
ABSTRACT
Viral infections are often accompanied by the induction of autophagy as an intrinsic cellular defense mechanism. Herpesviruses have developed strategies to evade autophagic degradation and to manipulate autophagy of the host cells to their benefit. Here we addressed the role of macroautophagy/autophagy in human cytomegalovirus replication and for particle morphogenesis. We found that proteins of the autophagy machinery localize to cytoplasmic viral assembly compartments and enveloped virions in the cytoplasm. Surprisingly, the autophagy receptor SQSTM1/p62 was also found to colocalize with HCMV capsids in the nucleus of infected cells. This finding indicates that the autophagy machinery interacts with HCMV already at the early nuclear stages of particle morphogenesis. The membrane-bound form of LC3 and several autophagy receptors were packaged into extracellular HCMV virions. This suggested that autophagic membranes were included during secondary envelopment of HCMV virions. To further address the importance of autophagy in HCMV infection, we generated an HCMV mutant that expressed a dominant-negative version of the protease ATG4B (BAD-ATG4BC74A). The proteolytic activity of ATG4B is required for LC3 cleavage, priming it for membrane conjugation. Surprisingly, both genome replication and virus release were enhanced in cells infected with BAD-ATG4BC74A, compared to control strains. These results show that autophagy operates as an antiviral process during HCMV infection but is dispensable for secondary HCMV particle envelopment.
Abbreviations: ATG: autophagy-related; BAC: bacterial artificial chromosome; BECN1: beclin 1; CPE: cytopathic effect; cVACs: cytoplasmic viral assembly compartments; d.p.i.: days post-infection; DB: dense body; EBV: Epstein-Barr virus; galK: galactokinase; HCMV: human cytomegalovirus; HFF: human foreskin fibroblasts; IE: immediate-early; IRS: internal repeat short; LC3: MAP1LC3A/B; m.o.i.; multiplicity of infection; MCP: major capsid protein; Pp: phosphoprotein; sCP/UL48a: smallest capsid protein; TRS: terminal repeat short; UL: unique long; US: unique short
Introduction
Autophagy is a physiological process that operates on different levels to maintain cell homeostasis and to foster immune defense [Citation1–3]. One specialized form of autophagy, termed xenophagy, serves to detect invading pathogens and to direct them to degradation [Citation4,Citation5]. However, many viruses have developed mechanisms during evolution to subvert autophagy to their benefit [Citation6]. Herpesviruses have a long history of co-evolution with their hosts and thus had ample time to develop a remarkable level of adaptation to their host’s defense mechanisms. Several human herpesviruses manipulate autophagy in host cells, by inhibiting either autophagy initiation or autophagic flux, to prevent viral degradation, or by utilizing elements from the autophagic machinery for virion assembly or transport [Citation6–11]. For example, the α-herpesvirus herpes simplex virus type 1 (HSV-1) protein ICP34.5 (infected cell protein 34.5) directly targets the cellular BECN1 (beclin 1), thereby disrupting the induction of autophagy upon viral entry [Citation12,Citation13]. Gamma-herpesviruses express BCL2 (B-cell lymphoma 2) homologs (vBCL-2), which also target BECN1, often more potently than their cellular counterpart [Citation4,Citation5,Citation14]. In addition, some gamma-herpesviruses inhibit the conjugation of LC3 to membrane lipids, thereby disrupting the elongation of the autophagic membrane [Citation15]. Other herpesviruses, like herpes simplex virus-2 (HSV-2) and varicella-zoster virus (VZV), appear to profit from a basal level of autophagy [Citation16,Citation17]. Furthermore, VZV and Epstein Barr virus (EBV) utilize autophagic membranes for their secondary envelopment and viral egress [Citation16,Citation18,Citation19].
Human cytomegalovirus (HCMV), a member of the betaherpesvirinae, infects up to 100% of the human population. Although both HCMV primary infection and reactivation are usually asymptomatic in immunocompetent individuals, the virus is frequently associated with morbidity and mortality in immunocompromised individuals, such as transplant recipients or acquired immune deficiency syndrome (AIDS) patients [Citation20,Citation21]. Furthermore, pre- and perinatal HCMV infections are a major cause of childhood disabilities and neuronal defects [Citation22,Citation23]. Like all herpesviruses, HCMV contains a large double-stranded DNA genome, located inside of an icosahedral capsid, which is stabilized by a layer of proteins forming the inner tegument [Citation24,Citation25]. Attached to the inner tegument of HCMV lies a layer of proteins forming the outer tegument, serving both structural as well as functional roles [Citation26–30]. The viral particle is covered by a double-layer envelope, containing protein complexes crucial for adsorption and penetration of host cells [Citation31].
Little is known about the interplay between HCMV and autophagy. HCMV induces autophagy shortly after infection but has developed strategies to prevent autophagic degradation. One study reported a complete inhibition of autophagy 24 h post-infection (h.p.i.) of human primary fibroblasts with HCMV [Citation32], identifying the protein TRS1 as a viral inhibitor of BECN1. However, others did not observe an autophagy inhibition upon infection with HCMV, but rather report a stable increase of autophagy upon infection in various cell types [Citation33]. In general, the induction of autophagy was shown to impair viral replication, demonstrating the antiviral effect of autophagy and supporting the assumption that autophagy serves as a cellular defense against HCMV [Citation33,Citation34]. There is evidence that the exogenous induction of autophagy has an antiviral effect on HCMV replication [Citation35]. On the other hand, some data have been published, telling that the virus has developed mechanisms to limit autophagy [Citation32]. An intricate balance appears to exist between HCMV and its host in the regulation of autophagy in infected cells. This balance is reminiscent of the converse mechanisms HCMV and its host have developed to control immune recognition or host cell death [Citation36–39]. In contrast to the wealth of knowledge that has been gathered about the molecular mechanisms governing these processes, the interactions between HCMV and its host cell concerning autophagy are poorly understood. This work aimed to increase our understanding of how autophagy influences HCMV replication and morphogenesis. We provided data indicating that autophagy is another level of competition between HCMV and its host to support host cell survival on the one hand and to guarantee successful viral replication on the other.
Results
Components of the autophagy machinery localize to HCMV virion assembly sites and virus particles
The interplay between HCMV and autophagy is poorly understood. Conflicting results have been published concerning the impact of inhibition of autophagy on HCMV replication [Citation33,Citation40]. As there is evidence for a role of autophagy in the particle morphogenesis of HCMV, comparable to other herpesviruses [Citation20,Citation41,Citation42], we followed that lead and looked for the specific accumulation of proteins of the autophagy machinery at sites of HCMV particle morphogenesis, termed cytoplasmic viral assembly compartments (cVACs) [Citation43].
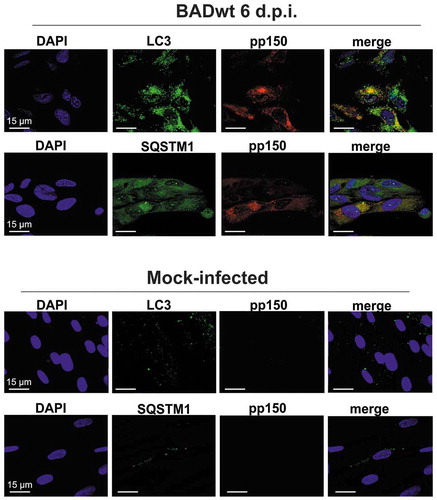
We infected human foreskin fibroblasts (HFF) with HCMV laboratory strain BADwt (AD169) at an m.o.i. of 0.2. Cells were analyzed by laser scanning microscopy at 6 d.p.i. Staining was done with antibodies against LC3 and SQSTM1. LC3, in its lipidated form, is covalently linked to membranes and mediates autophagic membrane formation. In addition, it is involved in the recruitment of cargo by interacting with receptor molecules such as SQSTM1. Both LC3 and SQSTM1 accumulated in juxtanuclear regions of HCMV–infected cells, identical in their shape with cVACs (). We performed co-staining using an antibody directed against the inner tegument protein pp150 (pUL32) of HCMV to confirm the recruitment of LC3 and SQSTM1 to cVACs. This protein is an established marker for cVACs of HCMV [Citation43]. The results showed that both LC3 and SQSTM1 localized to cVACs. In some instances, we found direct colocalization of LC3 or SQSTM1 with pp150.
Figure 1. LC3 and SQSTM1 localized to viral assembly compartments. Confocal immunofluorescence analysis of LC3 and SQSTM1, respectively (Alexa Fluor 488, green), and of the viral protein pp150 (Alexa Fluor 546, red). Cells were infected for 6 d with the laboratory strain BADwt at an m.o.i. = 0.2. Uninfected cells (mock) were carried along for control. Cells were then pre-incubated with goat antiserum to block unspecific binding of the antibodies. Rabbit monoclonal anti-LC3 or anti-SQSTM1 antibodies were used for staining. The mouse monoclonal pp150 antibody was used for staining of the cVACs. Both LC3 and SQSTM1 localize to cytoplasmic virus assembly compartments and colocalize with pp150
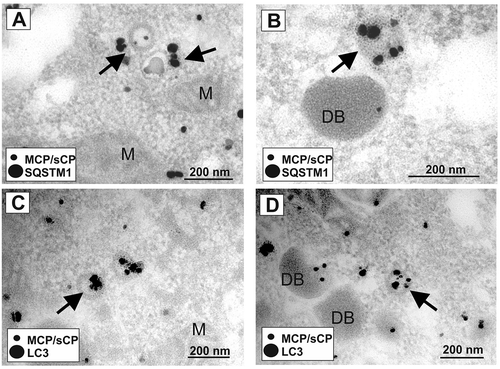
The recruitment of both LC3 and SQSTM1 to cVACs suggested that autophagy indeed played a role in HCMV morphogenesis. We thus tested if there was a direct juxtaposition of LC3 and SQSTM1 with viral particles, using immunoelectron microscopy. We again infected HFF with HCMV at an m.o.i. of 1 for 6 d. Samples were then processed for immunoelectron microscopy at 6 d of infection, using silver-enhanced immunogold labeling of the viral capsid (major capsid protein, MCP, and smallest capsid protein sCP/UL48a; MCP/sCP) of HCMV, and SQSTM1, respectively. As expected, MCP/sCP localized primarily to the nucleus of infected HFFs, particularly accumulating in the periphery of the viral replication compartments and the newly synthesized viral capsids in the nucleus (,). Surprisingly, we also found SQSTM1 at intranuclear viral capsids and close to the viral replication compartments (-). The data show that components of the autophagy machinery interact with HCMV particles already in the nucleus before viral envelopment.
Figure 2. SQSTM1 localized to intranuclear viral capsids. Immunoelectron micrographs of late-stage-infected fibroblasts. Fixed samples of HFF cells, infected with BADwt at an m.o.i. = 0.8 for 6 d were incubated with a 1:1 mix of two mouse monoclonal antibodies directed against MCP and sCP (A, B), respectively, or with a rabbit monoclonal antibody directed against anti-SQSTM1 (C-F), respectively. A secondary antibody, conjugated with gold particles and silver-enhanced, was used. Nucleus (N) and cytoplasm (C) are indicated. (A) Gold particles accumulate in the nucleus, labeling the viral capsids present in the nucleus of an HCMV–infected fibroblast (indicated by arrows). (B) is an enlarged version of the indicated field from (A). (C-F) SQSTM1 localizes to intranuclear viral capsids (indicated by arrows). (D, E, and F) display enlarged sections of (C)

We performed immunoelectron microscopy using a co-staining approach to address the interaction of SQSTM1 and LC3 with viral particles in the cytoplasm. We performed double staining with antibodies against MCP/sCP (smaller grains) and antibodies against SQSTM1 (larger grains) and revealed that SQSTM1 also localized to viral particles in the cytoplasm of infected cells (,). Similar to SQSTM1, LC3 also localized to viral particles in the cytoplasm (,). The LC3-specific grains were mainly found at or close to the viral envelope, suggesting that LC3 incorporated into the membranes of these particles. Together, these results showed that SQSTM1 localized to nuclear capsids and that both LC3 and SQSTM1 localized to cytoplasmic virions in HCMV–infected cells.
Figure 3. LC3 and SQSTM1 localized to cytoplasmic viral particles. Immunoelectron micrographs of late-stage-infected fibroblasts. HFF cells, infected with BADwt at an m.o.i. = 0.8 for 6 d were co-stained with a 1:1 mix of anti-MCP and anti-sCP mouse monoclonal antibodies and either an anti-SQSTM1 antibody (A, B) or an anti-LC3 monoclonal antibody (C, D), both derived from rabbit, respectively. A secondary antibody, conjugated with gold particles and silver-enhanced, was used. Secondary antibodies against mouse immunoglobulin (MCP/sCP) were conjugated to smaller gold particles, antibodies against rabbit immunoglobulin (LC3, SQSTM1) to larger gold particles. Arrows indicate HCMV virions in the cytoplasm of an infected fibroblast. Both SQSTM1 (A, B) and LC3 (C, D) colocalize with MCP/sCP at viral particles in the cytoplasm. Mitochondria (M) and subviral Dense Bodies (DB) are indicated
Components of the autophagy machinery are packaged into HCMV virions
To further test if both LC3 and SQSTM1 were incorporated into mature viral particles, extracellular virions were gradient purified from the cell culture supernatants of HFF, infected with different strains of HCMV. We detected SQSTM1 in purified virions from laboratory strains BADwt (AD169) and Towne-UL130-rep (Towne), as well as from the TB40 strain by mass spectrometry (not shown). We performed western blot analyses to corroborate these findings, confirming the presence of SQSTM1 within purified virions (). LC3 was not detectable in virions on repeated occasions, using mass spectrometry (not shown). We also performed immunoblot analyses on viral particles, using an LC3-specific antibody, to resolve this discrepancy to the EM-data. Using this assay format, LC3 was reproducibly detectable in considerable amounts in purified virions of HCMV strain BADwt (). Interestingly, only the lipidated form of LC3, LC3-II, was detected. Even when running 2–3 times the amount of purified particles out on an SDS-PAGE, it was the only LC3-II that was detectable, arguing against the possibility of cellular contamination during the purification process (not shown).
Figure 4. Lipidated LC3 is present in purified viral particles. Western blot analysis of whole-cell lysates of uninfected HFF and 30 µg of purified virions, respectively, and probed with antibodies against MCP, SQSTM1, and LC3B. Three different laboratory strains were tested. Only the lipidated form of LC3, LC3-II, was found in HCMV virions
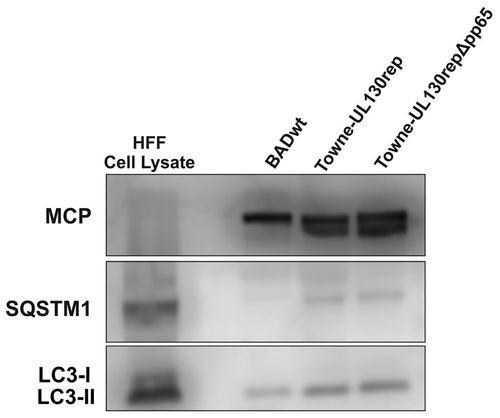
To ensure that this finding was not a strain-specific artifact, purified virions of another strain, we analyzed Towne-UL130rep, confirming the results with BADwt (). To finally show that the detection of SQSTM1 and LC3 was not related to possible contamination of the virion fraction by subviral dense bodies (DB), we analyzed purified virions of Towne-UL130rep∆pp65. This strain lacks the expression of subviral DBs because of the absence of pp65, which is the major component of the viral tegument [Citation44,Citation45]. Again, both SQSTM1 and LC3 were detectable (). The results confirmed that LC3-II, but not LC3-I, was present in purified virions of various strains of HCMV. These findings indicated that autophagosomal membranes contribute to the formation of the HCMV particle envelope and, in consequence, this suggested that autophagy was involved in the morphogenesis of HCMV viral particles.
SQSTM1 primarily associates with components of the HCMV capsid
Because the autophagy receptor SQSTM1 was localized to intranuclear viral capsids and sites of viral assembly in the cytoplasm, we investigated whether SQSTM1 recognizes viral proteins and whether specific components of the virion were preferred targets of SQSTM1. To address this, we performed immunoprecipitation with lysates from late-stage-infected HFF, using an antibody directed against SQSTM1. We subsequently subjected the samples to quantitative mass spectrometry. In total, we identified 43 virus protein groups (Table S1), 28 of which could be quantified in two independent, biological replicates (,). A substantial number of viral proteins co-precipitated with SQSTM1.
Figure 5. SQSTM1 co-purified with the viral capsid and components of the viral tegument. Immunoprecipitation of BADwt-infected HFF with mouse anti-SQSTM1 antibody and empty magnetic beads as control, and protein quantification by mass spectrometry. We identified 41 HCMV protein groups, 28 of which could be quantified in both IPs and technical replicates. (A) Log2 transferred average fold-change of SQSTM1, and viral component proteins are plotted with the standard error of the mean (SE), indicated as error bars. Values are calculated for two independent IPs (biological replicates) with two technical replicates in each case. (B) A Welch’s t-test was performed between the mean of all viral proteins and all human background proteins, with a fold-change between 0.5 and 2. The bar chart represents the average of all viral proteins and human background proteins. Error bars indicating the standard deviation (SD). (C) Immunoprecipitation of BADwt-infected HFF with mouse anti-SQSTM1 antibody and mouse IgG as control. Eluates and supernatants were subjected to SDS protein-gel separation, and western blot probed with antibodies against SQSTM1, pp28, and sCP/pUL48a. sCP/pUL48a specifically precipitates with SQSTM1, while pp28 does not. Unbound sCP/pUL48a and pp28 were still present in the cell lysates after immunoprecipitation. SQSTM1 was no longer detectable by western blot in the supernatant of the specific SQSTM1 precipitate. In the control supernatant, the amounts of SQSTM1 were comparable to input levels
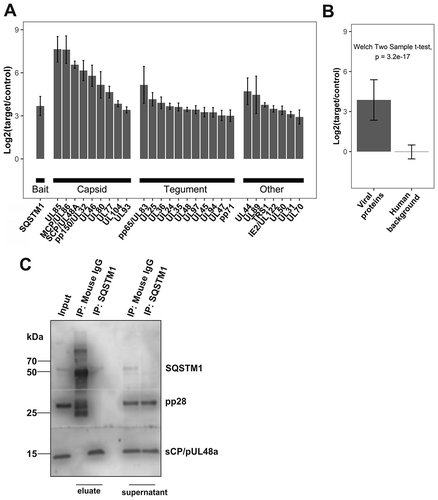
Interestingly, we reproducibly found all components of the viral capsid in the co-immunoprecipitates with SQSTM1. Besides abundant capsid components, such as the major capsid protein MCP (UL86) or the capsid-associated protein pp150, relatively large amounts of minor capsid constituents, like the portal protein UL104, co-immunoprecipitated with SQSTM1. In contrast, while abundant tegument proteins, such as pp65 or pUL25, were co-precipitated with an SQSTM1 antibody, other components of the viral tegument were not. Two examples of this are the tegument proteins pp28 (pUL99) and pUL26, which are also present in large amounts in infected cells and mature virions [Citation46]. This example, and the fact that only little variation was seen between biological replicates, argues in favor of a specific interaction of SQSTM1 with either single viral proteins or, more likely, viral protein complexes within late-infected cells. We confirmed the co-precipitation of viral capsids with SQSTM1 by western blot probed against the smallest capsid protein (sCP/UL48a). SCP was present in the precipitate of anti-SQSTM1 antibody-coated magnetic beads, but not with beads coated with a mouse IgG control antibody (). Furthermore, pp28 was present only in the whole cell lysate and the supernatant of the co-IP samples, but not in the precipitate of anti-SQSTM1, confirming that pp28 does not interact with SQSTM1.
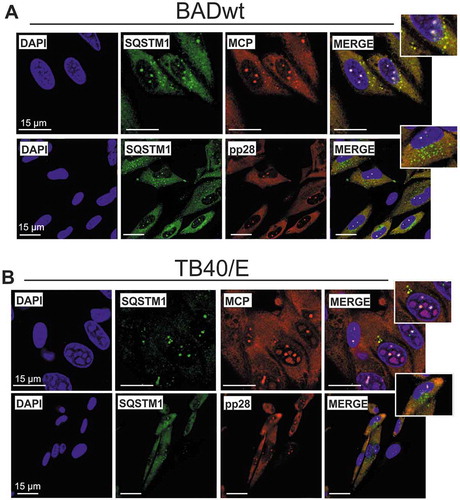
To confirm these results, We performed co-immunostaining of SQSTM1 with either the major capsid protein MCP or the tegument protein pp28 in HFF, infected at late stages (6 d.p.i.) with either BADwt or a clinical strain TB40/E, respectively, using confocal laser scanning microscopy. We found a clear colocalization of MCP with SQSTM1 in defined punctate structures at cVACs (,, upper panel). These punctate structures were not stained with a pp28-specific antibody (,, lower panel), indicating that there was no colocalization of SQSTM1 with pp28. Both the SQSTM1 antibody and the pp28 antibody showed diffuse cytoplasmic background staining. Together, these data showed that SQSTM1 co-precipitates with components of the viral capsid and colocalizes with the major capsid component MCP in cVACs. In addition, an SQSTM1 antibody appears to precipitate tegument proteins or tegument protein complexes. SQSTM1 appears to target these viral components before completed virion maturation.
Figure 6. SQSTM1 colocalized with the major capsid protein. Confocal immunofluorescence analysis of late-stage-infected HFF. Cells were infected with laboratory strain BADwt (A) or with strain TB40/E (B) at an m.o.i. of 0.2. After 6 d, cells were then pre-incubated with goat antiserum to block unspecific binding of the antibodies. Staining was performed with antibodies against SQSTM1 (Alexa Fluor 488, green) and antibodies against the viral proteins MCP or pp28, respectively (Alexa Fluor 546, red). SQSTM1 colocalizes with viral MCP (yellow), but not with the tegument protein pp28 (control)
Inhibition of autophagy enhances HCMV replication
At this point, we were left with controversial findings. On the one hand, the biochemical and microscopic analyses suggested a contribution of autophagy to viral particle morphogenesis and envelopment. On the other hand, precipitation of capsid and tegument components with an SQSTM1 antibody indicated that capsids and/or complexes of viral structural proteins were tagged by SQSTM1 and may thereby be fed into autophagic degradation. This notion left the question unanswered whether autophagy was either pro- or antiviral. Previous experiments using chemicals to investigate the impact of stimulation or inhibition of autophagy on HCMV replication have provided controversial results [Citation33,Citation47]. Such analyses, however, cannot easily control for off-target effects of the chemical substances used. The low transfection efficiency of HFF complicates the transfection of siRNAs to knockdown essential autophagy components. We thus decided to generate a mutant of HCMV expressing a dominant-negative version of the protease ATG4B (BAD-ATG4BC74A). ATG4B cleaves pro-LC3 at a C-terminal glycine to prime it for conjugation to membrane lipids [Citation48] (). The dominant-negative version ATG4BC74A contains a cysteine-to-alanine substitution at position 74 and prevents lipidation of LC3, reportedly by blocking its enzymatic activity and, also, inhibiting the interaction between LC3-I with ATG7 [Citation49,Citation50].
Figure 7. Infection with HCMV strain BAD-ATG4BC74A eliminated the lipidation of LC3. (A) Prior to lipidation, a precursor form of LC3, pro-LC3, is cleaved by the protease ATG4B. This step is essential for the subsequent membrane conjugation event of LC3. A dominant-negative version of ATG4B does not cleave pro-LC3, and also inhibits the interaction of LC3-I with ATG7, thereby preventing the lipidation of LC3-I. (B) Schematic presentation of an HCMV strain, expressing a dominant-negative version of ATG4B, termed BAD-ATG4BC74A. The generation of this virus was performed using the galK recombination method, producing a 2 USD-11-deleted intermediate BACmid. In this BACmid, the gene region 2 USD-11 of the parental strain BADwt was replaced by a DNA fragment, encoding the bacterial galK gene, required for positive selection in bacteria. In a second step, a DNA fragment encoding a hemagglutinin antibody epitope (HA)-tagged version of ATG4BC74A (HA-ATG4BC74A) was used to replace the galK gene. Both this BAC and the intermediate BAC-∆2 USD-11 were reconstituted, resulting in the viruses BAD-∆2 USD-11 (control) and BAD-ATG4BC74A. In the latter virus, the ATG4BC74A construct is driven by a modified version of the major immediate-early promoter of HCMV, containing a nonfunctional cis repressive sequence (crs). (C) Western blot analysis of HFF infected with BAD-ATG4BC74A for different times. The expression of ATG4BC74A starts at day 1 p.i. and also continues at later stages of infection. (D) Western blot analysis of LC3 expression in HFF infected with BADwt, BAD-∆2 USD-11, or BAD-ATG4BC74A, respectively, for different times. Mock-infected cells were taken along for reference. While BADwt and BAD-∆2 USD-11 induce the lipidation of LC3 (LC3-II) to a similar extent at 24 h.p.i., LC3-II is reduced and the amounts of non-lipidated LC3-I increase on day 2 post-infection onwards with BAD-ATG4BC74A. (E) Western blot analysis of purified viral particles of BADwt and BAD-ATG4BC74A. Virions of BAD-ATG4BC74A contain neither form of LC3, as opposed to purified virions of the parental strain BADwt, which display the membrane-bound form LC3-II. Both viruses contain similar amounts of SQSTM1
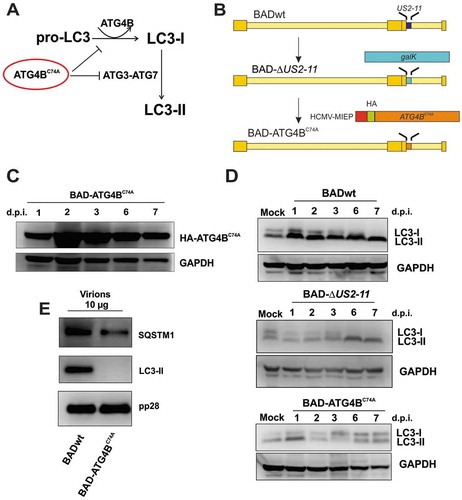
We inserted the coding sequence for an N-terminally HA-tagged version of ATG4BC74A into the genome of strain BADwt, using the galK BAC-recombination technology [Citation51] (). The construct was driven by the HCMV major immediate early promoter (MIEP). While the MIEP is a widely used and very potent promoter in various expression systems, it is usually silenced at the early stages of HCMV infection by the binding of the immediate-early protein 2 (IE2) to a cis-repressing sequence (crs) close to the transcription initiation site [Citation52–55]. The crs was mutated to prevent IE2-binding to ensure a continuous expression of HA-ATG4BC74A (). The ATG4BC74A expression cassette replaced the HCMV 2 USD-11 region of the genome. We chose this site as it is well established that HCMV replication in the HFF cell culture is not affected by 2 USD-11 deletion [Citation56–60]. The strain BAD-ATG4BC74A successfully reconstituted on HFF, following the transfection of the recombinant BAC-construct. We also reconstituted the precursor BAC containing the galK cassette (BAD-∆2 USD-11) as control. Genomic DNA from purified virions of BAD-ATG4BC74A, BAD-∆2 USD-11, and BADwt was subsequently isolated and analyzed by restriction endonuclease digestion and agarose gel electrophoresis. The banding patterns of the genomes were identical, excluding that larger deletions occurred in the course of BAC reconstitution (data not shown). The fact that BAD-ATG4BC74A reconstituted already provided a hint that suppression of autophagy did not inhibit viral replication of the strain used in this study and that autophagosomal membranes were likely not essentially required for viral morphogenesis and progeny production.
To verify the expression of HA-ATG4BC74A, we infected HFF with BAD-ATG4BC74A. We collected cells at different times after infection. We found HA-ATG4BC74A to be expressed from as early as 1 d.p.i. up to 7 d.p.i. (). We also found a substantial reduction in the levels of lipidated LC3 from day 2 p.i. onwards in BAD-ATG4BC74A-infected cells. The parental strain BADwt, as well as BAD-∆2 USD-11, analyzed for control, clearly induced autophagy from 1 d.p.i. onwards, as evidenced by the induction of LC3-II ().
To verify that the downregulation of ATG4B resulted in the loss of LC3-II packaging into extracellular HCMV virions, we infected cells with the mutant virus. Virions were purified from cell culture supernatants and subjected to immunoblot analysis. As expected, purified virions of BAD-ATG4BC74A lacked LC3. The levels of SQSTM1 in those particles remained comparable to particles of the parental strain. The levels of pp28 (control) did not differ in BAD-ATG4BC74A virions compared to particles from BADwt ().
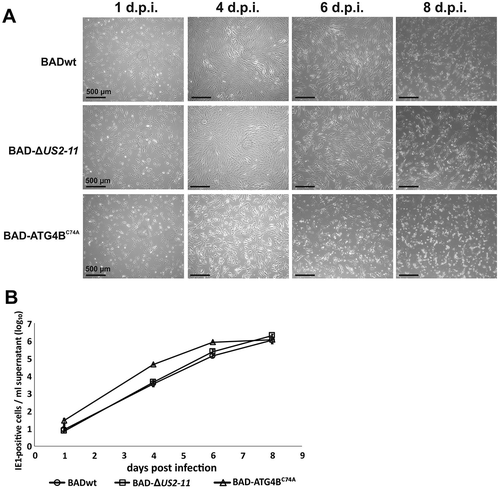
Having shown that infection of HFF with BAD-ATG4BC74A led to a distinct reduction of LC3-II lipidation, and thus inhibition of phagophore formation, we addressed the impact on HCMV replication. Regular inspection of infected cell cultures already indicated that, despite the reduced lipidation of LC3, BAD-ATG4BC74A replication did not seem to be impaired, since a marked cytopathic effect (CPE) was induced. In contrast, on day 4 and onwards, the CPE of BAD-ATG4BC74A-infected cells was substantially stronger than the CPE of control viruses BADwt and BAD-∆2 USD-11, suggesting that BAD-ATG4BC74A spread more easily in the culture. (). This effect remained until 6 d.p.i. At 8 d.p.i., the cell culture infected with BAD-ATG4BC74A displayed mostly dead cells, in contrast to the control cultures, which still contained a substantial number of adherent cells.
Figure 8. Cytopathic effect and progeny release are enhanced in BAD-ATG4BC74A-infected HFF. (A) Transmission microscopy of HFF infected with 0.25 genomes/cell of either BADwt, BAD-∆2 USD-11, or BAD-ATG4BC74A, respectively. The CPE of infected cells was documented at various time points. (B) Kinetics of virus release from HFF used in (A), infected with 0.25 genomes/cell of either BADwt, BAD-∆2 USD-11, or BAD-ATG4BC74A. Culture supernatants were collected at the indicated time points and analyzed by 8 technical replicates, using serial dilution and counting numbers of indicator cells, stained for the immediate-early 1 protein (IE1) of HCMV
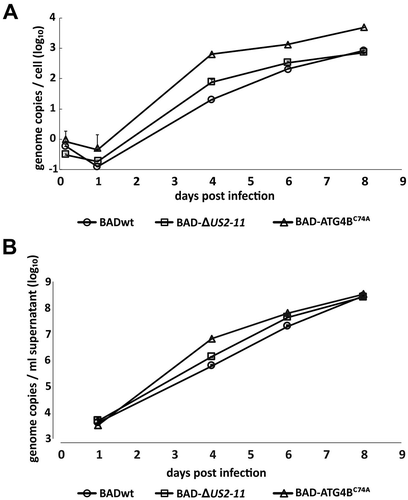
To approach this closer, supernatants from infected cells were collected and subjected to titration for infectivity on HFF, using IE1-staining as a read-out. These analyses confirmed the surprising result that cells infected with BAD-ATG4BC74A released more infectious virus than the parental strains BADwt and BAD-∆2 USD-11 until 6 d.p.i. (). At 8 d.p.i. the amount of infectious virus in the culture supernatant of BAD-ATG4BC74A-infected cells became comparable to that of the control strains, likely due to the reduction of viable cells capable of releasing viral progeny. To analyze the production of intracellular viral genomic DNA as well as its release into the cell culture supernatant, we performed quantitative PCR analyses with samples collected at different times after infection (). We detected considerably higher levels of HCMV genomes within cells expressing ATG4BC74A from 1 d.p.i. onwards (), indicating that viral DNA replication became enhanced in BAD-ATG4BC74A-infected cells compared to controls. Consistent with the results using IE1 titration, the supernatants from BAD-ATG4BC74A-infected cells contained higher levels of viral DNA at 4 d.p.i., but these levels converged with those of the control cultures at later times of infection (). We confirmed these experiments in an independent experiment. Together, these results showed that viral genome replication enhanced in cells with impaired autophagy. Virus release was initially also increased, but both the amount of infectious virus and viral DNA in the culture supernatant converged with the respective levels of the control cultures at later times of infection.
Figure 9. Viral DNA replication and release of progeny genomes are enhanced in BAD-ATG4BC74A-infected HFF. (A) Quantitative PCR analysis of the levels of viral genomes present in infected cells at different times after infection. HFF cells were infected with BADwt, BAD-∆2 USD-11, or BAD-ATG4BC74A with 0.25 genome copies/cell, respectively. Infection conditions were chosen in a way to ensure that equal genome copy numbers were initially present in the cells (at 4 h.p.i.). The values of the three technical replicates are shown. Cells were collected at the indicated time points, lysed, and subjected to PCR analysis. Copy numbers are indicated at selected time points. (B) Quantitative PCR analysis of viral genomes present in the cell media of the cultures used in (A). Samples were taken at the indicated time points and subjected to PCR analysis. Copy numbers are indicated at selected time points
Discussion
Autophagy is an important strategy for the cell to eliminate invading pathogens. Consequently, many pathogens have developed strategies to avoid autophagosomal clearance [Citation4,Citation5]. While some viruses and bacteria inhibit autophagy at various stages, others make use of components of the autophagy machinery, thereby converting the process and using it for their benefit [Citation6,Citation61]. Previous data provided conflicting results about the impact of HCMV on autophagy progression. We found induction of autophagy, suggesting that HCMV is initially targeted by autophagy upon infection. In our hands, autophagy levels remained elevated at later stages in HCMV–infected HFF. At this time, autophagy proteins SQSTM1 and LC3 localized to cytoplasmic viral assembly compartments and colocalized with pp150, a stabilizer of the viral capsid and component of the inner tegument of HCMV. Both SQSTM1 and LC3 were present at intracellular as well as in purified viral particles, suggesting that they were packaged in the process of HCMV morphogenesis and envelopment. The packaging of autophagy components into HCMV particles has recently also been shown by others [Citation42].
HCMV is covered by a phospholipid bilayer envelope, the origin of which has been the subject of discussion. During viral assembly, a broad rearrangement of membrane structures derived from different sources occurs at cVACs [Citation43,Citation62–67]. The presence of lipidated LC3-II and the simultaneous absence of non-phosphatidylethanolamine (PE)-conjugated LC3-I in purified viral particles showed that the viral envelope, at least in part, derives from autophagosomal membranes. This notion implies that the autophagy machinery recognizes one or several HCMV proteins in the course of secondary envelopment. It remains unclear at this point whether autophagic membranes contribute to viral envelopment or captured in the process of an evasion process that HCMV may initiate to subvert autophagy. However, our results clearly show that autophagic membranes are not essential components of the HCMV envelope and that membrane sources other than autophagosomal membranes, like those from the endosomal and ER-Golgi compartments, are sufficient to mediate HCMV secondary envelopment [Citation63].
There are several reports of the pro-viral effects of autophagy on viral morphogenesis and envelopment [Citation17,Citation19]. A recent study, for example, reported a pro-viral effect of autophagy during Varicella-Zoster virus envelopment [Citation41]. The presence of LC3-II-positive viral membranes led us to expect a similar effect for HCMV and, consequently, an antiviral effect mediated by the inhibition of autophagy. Surprisingly, our HCMV mutant expressing a dominant-negative version of the protease ATG4B displayed even enhanced viral genome replication until 8 d.p.i. and enhanced viral release up to 6 d.p.i. This result indicates that the induction of autophagy following HCMV infection has an antiviral effect at the early and early-late stages of infection. This notion contrasts with results presented in a recent report where the inhibition of autophagy led to a decrease in viral release at 10 d.p.i. We have not looked at this very-late stage after infection, as we noticed a high level of cell mortality in autophagy-suppressed, infected cultures already at 8 d.p.i. Indeed, the convergence of the levels of virus release to the controls at 8 d.p.i. (see and ) might be the result of an inability of the host cell to support progeny release further. Additional studies are required to elucidate this. Still, the fact that BAD-ATG4BC74A replicated to higher titers compared to the parental strains suggests an initial antiviral effect of autophagy on HCMV infection, despite the involvement of autophagy in the formation of viral particles.
While this paper was under revision, Taisne et al. published that LC3 localizes to cVACs in HCMV–infected cells and that components of the autophagy machinery, namely autophagic membranes, were associated with extracellular virions [Citation42], which agrees with our results. These authors also found that inhibition of autophagy in cell lines stably transfected with either a plasmid expressing a dominant version of ATG4B or siRNAs targeting key components of the autophagy machinery reduced the release of viral progeny, following HCMV infection. This observation contrasts with our findings, which clearly show that inhibition of autophagy by a virus expressing ATG4BC74A enhances both viral DNA replication and progeny release. One difference between our experimental strategy and that of Taisne et al. is that, in their approach, autophagy is permanently suppressed. This approach might have created a cellular environment, e.g., by the induction of antiviral interferon-responsible genes that might be unfavorable to HCMV infection. A crosstalk between autophagy and the antiviral interferon response has been reported [Citation68]. We used an approach where autophagy was suppressed upon infection. These conditions clearly supported viral replication. Surprisingly, our results are in line with findings also from the Esclatine laboratory, where they show that HCMV expresses two proteins, pTRS1 and pIRS1, that interfere with autophagy, and that suppression of autophagy by these viral proteins is proviral [Citation47]. These findings are also in agreement with data by Spector and colleagues who reported that stimulation of autophagy was inhibitory to HCMV infection [Citation33,Citation34].
The controversial results are interesting, as they are in line with our findings indicating a complex interaction between antiviral effects mediated by autophagy and the apparent contribution of autophagosomal membranes to HCMV particle envelopment. Thus, divergent results on that matter may simply be the results of different experimental conditions in different laboratories, reflecting a complex interaction of the virus with autophagy, possibly depending on the time, intensity, and onset of infection.
Proteomic analyses revealed that SQSTM1 interacts with viral capsid proteins in HCMV–infected fibroblasts. Electron microscopy confirmed this and provided the surprising finding that the autophagy receptor associated with non-enveloped capsids in the nucleus. This finding indicates that SQSTM1-capsid interaction is already established at this stage of the viral life cycle. Whether the interaction of SQSTM1 with the viral capsid in the nucleus is due to an orchestrated cellular defense, guiding viral capsids out of the nucleus and into autophagosomal degradation, or whether HCMV utilizes SQSTM1 for nuclear egress, will be addressed in further detail in future studies.
Both SQSTM1 and the lipidated form of LC3 were found in purified virions (this study and [Citation42]). This observation indicates that LC3 is a component of the viral envelope. Both LC3 and SQSTM1 were found in close proximity to cytoplasmic viral particles. Finally, we also found that SQSTM1 co-immunoprecipitated with viral tegument proteins from lysates of wt-infected cells. Current studies aim to identify viral proteins that specifically mediate the interaction with SQSTM1 and LC3.
Autophagy and HCMV viral morphogenesis may be simultaneous competitive processes
The data of this study confirm that inhibition of autophagy in HFF supports HCMV replication. The use of an HCMV mutant expressing a dominant-negative version of ATG4B provided, for the first time, the opportunity to analyze the effect of autophagy inhibition immediately after infection and in every cell, without side effects of a biochemical intervention. This approach revealed the ability of HCMV to avoid cellular defense by counteracting autophagy. This is consistent with reports by others, identifying the viral proteins TRS1 and IRS1 as inhibitors of autophagy via their interaction with BECN1 [Citation32,Citation47]. We propose a hypothetical model in which the degradation of newly synthesized viral capsids and immature virions on one the hand and the assembly of HCMV virions and DBs, on the other hand, occur simultaneously in infected cells (). According to our model, this interaction with autophagy components appears to be mediated by one or more proteins of the outer viral tegument. It remains unclear whether HCMV simply captures essential components of the autophagy machinery to evade autophagy or concomitantly uses autophagic membranes as a redundant, additional source for viral envelopment. In parallel, the autophagy machinery of the cell appears to recognize capsids in the nucleus and possibly also immature virions in the cytoplasm and might divert them into autophagic degradation. Based on our results, we cannot exclude those mature virions that are also recognized by autophagy.
Figure 10. Hypothetical model of autophagy and viral morphogenesis as simultaneous, competing processes. HCMV capsids are assembled in the cell nucleus. The inner tegument protein pp150 covers the viral capsid in the nucleus to stabilize it. SQSTM1 is present in the nucleus and interacts with capsids (A). Capsids are transported through the nuclear membrane by the process of envelopment and de-envelopment (B). Capsids are transported to cVACs, where they are decorated with proteins of the outer tegument (C). One or more of these tegument proteins may interact with SQSTM1 or other autophagy receptor proteins, possibly via ubiquitin or other small modifiers, enabling interaction with LC3-II-conjugated membranes to foster envelopment (D). How membranes of other sources merge into this process is unclear at this point. Vesicles containing one or more viral particles are then transported to the cell periphery (E), where they fuse with the plasma membrane to release viral particles (F). The findings that capsid proteins precipitate with SQSTM1 and that SQSTM1 interacts with viral capsids already in the nucleus, foster the hypothesis that capsids and attached inner tegument proteins are tagged with autophagy receptor proteins (G) to label them for autolysosomal degradation (H, I). The inhibition of autophagy may lead to an increase in “tegumented” capsids, and other membrane sources may compensate for the loss of LC3-positive membranes in the process of HCMV secondary envelopment
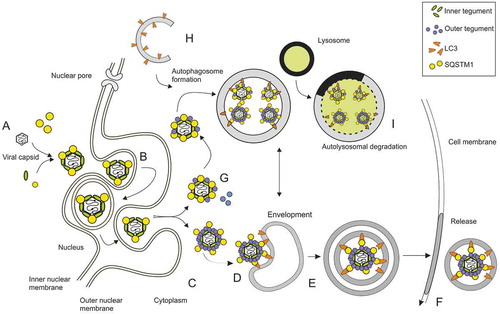
In summary, competitive autophagy processes appear to be active in HCMV–infected cells. Induction of autophagy appears to interfere with viral DNA replication and viral progeny release at early and early-late times after infection. In later stages, the virus may make use of autophagy for its own benefit by using autophagy membranes during particle morphogenesis. The results presented here provide the basis to elucidate the molecular mechanisms that mediate these effects but may also provide additional information about the basic mechanisms of autophagy.
Materials and methods
Cells and viruses
Primary human foreskin fibroblasts (HFF) were cultured, as described previously [Citation56]. The following strains were used in this study: BADwt [Citation69], kindly provided by Thomas Shenk (Lewis Thomas Laboratory, Princeton, NJ, USA); Towne-UL130-repaired (rep) [Citation70], derived from Towne-BAC [Citation71], kindly provided by Hua Zhu, Fenyong Liu, and Edward Mocarski (Emory University School of Medicine, Atlanta, GA, USA), and TB40/E [Citation72], kindly provided by Christian Sinzger (University Medical Center, Ulm, Germany).
Virus reconstitution from BAC-clones was performed by transfecting BAC DNA into HFF, as described previously [Citation73]. The genomes of BADwt and its derivatives contain a self-excisable BAC vector, leading to automatic deletion of the BAC sequence upon transfection into HFF through the expression of the CRE-recombinase [Citation69]. Analysis of intracellular viral DNA replication was performed by quantitative PCR-analysis, as previously described [Citation73].
Virus titers were determined either by quantitation of viral genomes in cell culture supernatants by quantitative PCR or by counting IE1-positive cells [Citation74].
To create BAD-ATG4BC74A, the genomic 2 USD-11 region of the parental strain BADwt was replaced by a DNA fragment encoding the bacterial galactokinase galK cassette as previously described [Citation51]. In the next step, the galK cassette was replaced by a DNA fragment encoding HA-ATG4BC74A (kindly provided by Reinhild Prange [University Medical Center, Mainz, Germany]) [Citation49]. The resulting protein HA-ATG4BC74A contains a hemagglutinin antibody epitope (HA), fused to ATG4BC74A. The construct is driven by a modified version of the major immediate-early promotor of HCMV, containing a nonfunctional cis repressive sequence (crs). The original crs sequence of the CMV MIEP 3ʹ-TCGTTTAGTGAACC-5ʹ was mutated to 3ʹ-CTAGGTAGGGAGAA-5ʹ.
Immunofluorescence
For confocal laser scanning immunofluorescence microscopy, HFF cells (2x105) were grown on coverslips in 6-well plates. On the next day, cells were infected with HCMV (strains BADwt or BAD-ATG4BC74A, respectively) at a multiplicity of infection (m.o.i.) of 0.2. After 6 d, cells were washed with PBS (Sigma, D8537). For blocking of unspecific binding, samples were treated with normal goat serum (Merck, S26)) for 2 h at 37°C, then washed again. The samples were further handled, as previously described [Citation74]. For detection of viral pp150, pp28, or MCP, specific primary mouse antibodies were used (anti-pp150, anti-pp28, and anti-MCP kindly provided by William Britt [University of Alabama, Birmingham, AL, USA], and anti-pp28, Santa Cruz Biotechnology, sc-56975, respectively). For the fluorescent staining of autophagy proteins, rabbit antibodies were purchased from Cell Signaling Technology (SQSTM1, 8025 and LC3B, 3868). Fluorescent secondary antibodies were purchased from Invitrogen, (A11003; R37118)
Immunoprecipitation and western blot
HFF were infected with the respective HCMV strains at an m.o.i. of 1. Infected HFF cells were harvested after 6 d, washed and resuspended in lysis buffer (0.5 M NaCl, 0.05 M Tris-HCl, pH = 8.5, 0.5% NP-40 Alternative [Calbiochem, 492018], 10 mM DTT [Thermo Fisher, R0862]). Cell lysates were sonicated (Branson Sonifier 250, 1 × 10 s, 30% output), and proteins were subsequently bound to specific antibodies (anti-SQSTM1, Cell Signaling Technology, 8025) overnight at 4°C in a rotator. Antibody-protein complexes were then collected by a 1:1 mix (20 µL each) of IgG protein A and protein G Dynabeads (NEB, S1425 S; Novex, 10004D) for 2 h at RT. Magnetic beads were washed 3 times with lysis buffer and subsequently resuspended with 50 µL elution buffer (1 M glycine, pH 2.5), and/or 50 µL Laemmli sample buffer, respectively.
Purified viral particles were lysed in 2x Laemmli sample buffer to a total volume of 20 µL. Protein samples were separated on 10% SDS-PAGE (Life Technologies, NP0301BOX) and transferred to PVDF membranes (Immobilon, ISEQ00010). The filters were probed with specific primary antibodies LC3B (Cell Signaling Technology, 3868), SQSTM1/p62 (Abcam, ab56416), MCP, and pp28 (kindly provided by William Britt). HRP-conjugated secondary antibodies were used for detection (Dako; P0260, P0217).
Virion purification
Virions were produced and purified by glycerol-tartrate centrifugation, according to Irmiere and Gibson, and as previously described [Citation74,Citation75]. For this, 1.8 × 106 HFF were seeded in 10 × 175 cm2 tissue culture flasks and incubated. The cells were infected the following day. After 7 d, when the cultures showed late cytopathic effects, the culture supernatants were collected. Supernatants of 10 or 20 flasks each were combined and centrifuged (1300x g, 10 min, RT). After that, the supernatant was collected and centrifuged at 100,000x g (70 min, 10°C) in an SW32Ti rotor in a Beckman Optima L-90 K ultracentrifuge. Meanwhile, the gradients were prepared by mixing 4 mL 35% Na-tartrate solution with 5 mL 15% Na-tartrate, 30% glycerin-solution in 0.04 M sodium-phosphate buffer, pH 7.4, using a gradient mixer and Beckman Ultra-clearTM centrifuge tubes (14x89 mm). Following centrifugation, the pellets were resuspended in 1 mL 1x PBS. The suspension was applied on top of one gradient. Centrifugation was performed at 91,000x g (60 min, 10°C) in an SW41 rotor. After centrifugation, the bands, corresponding to noninfectious enveloped particles (NIEPs), virions and DBs were visualized by light scattering and collected from the gradient, using a syringe and an 80Gx1,5”-gauge needle (BD Microlance, 304622). Each sample was supplemented with 1x PBS to give a total volume of 10 mL. Centrifugation was then performed at 99,000x g (90 min, 10°C) in an SW41 rotor. Following centrifugation, the pellets were resuspended in 50 µL (virions, DBs) or 100 µL (NIEPs) 1x PBS. 15 µL were taken for the determination of the protein content, and the other samples were stored in aliquots at −80°C until further use. The protein concentrations in the samples were evaluated by using the Pierce BCA protein assay kit (Thermo Scientific, 23225) according to the manufacturer’s instructions. 10% SDS-polyacrylamide gel was used for the separation of the proteins in the samples; 2 µg of each sample was used. Silver staining of the proteins was done using the Roti®-Black P-Silberfärbungskit für Proteine (Roth, L533.1) according to the manufacturer’s instructions.
Analysis of genome replication kinetics and DNA quantification
HFF (2 × 105) were seeded in each 30 cm2 culture dish. Appropriate culture medium (5 mL) was added. Incubation was overnight at 37°C under humid conditions. On the following day, cells were infected with the different HCMV strains with 0.25 genomes/cell. At 1.5 h after the addition of virus, the inoculum was removed. One dish each was taken at 4 h after infection to normalize the amount of DNA that had entered the cells. Cells and supernatants were harvested at the indicated time points, and DNA was extracted and quantified as described recently [Citation73].
Transmission electron microscopy (TEM)
Two flasks of HFF (1.8 million cells per flask) were infected at an m.o.i. of 0.8. Cells were incubated for 6 d and were subsequently detached from the support using trypsin. The cells from the two flasks were pooled and centrifuged at 270 × g, 5 min.
For conventional TEM analysis, cells were then fixed by adding 1 mL fixative (2.5% glutaraldehyde in 0.1 M cacodylate buffer containing 0.1 M sucrose) and resuspending the cells carefully. Following a centrifugation step for 5 min at 270 × g, the cell pellet was again resuspended in 1 mL fixative. The cells were subsequently incubated for 1 h at room temperature and then again centrifuged for 5 min at 270 × g. The cells were then resuspended in washing buffer (0.1 M cacodylate buffer [cacodylic acid sodium salt trihydrate; Roth, Karlsruhe, Germany 5169.1] containing 0.1 M sucrose [D{+}-sucrose Roth, Karlsruhe, Germany, 4621.1]) and centrifuged. The procedure was repeated twice. After the final washing step, the cells were not centrifuged but incubated for 10 min at room temperature. Cells were then transferred in 1.5 mL reaction cups. Samples were then further processed for conventional TEM, as previously described [Citation74].
Immunotem sample preparation
For immunoelectron microscopy, cells were infected, incubated, and harvested as outlined above. After centrifugation, cells pooled from two flasks were fixed by adding 1 mL immunoTEM fixative (3% paraformaldehyde, 0.1% glutaraldehyde in 0.1 M Na2HPO4/KH2PO4 Soerensen phosphate buffer [MerckMillipore, 1197530250; 1051041000], pH = 7,3). After fixation for immunoTEM, cells were washed three times in Soerensen phosphate as described for conventional fixation. After the final washing, step cells were embedded in LR White (hard) resin (Science Services, E14383-UC) and further processed for immunoelectron microscopy as previously described [Citation76]. For post-embedding labeling, LR White ultrathin sections were incubated with primary antibodies (1:1 volume mix of anti-MCP/anti-sCP [William Britt, UAB, Birmingham, AL, USA]), and anti-SQSTM1/p62, (Cell Signaling Technology). For single immunolabeling, the primary antibodies were detected with anti-mouse-Fab or anti-rabbit-Fab secondaries coupled to Nanogold™-(gold particle size: 1.4-nm; Nanoprobes, NMF 2002 or NRF 2004, respectively). For double labeling, we combined the anti-mouse-Fab Nanogold™-labeling with anti-rabbit IgG antibodies coupled with colloidal gold GAR 10 nm (Aurion, Wageningen
NL, Aurion code 810.011) [Citation76]. In all cases, we subsequently silver-enhanced immunogold labeling [Citation76]. In controls that were incubated only with the secondary antibody, no labeling was observed. Counterstained ultrathin sections were analyzed in an FEI Tecnai 12 Omega TEM (FEI, Eindhoven, NL). Images were obtained with a CCD camera (SIS MegaView3, Surface Imaging Systems, Herzogenrath, Germany).
Mass spectrometry sample preparation
Immunoprecipitation was performed with lysates from late-stage infected HFF, using an antibody directed against SQSTM1 (Cell Signaling Technology, 8025) and control magnetic beads under the same conditions as described for immunoprecipitation. Washed magnetic beads with bound viral proteins and control beads were boiled for 10 min at 70°C in 1x NuPAGE LDS Sample Buffer (Life Technologies, NP0007) supplemented with 100 mM DTT and separated on a 10% NuPAGE Bis-Tris gel (Life Technologies, NP0301BOX) for 10 min at 180 V in 1x MES running buffer (Life Technologies, NP0002). The proteins in the gel were fixated for 15 min in a solution of 7% acetic acid and 40% methanol and subsequently stained for 15 min with a solution of 0.25% Coomassie Brilliant Blue G-250 (Biozym, HS-605), 7% acetic acid and 45% ethanol. The gel was afterward washed several times with deionized water to remove excess dye. In-gel digestion was performed in principle as described previously [Citation77] and detailed as follows. Each gel lane was cut separately, minced, and transferred to a microcentrifuge tube. Gel pieces were destained (50% ethanol in 25 mM NH4HCO3 [Sigma Aldrich, 90830], pH 8.0) with the tubes rotating at room temperature for 15 min. The supernatant was removed and the gel pieces dehydrated by adding pure acetonitrile (Merck Millipore, 1000302500) for 10 min with the tubes rotating in a rotator. The acetonitrile was removed first by pipetting, and the samples afterward dried to completion using a vacuum evaporator (Eppendorf). Vacuum dried samples were rehydrated and disulfide bonds were reduced using reduction buffer (10 mM DTT in 50 mM NH4HCO3, pH 8.0) for 1 h at 56°C. Buffer was removed by pipetting and cysteine residues of proteins were subsequently alkylated with 50 mM iodoacetamide (Sigma Aldrich, I1149) dissolved in 50 mM NH4HCO3, pH 8.0 for 45 min at room temperature in the dark. Samples were dehydrated again by adding pure acetonitrile which was removed by pipetting, and vacuum evaporation. The vacuum dried gel slices were incubated with 1 µg trypsin per tube in 50 mM triethylammonium bicarbonate buffer (VWR, SASFT7408) pH 8.0 at 37°C overnight. Digested peptides were extracted by adding twice 150 µL with 30% acetonitrile and once 150 µL pure acetonitrile for 15 min at 25°C agitating at 1400 rpm in a Thermo shaker (Eppendorf). As a next step, we performed reductive dimethylation as described previously [Citation78,Citation79] for each sample pair including a replicate with switched dimethyl labels. The SQSTM1-IP1 samples were marked with the heavy label (+36 Da) mass shift for a single labeled peptide, the SQSTM1-IP2 with the “medium” label (+32 Da) and the control IP was incubated with the light (+28 Da). Equal amounts of peptides from all three labeled samples were mixed. For the technical replicate, we performed a label switch in which the control was labeled heavy, SQSTM1-IP1 was labeled “medium,” SQSTM1-IP2 labeled light, and also mixed at equal peptide amounts. Purification and desalting were done using C18 material (M3 company, Agilent Technologies, 12145004) stage tips as previously described [Citation80]. The eluted peptides were loaded on a column of 75 µm inner diameter (New Objective, FS360-75-8-N-5-C30) packed to 25-cm length with 1.9 µm Reprosil beads (Dr. Maisch GmbH, 11-1-9. aq.0001) using the EasyLC1000 (Thermo).
Mass spectrometry measurement and data analysis
Peptides were separated on an EasyLC1000 HPLC (Thermo Fisher Scientific) with the following reversed-phase chromatography gradient: 0–67 min 0-22% mixture B, 67–88 min 22-40% mixture B, 89–92 min 40-95% mixture B (mixture A: 0.1% formic acid, mixture B: 80% acetonitrile containing 0.1% formic acid) and directly sprayed into a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was operating in positive scan mode with a full scan resolution of 70,000; AGC target 3x106; max. IT = 20 ms; Scan range 300–1650 m/z with a top10 MS/MS DDA method. Normalized collision energy was set to 25, and MS/MS scan mode operated at a resolution of 17,000; AGC target 1 × 105 and max IT of 120 ms. Database search was performed using MaxQuant Version 1.5.2.8 (publicly available at maxplanck.org) [Citation79] with the Homo sapiens Uniprot database (199,590 entries) combined with the Uniprot database of Human cytomegalovirus (13,760 entries), HCMV strain AD169 (369 entries) and HCMV strain Towne (305 entries) downloaded on 16 January 2019. Trypsin/P was set as digestion mode allowing for 2 missed cleavages. Further settings were variable modification: acetyl (Protein N-term), oxidation (M), fixed modifications: carbamidomethyl (C), FDR of 1% on peptide and protein level was applied.
For quantification, light label, medium label, and heavy label were assigned on either lysine residues or N-terminal amino groups with a maximum allowance for 3 labeled amino acids per peptide. Proteins with at least two peptides, one of them unique, were considered as identified. We included MCP/UL86 (C8CFC5), which was identified with 84 peptides but none unique and TRS1 (V9LRY4) with 22 non-unique peptides. The MaxQuant software reported five additional MCP protein variants with minor amino acid changes (represented by the Uniprot entries: A0A2S0DCH4, A0A0G2U786, A0A126N9X5, A0A0G2TAF1, A0A126NCF7), which we filtered out for simplicity. Equally, we excluded the TRS1 variant with the Uniprot accession number A0A2H4GMV9. Proteins matching the reverse database or a common contamination list, as well as proteins containing only peptides with a modification, were removed. A filtered MaxQuant protein group list is available as Table S2. The quantification ratios of the dimethyl labels were log2-transformed. They follow a bimodal distribution. Background maximum was used to normalize all protein ratios, as described in Rai et al. 2018 [Citation81] (Fig. S1). Proteins quantified in both IPs and their technical replicates were selected for further analysis. Protein averages and standard error of the mean (SE) were plotted for the bait protein SQSTM1 and all viral proteins using R-3.5.1 and the following packages (“ggplot2ʹ,”tidyr,”dplyr,”xlsx,’ ’plotrix’).
The Welch’s t-test (function “t.test”) was performed between the log2 mean of all viral proteins quantified at least 2 times and all human proteins with a log2 fold-change between −1 and 1 (quantified two to four times), which are referred to as human background proteins. The average log2 protein fold-change was visualized as a bar graph with indicated standard deviation (function “sd”).
Supplemental Material
Download Zip (2.5 MB)Acknowledgments
We appreciate the donation of monoclonal antibodies by William Britt. We are grateful for receiving viruses, plasmids, and BAC clones from Reinhild Prange, Eva Borst, Martin Messerle, Hua Zhu, Fenyong Liu, Edward Mocarski, and Thomas Shenk.
Disclosure Statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109.
- Munz C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev. 2016;272(1):17–27.
- Munz C. The macroautophagy machinery in endo- and exocytosis. J Mol Biol. 2017;429(4):473–485.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939.
- Sinha S, Colbert CL, Becker N, et al. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4(8):989–997.
- Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354.
- Cavignac Y, Esclatine A. Herpesviruses and autophagy: catch me if you can! Viruses. 2010;2(1):314–333.
- Cirone M. EBV and KSHV infection dysregulates autophagy to optimize viral replication, prevent immune recognition and promote tumorigenesis. Viruses. 2018;10(11). DOI:10.3390/v10110599.
- Graybill C, Morgan MJ, Levin MJ, et al. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology. 2018;522:220–227.
- Lussignol M, Esclatine A. Herpesvirus and autophagy: “All right, everybody be cool, this is a robbery!” Viruses. 2017;9(12). DOI:10.3390/v9120372.
- Smith JD, de Harven E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J Virol. 1978;26(1):102–109.
- Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23–35.
- Orvedahl A, Levine B. Autophagy and viral neurovirulence. Cell Microbiol. 2008;10(9):1747–1756.
- Liang C. Herpesviral interaction with autophagy. J Bacteriol Virol. 2011;41(4):213–223.
- Lee JS, Li Q, Lee JY, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355–1362.
- Buckingham EM, Carpenter JE, Jackson W, et al. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J Virol. 2014;88(2):890–902.
- Yakoub AM, Shukla D. Basal autophagy is required for herpes simplex virus-2 infection. Sci Rep. 2015;5:12985.
- Munz C. The autophagic machinery in viral exocytosis. Front Microbiol. 2017;8:269.
- Nowag H, Guhl B, Thriene K, et al. macroautophagy proteins assist epstein barr virus production and get incorporated into the virus particles. EBioMedicine. 2014;1(2–3):116–125.
- Drew WL. Diagnosis of cytomegalovirus infection. Rev Infect Dis. 1988;10(Suppl 3):S468–76.
- Rubin RH. Impact of cytomegalovirus infection on organ transplant recipients. Rev Infect Dis. 1990;12(Suppl 7):S754–66.
- Fowler KB, Boppana SB. Congenital cytomegalovirus infection. Semin Perinatol. 2018;42(3):149–154.
- Gaytant MA, Steegers EA, Semmekrot BA, et al. Congenital cytomegalovirus infection: review of the epidemiology and outcome. Obstet Gynecol Surv. 2002;57(4):245–256.
- Dai X, Yu X, Gong H, et al. The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus. PLoS Pathog. 2013;9(8):e1003525.
- Yu X, Shah S, Lee M, et al. Biochemical and structural characterization of the capsid-bound tegument proteins of human cytomegalovirus. J Struct Biol. 2011;174(3):451–460.
- Kalejta RF. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr Top Microbiol Immunol. 2008;325:101–115.
- Phillips SL, Cygnar D, Thomas A, et al. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J Virol. 2012;86(18):9995–10005.
- Schierling K, Stamminger T, Mertens T, et al. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J Virol. 2004;78(17):9512–9523.
- Smith RM, Kosuri S, Kerry JA. Role of human cytomegalovirus tegument proteins in virion assembly. Viruses. 2014;6(2):582–605.
- Tomtishen JP 3rd. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol J. 2012;9:22.
- Schauflinger M, Villinger C, Mertens T, et al. Analysis of human cytomegalovirus secondary envelopment by advanced electron microscopy. Cell Microbiol. 2013;15(2):305–314.
- Chaumorcel M, Lussignol M, Mouna L, et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol. 2012;86(5):2571–2584.
- Belzile JP, Sabalza M, Craig M, et al. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J Virol. 2016;90(3):1259–1277.
- Clark AE, Sabalza M, Gordts P, et al. Human cytomegalovirus replication is inhibited by the autophagy-inducing compounds trehalose and SMER28 through distinctively different mechanisms. J Virol. 2018;92(6). DOI:10.1128/JVI.02015-17.
- Nevels M, Nitzsche A, Paulus C. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev Med Virol. 2011;21(3):154–180.
- Guo H, Kaiser WJ, Mocarski ES. Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol. 2015;204(3):439–448.
- Noriega V, Redmann V, Gardner T, et al. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res. 2012;54(1–3):140–151.
- Scherer M, Stamminger T. Emerging role of PML nuclear bodies in innate immune signaling. J Virol. 2016;90(13):5850–5854.
- Tavalai N, Stamminger T. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 2011;157(2):128–133.
- McFarlane S, Aitken J, Sutherland JS, et al. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol. 2011;85(9):4212–4221.
- Buckingham EM, Jarosinski KW, Jackson W, et al. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016;90(19):8673–8685.
- Taisne C, Lussignol M, Hernandez E, et al. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci Rep. 2019;9(1):4560.
- Sanchez V, Greis KD, Sztul E, et al. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol. 2000;74(2):975–986.
- Chevillotte M, Landwehr S, Linta L, et al. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J Virol. 2009;83(6):2480–2490.
- Schmolke S, Kern HF, Drescher P, et al. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J Virol. 1995;69(10):5959–5968.
- Reyda S, Tenzer S, Navarro P, et al. The tegument protein pp65 of human cytomegalovirus acts as an optional scaffold protein that optimizes protein uploading into viral particles. J Virol. 2014;88(17):9633–9646.
- Mouna L, Hernandez E, Bonte D, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 2016;12(2):327–342.
- Marino G, Uria JA, Puente XS, et al. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278(6):3671–3678.
- Doring T, Zeyen L, Bartusch C, et al. Hepatitis B virus subverts the autophagy elongation complex Atg5-12/16L1 and does not require Atg8/LC3 lipidation for viral maturation. J Virol. 2018;92(7). DOI:10.1128/JVI.01513-17.
- Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19(11):4651–4659.
- Warming S, Costantino N, Court DL, et al. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33(4):e36.
- Arlt H, Lang D, Gebert S, et al. Identification of binding sites for the 86-kilodalton IE2 protein of human cytomegalovirus within an IE2-responsive viral early promoter. J Virol. 1994;68(7):4117–4125.
- Cherrington JM, Khoury EL, Mocarski ES. Human cytomegalovirus ie2 negatively regulates alpha gene expression via a short target sequence near the transcription start site. J Virol. 1991;65(2):887–896.
- Lang D, Stamminger T. The 86-kilodalton IE-2 protein of human cytomegalovirus is a sequence-specific DNA-binding protein that interacts directly with the negative autoregulatory response element located near the cap site of the IE-1/2 enhancer-promoter. J Virol. 1993;67(1):323–331.
- Liu B, Hermiston TW, Stinski MF. A cis-acting element in the major immediate-early (IE) promoter of human cytomegalovirus is required for negative regulation by IE2. J Virol. 1991;65(2):897–903.
- Besold K, Wills M, Plachter B. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology. 2009;391(1):5–19.
- Jones TR, Muzithras VP. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J Virol. 1992;66(4):2541–2546.
- Jones TR, Muzithras VP, Gluzman Y. Replacement mutagenesis of the human cytomegalovirus genome: US10 and US11 gene products are nonessential. J Virol. 1991;65(11):5860–5872.
- Kollert-Jons A, Bogner E, Radsak K. A 15-kilobase-pair region of the human cytomegalovirus genome which includes US1 through US13 is dispensable for growth in cell culture. J Virol. 1991;65(10):5184–5189.
- Falk CS, Mach M, Schendel DJ, et al. NK cell activity during human cytomegalovirus infection is dominated by US2-11-mediated HLA class I down-regulation. J Immunol. 2002;169(6):3257–3266.
- Munz C. Macroautophagy–friend or foe of viral replication? EMBO Rep. 2013;14(6):483–484.
- Close WL, Anderson AN, Pellett PE. Betaherpesvirus virion assembly and egress. Adv Exp Med Biol. 2018;1045:167–207.
- Close WL, Glassbrook JE, Gurczynski SJ, et al. Infection-induced changes within the endocytic recycling compartment suggest a roadmap of human cytomegalovirus egress. Front Microbiol. 1888;9(2018). DOI:10.3389/fmicb.2018.01888.
- Das S, Ortiz DA, Gurczynski SJ, et al. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol. 2014;88(16):9086–9099.
- Das S, Vasanji A, Pellett PE. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol. 2007;81(21):11861–11869.
- Severi B, Landini MP, Govoni E. Human cytomegalovirus morphogenesis: an ultrastructural study of the late cytoplasmic phases. Arch Virol. 1988;98(1–2):51–64.
- Tooze J, Hollinshead M, Reis B, et al. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur J Cell Biol. 1993;60(1):163–178.
- Jin S. The cross-regulation between autophagy and type I interferon signaling in host defense. Adv Exp Med Biol. 2019;1209:125–144.
- Yu D, Smith GA, Enquist LW, et al. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol. 2002;76(5):2316–2328.
- Lehmann C, Falk JJ, Buscher N, et al. Dense bodies of a gH/gL/UL128/UL130/UL131 pentamer-repaired towne strain of human cytomegalovirus induce an enhanced neutralizing antibody response. J Virol. 2019;93(17). DOI:10.1128/JVI.00931-19.
- Marchini A, Liu H, Zhu H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol. 2001;75(4):1870–1878.
- Sampaio KL, Weyell A, Subramanian N, et al. A TB40/E-derived human cytomegalovirus genome with an intact US-gene region and a self-excisable BAC cassette for immunological research. Biotechniques. 2017;63(5):205–214.
- Zimmermann C, Buscher N, Krauter S, et al. The abundant tegument protein pUL25 of human cytomegalovirus prevents proteasomal degradation of pUL26 and supports its suppression of ISGylation. J Virol. 2018;92(24). DOI:10.1128/JVI.01180-18.
- Krommelbein N, Wiebusch L, Schiedner G, et al. Adenovirus E1A/E1B transformed amniotic fluid cells support human cytomegalovirus replication. Viruses. 2016;8(2):37.
- Irmiere A, Gibson W. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology. 1983;130(1):118–133.
- Wolfrum U, Schmitt A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil Cytoskeleton. 2000;46(2):95–107.
- Shevchenko A, Tomas H, Havlis J, et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–2860.
- Boersema PJ, Raijmakers R, Lemeer S, et al. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4(4):484–494.
- Hsu JL, Huang SY, Chow NH, et al. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem. 2003;75(24):6843–6852.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2(8):1896–1906.
- Rai AK, Chen JX, Selbach M, et al. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 2018;559(7713):211–216.