
ABSTRACT
SNCA/α-synuclein is a major component in the Lewy body (LB), a pathological hallmark of Parkinson disease (PD) and dementia with Lewy body (DLB), collectively known as synucleinopathies. SNCA/α-synuclein can be secreted from neurons and transmitted to neighboring cells including neurons and glia, which underlie the spreading of LB pathology as described by Braak and colleagues. We recently have investigated the mechanism and significance for microglia, a prototypic phagocyte in the brain, in engulfing and controlling SNCA/α-synuclein homeostasis in the brain. Using microglia-specific autophagy-deficient mice, we demonstrated that microglia ingest and degrade neuron-released SNCA/α-synuclein through SQSTM1/p62-mediated selective autophagy both in vivo and in vitro. This process requires the presence of TLR4 (toll like receptor 4), which interacts with SNCA/α-synuclein to induce the transcriptional upregulation of Sqstm1/p62 through the NFKB/NF-κB pathway. We term the selective autophagy of SNCA/α-synuclein as “synucleinphagy”. We showed that the disruption of microglial autophagy causes accumulation of misfolded SNCA/α-synuclein and loss of dopaminergic neurons, two hallmarks of PD. Hence, our study reveals a neuroprotective role of microglia through an autophagy-mediated “community cleanup program”.
KEYWORDS:
A hallmark of PD is the accumulation of neuronal LBs which contain aggregated SNCA/α-synuclein. Postmortem neuropathology studies suggested that aggregated SNCA/α-synuclein spreads by “prion-like” cell-to-cell transmission from the subcortical to cortical areas, a phenomenon known as the “Braak hypothesis”. This hypothesis also implies that controlling the extracellular levels of SNCA/α-synuclein produced in neurons is critical for preventing PD progression. Several studies have indicated that neurons and glial cells can uptake and degrade SNCA/α-synuclein in vitro. Importantly, microglia become activated in response to SNCA/α-synuclein in vitro. However, whether and how microglia can engulf and degrade SNCA/α-synuclein in vivo is unclear.
To investigate the role of microglia in SNCA/α-synuclein clearance, we adopted two different PD animal models: an AAV-mediated human SNCA/α-synuclein overexpression model (AAV-hα-Syn) and a transgenic model expressing a moderate level of human SNCA/α-synuclein (hα-Syn-Tg) using neuron-specific promoters, Syn (synapsin) and Thy1, respectively [Citation1]. In both PD models, microglia become activated as evidenced by shortened processes, reduced branch complexity, and increased cell number. Remarkably, we confirmed the presence of human SNCA/α-synuclein through western blot in freshly isolated microglia (ITGAM/CD11bhigh and PTPRC/CD45Intermediate cells) from the above models, which is the first line of direct evidence showing microglial engulfment of neuron-derived SNCA/α-synuclein in vivo.
We then asked how activated microglia clear up the ingested human SNCA/α-synuclein. It has been suggested that wild-type SNCA/α-synuclein is degraded by macroautophagy (hereafter referred to as autophagy), chaperone-mediated autophagy, and the proteasome. Early evidence for autophagic degradation was limited due to the use of autophagy inhibitors with poor specificity (e.g., 3-methyladenine) or cultured cells poorly linked to its original function (e.g., the PC12 cell line). In our study, we observed colocalization and puncta formation of ingested human SNCA/α-synuclein with autophagy machinery including ubiquitin, GFP-LC3, and SQSTM1, both in vivo and in vitro. Moreover, treatment with recombinant human SNCA/α-synuclein protein causes transcriptional upregulation of Sqstm1 mRNA and SQSTM1 protein production. Notably, SQSTM1, but not other autophagy receptors, such as CALCOCO2/Ndp52, OPTN, or NBR1, was found to be increased, suggesting specificity of SQSTM1 in acting as a receptor for SNCA/α-synuclein recruitment. Direct in vivo evidence also shows an increase of SQSTM1-positive puncta in microglia using the two above PD models. Mechanistically, it is known that SQSTM1, a selective autophagy receptor, interacts and recruits its target through oligomerization and connects this complex to phagophores for degradation. Consistent with this, we observed an interaction between ingested human SNCA/α-synuclein and high-molecular-weight oligomeric SQSTM1. Finally, we determined that SQSTM1 is required for human SNCA/α-synuclein and ubiquitin double-positive puncta using SQSTM1 knockout (KO) microglia. These findings highlight the key role of SQSTM1 as a receptor for SNCA/α-synuclein in synucleinphagy.
Interestingly, SQSTM1 recognizes and bind ubiquitinated targets through its ubiquitin-binding domain. Because we observed the colocalization of ingested SNCA/α-synuclein with ubiquitin, an important question that has arisen is whether SNCA/α-synuclein in microglia becomes ubiquitinated, and which E3 ubiquitin ligases may be responsible for this process. Further investigation is warranted to determine the role of ubiquitination in synucleinphagy.
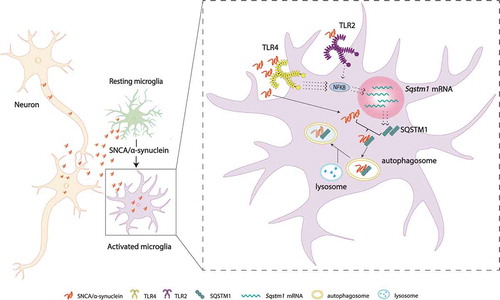
Our next question became, what was responsible for SQSTM1 upregulation in response to neuron released SNCA/α-synuclein? SQSTM1 can be upregulated by the transcription factor NFKB, downstream of TLRs. Interestingly, it was suggested that TLR4 and TLR2 may interact with SNCA/α-synuclein for microglial activation. Using TLR4 KO microglia we observed that SNCA/α-synuclein failed to induce upregulation of Sqstm1 mRNA and protein. However, Tlr4 KO microglia still show a minimal increase of Il1b and Tnf mRNA in response to SNCA/α-synuclein, which is completely blocked using a TLR2-blocking antibody, indicating that SNCA/α-synuclein also activate TLR2. Separately, using a TLR-NFKB-luciferase system established in HEK293 T cells, we showed that SNCA/α-synuclein can activate both TLR4 and TLR2. We also found that this system exhibited a binding preference for TLR4, further supporting the dominant role of TLR4 in binding SNCA/α-synuclein and causing SQSTM1 upregulation.
Despite our observations, many questions remain unanswered. What structural forms of SNCA/α-synuclein protein are required for TLR binding? For example, our study used primarily the human recombinant SNCA/α-synuclein, which is mostly in a soluble form. Another form of SNCA/α-synuclein that has been widely applied to modeling PD is SNCA/α-synuclein preformed fibrils (PFF). Injection of PFF into mice and rats recapitulates some characteristics of human PD including loss of dopaminergic neurons and the appearance and spreading of LB-like structures. Whether PFF can induce a similar synucleinphagy pathway in microglia remains unknown.
Finally, we showed that, despite the lack of neurodegeneration in microglia-specific autophagy-deficient mice (Atg7-cKO) or hα-Syn-Tg mice, the combination of the two genetic alterations by intercross of these two mice causes remarkable accumulation of insoluble and p-S129 SNCA/α-synuclein in lateral substantia nigra and dorsal striatum. Furthermore, the compound mice display a significant loss of TH-positive cells in the substania nigra pars compacta (SNpc). Therefore, our study demonstrates a critical role for microglia-specific autophagy in neuroprotection by clearing neuron-released SNCA/α-synuclein (). Given the results from genome-wide association study/GWAS showing the convergence of PD pathways with autophagy-lysosomes, it would be interesting to investigate how PD-risk factors affect synucleinphagy in glial cells (e.g. LRRK2G2019S). In summary, our findings reveal a novel protective function of microglia against neurodegeneration through a “community cleanup program”, implicating microglial synucleinphagy as a potential therapeutic target for PD or DLB.
Figure 1. A hypothetical model of “synucleinphagy”. Neuron-released SNCA/α-synuclein is degraded through SQSTM1-mediated selective autophagy in microglia. SNCA/α-synuclein preferentially binds TLR4 over TLR2 and activates the NFKB pathway, which leads to transcriptional upregulation of Sqstm1 mRNA. SQSTM1 then binds and recruits the SNCA/α-synuclein to the phagophore for degradation through a lysosome. TLR4, toll like receptor 4; TLR2, toll like receptor 2.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
Reference
- Choi I, Zhang Y, Seegobin SP, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386. PMID: 32170061.