
ABSTRACT
Alternative autophagy is an ATG5 (autophagy related 5)-independent, Golgi membrane-derived form of macroautophagy. ULK1 (unc-51 like kinase 1) is an essential initiator not only for canonical autophagy but also for alternative autophagy. However, the mechanism as to how ULK1 differentially regulates both types of autophagy has remained unclear. Recently, we identified a novel phosphorylation site of ULK1 at Ser746, which is required for alternative autophagy, but not canonical autophagy. We also identify RIPK3 (receptor-interacting serine-threonine kinase 3) as the kinase responsible for genotoxic stress-induced ULK1 S746 phosphorylation. These findings indicate that RIPK3-dependent ULK1 S746 phosphorylation plays a pivotal role in genotoxic stress-induced alternative autophagy.
The molecular mechanism of macroautophagy/autophagy was clarified mainly via studies on starvation-induced autophagy, in which autophagic membranes originate from the endoplamic reticulum (ER) or mitochondria-associated ER membranes, and functional complexes containing autophagy-related (ATG) proteins drive the formation of autophagosomes. However, different stimuli, including genotoxic stress, induce not only canonical autophagy but also a different type of autophagy, namely, ATG5-independent alternative autophagy or Golgi membrane-associated protein degradation (GOMED). In alternative autophagy, autophagosomal membranes originate from the trans-Golgi membranes, instead of ER membranes. Furthermore, most core effector molecules of canonical autophagy, such as ATG5, ATG9, and LC3, are not involved in alternative autophagy. Importantly, the degraded substrates are also different, indicating that these two autophagic pathways function differently in cells.
Although the molecules involved in these two autophagic pathways are mostly different, ULK1, a serine/threonine kinase, is commonly used in both pathways at the initial step. Therefore, elucidation of the mechanism by which ULK1 activates the different autophagic pathways is important in understanding autophagy, and we recently characterized this mechanism [Citation1]. We first focused on the ULK1 modifications observed during alternative autophagy. Using mass spectrometry analysis, we identified the specific phosphorylation of ULK1 at Ser746 (p-S746 ULK1) in ATG5-deficient cells upon etoposide treatment. Importantly, genotoxic stress-induced alternative autophagy is induced in ATG5 and ULK1-deficient cells expressing wild-type ULK1, but not in cells expressing a phosphodeficient ULK1 mutant (S746A). This ULK1 mutation does not affect canonical autophagy, indicating that the phosphorylation of ULK1 at Ser746 is the key to the different modes of activation of the two autophagic pathways.
We next made a polyclonal antibody against p-S746 ULK1 for the analysis of alternative autophagy. This antibody is applicable to immunofluorescence and immunoprecipitation, but not to immunoblotting. Using this antibody, we obtained the following results: (1) p-S746 ULK1 is generated at the early time points of genotoxic stress-induced alternative autophagy, and (2) p-S746 ULK1 signals are observed only on the Golgi apparatus, where phagophores originate. From these results and other detailed spatiotemporal analyses, we concluded that ULK1 is phosphorylated in the cytosol, then dissociates from the ULK1 complex together with RB1CC1/FIP200 and ATG13, translocates to the Golgi apparatus, and induces the generation of phagophores.
We and others previously reported the crucial role of ULK1 dephosphorylation at Ser637 in canonical autophagy. Interestingly, we found an essential role of this dephosphorylation also in alternative autophagy, because the phosphomimetic mutant of ULK1 (S637D) does not induce either canonical or alternative autophagy. Consistently, the S637A S746D mutant of ULK1 is sufficient to induce alternative autophagy. These data indicated that dephosphorylation at Ser637 determines the induction of autophagy, and phosphorylation at Ser746 determines the type of autophagy that is induced ().
Figure 1. Schematic model of alternative autophagy. Dephosphorylation of ULK1 at Ser637 determines the induction of autophagy, and further phosphorylation at Ser746 by RIPK3 determines the type of autophagy that is induced.
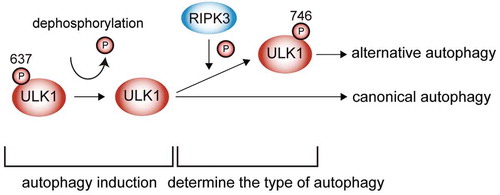
We further searched for the upstream kinase that phosphorylates ULK1 at Ser746, and identified RIPK3 (receptor-interacting serine-threonine kinase 3). This kinase was identified from the observations that (1) RIPK3 phosphorylates ULK1 at Ser746 in vitro, and (2) p-S746 ULK1 and alternative autophagy are not induced by genotoxic stress in RIPK3-deficient cells. Unlike alternative autophagy, RIPK3 does not affect canonical autophagy. Taken together, these data indicate that RIPK3 plays a pivotal role in genotoxic stress-induced alternative autophagy. RIPK3 is a well-known kinase involved in necroptosis, in which RIPK3 phosphorylates MLKL (mixed lineage kinase domain-like). Interestingly, the mechanism of phosphorylation of ULK1 and MLKL by RIPK3 is completely different. In the necroptosis signal, RIPK1 recruits RIPK3 using their RIP homotypic interaction motif (RHIM) domains, eventually inducing RIPK3 homo-oligomerization and generation of p-T231,S232 RIPK3, leading to MLKL activation. In contrast, RIPK3, but not RIPK1 and MLKL, is required for alternative autophagy. Furthermore, neither the RHIM domain, RIPK3 homo-oligomerization, nor p-T231,S232 RIPK3 generation is required for p-S746 ULK1 generation and alternative autophagy.
To date, several crosstalk pathways between cell death and autophagy have been reported. For example, the anti-apoptotic molecule BCL2 negatively regulates BECN1, and represses autophagy. We previously found that the pro-apoptotic factor PMAIP1/Noxa interacts with LC3 and is degraded by canonical autophagy upon genotoxic stress. In addition, our new findings suggest a crosstalk between necroptosis and alternative autophagy via sharing of the same molecule, namely, RIPK3. Because autophagy mainly contributes to cell survival, survival and death signals may act on RIPK3 and regulate each other.
In conclusion, we showed the following three findings: First, we identified a new phosphorylation site of ULK1 at Ser746, which determines the type of autophagy that is induced. Second, RIPK3 was identified as the kinase of p-S746 ULK1. Third, although RIPK3 phosphorylates MLKL for the induction of necroptosis, and ULK1 for the induction of alternative autophagy, their mechanisms are completely different. Our data also provide a marker of alternative autophagy, which is p-S746 ULK1. When alternative autophagy-inducing stimuli enter downstream of ULK1, p-S746 ULK1 will not be observed, and hence it is not a requisite marker. However, if this phosphorylation is observed, it means alternative autophagy is induced. We think our findings will be useful for further analyses of alternative autophagy in the future.
Disclosure statement
The authors declare no competing interests.
Reference
- Torii S, Yamaguchi H, Nakanishi A, et al. Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat Commun. 2020;11:1754.