
ABSTRACT
Macroautophagy/autophagy, a eukaryotic homeostatic process that sequesters cytoplasmic constituents for lysosomal degradation, is orchestrated by a number of autophagy-related (ATG) proteins tightly controlled by post-translational modifications. However, the involvement of reversible ubiquitination in the regulation of autophagy remains largely unclear. Here, we performed a single-guide RNA-based screening assay to investigate the functions of deubiquitinating enzymes (DUBs) in regulating autophagy. We identified previously unrecognized roles of several DUBs in modulating autophagy at multiple levels by targeting various ATG proteins. Mechanistically, we demonstrated that STAMBP/AMSH (STAM-binding protein) promotes the stabilization of ULK1 by removing its lysine 48 (K48)-linked ubiquitination, whereas OTUD7B mediates the degradation of PIK3 C3 by enhancing its K48-linked ubiquitination, thus positively or negatively affects autophagy flux, respectively. Together, our study elaborated on the broad involvement of DUBs in regulating autophagy and uncovered the critical roles of the reversible ubiquitination in the modification of ATG proteins.
Abbreviations: ATG: autophagy-related; Baf A1: bafilomycin A1; DUB: deubiquitinating enzyme; EBSS: Earle’s balanced salt solution; KO: knockout; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; OTUD7B: OTU domain-containing protein 7B; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; sgRNA: single-guide RNA; SQSTM1/p62: sequestosome 1; STAMBP/AMSH: STAM-binding protein; ULK1: unc-51 like autophagy activating kinase 1; USP: ubiquitin specific peptidase.
Introduction
Macroautophagy, hereafter referred to as autophagy, is a conserved lysosomal degradation system, which breaks down intracellular material through the formation of double-membrane autophagosomes in eukaryotic cells [Citation1–3]. The autophagic process orchestrated by a series of autophagy-related (ATG) proteins plays an essential role in tissue homeostasis, adaptation to stress situations, immune responses, and the regulation of inflammatory responses [Citation4–8]. Upon autophagy induction, the ULK1 (unc-51 like autophagy activating kinase 1) complex is activated, which in turn leads to the activation of the class III phosphatidylinositol 3-kinase complex I (PtdIns3 K-C1), thus promoting autophagosome nucleation [Citation2,Citation3]. Subsequently, Atg8-family proteins, including the most common autophagosomal marker MAP1LC3/LC3 (microtubule associated protein 1 light chain 3), are lipidated to complete the formation of autophagic membranes [Citation9].
Given the functional importance of the autophagy machinery, autophagy must be stringently controlled to avoid cellular disorder. It has been well studied that post-translational modification (PTM), especially reversible ubiquitination, plays central roles in the regulation of autophagy. A recent report suggested that a large fraction of tripartite motif (TRIM) proteins serve as platforms for the assembly of activated ULK1 and BECN1 to initiate autophagy, indicating the critical involvement of E3 ubiquitin ligases in autophagy [Citation10]. The ubiquitin ligase, TRAF6 mediates the K63-linked ubiquitination of BECN1 to disrupt the interaction between BECN1 and BCL2, whereas the DUB TNFAIP3/A20 can reverse this process [Citation11]. Furthermore, ubiquitination is associated with the proteasomal degradation of ATGs. RNF216 (ring finger protein 216) promotes the ubiquitination of BECN1, leading to its degradation in a proteasome-dependent manner [Citation12]. Degradative ubiquitination of BECN1 is counteracted by various deubiquitinases, including ubiquitin-specific peptidases, USP10 and USP13 [Citation13], while AMBRA1 blocks the degradation of MTOR inhibitor DEPTOR through interfering with the E3 ligase CUL5/CULLIN-5, thus indirectly promoting ULK1 activity [Citation4]. Our previous study revealed that USP19 also stabilizes BECN1 by removing the K11-linked ubiquitin chains on BECN1 at Lys (K) 437 [Citation14,Citation15]. Despite the importance of several reported DUBs, the precise regulation of autophagy mediated by a large portion of other DUBs still remains elusive, and a systematic analysis is needed to identify the involvement of DUBs in autophagy.
Here, we employed a screening system based on clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 (CRISPR-associated protein 9) to identify functional DUBs in autophagy and demonstrated their multi-layer regulations of autophagy through diverse mechanisms. Our study found that STAMBP/AMSH stabilized ULK1, while OTUD7B exerted an opposite influence on PIK3C3. The altered K48-linked ubiquitination of ULK1 and PIK3C3 mediated by STAMBP and OTUD7B confer the positive and negative regulation of autophagy flux, respectively. Together, we have identified the broad, unprecedented involvement of DUBs in regulating autophagy. By revealing the modification imposed by STAMBP and OTUD7B, we prove that deubiquitination is critical for the stability of ATG proteins through complicated regulatory mechanisms, thus providing novel targets for clinical therapy based on autophagy.
Results
DUB family members participate in the regulation of autophagy flux
We screened the functions of a panel of DUBs in autophagy by using a validated single-guide RNAs (sgRNAs) screening system. 170 sgRNAs targeting 85 individual DUB family genes were designed and constructed into the lentiviral vectors expressing Cas9 as previously described [Citation16]. HeLa cells stably expressing GFP-LC3B (HeLa-GFP-LC3B) were then infected with lentiviruses delivering Cas9 and sgRNA to generate sg_DUB HeLa-GFP-LC3B cells. We next confirmed the knockdown efficiency of sg_DUB HeLa-GFP-LC3B cells using quantitative polymerase chain reaction (qPCR) analysis (Fig. S1). sgRNA sequences with low knockdown efficiency were excluded from subsequent analysis. To determine the function of DUBs in autophagy, the numbers of GFP-LC3B puncta were examined in all sg_DUB HeLa-GFP-LC3B cells at basal level or after autophagy inducer, rapamycin treatment. Over 100 cells for each target were selected randomly and images were analyzed using imageJ software (https://imagej.nih.gov/ij) to calculate the numbers of GFP-LC3B puncta ()). We confirmed the feasibility of this system by testing several DUBs, including USP19 and TNFAIP3, whose functions in autophagy have been elucidated [Citation11,Citation14]. Consistent with previous studies, the numbers of GFP-LC3B puncta significantly decreased in the sg_USP19 HeLa-GFP-LC3B cells but increased in sg_TNFAIP3 HeLa-GFP-LC3B cells compared to the control cells ()).
Figure 1. DUBs participate in the regulation of autophagy. (a) Schematic overview of the GFP-LC3B puncta screening assay. HeLa-GFP-LC3B cells were transduced with lentiviruses expressing Cas9 and sgRNAs targeting individual DUBs. After puromycin selection twice, the cells were seeded in glass bottom dishes and imaged for eGFP at basal level or after rapamycin (250 nM) treatment for 18 h using fluorescence microscope. (b-d) Representative images (b) of GFP-LC3B puncta in sg_USP19/sg_TNFAIP3 HeLa-GFP-LC3B cells at basal level or after rapamycin treatment. Scale bar: 20 μm. Quantification of GFP-LC3B puncta per cell at basal level (c) or after rapamycin treatment (d). (e-f) Average number of GFP-LC3B puncta per cell at basal level (e) or after rapamycin treatment (f). NT, non-treated. Data in (c-f) are expressed as means ± SEM of three independent biological experiments. (20 cells per sample). *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (two-tailed Student’s t-test)
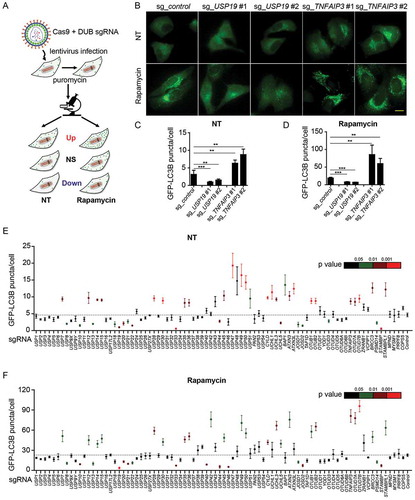
By screening 85 DUBs, we found approximately 41% of DUBs could modulate autophagy to varying degrees. Nearly 30% of sg_DUB cells enhanced the numbers of GFP-LC3B puncta, while approximately 17% of them decreased the GFP-LC3B puncta under normal conditions ()). Additionally, after rapamycin treatment, 26% of sg_DUB cells displayed an enhanced accumulation of GFP-LC3B puncta, while 20% of sg_DUBs cells exhibited a decreased GFP-LC3B puncta formation ()). Among them, several DUBs, including USP10, USP13, and USP33, were reported previously as important positive regulators in autophagy, thus validating the reliability of our screening system [Citation13,Citation17].
Function validation of selected DUBs in autophagy
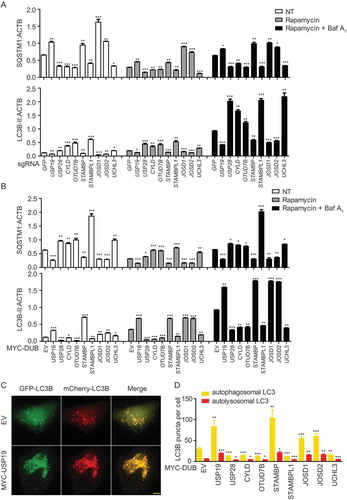
To confirm the functions of DUBs in autophagy, we chose several candidate DUBs for further study. After excluding those DUBs reported as the autophagy regulators previously [Citation11,Citation15,Citation17–21], 8 representative deubiquitinases (USP28, CYLD, OTUD7B, STAMBP, STAMBPL1, JOSD1, JOSD2, and UCHL3) from five different DUB subfamilies were selected to study their mechanisms in regulating autophagy.
HeLa cells were transfected with vectors encoding sg_GFP (as a control) or selected sg_DUBs and the cells were collected for immunoblot after treatment with rapamycin or bafilomycin A1 (Baf A1, a vacuolar-type H+-translocating ATPase inhibitor which can block autophagosome turnover). We then examined the protein levels of SQSTM1/p62 (sequestosome 1) and LC3B to monitor the autophagy flux () and S2A-I). Deficiency of USP19, STAMBP, JOSD1, and JOSD2 leads to the decreased SQSTM1 consumption and LC3B-II accumulation, while depletion of USP28, CYLD, OTUD7B, STAMBPL1, and UCHL3 showed the opposite results. To better confirm the functions of these selected DUBs in autophagy, we employed the overexpression system to examine the SQSTM1 level and LC3B as well () and S2J-R). Consistently, overexpression of USP19, STAMBP, JOSD1, and JOSD2 leads to the decreased SQSTM1 and enhanced LC3B-II level, while overexpression of USP28, CYLD, OTUD7B, STAMBPL1, and UCHL3 resulted in the opposite results. Additionally, we observed the similar pattern using Baf A1 to inhibit the formation of autolysosome in both depletion and overexpression systems () and S2A-R). These results suggested that the selected DUBs mainly affect the autophagy initiation but not the autophagosome fusion. We further employed a tandem fluorescent image-based system to quantitate the redistribution of the autophagosomal membrane protein LC3 in the mCherry-GFP-LC3-stably-expressing HeLa cells ()). Since GFP is unstable in acidic environment, the yellow puncta (co-localization of GFP and mCherry) would indicate completed autophagosomes, while red puncta indicate autolysosomes. The quantification data of yellow and red puncta further demonstrated the functions of our identified DUBs in autophagy. Together, we identify STAMBP, JOSD1, and JOSD2 as positive regulators of autophagy, whereas USP28, CYLD, OTUD7B, STAMBPL1, and UCHL3 as negative regulators of autophagy.
Figure 2. Function validation of selected DUBs in autophagy. (a) HeLa cells were transfected with sg_GFP (control) or sg_USP19, sg_USP28, sg_CYLD, sg_OTUD7B, sg_STAMBP, sg_STAMBPL1, sg_JOSD1, sg_JOSD2 or sg_UCHL3 and treated with rapamycin (250 nM) for 12 h alone or with Baf A1 (0.2 μM) as combination for 6 h. Cell lysates were collected for subsequent immunoblot assay. Quantification data of the protein level of LC3B and SQSTM1 compared with ACTB/β-actin. The corresponding data are shown in Fig. S2A-I. Data are normalized by the protein levels of LC3B-II or SQSTM1 in control cells. (b) HeLa cells were transfected with MYC-EV (control) or MYC-USP19, MYC-USP28, MYC-CYLD, MYC-OTUD7B, MYC-STAMBP, MYC-STAMBPL1, MYC-JOSD1, MYC-JOSD2 or MYC-UCHL3 and treated with rapamycin (250 nM) for 12 h alone or with Baf A1 (0.2 μM) as combination for 6 h. Cell lysates were collected for subsequent immunoblot assay. Quantification data of the protein level of LC3B and SQSTM1 compared with ACTB. The corresponding data are shown in Figure S2 J-R. Data are normalized by the protein levels of LC3B-II or SQSTM1 in control cells. (c-d) Representative fluorescence images (c) and quantification (d) of yellow and red puncta for LCB expression in mCherry-GFP-LC3B stably expressing HeLa cells transfected with indicated DUBs and treated with rapamycin (250 nM) for 12 h. Data in (a-d) are expressed as means ± SEM of three independent biological experiments, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. (two-tailed Student’s t-test)
DUBs target different key components of autophagy
To further investigate the roles of DUBs in autophagy, we next performed co-immunoprecipitation (co-IP) experiments to identify the interaction between DUBs and various key molecules in autophagy, including ULK1, PIK3C3, BECN1, ATG5, LC3B, and SQSTM1. We found six DUBs (USP19, CYLD, OTUD7B, STAMBP, STAMBPL1, and JOSD1) targeted ULK1, which is associated with autophagy initiation ()). USP19, USP28, CYLD, and OTUD7B interacted PIK3C3, the essential protein in autophagosome formation ()). Meanwhile, another four DUBs (USP19, CYLD, OTUD7B, and JOSD1) could associate with BECN1 (Fig. S3A), which is an important component of PIK3C3 complex. At the vesicle elongation stage, the ATG12–ATG5 covalent conjugates could be targeted by CYLD (Fig. S3B). Subsequently, USP19 also participated in the process of autophagosome maturation by targeting LC3B, while the weak interactions between LC3B and CYLD, OTUD7B or JOSD1 could also be detected (Fig. S3C). Additionally, USP19, CYLD, OTUD7B, and JOSD1 were also involved in selective autophagy by associating with the cargo receptor SQSTM1 (Fig. S3D).
Figure 3. DUBs target different key components of autophagy. (a-b) Immunoblot analysis of extracts of HEK293 T cells transfected with plasmids encoding HA-ULK1 (a) or HA-PIK3C3 (b) and MYC-DUBs chosen in Figure 2, followed by IP with anti-MYC beads and immunoblot detection with anti-HA. (c) Summary of interactions between DUBs and key components in autophagy. The corresponding data are shown in Fig. S3A-D. Data are representative of three independent biological experiments
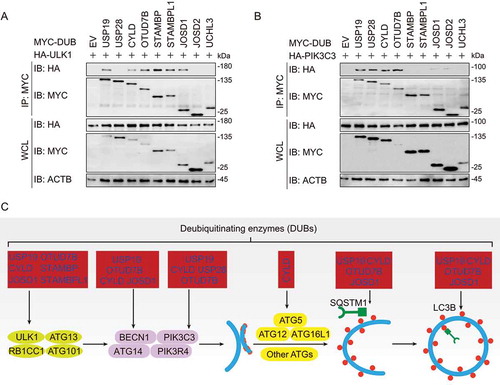
In summary, several DUBs, including USP19, OTUD7B, CYLD, and JOSD1, might have a global regulatory function while the other DUBs such as STAMBP, STAMBPL1, and USP28 specifically target to certain ATGs. Though deletion of JOSD2 and UCHL3 remarkably affected autophagy, no interaction between them with ATG proteins was detected () and S2). We speculated that they might affect the upstream MTOR (mechanistic target of rapamycin kinase) complex, or the downstream lysosomal degradation. Collectively, our findings reveal a panoramic view that diverse DUBs are involved in autophagy at different levels ()).
STAMBP positively regulates autophagy via stabilizing ULK1
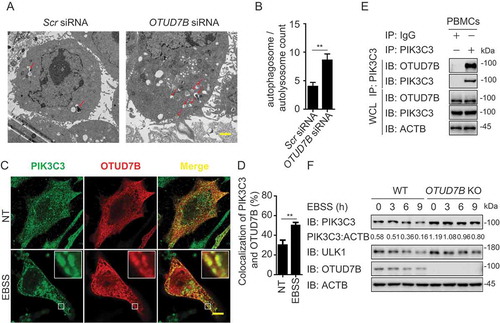
Among the DUBs which target ULK1 complex, STAMBP showed the strongest binding ability with ULK1 ()). Previous studies in patient lymphoblast cell lines indicated the potential connection between STAMBP and autophagy as well [Citation22]. Therefore, we chose STAMBP to investigate the influence of DUBs on the initiation of autophagy. We first confirmed the effect of STAMBP deficiency on autophagy using electron microscopy. Less autophagosomes and autolysosomes were observed in STAMBP knockdown HeLa cells compared with the control cells ()). To understand the detailed mechanism employed by STAMBP, we examined the co-localization between STAMBP and ULK1 at basal level or after Earle’s balanced salt solution (EBSS; an autophagy agonist) treatment in HeLa cells. The co-localization between STAMBP and ULK1 was markedly enhanced after EBSS treatment ()). We further observed the endogenous interaction between STAMBP and ULK1 in A549 cells (Fig. S4A) and human peripheral blood mononuclear cells (PBMCs) ()). The interaction between STAMBP and ULK1 was also enhanced after EBSS treatment (Fig. S4A and 4E). Additionally, we examined the interaction between STAMBP and other components of ULK1 or PIK3C3 complex. STAMBP mainly interacted with ULK1, together with other ULK1 complex members, including ATG13, RB1CC1/FIP200, and ATG101 (Fig. S4B), but not PIK3C3 complex (Fig. S3C), suggesting STAMBP affects autophagy by targeting ULK1 complex.
Figure 4. STAMBP positively regulates autophagy via stabilizing ULK1. (A-B) Electron microscopy observation (a) of ultra-structure of the cells. HeLa cells transfected with scramble or STAMBP siRNA are treated with rapamycin (250 nM) for 18 h before analysis. Arrows indicate autophagosomes or autolysosomes. Scale bar, 2 μm. Quantification (b) of autophagosomes and autolysosomes per cell after rapamycin treatment. (c-d) Confocal microscopy (c) and quantitative data (d) of HeLa cells at basal level or incubated in EBSS medium for 1 h. Scale bar, 20 μm. (e) co-IP and immunoassay of extracts of PBMCs treated with EBSS for various times (above lane). (f) Immunoassay of extracts of wild-type (WT) and STAMBP knockout (KO) HEK293 T cells cultured in EBSS medium for the various times (above lane). Data are representative of three independent biological experiments. Data in (a-d) are expressed as means ± SEM of three independent biological experiments (20 cells per sample). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. (two-tailed Student’s t-test)
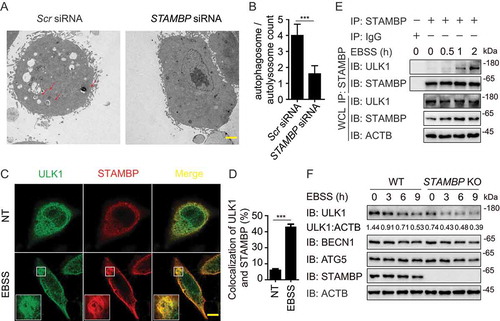
We next explored how STAMBP promoted autophagy through ULK1. We found that enforced expression of STAMBP stabilized ULK1, but not other components from ULK1 complex (Fig. S4D). qPCR analysis showed that the abundance of ULK1 mRNA did not change with increasing expression of STAMBP (Fig. S4E), which indicated STAMBP promoted the stabilization of ULK1 at the protein level. Moreover, STAMBP overexpression blocked the EBSS-induced degradation of ULK1 (Fig. S4F), while knockout (KO) of STAMBP in A549 cells resulted in the accelerated degradation of ULK1 after EBSS treatment ()). Therefore, STAMBP upregulates autophagy by promoting the stabilization of ULK1 at the protein level.
STAMBP stabilizes ULK1 through cleaving its K48-linked ubiquitin chains
To investigate the detailed mechanism employed by STAMBP to mediate the stabilization of ULK1, we next exploited the inhibitors of different degradation pathways. We found that the stabilization of ULK1 by STAMBP was abrogated by MG132 (a proteasome inhibitor) but not Baf A1, suggesting that STAMBP stabilizes ULK1 by blocking its degradation through the proteasome pathway ()). Collectively, STAMBP targets ULK1 and reduces its proteasomal degradation.
Figure 5. STAMBP stabilizes ULK1 through cleaving its K48-linked ubiquitin chains. (a) Immunoassay of extracts of HEK293 T cells transfected with plasmids for MYC-tagged EV or STAMBP and treated with dimethyl sulfoxide (DMSO; vehicle), MG132 (10 μM) or Baf A1 (0.2 μM) for 6 h. (b) Immunoassay of extracts of HEK293 T cells transfected with plasmid encoding MYC-tagged EV or STAMBP and treated with MG132 for 6 h, followed by denatured IP with anti-ULK1 antibody and immunoblot detection with anti-Ub antibody. (c) Immunoassay of extracts of HEK293 T cells transfected with plasmid encoding MYC-tagged EV or STAMBP and cultured in EBSS medium for the various times (above lane), together with MG132 treatment for 6 h, followed by denatured IP with anti-ULK1 antibody and immunoblot detection with anti-K48-Ub antibody. (d) Purified ubiquitinated ULK1 was incubated with immunopurified Flag-STAMBP in vitro in deubiquitinating buffer. The immunoblot was detected with anti-HA. Data are representative of three independent biological experiments
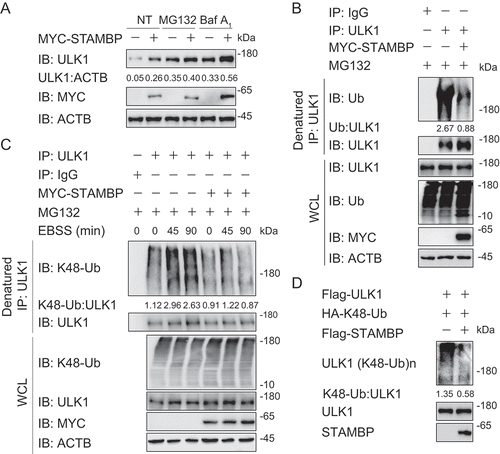
Since ubiquitination is important for proteasomal degradation, we next assessed whether STAMBP affects the ubiquitination of ULK1. We found STAMBP cleaved the poly-ubiquitin chains on ULK1 ()). Considering the well-studied roles of K48-linked ubiquitination in proteasomal degradation [Citation23,Citation24], we then checked the endogenous K48-linked ubiquitination of ULK1. The K48-linked ubiquitination of ULK1 was strongly enhanced after EBSS treatment while gradually decreased after 2 h of EBSS treatment (Fig. S5A). We then overexpressed MYC-tagged STAMBP in HEK293 T cells, and treated the cells with MG132 for 6 h to ensure that the protein level of ULK1 in control group and STAMBP-overexpressing group are comparable. We next examined the endogenous K48-linked ubiquitination of ULK1 upon starvation and found that STAMBP specifically reduced K48-linked ubiquitination of ULK1 ()). We also employed in vitro deubiquitination assay using purified STAMBP and K48-linked ubiquitinated ULK1 and observed that STAMBP also removed K48-linked ubiquitin chains from the purified ULK1 in the cell-free deubiquitination buffer ()). The K63-linked ubiquitination of ULK1 has also been reported to promote ULK1 stability and function [Citation25]. However, we found that STAMBP did not affect the K63-linked ubiquitination of ULK1 (Fig. S5B). Together, these findings indicate that STAMBP positively regulates autophagy by promoting the stabilization of ULK1 through removing its K48-linked ubiquitin chains.
OTUD7B targets PIK3C3 to promote its degradation
Among these chosen DUBs, OTUD7B has emerged a significant influence and broad involvement in autophagy regulation as it can target autophagy-associated proteins at multiple layers. We further studied the detailed mechanism of OTUD7B in autophagy. We employed electron microscopy and observed that the formation of autophagosomes and autolysosomes were significantly decreased in OTUD7B-depleted HeLa cells ()), indicating the negative regulatory role of OTUD7B in the formation of autophagosomes. Since OTUD7B showed a robust interaction with PIK3C3 complex () and S3A), we first performed a confocal microscopic analysis and observed the co-localization between PIK3C3 and OTUD7B ()). The co-localization was increased after EBSS treatment in HeLa cells ()). These results were furtherly confirmed by endogenous co-IP experiments in A549 cells (Fig. S6A) and human PBMCs ()).
Figure 6. OTUD7B targets PIK3C3 to promote its degradation. (a-b) Electron microscopy observation (a) of ultra-structure of the cells. HeLa cells transfected with scramble or OTUD7B siRNA are treated with rapamycin (250 nM) for 18 h before analysis. Arrows indicate autophagosomes or autolysosomes. Scale bar, 2 μm. Quantification (b) of autophagosomes and autolysosomes per cell after rapamycin treatment. (c-d) Confocal microscopy (c) and quantitative data (d) of HeLa cells treated with MG132 at basal level or incubated in EBSS medium for 1 h. Scale bar, 20 μm. (e) co-IP and immunoassay of cell extracts of PBMCs treated with EBSS for 1 h. (f) Immunoassay of extracts of WT and OTUD7B KO HEK293 T cells cultured in EBSS medium for the indicated time points (above lane). Data are representative of three independent biological experiments. Data in (a-d) are expressed as means ± SEM of three independent biological experiments (20 cells per sample). *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (two-tailed Student’s t-test)
To figure out how OTUD7B inhibits autophagy, we investigated the protein abundance of ATGs in PIK3C3 complex with the existence of OTUD7B and found enforced expression of OTUD7B specifically decreased the protein level of PIK3C3, but not other members of PIK3C3 complex (Fig. S6B). We also checked the PIK3C3 mRNA level with increasing OTUD7B overexpression and observed that the transcription levels of PIK3C3 remained unchanged (Fig. S6C). Meanwhile, overexpression of OTUD7B promoted endogenous PIK3C3 degradation (Fig. S6D). Consistently, knockout of OTUD7B blocked the degradation of PIK3C3 under both the basal and autophagy-inducing conditions ()). These data validate the results of our screening and indicate that OTUD7B affects autophagy by degrading PIK3C3.
OTUD7B mediates the K63-K48 type ubiquitination type transition of PIK3C3
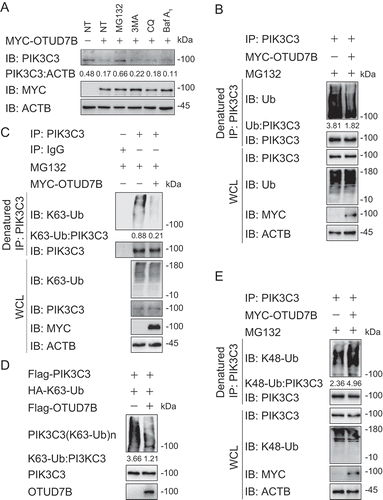
To distinguish which degradation system dominantly regulates the stability of PIK3C3 mediated by OTUD7B, we examined the protein amount of PIK3C3 in the presence of OTUD7B using pharmacologic approaches and found that OTUD7B-mediated PIK3C3 degradation could only be rescued by MG132 treatment, suggesting that OTUD7B promoted the proteasomal degradation of PIK3C3 ()). Since ubiquitination is indispensable for proteasomal degradation, we next assessed whether OTUD7B affects the stabilization of PIK3C3 by altering its ubiquitination. We overexpressed OTUD7B in HEK293 T cells and treated cells with MG132 for 6 h. We then checked the endogenous ubiquitination of PIK3C3, and surprisingly found that OTUD7B decreased the total ubiquitination of PIK3C3 () and S7A). It was reported that K63-linked ubiquitination of PIK3C3 can promote PIK3C3 stability [Citation26]. We next checked the K63-linked ubiquitination of PIK3C3 in the presence of OTUD7B and found that OTUD7B overexpression significantly reduced the K63-linked ubiquitination of PIK3C3 ()). To confirm our results, we performed in vitro deubiquitination assay by using purified OTUD7B and purified ubiquitinated PIK3C3 and observed that OTUD7B mediated the deubiquitination of purified PIK3C3 with K63 linkage in cell-free system ()). However, there is no direct evidence that K63-linked ubiquitination of PIK3C3 mediates its proteasomal degradation. We decided to check the K48-linked ubiquitination of PIK3C3, which is a universal recognition signal for the proteasome. Interestingly, we found that OTUD7B overexpression resulted in the upregulation of K48-linked ubiquitination of PIK3C3 ()), while knockdown of OTUD7B showed the decreased K48-linked ubiquitination of PIK3C3 (Fig. S7B). Considering the role of K48-linked ubiquitin in the proteasome, the increased K48-linked ubiquitination of PIK3C3 might be the direct reason that leads PIK3C3 to degradation. Together, these results indicate that OTUD7B might remove the K63-linked ubiquitin chains on PIK3C3, thus leaving the space for subsequent K48-linked ubiquitination to mediate its proteasomal degradation to inhibit autophagy flux.
Figure 7. OTUD7B modifies the K63-K48 type ubiquitination transition of PIK3C3. (a) Immunoassay of extracts of HEK293 T cells transfected with plasmids for MYC-tagged EV or OTUD7B, and then treated with dimethyl sulfoxide (DMSO; vehicle), MG132 (10 μM), 3 MA (10 mM), CQ (50 μM) or Baf A1 (0.2 μM) for 6 h. (b) Immunoassay of extracts of HEK293 T cells transfected with plasmids encoding MYC-tagged EV or OTUD7B and then treated with MG132 for 6 h, followed by denatured IP with anti-PIK3C3 and immunoblot analysis with anti-Ub. (c) Immunoassay of extracts of HEK293 T cells transfected with plasmids encoding MYC-tagged EV or OTUD7B and then treated with MG132 for 6 h, followed by denatured IP with anti-PIK3C3 and immunoblot analysis with anti-K63-Ub. (d) Purified ubiquitinated PIK3C3 was incubated with immunopurified Flag-OTUD7B in vitro in deubiquitinating buffer. The immunoblot was detected with anti-HA. (e) Immunoassay of extracts of HEK293 T cells transfected with plasmids encoding MYC-tagged EV or OTUD7B and then treated with MG132 for 6 h, followed by denatured IP with anti-PIK3C3 and immunoblot analysis with anti-K48-Ub. Data are representative of three independent biological experiments
Discussion
Since autophagy is a key process to maintain homeostasis and control protein quality [Citation9], its dysregulation is often associated with various diseases, such as Alzheimer disease, Paget disease and cancer [Citation6]. Thus, the autophagic process must be tightly regulated with multiple PTMs to avoid cellular disorders. Ubiquitination serves as one of the critical PTMs in autophagy regulation [Citation27]. It has been widely reported that plenty of E3 ligases, especially TRIM family proteins, function as crucial regulators during the autophagy process [Citation10,Citation28]. However, the importance of its reverse process – deubiquitination, is largely underestimated in autophagy.
Previous researches have revealed that DUBs employ diverse mechanisms to regulate various critical cell signaling, such as DNA repair [Citation29,Citation30], innate immune signaling pathways [Citation16], and cell cycle [Citation31]. From our screening, at least 30% of known DUBs are identified to influence autophagy to certain degree. The broad involvement of DUBs in autophagy and other cellular processes indicates the complicated coordination of autophagy with other critical cellular signaling. Based on our previous report, a large proportion of DUBs also participated in regulating antiviral immune responses [Citation16], implying that there is cross-regulation between autophagy and antiviral immune responses mediated by DUBs. Previously, we also demonstrated that USP19 functions as a positive regulator in autophagy, but a negative regulator in DDX58/RIG-I-induced type I interferon signaling [Citation14,Citation15]. This research may provide reference basis to explain the crosstalk between autophagy and antiviral immune responses.
To better uncover the roles of these DUBs in autophagy, we employed a co-IP screen system to investigate the relationship between the ATGs and DUBs. We discovered several previously unidentified DUBs as novel functional regulators in autophagy and revealed their involvement in regulating various key ATG proteins. We found that several ATGs, including ULK1 and PIK3C3, could be targeted by more than one DUB. Since the stability and activity of those ATGs are tightly linked to the induction and termination of autophagy, they should be strictly controlled and maintained at a reasonable level. Ubiquitination, as an important post-translational modification, plays a critical role in regulating the stability and activity of ATGs. For example, BECN1 has been reported to be regulated by several DUBs and E3 ligases. TRAF6 mediates the K63-linked ubiquitination of BECN1 to disrupt the interaction between BECN1 and BCL2, whereas the DUB TNFAIP3 can reverse this process [Citation11]. USP14 can also regulate autophagy by negatively controlling K63-linked ubiquitination of BECN1 [Citation18]. RNF216 promotes the ubiquitination and degradation of BECN1 [Citation12]. On the contrary, USP19 stabilized BECN1 by removing its K11-linked ubiquitin chains at Lys (K) 437 [Citation14,Citation15]. Collectively, different DUBs may exert different influences on the same ATG complex members to decide whether to enhance or inhibit autophagy and even the on-or-off of autophagy. Thus, we propose that the involvement of multiple DUBs generates a complicated and subtle network to fine-tune autophagy through regulating certain ATGs.
On the basis of the ubiquitin-modulating functions of STAMBP and OTUD7B, we proposed a working model to illustrate how they modulate autophagy at different levels, which might help us understand the diverse function of DUBs in autophagy. On the one hand, upon autophagy induction, STAMBP cleaves the K48-linked ubiquitin chains of ULK1 to block its proteasomal degradation, thus enhancing autophagy initiation. Other the other hand, OTUD7B deubiquitinates PIK3C3, which allows its subsequent K48-linked poly-ubiquitination for proteasomal degradation to prevent excessive activation of autophagy. Previously, many ubiquitination signals were reported to mediate the degradation of ATGs. KLHL20-CUL3 mediates the K48-linked ubiquitination of ULK1 and promotes its degradation [Citation32]. NEDD4 L is identified to enhance the K27- and K29-linked ubiquitination of ULK1, thereby leading to its proteasomal degradation [Citation25]. FBXL20 controls the ubiquitination and proteasomal degradation of PIK3C3 [Citation33]. Here, we also identified another DUB – STAMBP, which can affect ULK1 stability by removing its K48-linked ubiquitination. However, other roles of ubiquitination in autophagy remain largely unclear. In previous report, UBE2 N–UBE2V1–CHN1 can mediate the K63-linked ubiquitination of PIK3C3. Distinct from the previously reported ubiquitin-mediated proteasome degradation of PIK3C3 [Citation32,Citation33], this K63-linked ubiquitination promotes PIK3C3 stability [Citation26]. In another research on ULK1, the K63 linkage has also been found to promote ULK1 stability and function [Citation34]. These works initiate the study of other roles of ubiquitination of ATGs. However, it remains unsolved why K63-linked ubiquitination could enhance the stability of PIK3C3. Here, we found that the decrease of K63-linked ubiquitination of PIK3C3 eventually led to its increased K48-linked ubiquitination, which will mediate it proteasomal degradation. Some key proteins in immune signaling are reported to be ubiquitinated with linkage type transition. TBK1 undergoes a similar ubiquitination type transition (from K33 to K48) mediated by USP38 which is important for negatively regulating type I interferon signaling [Citation35]. TNFAIP3 also mediates the K63-K48 ubiquitination transition of RIPK1 (receptor interacting serine/threonine kinase 1) to affect NFKB signaling [Citation36]. The transition of ubiquitination of PIK3C3 might present us a different mechanism of autophagy regulation. The cleavage of one type of ubiquitin chain and the linkage of another type of ubiquitin chains might lead to the switch of signals in the autophagy initiation or termination. Overall, we reveal the distinct deubiquitination patterns mediated by DUBs tightly control the homeostasis of autophagic flux through manipulating the stability of ATGs.
Collectively, our findings create new insights into how DUBs regulate autophagy. Using a high-throughput screen, we found a large number of DUBs modulate autophagy, which is essential for the tight and subtle regulation of autophagy. Additionally, we report the detailed mechanisms employed by STAMBP and OTUD7B, which uncover the important roles of ubiquitination in the modification of ATG proteins. By revealing the stringent regulation of autophagy mediated by various DUBs, our study might yield novel diagnostic and therapeutic targets in the future.
Materials and methods
Cell lines and culture conditions
HEK293 T (ATCC, CRL-11,268), HeLa (ATCC, CCL-2), and A549 (ATCC, CCL-185) cells were cultured in DMEM medium (Gibco, 10,566,016) with 10% (vol:vol) fetal bovine serum (Gibco, 10,270,160) and 1% glutamine (Gibco, 25,030,081). PBMCs was maintained in RPMI-1640 medium (Gibco, 11,875–085) 10% fetal bovine serum. To induce starvation, cells were washed with phosphate-buffered saline (PBS; Gibco, 10,010,049) and incubated in EBSS (Gibco, 24,010,043). All cells were incubated in a 37°C incubator with 5% CO2.
Antibodies
Anti-ULK1 (sc-33,182), anti-STAMBP (sc-398,480), goat anti-mouse IgG-HRP (sc-2005) and anti-ubiquitin (sc-8017) were purchased from Santa Cruz Biotechnology; goat anti-rabbit IgG-HRP (31,460) was purchased from Thermo Fisher Scientific; anti-OTUD7B (66,276-1-Ig) were purchased from Proteintech; anti-BECN1 (3738), anti-LC3B (PM036), anti-K63-Ub (12,930) and anti-K48-Ub (4289) were purchased from Cell Signaling Technology; horseradish peroxidase (HRP)-anti-Flag (M2) (A8592) and anti-ACTB/β-actin (A1978) were purchased from Sigma; HRP-anti-hemagglutinin (clone 3F10), anti-MYC/c-Myc-HRP (11,814,150,001) were purchased from Roche Applied Science; unlabeled anti-MYC/c-Myc (9211) was purchased from TransGen; anti-PIK3C3 (ab5451) was purchased from Abcam The secondary antibodies for immunofluorescence were purchased from Biotium. Rabbit and mouse IgG were from Beyotime. Protein G agarose and Protein A agarose were purchased from Pierce.
Reagents
Puromycin (P9620), MG132 (C-2211-5 MG), rapamycin (37,094–10 MG), bafilomycin A1 (Baf A1; H2714), chloroquine (CQ; PHR1258-1 G), and 3-methyladenine (3 MA; M9281-100 MG) were purchased from Sigma. StarFect High-efficiency Transfection Reagent (GenStar, C101) was used to transfect expression plasmids.
RNA interference
Chemically synthesized 21-nucleotide siRNA duplexes were obtained from TranSheepBio and transfected using RNAiMAX (Invitrogen, 13,778,150) according to the manufacturer’s instructions. RNA oligonucleotides used in this study are as follows:
NC: 5′-GUUAUCGCAACGUGUCACGUA-3′;
STAMBP siRNA #1: 5′- GCAATATGAATGGAGCTTA-3′;
STAMBP siRNA #2: 5′- CAGAGTCAGTAGCCATTGT-3′;
OTUD7B siRNA #1: 5′-GGAUGACAUCGUUCAAGAATT-3′;
OTUD7B siRNA #2: 5′-CCCAUCCCUUGGAACGUAATT-3′.
Immunoassay and immunoprecipitation
To perform immunoprecipitation, whole-cell extracts were collected after transfection or stimulation, followed by incubation overnight at 4°C with the indicated primary antibodies plus Protein A/G beads (Pierce, 20,423), anti-Flag (Sigma, A2220) or anti-MYC agarose gels (Pierce, 20,168). Beads were then washed four times with low salt lysis buffer. For denatured deubiquitination assays in cultured cells, the cells were lysed with low-salt lysis buffer (50 mM HEPES [Gibco, 15,630,080], 150 mM NaCl [Guangzhou Chemical Reagent Factory], 1 mM EDTA [Vetec, V900106], 10% glycerol [Thermo Scientific, 17,904], 1.5 mM MgCl2 [Vetec, V900020], and 1% Triton X-100 [Invitrogen, HFH10]) and the supernatants were denatured at 95°C for 5 min in the presence of 1% SDS (Amresco, 0227). The denatured lysates were diluted with lysis buffer to reduce the concentration of SDS below 0.1% followed by immunoprecipitation (denature-IP) with the indicated antibodies. For immunoassay, immunoprecipitates or whole-cell lysates were resolved by SDS-PAGE. Proteins were then transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, 1,620,177) and immunoblotted with the indicated antibodies. Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0500) was used for protein detection.
In vitro deubiquitination assay
Ubiquitinated PIK3C3 or ULK1 was isolated from 293 T cells transfected with expression vectors for HA-Ub and Flag-PIK3C3 or Flag-ULK1. Ubiquitinated PIK3C3 or ULK1 was purified from the cell extracts with anti-Flag agarose gels (Sigma, A2220) in low salt lysis buffer with fresh protease inhibitor (Roche, 11,697,498,001). After extensive washing with the low salt lysis buffer, the proteins were eluted with Flag peptides (Sigma, F3290). The recombinant Flag-OTUD7B or Flag-STAMBP was expressed in 293 T cells and purified using anti-Flag agarose gels and eluted with Flag peptide. For in vitro deubiquitination assay, ubiquitinated PIK3C3 or ULK1 protein was incubated with recombinant OTUD7B or STAMBP in the deubiquitination buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM DTT [Sigma, 10,197,777,001], 5% glycerol) for 2 h at 37°C.
Fluorescence microscopy
HeLa cells were seeded on a glass-bottomed dish. For immunofluorescence, the cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized using methyl alcohol for 10 min at −20°C. Cells were then blocked in 5% fetal goat serum (Boster, AR0009) for 1 h and then stained with specific antibodies overnight. After washed with PBS for 3 times, cells were incubated with fluorescently labeled secondary antibodies (Alexa Fluor 488-conjugated antibodies against rabbit IgG [Biotium, CF®488], or Alexa Fluor 568-conjugated antibodies against mouse IgG [Biotium, CF®568]). Colocalization images were examined with a confocal microscope (Leica TCS SP8) equipped with a × 100 (NA 1.4) oil-immersion objective. For LC3B puncta quantification, cells were imaged by Leica DMI3000 B with a × 100 oil-immersion objective.
Quantitative real-time PCR
Total RNA was extracted from cells using the TRIzol reagent (Invitrogen, 10,296,010) according to the manufacturer’s instructions. For RT-PCR analysis, cDNA was generated from total RNA with HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R223-01) and was analyzed by quantitative real-time PCR using the SYBR Green qPCR Mix (GenStar, A301). All data were normalized to RPL13A expression. The primers used above are as follows:
STAMBP F: 5′-TCGATGGATTGCGCCATGT-3′;
STAMBP R: 5′-GTGTTGCAGTAATCAGACCCA-3′;
ULK1 F: 5′-GGCAAGTTCGAGTTCTCCCG-3′;
ULK1 R: 5′-CGACCTCCAAATCGTGCTTCT-3′;
OTUD7B F: 5′-GACAGAGAGCCTACTCGCC-3′;
OTUD7B R: 5′-CACAGATGGGCATTTCCAGG-3′;
PIK3C3 F: 5′-CCTGGAAGACCCAATGTTGAAG-3′;
PIK3C3 R: 5′-CGGGACCATACACATCCCAT-3′;
RPL13A F: 5′-GCCATCGTGGCTAAACAGGTA-3′;
RPL13A R: 5′-GTTGGTGTTCATCCGCTTGC-3′.
Generation of knockout cells
For sg_DUB HeLa-GFP-LC3B cells, lentiviral particles were produced by transfecting HEK293 T cells with target sequences cloned into pLentiCRISPRv2 (Addgene, 52,961). The medium was changed the following day and the viral containing supernatant was collected 48 h after transfection, filtered through a 0.45-μm filter (Millipore, SLHV033RB) and subsequently used to infect cells with polybrene (8 μg/ml; Sigma, TR-1003-G). HeLa-GFP-LC3B cells were infected by incubation with lentivirus-containing supernatant for 48 h. Transduced cells were purified by puromycin (Gibco, A1113803) selection.
For OTUD7B and STAMBP knockout cells, monoclonal cells were generated by standard CRISPR/Cas9 technology as previously described. The target sequences are as follows:
STAMBP sgRNA #1: 5′-GCCAGAGCGGAAGTACCGACG-3′;
STAMBP sgRNA #2: 5′-GCCCCGTCGGTACTTCCGCTC-3′;
OTUD7B sgRNA #1: 5′-GAGGAGATCTCGCGCTAGCCC-3′;
OTUD7B sgRNA #2: 5′-GTCAGATTTTGTCCGTTCCAC-3′.
Statistical analysis
Data are represented as mean ± SEM unless otherwise indicated, and Student’s t-test was used for all statistical analyses with the GraphPad Prism 5 software. Differences between two groups were considered significant when P value was less than 0.05.
Supplemental Material
Download MS Word (2.4 MB)Acknowledgments
We would like to thank Hongmei Li (School of Life Sciences, Sun Yat-sen University) for technical help for electron microscopy analysis. We are grateful to Daijia Huang (Sun Yat-sen University Cancer Center) for the kind gift of PBMCs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: How does it work? Annu Rev Biochem. 2016 Jun 2;85:685–713.
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate [Review]. Cell. 2014 Mar;157(1):65–75.
- Morel E, Mehrpour M, Botti J, et al. Autophagy: a druggable process. Annu Rev Pharmacol Toxicol. 2017;57:375–398.
- Antonioli M, Albiero F, Nazio F, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. 2014 Dec 22;31(6):734–746.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013 Dec;14(12):759–774.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013 May 9;368(19):1845–1846.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737.
- Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016 Jul 14;166(2):288–298.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues [Review]. Cell. 2011 Nov;147(4):728–741.
- Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014 Aug 25;30(4):394–409.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010 May 25;3(123):ra42.
- Xu C, Feng K, Zhao X, et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. 2014;10(12):2239–2250.
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011 Sep 30;147(1):223–234.
- Cui J, Jin S, Wang RF. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy. 2016 Jul 2;12(7):1210–1211.
- Jin S, Tian S, Chen Y, et al. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. Embo J. 2016 Apr 15;35(8):866–880.
- Liu Q, Wu Y, Qin Y, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv. 2018 May;4(5):eaar2824.
- Simicek M, Lievens S, Laga M, et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat Cell Biol. 2013 Oct;15(10):1220–1230.
- Xu D, Shan B, Sun H, et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016 Aug 1;30(15):1718–1730.
- Taillebourg E, Gregoire I, Viargues P, et al. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy. 2012 May 1;8(5):767–779.
- Kim JH, Seo D, Kim SJ, et al. The deubiquitinating enzyme USP20 stabilizes ULK1 and promotes autophagy initiation. EMBO Rep. 2018 Apr;19(4). DOI:10.15252/embr.201744378.
- Huang L, Hu C, Cao H, et al. MicroRNA-29c increases the chemosensitivity of pancreatic cancer cells by inhibiting USP22 mediated autophagy. Cell Physiol Biochem. 2018;47(2):747–758.
- McDonell LM, Mirzaa GM, Alcantara D, et al. Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly-capillary malformation syndrome. Nat Genet. 2013 May;45(5):556–562.
- Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989 Mar 24;243(4898):1576–1583.
- Finley D, Sadis S, Monia BP, et al. Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol. 1994 Aug;14(8):5501–5509.
- Nazio F, Carinci M, Valacca C, et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol. 2016 Dec 19;215(6):841–856.
- Liu J, Li M, Li L, et al. Ubiquitination of the PI3-kinase VPS-34 promotes VPS-34 stability and phagosome maturation. J Cell Biol. 2018 Jan 2;217(1):347–360.
- Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322.
- Pan JA, Sun Y, Jiang YP, et al. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. 2016 Apr 7;62(1):149–151.
- Moyal L, Lerenthal Y, Gana-Weisz M, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell. 2011 Mar 4;41(5):529–542.
- Nijman SM, Huang TT, Dirac AM, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005 Feb 4;17(3):331–339.
- Bonacci T, Suzuki A, Grant GD, et al. Cezanne/OTUD7B is a cell cycle-regulated deubiquitinase that antagonizes the degradation of APC/C substrates. Embo J. 2018 Aug 15;37:16.
- Hu MM, Yang Q, Xie XQ, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus [Article]. Immunity. 2016 Sep;45(3):555–569.
- Xiao J, Zhang T, Xu D, et al. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev. 2015 Jan 15;29(2):184–196.
- Nazio F, Strappazzon F, Antonioli M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013 Apr;15(4):406–416.
- Lin M, Zhao Z, Yang Z, et al. USP38 inhibits type i interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol Cell. 2016 Oct 20;64(2):267–281.
- Wertz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004 Aug 5;430(7000):694–699.