
ABSTRACT
Caloric restriction mimetics (CRMs) are nontoxic macroautophagy/autophagy enhancers that act through the stimulation of cytoplasmic protein deacetylation reactions. Thus far, three functional classes of CRMs have been described: inhibitors of acetyltransferases (such as spermidine), inhibitors of acetyl coenzyme (AcCoA) synthesis (such as hydroxycitrate) and activators of deacetylases/sirtuins (such as resveratrol). Triethylenetetramine (also called trientine, abbreviated TETA) is a synthetic polyamine with resemblance in its structure to spermidine, a natural polyamine reputed for its pro-autophagic, anti-obesity and anti-aging effects. TETA, which is approved for the treatment of Wilson disease, has no effects on the longevity of mice, yet does induce autophagy and reduces weight gain in mice fed a high-fat diet (HFD). Mechanistically, these effects of TETA involve an increased activity of the TETA-metabolizing enzyme, SAT1 (spermidine/spermine N1-acetyltransferase 1). SAT1 overactivation ultimately results in the depletion of intracellular AcCoA with a consequent de-acetylation of cytoplasmic proteins and induction of autophagy. Accordingly, TETA fails to induce autophagy or to control HFD-induced weight gain in SAT1-deficient mice. Altogether, these findings indicate that TETA induces autophagy through a novel mode of action, namely, by the activation of an AcCoA-depleting enzyme.
There is large body of evidence in support of the notion that the overstimulation of nutrient-responsive circuits precipitates the decline in cellular fitness that characterizes the aging process, thus reducing the longevity of model organisms. Likewise, a “westernized” lifestyle (i.e., sedentarism coupled to excessive calorie intake) represents the principal risk factor for the onset of age-associated pathologies (e.g., type 2 diabetes, neurodegeneration, cardiovascular diseases and cancer) in humans. Conversely, non-pharmacological interventions that entail the general or selective shortage of nutrients (such as fasting or methionine restriction, respectively), as well as an enhanced oxidation of anabolic intermediates (i.e., physical activity), yield tremendous benefits in terms of lifespan and healthspan, regardless of the species tested.
While deciphering the mechanistic bases underlying the anti-aging properties of caloric restriction (CR) remains a challenging task, it appears that the ignition of cytoprotective autophagy is propaedeutic for the translation of the widespread cellular effects of CR into tangible therapeutic advantages. The deprivation of nutrients is in fact sufficient to elevate the rate of autophagosome formation beyond the physiological threshold in all tissues. In contrast, settings of autophagy deficiency (as achieved by the genetic deletion of the essential genes Atg5 or Atg7 late in life) precipitate the aging process and neutralize the anti-aging actions of CR.
Recently our group endeavored to provide a systematic definition of “caloric restriction mimetics” (CRMs), to be referred to as compounds that are able to recapitulate the major biochemical features of CR. Agents that fall under this classification must invariably elicit a triad of effects, namely: the absence of detectable toxicity, the reduction in the overall levels of protein acetylation, and the induction of autophagy. Based on these criteria, we identified three classes of CRMs:
Class 1 CRMs: Inhibitors of protein acetyltransferases, the autophagy repressor EP300 (E1A binding protein p300) being the prototypical one. Natural compounds (e.g., spermidine), established drugs (e.g., aspirin) and synthetic EP300 antagonists (e.g., C646) belong to this class.
Class 2 CRMs: Inhibitors of the acetyl co-enzyme A (AcCoA) biosynthetic pathways (e.g., the ACLY [ATP citrate lyase] inhibitor hydroxycitrate), which indirectly halt acetyltransferase enzymatic activity by reducing substrate availability.
Class 3 CRMs: Activators of protein (de)acetylases (in particular, sirtuins), such as resveratrol and NAD+-replenishing agents.
Noteworthy, several molecules annotated as bona fine CRMs display preclinical efficacy in numerous pathological settings, including (but not limited to) cancer and metabolic diseases. In particular, the natural polyamine spermidine is now in the spotlight for its wide positive effects on health that span from an extension of longevity that is evolutionarily conserved (from yeast to humans) to the prevention of cardiovascular, metabolic and neurodegenerative disorders. In our view, spermidine should be considered as a class I CRM, by virtue of its inhibitory action on EP300 and its capacity to decrease protein acetylation and to induce autophagy; an additional mechanism (i.e., the EIF5A [eukaryotic translation initiation factor 5A]-dependent translation of the pro-autophagic TFEB [transcription factor EB]) is proposed to explain the rejuvenating effect of spermidine in old B cells, leaving open the possibility that spermidine’s mode of action is tissue/cell-type specific.
Recently, we elucidated the mode of action of triethylenetetramine (trientine or TETA), an FDA-approved copper-chelating agent used as a second intention treatment in Wilson disease. Intrigued by its structural resemblance to spermidine, we decided to assess the efficacy of TETA to improve systemic glucose homeostasis in mice challenged by high-calorie dietary regimens (high-fat diet [HFD] or 30% sucrose) or in LEP (leptin)-deficient ob/ob animals. We found that TETA displays a prominent anti-metabolic syndrome effect under obsesogenic conditions, as indicated by improved glucose tolerance, increased INS (insulin) sensitivity and reduced adiposity, with minimal/absent signs of systemic toxicity. At the hepatic level, TETA mitigates the rate of steatosis, suggesting that this agent may be safely employed to counteract aberrant lipid accumulation in nonalcoholic fatty liver disease (NAFLD). Of note, these effects cannot be attributed to the chelation of bivalent ions, as the levels of Cu2+, Fe2+ and Zn2+ remain unaffected, even upon continuous treatment with TETA. Untargeted metabolomics analysis performed on the liver of TETA- or spermidine- treated animals revealed that TETA elicited significant disturbance of endogenous spermidine metabolism, as revealed by the heightened N1-acetylspermidine:spermidine ratio; this result (that was further confirmed by metabolic flux analysis using 13 C-labeled spermidine) indicates that TETA can indeed enhance intracellular polyamine metabolism.
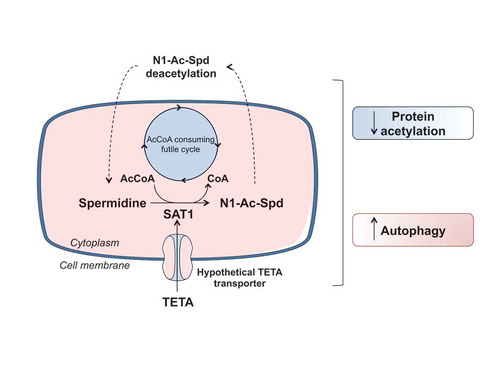
The catabolism of endogenous spermidine (which is present in the cells at an order of magnitude of 1 mM) mainly relies on the activity of SAT1, a rate-limiting enzyme that acetylates intracellular spermidine and promotes its secretion, at the expense of AcCoA. Importantly, TETA stabilizes and increases the activity of SAT1 at the post-translational level, instigating an AcCoA-consuming futile cycle. Accordingly, SAT1 activation by TETA is sufficient to reduce protein acetylation in the liver and to ignite autophagy in hepatocytes. Conversely, knockout of the gene coding for SAT1 abolishes the TETA effects that define it as a CRM (in particular, cytoplasmic protein deacetylation and autophagy induction), lending further credibility to the hypothesis that SAT1 is the main target of TETA. Importantly, sat1−/-, but not partially autophagy-defective atg4b−/-, mice are resistant to the anti-diabetic effects of TETA, highlighting SAT1 as a promising candidate for the treatment of age-associated metabolic disorders. More in-depth investigation will be needed to clarify the organ-specific contribution of SAT1 to the systemic benefits of TETA.
Collectively, our data designate TETA as a novel CRM that does not fall in any of the categories previously described; the ability of TETA to induce autophagy appears to be tied to the stabilization/activation of the AcCoA-consuming enzyme SAT1, which acetylates endogenous spermidine, therefore reducing the availability of AcCoA for further protein acetylation reactions (). In the near future it will be important to solve the conundrum of whether the pharmacological or genetic activation of enzymes that consume AcCoA could be considered as a general strategy to recapitulate the beneficial effects of CR. In that case, TETA would represent the prototypical example of an AcCoA-depleting class 4 CRM.
Figure 1. Mechanism of autophagy induction by TETA. The activation of SAT1 (spermidine/spermine N1-acetyltransferase 1) results into the generation of N1-acetyl spermidine (N1-Ac-Spd), which is extruded from the cell, deacetylated in the extracellular space and then re-imported into the cell. This creates a futile cycle leading to the consumption of acetyl co-enzyme A (AcCoA). Because the cytosolic AcCoA concentration is in an equilibrium state with lysine acetylation of cytoplasmic protein, this results in protein deacetylation and induction of autophagy.
Acknowledgments
F.M. is grateful to the Austrian Science Fund FWF (SFBLIPOTOX F3007&F3012, W1226, P29203, P29262, P27893) and the Austrian Federal Ministry of Education, Science and Research and the University of Graz for grants “Unkonventionelle Forschung” and “flysleep” (BMWFW-80.109/0001-WF/V/3b/2015). F.M. acknowledges the funding of DK Metabolic and Cardiovascular Disease (FWF) and the Doctoral College “Metabolic and Cardiovascular Disease” (FWFW1226) as well as support from NAWI Graz and the BioTechMed-Graz flagship project “EPIAge”. GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Le Cancer du Sein, Parlons-en!”; Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a dona-tion by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Pro-ject Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085), Institut National du Cancer (INCa); Inserm (HTE); Institut Univer-sitaire de France; LeDucq Foundation; the LabEx Immu-no-Oncology (ANR-18-IDEX-0001); the RHU Torino Lu-mière; the Seerave Foundation; the SIRIC Stratified On-cology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). F.P. is supported by a Karolinska Institute Starting Grant; Starting Grant from the Swedish Research Council (2019_02050_3).
Disclosure statement
F. M. and G.K. are the scientific co-founders of Samsara Therapeutics, a company that develops novel pharmacological autophagy inducers. F.M. is a scientific cofounder of The Longevity Labs, a company that develops natural food extracts. GK consults for The Longevity Labs and is a co-founder of Therafast Bio, a company that develops caloric restriction mimetics for the treatment of cancer. G.K. and F.P. hold a patent protecting new medical use for TETA.
Reference
- Castoldi F, Hyvönen M, Durand S, et al. Chemical activation of SAT1 corrects diet-induced metabolic syndrome. Cell Death Differ. 2020 May 6. doi:10.1038/s41418-020-0550-z. Online ahead of print.