy was crucial for its attenuated innate immune response.
PB1-F2 acts as a potential receptor of mitophagy to interact with LC3B via the LIR motif
Mitophagy is a type of autophagy that selectively sequesters the damaged mitochondria by autophagosome via the interaction of mitochondrial receptors with LC3B [Citation4]. Given that PB1-F2 was able to colocalize well with GFP-LC3 puncta (), suggesting that PB1-F2 may physically interact with LC3B. As expected, results of coIP experiments showed that GFP-tagged-LC3 did physically interact with HA-tagged-PB1-F2PR8 () and Flag-tagged-PB1-F2PR8 (). Furthermore, we also coimmunoprecipitated the endogenous LC3B with HA-tagged-PB1-F2 in HEK 293T cells (). ATG5 and SQSTM1 can translocate to mitochondria and involved in mitophagy induction [Citation73–75]. So, we performed coIP assays to preclude other autophagy-related genes involved in regulating PB1-F2 and LC3B interaction. Results showed that the HA-tagged PB1-F2PR8 could not bind with the endogenous ATG5, BECN1, ATG7, and SQSTM1 (Fig. S6A) suggesting that PB1-F2 acted as a potential receptor of mitophagy by especially binding to LC3B.
Figure 6. PB1-F2 acts as a receptor of mitophagy to interact with LC3B. (A and B) HEK 293T cells were transfected with GFP-LC3 and HA-PB1-F2PR8. Cell lysates were immunoprecipitated with anti-GFP antibody (A) or anti-HA antibody (B) and then analyzed by western blot. (C) HEK 293T cells were transfected with GFP-LC3 and Flag-PB1-F2PR8. Cell lysates were immunoprecipitated with anti-GFP antibody and then were subjected to western blot analysis. An asterisk next to the blot indicated the protein. (D) HEK 293T cells were transfected with HA-PB1-F2PR8, and lysates were subjected to IP and analyzed to detect endogenous LC3B via western blot. (E) The sequences of the LIR motif (W/Y××I/L) in PB1-F2 were aligned manually with typical autophagy receptors. (F) HEK 293T cells were co-transfected with each HA-tagged WT PB1-F2PR8 and its LIR mutants (PB1-F2PR8W61A, PB1-F2PR8W61A,L64A, and PB1-F2PR8∆LIR) with GFP-LC3. Cell lysates were immunoprecipitated with anti-GFP antibody and then analyzed by western blot. An asterisk next to the blot indicated the protein. (G) HEK 293T cells were transfected with HA-WT PB1-F2PR8 and its LIR mutants for 24 h. Cell lysates were harvested for western blot analysis
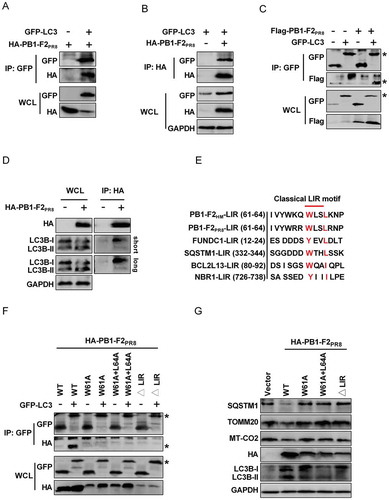
Some mitochondrial proteins, such as SQSTM1, NBR1, TAX1BP1, BNIP3L, and FUNDC1 that interact with LC3B and function as autophagy receptors to regulate mitophagy, normally contain a classical LIR (LC3-interacting region) motif with a conserved sequence of W/Y××L/I [Citation5,Citation6,Citation8,Citation76–79]. Intriguingly, we observed that both PB1-F2PR8 and PB1-F2HM carried a typical conserved LIR motif W(61)××L(64) at its C-terminal region (), with the motif exposed to the cytosolic site of the mitochondrial membrane. To test whether the LIR motif is critical for PB1-F2 interaction with LC3B, we constructed a series of mutant forms of PB1-F2PR8: the PB1-F2W61A point mutant, the PB1-F2W61A,L64A double-point mutant, and the LIR-deleted form, PB1-F2∆LIR. Results of coIP assays indicated that mutations in the LIR motif impaired the binding of PB1-F2PR8 to GFP-LC3 (), suggesting that the LIR motif was essential for PB1-F2 interaction with LC3B. Next, we determined whether the PB1-F2-induced mitophagy functionally requires the LIR motif of PB1-F2. As expected, the LIR mutants of PB1-F2PR8 failed to induce mitophagy ( and S6B), but still was able to bind to TUFM (Fig. S6C). Furthermore, we also showed that the interaction of TUFM with LC3B only occurred in the presence of HA-PB1-F2PR8 but not HA-PB1-F2PR8∆LIR (Fig. S6D). These results indicated that PB1-F2-induced mitophagy required its interaction with both LC3B and TUFM. Next, we also determined the effect of PB1-F2PR8 LIR mutants on the IFNB promoter activity. We showed that the PB1-F2PR8 LIR mutants eliminated the inhibitory effect of WT PB1-F2PR8 on the IFNB promoter activity triggered by both MAVS (Fig. S7A) and SEV (Fig. S7B). In addition, PB1-F2PR8 LIR mutants reversed the reduction of the MAVS level by PB1-F2PR8 (Fig. S7A). To further test whether PB1-F2-mediated suppression of innate immunity specifically via its interaction with TUFM, we performed rescue experiments with or without PB1-F2PR8 or PB1-F2∆LIR in TUFM-KO cells. Results of luciferase assays indicated that WT TUFM significantly suppressed the IFNB promoter activity but not TUFM∆domain I both in PB1-F2PR8- and PB1-F2∆LIR-transfected cells under SEV stimulation, and knockout TUFM reversed those effects (Fig. S7C). Together, these results indicated that the LIR motif in PB1-F2 was critical for the activation of mitophagy signaling, which required its interaction with TUFM and binding with LC3B.
Discussion
Mitophagy, the selective engulfment, and clearance of mitochondria are essential for the homeostasis of a healthy network of functioning mitochondria and prevent excessive production of cytotoxic ROS (reactive oxygen species) from damaged mitochondria. Mitochondrial dynamics and mitophagy are closely related [Citation67]. Many viruses impair mitochondrial networks and induce mitophagy [Citation3,Citation11,Citation14,Citation15,Citation18,Citation44]. Several viral proteins can serve as viral components that directly mediate mitophagy induction [Citation3,Citation10,Citation11]. Of note, the A/PR8/H1N1 virus infection induces mitophagy [Citation20], and its PB1-F2 protein leads to a loss of the MMP, which results in abnormal fragmented mitochondria [Citation32,Citation42], suggesting that it is most likely to be a viral factor that directly involved in IAV-mediated mitophagy. Thus, in this study, we confirmed that PB1-F2 proteins from both H5N1/HM and A/PR8/H1N1 viruses mediated IAV–infection-induced mitophagy by both western blot and immunofluorescence analysis. Because IAV infection induces incomplete autophagic process [Citation80], we further determined which type of mitophagy PB1-F2 induced, and showed that PB1-F2 protein triggered complete mitophagy. The mitochondrially targeted PINK1 and the PRKN are well-established synergistic mediators of mitophagy of dysfunctional mitochondria, which relies on the ubiquitination of a number of mitochondrial outer membrane substrates and subsequent docking of autophagy receptors to selectively clear mitochondria [Citation71,Citation81]. Because IAV infection induces mitophagy independent on the PRKN pathway [Citation20], indicating that PRKN is not involved in IAV proteins-mediated mitophagy process. Thus, we did not further detect whether PB1-F2PR8-induced mitophagy was dependent on PRKN. Different mitophagy effectors have been characterized, such as the receptors BNIP3L, FUNDC1, and BCL2L13 in mammals, to mediate engulfment of mitochondria into autophagosome through their association with LC3B [Citation82]. The A/PR8/H1N1 virus PB1-F2 also translocates into mitochondria relying on the TOMM40 channels [Citation32]. However, it cannot exclude the possibilities that other proteins in mitochondria may also facilitate the recruitment of PB1-F2 to mitochondria. Intriguingly, our study provided compelling evidence showing that TUFM, a mitochondrial protein, acted as an adaptor protein that bound to PB1-F2 to bridge the interaction of PB1-F2 with LC3B on the mitochondria, thereby recruiting mitochondria to the autophagosome, thus resulting in the induction of mitophagy. Similar studies also showed that both M protein of HPIV3 and Gn of HTNV translocate into mitochondria via their interaction with TUFM. Then, they mediate mitochondrial sequestration by autophagosome via their interaction with LC3B, leading to mitophagy [Citation10,Citation12].
It has been well-established that the late stage of autophagy, the process of autolysosome formation, is involved in the removal of the aggregated proteins such as MAVS aggregates by the lysosome enzyme [Citation70]. The IAV PB1-F2 protein also decreases MMP and accelerates mitochondrial fragmentation, which suppresses the induction of type I interferon, such as the DDX58 signaling pathway by binding to MAVS [Citation29,Citation32,Citation70]. It promotes the activation of NLRP3 inflammasomes [Citation42]. However, the detailed mechanisms remain unclear. TUFM can interact with ATG12–ATG5-ATG16L1 to form a molecular complex that augments autophagy and reduces DDX58-activated cytokines [Citation62,Citation63]. Herein, we demonstrated that PB1-F2 triggered the TUFM-dependent mitophagy, which further promoted the MAVS degradation in the fusion process of mitophagosome with the lysosome, thus leading to the PB1-F2-attenuated innate immune response. Mitophagy promotes viral replication by inhibiting the innate antiviral immunity [Citation44]. PB1-F2 protein is a virulent gene of IAV which influences viral pathogenicity, promotes lung inflammation, and increases the mortality and morbidity by modulating the host innate immune response [Citation35,Citation83]. The C-terminal of PB1-F2 is critical for its mitochondria translocation, in which the amino acids 61–87 contributes to IAV pathogenesis and severe inflammation [Citation35].
Moreover, the N66S substitution in PB1-F2 protein is associated with enhanced virulence in mice, as shown previously [Citation38]. In this study, we provided evidence for the first time that the PB1-F2 protein acted as a mitophagy receptor that was recognized specifically by LC3B. First, like other mitophagy receptors, PB1-F2 has a typical LIR motif W(61)××L(64) at its C-terminal region, which is essential for its interaction with LC3B. Second, mutations in or deletion of the LIR motif in PB1-F2 disrupted its ability to induce mitophagy signaling and inhibit the IFN production. Third, PB1-F2-induced mitophagy appeared to depend on its mitochondrial localization, as TUFM knockout not only decreased the localization of PB1-F2 to mitochondria but also abolished its capacity to induce autophagic and subsequent mitophagy signaling. These results indicated that PB1-F2 protein-induced mitophagy was possibly associated with IAV pathogenesis. Thus, we tried to assure that the LIR motif was responsible for PB1-F2-induced mitophagy and attenuated innate immunity in infection. Still, we failed to rescue the IAV containing PB1-F2 LIR mutations, and it might be due to the abolishment of the polymerase activity when we mutated the corresponding LIR motif in the IAV PB1 gene (data not shown).
In conclusion, our study proposed a model that IAV PB1-F2 protein acted as an autophagy receptor to mediate mitophagy by interacting with both TUFM and LC3B, thereby leading to the inhibition of the antiviral innate immune response. Thus, improved understanding of mitophagy induction during IAV infection will facilitate the development of antiviral therapeutics strategies to combat influenza viral infection and pathogenesis.
Materials and methods
Antibodies and reagents
Mouse monoclonal anti-ATG5 (Arigo biolaboratories, ARG54822), HA-tag (MBL, M180-3), GST-tag (HUABIO, EM80701), GFP-tag (CWBIO, CW0258A) and GAPDH (California Bioscience, CB100127), rabbit polyclonal anti-SQSTM1 (Proteintech, 18420-1-AP), TOMM20 (Proteintech, 11802-1-AP), MAVS (Proteintech, 14341-1-AP), LAMP1 (Proteintech, 21997-1-AP), HSPA5/GRP78 (HuaAn Biotechnology, ER40402), IAV NP (Gene Tex, GTX30852), TUFM (ABclonal Biotechnology, A6423), MT-CO2 (ABclonal Biotechnology, A11522), BECN1 (ABclonal Biotechnology, A7353), rabbit monoclonal anti-ATG7 (8558S, Cell Signaling Technology), rabbit control IgG (ABclonal Biotechnology, AC005), rabbit anti-MAP1LC3B/LC3B (Novus Biologicals, NB100-2220), mouse anti-Flag M2 (Sigma-Aldrich, F1804) were incubated overnight at 4°C. FITC-goat anti-mouse (Sungene Biotech, GM200G-02C), FITC-goat anti-rabbit (Sungene Biotech, GR200G-02C), 594-conjugated goat anti-mouse (Sungene Biotech, GM200G-43C), 594-conjugated goat anti-rabbit (Sungene Biotech, GR200G-43C) or 405-goat anti-mouse (Sungene Biotech, GM200G-46C) secondary antibodies were incubated 1 h at room temperature. Horseradish peroxidase (HRP)-conjugated anti-mouse (Beijing Biodragon Inmmunotechnologies, BF03001) and anti-rabbit (Beijing Biodragon Inmmunotechnologies, BF03008) secondary antibodies were incubated 1 h at room temperature.
Chloroquine (CQ; Sigma-Aldrich, C6628) and DAPI (Invitrogen, 00-4959-52) incubated cells at the corresponding concentrations.
Cells and viruses
Human lung epithelial cell line A549 cells (ATCC CCL-185™), human embryonic kidney (HEK) 293T cells (ATCC, CRL-3216™) and Madin–Darby canine kidney (MDCK) cells (ATCC, ATCCPTA-7909) were cultured in HAM’S/F-12 (HyClone, SH30026.01), RPMI 1640 medium (HyClone, SH30809.01) and DMEM/HIGH GLUCOSE (HyClone, SH30022.01), respectively, and supplemented with 10% heat-inactivated fetal bovine serum (FBS; PAN biotech, P30-3302) at 37°C with 5% CO2.
A/duck/Hubei/Hangmei01/2006(H5N1) (H5N1/HM) [Citation84] and A/PR8/H1N1 viruses were stored in our laboratory. Viral titers were determined by 50% tissue culture infectious dose (TCID50) analysis on MDCK cells. Briefly, A549 cells were infected with A/PR8/H1N1 virus at a multiplicity of infection (MOI) of 0.1. After 1 h of adsorption, cells were washed once with warm phosphate-buffered saline (PBS) and then incubated in serum-free HAM’S/F-12 medium with 0.25 mg/ml L-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma, 4352157-1KT) at 37°C. Virus stocks were collected at 24 h post-infection (hpi) and serially diluted in DMEM and adsorbed onto confluent MDCK cells for 1 h at 37°C. Later, the inoculum was removed, and cells were washed twice with PBS and cultured with serum-free DMEM medium with 0.5 mg/ml TPCK. After 3 d of incubation, virus titers were determined by calculating log10TCID50/milliliter on MDCK cells using the method developed by Reed and Muench [Citation85]. All experiments with the H5N1/HM were performed in the Biosafety Level 3 laboratory at Huazhong Agricultural University.
SDS-PAGE and western blot
Cells were lysed with cell lysis buffer for western blotting and IP (Beyotime, P0013) or mammalian cell lysis buffer (CWBIO, CW0889) containing protease inhibitor (Roche, 04693132001). The supernatants were collected, and equal amounts of proteins were separated by SDS-PAGE, and then were transferred to a nitrocellulose membrane (GE Healthcare Life sciences, 10,600,001 and 10,600,002). After blocking with 2% bovine serum albumin (BSA; Sigma, A1933) in tris-buffered saline with Tween (TBST; Sigma, SRE0031), the membrane were incubated with the indicated primary antibodies, followed by HRP-conjugated secondary antibodies, and signals were detected by using a WesternBrightTM ECL detection kit (advansta, K-12,045-D50) in an ECL detection system (Amersham Biosciences, Piscataway, NJ, USA). All bands of western blots were detected within the linear range.
Plasmids and mutagenesis
Full-length cDNA encoding H5N1/HM and A/PR8/H1N1 PB1-F2 and TUFM proteins were amplified by RT-PCR from the total RNA of A549 cells infected with H5N1/HM or A/PR8/H1N1 virus using specific primers (available upon request) and cloned into p3xFlag-CMV (Flag-PB1-F2 and Flag-TUFM), pGEX-6P-1 (GST-TUFM), HA-pCAGGS (HA-PB1-F2), and pEGFP-N1 (GFP-PB1-F2), respectively. The 4 empty vectors (p3xFlag-CMV [Sigma, E7908], pGEX-6P-1 [GE Healthcare Life sciences, 27-4597-01], HA-pCAGGS [YouBia, VT1076] and pEGFP-N1 [YouBia, VT1110]) were stored in our laboratory. The tandem fluorescent monomeric red fluorescent protein (mRFP)-GFP-LC3 (ptfLC3) [Citation69] was obtained from Chan Ding (Shanghai Veterinary Research Institute, China). The constructs encoding LC3 (GFP-LC3), a mitochondrial-targeting signal fused to RFP gene (dsRed-mito), and pmRFP-GFP-Mito were stored in our laboratory. MAVS expression plasmid and luciferase (luc) reporter plasmids (IFNB promoter [IFNB-luc]) were kindly provided by Professor Zhengfan Jiang (Peking University, China). The A Renilla control plasmid (pGL4.75 hRluc/CMV, Promega, E6931) was used as a control. The PB1-F2PR8 mutants (PB1-F2PR8∆N37, PB1-F2PR8∆C50, PB1-F2PR8W61A, PB1-F2PR8W61A,L64A, PB1-F2PR8∆LIR) and TUFM mutants (TUFM∆domain I, TUFM∆domain II and TUFM∆domain III) were generated by using A QuickChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and overlap cloning techniques.
SiRNA oligonucleotides transfection
For the RNA interference knockdown experiments, siRNA (sense strand-only shown) against human ATG5 (5′-GATTCATGGAATTGAGCCA-3′) as described in our previous study [Citation86] and TUFM (5′-CAGCUUCCCUUGCGUUUAA-3′) as described in a previous study [Citation69] were synthesized from Suzhou GenePharma. HEK 293T cells were transfected with 60 nM (final concentration) siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13,778,030) following the manufacturer’s protocols. At 12 h post-transfection with siRNA, cells were further transfected with indicated plasmids with X-tremeGENE HP (Roche, 32,223,900) for another 24 h. Negative Control siRNA (sense, 5′-UUCUCCGAACGUGUCACGUTT -3′; antisense, 5′-ACGUGACACGUUCGGAGAATT-3′) from Suzhou GenePharma was used as the control.
Generation of TUFM-knockout (KO) A549 and HEK 293T cells
TUFM-KO A549 and HEK 293T cells were generated by using the CRISPR/Cas9 system, as described previously [Citation87,Citation88]. The specific oligonucleotides targeting the TUFM gene were designed using the Cas9 target design tools (https://crispr.cos.uni-heidelberg.de/index.html?tdsourcetag=s_pctim_aiomsg). In brief, the single guide RNA (sgRNA) sequence targeting the human TUFM gene (5′-TGTGTGGGCGTAGTGGCGGG-3′) was cloned into LentiGuide-puro (Addgene, Feng Zhang, 52,963) and lentiCRISPR v2 (Addgene, Feng Zhang, 52,961) vectors, respectively. For the generation of TUFM-KO A549 cells, the TUFM-specific lentiGuide-Puro plasmid, lentivirus packaging plasmid psPAX2 (Addgene, Didier Trono, 12,260), and envelope plasmid pMD2.G (Addgene, Didier Trono, 12,259) were co-transfected into HEK293T cells using Lipofectamine8000TM Transfection Reagent (Beyotime, C0533). The medium was replaced 12 h later. Viral particles were collected 60 h post-transfection and filtered with low-protein-binding filters (Millex-HV, 0.45-mm polyvinylidene difluoride; EMB Millipore). When the A549-Cas9-Blast cells seeded on six-well plates were ∼30% confluent, they were infected with the TUFM-specific lentiGuide-puro lentivirus-containing fresh medium with 8 μg/mL polybrene (Sigma, TR-1003-G) or the empty vector lentiGuide-puro lentivirus-containing fresh medium with 8 μg/mL polybrene. After 48 hpi, the lentivirus-infected cells were selected in 1.5 μg/mL puromycin (Sigma, P9620-10ML) for 3 d with the medium changed daily to select the positive clones. For the generation of TUFM-KO HEK 293T cells, HEK 293T cells seeded on six-well plates with ∼60% confluent was transfected with the TUFM-specific lentiCRISPR v2 or the empty vector lentiCRISPR v2 plasmid using Lipofectamine8000TM Transfection Reagent (Beyotime, C0533). After 24 h post-transfection, the cells were selected in 3 μg/mL puromycin for 3 d with the medium changed daily to select the positive clones. Finally, the monoclonal cells were acquired by using the limiting dilution method in 96-well plates. The TUFM-KO cell lines were obtained from these enlarged monoclonal cells and confirmed by immunoblotting. The A549-Cas9-Blast cells were kindly provided by Professor Gang Cao (Huazhong Agricultural University, Wuhan, China).
Immunofluorescence analysis
A549 cells plated on coverslips in 12-well plates were infected with A/PR8/H1N1 virus (or transfected with indicated plasmids) for 24 h, and washed with phosphate-buffered saline (PBS [PH 7.2]; HyClone, SH30256.01) and fixed with 4% paraformaldehyde (biosharp, BL539A) for 15 min. Cells were then washed with PBS and incubated with 0.1% Triton X-100 (Sigma-Aldrich, X100) for 10 min. Next, Cells were blocked with 2% BSA for 1 h at room temperature and then incubated with corresponding antibodies at 4°C overnight, followed by Alexa Fluor FITC- 405- or 594-conjugated goat anti-mouse or anti-rabbit as a secondary antibody for 2 h. All cells were washed with PBS 5 times after each step and were observed by confocal microscopy (Carl Zeiss LSM 880 Confocal Microscope and ZEN 2.3 LITE software).
Luciferase reporter assay
HEK 293T cells grown in 24-well plates were co-transfected with 0.25 μg/well of reporter plasmids IFNB-luc, 0.01 μg/well of plasmid pGL4.75 (hRluc/CMV), 0.25 μg/well MAVS, respectively, and 0.5 μg/well of HA-WT PB1-F2PR8, HA-PB1-F2PR8W61A, HA-PB1-F2PR8W61A+L64A, PB1-F2PR8∆LIR expression plasmid or an empty vector for 24 h. When necessary, MAVS transfection was replaced by infecting with SEV to stimulate cells at 24 h post-transfection. Cells were lysed at 12 hpi, and firefly luciferase and Renilla luciferase activities were determined with the Dual-Luciferase reporter assay system (Promega, E1910) according to the manufacturer’s protocol. Data were presented as relative firefly luciferase activities normalized to Renilla luciferase activities and were representative of 3 independent experiments.
Subcellular fractionation
Pure mitochondrial fractionation was isolated by utilizing the Mitochondria/Cytosol Fractionation Kit (Millipore, MT1000) according to the manufacturer’s instructions. Briefly, HEK 293T cells were plated onto a 15-cm dish and transfected with 30 μg of HA-WT PB1-F2PR8 for 24 h. Cells were washed with ice-cold PBS twice and then detached with a cell scraper. Subsequently, the cell pellets were collected after centrifugation and resuspended with 800 μl of Isotonic Mitochondrial Buffer (Millipore, CS204255) with protease inhibitor cocktail at 1:100 dilutions. Cells were homogenized with a 1 ml Dounce homogenizer via 40 strokes on ice and were centrifuged at 600 × g for 10 min. The supernatants were recentrifuged at 10,000 × g for 30 min. All centrifugation was performed at 4°C. The mitochondrial fraction in the pellets was resuspended with 40 μl of the Mitochondrial Lysis Buffer (Millipore, CS204257) containing protease inhibitors. The supernatants were considered as the cytosolic fraction. Equivalent amounts of proteins (30 μg) from each fraction were analyzed by western blot using the indicated antibodies.
GST affinity-isolation assays
HA-PB1-F2PR8 was expressed in HEK293T cells for 24 h, and pure mitochondrial fractionation was isolated by utilizing the Mitochondria/Cytosol Fractionation Kit (Millipore, MT1000) according to the manufacturer’s instructions. The mitochondrial fraction in the pellets was resuspended with cell lysis buffer for western blotting and IP (Beyotime, P0013) containing protease inhibitors, and supernatants were collected via centrifugation at 10,000 rpm for 10 min at 4°C. Furthermore, GST-tagged proteins (GST and GST-TUFM) were purified using glutathione sepharose 4B (GE Healthcare). To determine the direct interaction between PB1-F2 and TUFM, an equal amount of GST-tagged proteins (10 μg) were respectively bound to 50 μl glutathione sepharose 4B and were incubated for 1 h at 4°C with rotation. The glutathione sepharose 4B were washed alternately with PBS containing 147 nM NaCl and PBS containing 500 nM NaCl, equal amounts of cytoplasmic/mitochondrial HA-PB1-F2PR8 lysates were added into the glutathione sepharose 4B and incubated for another 1 h at 4°C with rotation. After washed alternately with PBS containing 147 nM NaCl and PBS containing 500 nM NaCl, the purified proteins were eluted with 10 mM reduced glutathione, and then subjected to western blot analysis using both anti-GST (HUABIO, EM80701) and anti-HA (MBL, M180-3) antibodies.
Statistical analyses
Data were expressed as means ± standard deviations (SD). Statistical analysis was performed by a paired two-tailed Student’s t test. P value equal or lower to 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001). P value > 0.05 was considered statistically non-significant.
Supplemental Material
Download Zip (53.3 MB)Acknowledgments
We thank Ms. Xiao Xiao for her critically proof reading the manuscript, professor Chan Ding from Shanghai Veterinary Research Institute providing the tandem fluorescent monomeric red fluorescent protein (mRFP)-GFP-LC3 (ptfLC3) construct, professor Gang Cao from Huazhong Agricultural University providing the A549-Cas9-Blast cells, and professor Zhengfan Jiang from Peking University providing the luciferase reporter constructs including IFNB promoter (IFNB-luc) as well as the Sendai virus.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253.
- Jin SM, Youle RJ. PINK1-and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799.
- Kim SJ, Khan M, Quan J, et al. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9:e1003722.
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946.
- Gregoire IP, Richetta C, Meyniel-Schicklin L, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011;7:e1002422.
- Geisler S, Kmskujat H. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131.
- Aoki Y, Kanki T, Hirota Y, et al. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell. 2011;22:3206–3217.
- Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8:e1000298.
- Ding B, Zhang L, Li Z, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47 e4.
- Sin J, McIntyre L, Stotland A, et al. Coxsackievirus B escapes the infected cell in ejected mitophagosomes. J Virol. 2017;91:e01347–17.
- Wang K, Ma H, Liu H, et al. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 2019;27:2075–91 e5.
- Khan M, Syed GH, Kim SJ, et al. Mitochondrial dynamics and viral infections: a close nexus. Biochim Biophys Acta. 2015;1853:2822–2833.
- Gou H, Zhao M, Xu H, et al. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget. 2017;8:39382–39400.
- Li S, Wang J, Zhou A, et al. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget. 2016;7:56002–56012.
- Kim SJ, Syed GH, Khan M, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA. 2014;111:6413–6418.
- Teodorof-Diedrich C, Spector SA. Human immunodeficiency virus type 1 gp120 and tat induce mitochondrial fragmentation and incomplete mitophagy in human neurons. J Virol. 2018;92:e00993-18.
- Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013;9:e1003285.
- Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy. 2018;14:15548627.2018.1466014.
- Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14:480-+.
- Nguyen-Van-Tam JS. Influenza and other respiratory viruses. From the editor’s desk. Influenza Other Respir Viruses. 2015;9:99–100.
- Molinari NA, Ortegasanchez IR, Messonnier ML, et al. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine. 2007;25:5086–5096.
- Zhao YY, Sun XF, Nie XL, et al. COX5B regulates MAVS-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLoS Pathog. 2012;8:e1003086.
- Hsu SF, Su WC, Jeng KS, et al. A host susceptibility gene, DR1, facilitates influenza A virus replication by suppressing host innate immunity and enhancing viral RNA replication. J Virol. 2015;89:3671–3682.
- Iwai A, Shiozaki T, Kawai T, et al. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J Biol Chem. 2010;285:32064–32074.
- Graef KM, Vreede FT, Lau YF, et al. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol. 2010;84:8433–8445.
- Liedmann S, Hrincius ER, Guy C, et al. Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun. 2014;5:5645.
- Fernandez-Sesma A, Marukian S, Ebersole BJ, et al. Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol. 2006;80:6295–6304.
- Varga ZT, Ramos I, Hai R, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067.
- Chen W, Calvo PA, Malide D, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312.
- Chakrabarti AK, Pasricha G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology. 2013;440:97–104.
- Yoshizumi T, Ichinohe T, Sasaki O, et al. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun. 2014;5:4713.
- McAuley JL, Hornung F, Boyd KL, et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249.
- Weeks-Gorospe JN, Hurtig HR, Iverson AR, et al. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with Gram-positive respiratory pathogens. J Virol. 2012;86:9035–9043.
- Alymova IV, Amali S, Peter V, et al. A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J Virol. 2014;88:503–515.
- Conenello GM, Tisoncik JR, Rosenzweig E, et al. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol. 2011;85:652–662.
- Alymova IV, Green AM, van de Velde N, et al. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J Virol. 2011;85:12324–12333.
- Conenello GM, Zamarin D, Perrone LA, et al. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–1421.
- Gibbs JS, Malide D, Hornung F, et al. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003;77:7214–7224.
- Yamada H, Chounan R, Higashi Y, et al. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336.
- Dmitriy Z, Adolfo GS, Xiaoyao X, et al. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4.
- Varga ZT, Grant A, Manicassamy B, et al. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. 2012;86:8359–8366.
- Mohsin K, Hussain SG, Seong-Jun K, et al. Hepatitis B virus-induced parkin-dependent recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 2016;12:e1005693.
- Xia M, Gonzalez P, Li C, et al. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol. 2014;88:5152–5164.
- He K, Guo X, Liu Y, et al. TUFM downregulation induces epithelial–mesenchymal transition and invasion in lung cancer cells via a mechanism involving AMPK-GSK3β signaling. Cell Mol Life Sci. 2016;73:2105–2121.
- Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta. 2012;1819:1035–1054.
- Samec N, Jovcevska I, Stojan J, et al. Glioblastoma-specific anti-TUFM nanobody for in-vitro immunoimaging and cancer stem cell targeting. Oncotarget. 2018;9:17282–17299.
- Bellance N, Lestienne P, Rossignol R. Mitochondria: from bioenergetics to the metabolic regulation of carcinogenesis. Front Biosci (Landmark Ed). 2009;14:4015–4034.
- Dakubo GD. Mitochondrial genetics and cancer. Springer Verlag NY; 2010;59:10–16.
- He J, Cooper HM, Reyes A, et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012;40:6109–6121.
- Valente L, Tiranti V, Marsano RM, et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80:44–58.
- Smeitink JA, Elpeleg O, Antonicka H, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–877.
- Bestwick ML, Shadel GS. Accessorizing the human mitochondrial transcription machinery. Trends Biochem Sci. 2013;38:283–291.
- Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825.
- Parmeggiani A, Nissen P. Elongation factor Tu-targeted antibiotics: four different structures, two mechanisms of action. FEBS Lett. 2006;580:4576–4581.
- Antonicka H, Sasarman F, Kennaway NG, et al. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15:1835–1846.
- Bhargava K, Templeton P, Spremulli LL. Expression and characterization of isoform 1 of human mitochondrial elongation factor G. Protein Expr Purif. 2004;37:368–376.
- Suzuki H, Ueda T, Taguchi H, et al. Chaperone properties of mammalian mitochondrial translation elongation factor Tu. J Biol Chem. 2007;282:4076–4084.
- Wells J, Henkler F, Leversha M, et al. A mitochondrial elongation factor-like protein is over-expressed in tumors and differentially expressed in normal-tissues. FEBS Lett. 1995;358:119–125.
- Schmeing TM, Voorhees RM, Kelley AC, et al. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694.
- Spremulli LL, Coursey A, Navratil T, et al. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog Nucleic Acid Res Mol Biol. 2004;77:211–261.
- Lei Y, Wen H, Yu Y, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type 1 interferon and autophagy. Immunity. 2012;36:933.
- Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy. 2013;9:432–433.
- Lei Y, Kansy BA, Li J, et al. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene. 2016;35:4698–4707.
- Kuo SM, Chen CJ, Chang SC, et al. Inhibition of avian influenza A virus replication in human cells by host restriction factor TUFM is correlated with autophagy. MBio. 2017;8:00481-17.
- Gannage M, Dormann D, Albrecht R, et al. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380.
- Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13.
- Wang R, Zhu Y, Lin X, et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy. 2019;15:1163–1181.
- Sun YJ, Yu SQ, Ding N, et al. Autophagy benefits the replication of Newcastle disease virus in chicken cells and tissues. J Virol. 2014;88:525–537.
- Bjrky G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309-+.
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501.
- Mai S, Muster B, Bereiter-Hahn J, et al. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence life span. Autophagy. 2012;8:47–62.
- Tsai WT, Lo YC, Wu MS, et al. Mycotoxin patulin suppresses innate immune responses by mitochondrial dysfunction and p62/sequestosome-1-dependent mitophagy. J Biol Chem. 2016;291:19299.
- Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145.
- Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010;584:1379–1385.
- Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185.
- Kanki T, Wang K, Cao Y, et al. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109.
- Ren Y, Li C, Feng L, et al. Proton channel activity of influenza A virus matrix protein 2 contributes to autophagy arrest. J Virol. 2016;90:591–598.
- Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–4044.
- Vigie P, Camougrand N. [Role of mitophagy in the mitochondrial quality control]. Med Sci (Paris). 2017;33:231–237.
- Le Goffic R, Bouguyon E, Chevalier C, et al. Influenza A virus protein PB1-F2 exacerbates IFN-beta expression of human respiratory epithelial cells. J Iimmunol. 2010;185:4812–4823.
- Zou W, Yu ZJ, Zhou HB, et al. Genetic characterization of an H5N1 avian influenza virus with neurovirulence in ducks. Virus Genes. 2009;38:263–268.
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–497.
- Wang R, Zhu Y, Zhao J, et al. Autophagy promotes replication of influenza A virus in vitro. J Virol. 2019;93:e01984-18.
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784.
- Xia Z, Xu G, Yang X, et al. Inducible TAP1 negatively regulates the antiviral innate immune response by targeting the TAK1 complex. J Iimmunol. 2017;198:3690–3704.