
ABSTRACT
As one of the major, highly conserved catabolic pathways, autophagy delivers cytosolic components to lysosomes for degradation. It is essential for development, cellular homeostasis, and coping with stress. Reduced autophagy increases susceptibility to protein aggregation diseases and leads to phenotypes associated with aging. Of the three major forms of autophagy, macroautophagy (MA) can degrade organelles or aggregated proteins, and chaperone-mediated autophagy is specific for soluble proteins containing KFERQ-related targeting motifs. During endosomal microautophagy (eMI), cytoplasmic proteins are engulfed into late endosomes in an ESCRT machinery-dependent manner. eMI can be KFERQ-specific or occur in bulk and be induced by prolonged starvation. Its physiological regulation and function, however, are not understood. Here, we show that eMI in the Drosophila fat body, akin to the mammalian liver, is induced upon oxidative or genotoxic stress in an ESCRT and partially Hsc70-4-dependent manner. Interestingly, eMI activation is selective, as ER stress fails to elicit a response. Intriguingly, we find that reducing MA leads to a compensatory enhancement of eMI, suggesting a tight interplay between these degradative processes. Furthermore, we show that mutations in DNA damage response genes are sufficient to trigger eMI and that the response to oxidative stress is under the control of MAPK/JNK signaling. Our data suggest that, controlled by various signaling pathways, eMI allows an organ to react and adapt to specific types of stress and is thus likely critical to prevent disease.
Abbreviations: Atg: autophagy-related; CMA: chaperone-mediated autophagy; DDR: DNA damage repair; Df: deficiency (deletion); (E)GFP: (enhanced) green fluorescent protein; eMI: endosomal microautophagy; ER: endoplasmatic reticulum; ESCRT: endosomal sorting complexes required for transport; Eto: etoposide; FLP: flipase; Hsc: heat shock cognate protein; LAMP2A: lysosomal-associated membrane protein 2A; LE: late endosome; MA: macroautophagy; MI: microautophagy; MVB: multivesicular body; PA: photoactivatable; Para: paraquat; ROS: reactive oxygen species; SEM: standard error of means; Tor: target of rapamycin [serine/threonine kinase]; UPR: unfolded protein response; Vps: vacuolar protein sorting.
Introduction
Cellular homeostasis requires a well-controlled balance between continuous protein synthesis and degradation of damaged or unneeded peptides. Protein turnover is intricately connected to the cellular stress response and failure to react to adverse conditions including starvation, oxidative damage, or DNA damage can have detrimental consequences for an organism [Citation1–4]. Autophagy is one of the major proteolytic systems present in all eukaryotic organisms analyzed to date and contributes to the removal of long-lived, altered, nonfunctional proteins and organelles and is therefore critical to a cell’s response to cytotoxic stress [Citation3–7]. Indeed, alterations in autophagy are associated with a large array of pathologies including cardiomyopathies, neurodegeneration, cancer, and aging [Citation3,Citation7–11]. Three major types of autophagy have been described: macroautophagy (MA), chaperone-mediated autophagy (CMA) and (endosomal) microautophagy (eMI). Macroautophagy is initiated with the de novo formation of a double-membrane structure (phagophore) that grows to form a vesicle, the autophagosome, engulfing cytoplasmic material targeted for degradation upon fusion with lysosomes [Citation12–15]. Autophagy was initially thought to be a “bulk” process, but recently, selective degradation of pathogens, organelles or aggregated proteins via MA have been described [Citation16,Citation17]. CMA, on the other hand, selectively degrades soluble cytosolic proteins containing a motif biochemically related to KFERQ [Citation18]. During CMA, the KFERQ-motif of the substrate protein is recognized by the cytosolic chaperone HSPA8/Hsc70 (heat shock protein family A [Hsp70] member 8) [Citation19], which delivers substrates to LAMP2A (lysosomal associated membrane protein 2A) [Citation20], the limiting factor for CMA. Upon unfolding, the substrates translocate across the lysosomal membrane through a channel formed by LAMP2A and are degraded in the lysosomal lumen [Citation21].
Less is known about microautophagy, the engulfment of material directly into the lysosome (mainly described in yeasts), or in the case of endosomal microautophagy (eMI), by the late endosome (reviewed in [Citation21–23]). Biochemical and electron microscopic studies in mammalian cells showed that late endosomes (LEs) engulf cytosolic material in multivesicular bodies (MVBs), which is then degraded in late endosomes or, upon their fusion, in lysosomes [Citation24]. eMI can degrade cytosolic components in bulk by trapping as well as selectively in a KFERQ- and HSPA8-dependent manner. In contrast to CMA, which requires LAMP2A for HSPA8 binding to lysosomes, HSPA8 docks on phosphatidylserine lipids on LEs and substrate invagination is dependent on the endosomal sorting complexes required for transport (ESCRT) machinery [Citation24]. While CMA seems restricted to mammals and potentially birds due to the absence of the LAMP2A isoform in the genome of other species [Citation25], eMI has recently also been identified in Drosophila melanogaster [Citation26,Citation27]. As in mammals, eMI in flies requires Hsc70-4, a paralog of HSPA8 and members of the ESCRT I, II, and III complexes [Citation26,Citation27]. However, not much is known about the physiological role of eMI. It has been proposed that it may be involved in the degradation of oxidized proteins that accumulate in MVBs of old mice [Citation21,Citation28]. In Drosophila larvae, eMI controls the turnover of synaptic proteins in neuromuscular junctions in a Hsc70-4- and KFERQ-dependent manner [Citation27]: a faster protein turnover induced by overexpression of Hsc70-4 increases the readily releasable pool of neurotransmitters.
Autophagy often protects against cellular stress such as nutrient scarcity. Similar to CMA [Citation29–31], but distinct from eMI in mammals, where it was not induced by starvation [Citation24], eMI in the fly fat body that has functions akin to the liver is induced upon prolonged starvation [Citation26]. Our lab previously showed that genetic or pharmacological inhibition of Tor signaling is sufficient to induce eMI under nutrient-rich conditions. Starvation-induced eMI is dependent on the autophagy genes Atg1 and Atg13 [Citation26], homologs of two members of the ULK1 complex, suggesting that the Atg1-Atg13 complex, similar to its function in MA, also is required for Tor-inhibition-mediated activation of eMI.
To identify physiological roles of eMI, we investigated the induction of eMI upon exposure to different forms of cellular stress. In various organisms, both MA and CMA are induced not only by starvation but also by exposure to DNA-damaging agents or reactive oxygen species [Citation29,Citation30,Citation32–39]. Using a photoactivatable eMI sensor [Citation26], we found that, in addition to starvation, prolonged exposure to oxidative stress and DNA damage, but not ER stress, induce eMI in the larval fat body, revealing an unexpected selectivity. While strictly dependent on the ESCRT machinery, a significant fraction of the response seen is due to bulk eMI, as it is only partially dependent on Hsc70-4 and a KFERQ motif. Mechanistic studies show that the DNA-damage response kinases ATM or ATR are dispensable for induction of eMI upon DNA damage and that JNK signaling regulates ROS-mediated eMI. Our data thus suggest that eMI in Drosophila has broad, but selective functions in response to environmental stress.
Results
eMI in Drosophila is activated by DNA damage and oxidative stress, but not ER stress
Under physiological conditions, animals are confronted with DNA damage of various origins including radiation and oxidative stress mainly due to reactive oxygen species (ROS) coming from the mitochondrial respiratory chain under high energy demand [Citation37,Citation40]. DNA damage triggers MA [Citation37] and CMA [Citation41] in mammalian cells; thus, we tested whether DNA damage induced eMI. The Topoisomerase II inhibitor etoposide (Eto) induces double-strand breaks in cultured Drosophila cells [Citation42]. To measure the effect of Eto on the eMI pathway, we subjected late second/early third instar larvae expressing an eMI sensor (photoactivatable [PA] UAS-KFERQ-PA-mCherry) [Citation26] together with an (endo)lysosomal marker (GFP-HsLAMP1 [Citation43]) under the control of the fat body-specific cg-Gal4 driver to food containing heat-inactivated yeast and 50 µM Eto. An eMI response reflected by sensor puncta formation was detected upon treatment for 12 h that increased upon 25 h of treatment (; quantified in ). No sensor puncta were found at 4 h of treatment (). To confirm this response, we fed larvae doxorubicin, known to affect DNA replication and to deregulate the DNA repair response (DDR) [Citation44] and found a strong eMI sensor signal ().
Figure 1. DNA damage and oxidative stress induce eMI in the larval fat body. (A, B) Compared to untreated control (A), treatment of larvae with the DNA damaging agent Eto for 25 h robustly induces eMI puncta. (C, D) Oxidative stress induced by paraquat induces eMI. (C) Untreated control, (D) fat body of paraquat-treated larvae. eMI sensor is shown in red, (endo)lysosomes in green, nuclei are in blue. A’-D’ and A” to D”: single-channel image of indicated channels. Scale bar: 20 µm. (E) Quantification of eMI puncta per cell at indicated time points. Grey: untreated, blue: paraquat-, and magenta: Eto-treated. (F) Quantification of eMI puncta upon treatment with 50 µM doxorubicin (magenta). (G) Quantification of eMI puncta upon treatment with indicated concentrations of menadione bisulfite (blue). E: One-way ANOVA (Dunnett corrections) p < 0.05; F: Unpaired T-test ****, P < 0.0001. G: One-way ANOVA (Tukey corrections) p < 0.0001. *, P < 0.05; **, P < 0.01, ****, P < 0.0001. Unpaired T-test results are indicated as “t-values”
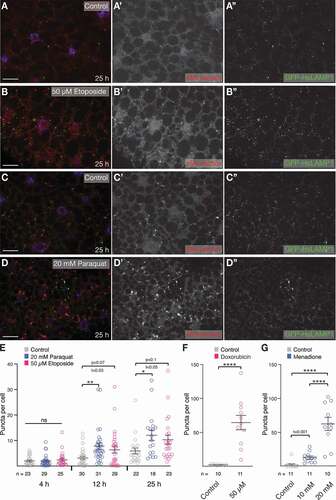
Oxidative stress is an additional activator of the MA and CMA pathways to reduce toxic or damaging agents as a way to cope with homeostatic imbalance [Citation35,Citation45]. In Drosophila, paraquat and its metabolites were shown to elicit a ROS response when provided in regular food, as measured by an increase in superoxide dismutase activity [Citation46]. Additionally, in larvae, MA is induced in the fat body and midgut by exposing animals to 100 mM paraquat [Citation47]. We find a robust eMI response caused by 20 mM paraquat after 12 h and 25 h of drug treatment (; quantified in ; dose-response in Fig. S1B). As an independent manner to induce oxidative stress, we treated larvae with menadione Na-bisulfite, which generates superoxide anion radicals and is known to increase superoxide dismutase activity in flies and to stimulate CMA in mammalian cells [Citation48,Citation49]. Menadione also strongly induced the eMI sensor (). Under the conditions tested, neither paraquat nor Eto induced a strong cell death response as quantified by staining for activated Dcp-1 (the fly ortholog of CASP3; Fig. S2). Thus, prolonged treatment of larvae with DNA damaging and oxidizing agents elicits a strong eMI sensor response similar to the induction of eMI upon starvation [Citation26].
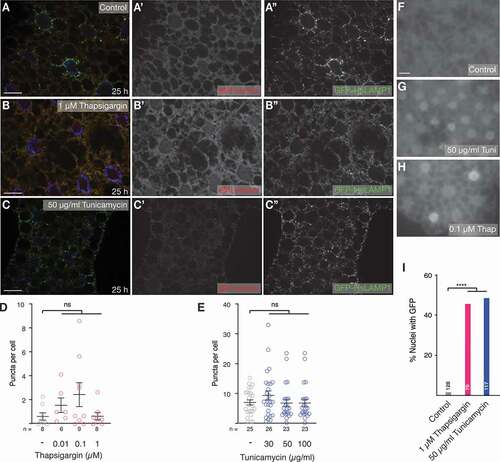
ER stress can be caused by accumulation of misfolded proteins, which activates the unfolded protein response (UPR) that can trigger MA in vertebrates [Citation50]. Thapsigargin inhibits ATP2A/SERCA, and tunicamycin blocks protein N-glycosylation, and both are commonly used to induce ER stress [Citation51]. To elucidate the effect of ER stress on eMI, we treated larvae with increasing concentrations of thapsigargin and tunicamycin, but failed to observe an effect on the eMI sensor (compare control in with ; quantification of dosage curve in ). It was, therefore, necessary to confirm that ER stress was indeed induced at the drug concentrations used. The UPR triggers non-canonical splicing of Xbp1 by Ire1, resulting in the removal of a frame-shift in Xbp1 and thus the formation of an active transcription factor. Using a transgene that indicates ER stress by nuclear localization of Xbp1-GFP upon UPR activation, we found that at concentrations used, both drugs strongly induced ER stress (; quantification in ). Our data thus suggest that eMI is selectively activated by starvation, DNA damage, and oxidative stress, but not by ER stress.
Figure 2. ER stress does not elicit an eMI response. (A-C) Compared to control (A, A’), treatment of larvae with 1 µM thapsigargin (B, B’) or 50 µg/ml tunicamycin (C, C’) does not cause the formation of sensor puncta in the fat body. (Endo)lysosomes are marked using GFP-hsLAMP1. Monochrome images show indicated channels. Nuclei are in blue. (D, E) Quantification of a dose-response curve of treatment with thapsigargin (D) and tunicamycin (E). One-way ANOVA (Dunnett corrections): ns: not significant. (F-I) At indicated concentrations thapsigargin (G) and (H) tunicamycin induce ER stress reflected by nuclear GFP signal of the Xbp1 sensor. (I) Percentage of nuclei with GFP of indicated samples; Fisher’s exact test (Bonferroni correction); ****, P < 0.0001. n: fields of view (fat body lobes), except in (I), where it reflects total number of cells. Scale bars: 20 µm
The eMI response is ESCRT-dependent and also occurs in bulk
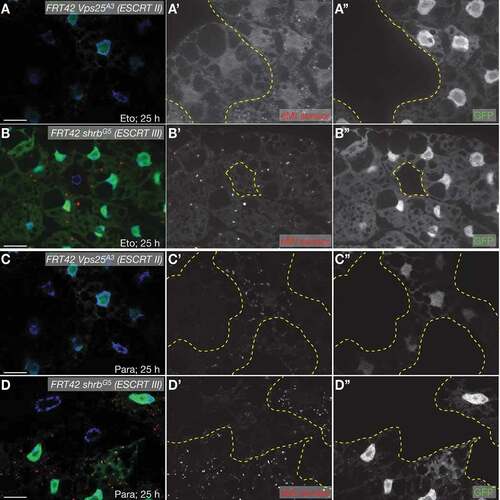
In contrast to CMA, eMI is dependent on MVB formation for uptake of substrates into late endosomes in mammalian cells and in Drosophila [Citation24,Citation26,Citation27]. Similar to starvation-induced eMI, sensor puncta formation upon DNA damage and oxidative stress is dependent on members of the ESCRT machinery. Compared to neighboring wildtype cells, cells homozygous mutant for the ESCRT II component Vps25 or the ESCRT III component shrb/Vps32 fail to form eMI puncta upon treatment with Eto () or paraquat (), respectively.
Figure 3. eMI sensor puncta formation requires ESCRTs cell-autonomously. Fat body cells lacking GFP (outlined in yellow in A’-D’) are homozygous mutant for the ESCRT II component Vps25 (A, B) and the ESCRT III component shrb (C, D) and fail to elicit an eMI response (red in composite image; compare with heterozygous or homozygous wildtype, GFP-positive cells). (A, B) 50 µM Eto treatment for 25 h. (C, D) 20 mM paraquat treatment for 25 h. Nuclei are in blue in merged images. A’- D’ and A”- D” are monochrome images of the indicated channels. Scale Bar: 20 µm
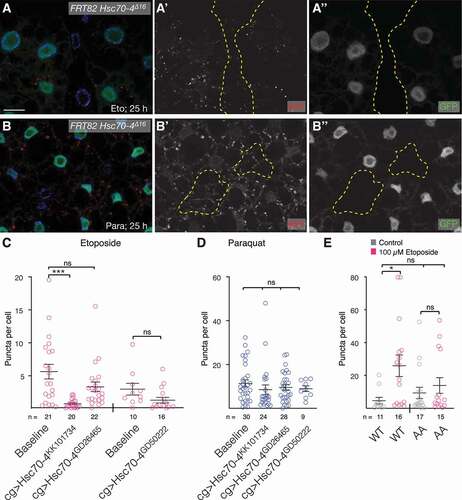
In mammalian cells, eMI can occur in bulk, but also in a manner dependent on a KFERQ motif, as knockdown of HSPA8 reduces eMI to 50% [Citation24]. Similarly, eMI in the neuromuscular junction is dependent on and can be stimulated by Hsc70-4, the most closely related homolog of HSPA8 [Citation27], and we originally showed a strict KFERQ dependence in starvation-induced eMI in the larval fat body (see discussion) [Citation26]. Homozygous mutant cells for the Hsc70-4Δ16 null allele show a reduction of eMI sensor puncta in larvae exposed to Eto and paraquat (). RNAi-mediated knockdown of Hsc70-4 showed a tendency toward reduction of eMI puncta upon treatment with Eto () but no significant effect by paraquat (). Consistent with the partial Hsc70-4-dependence, we find that a sensor in which the KFERQ motif was mutated to KFEAA [Citation26] shows a lower (and not significant) response to Eto treatment (). Overall, these data suggest that eMI induced by DNA damage or oxidative stress is due to a combination of selective and bulk uptake of cargo into MVBs.
Figure 4. DNA damage and ROS induced eMI is in part a bulk process. (A, B) Eto-treated (A) or paraquat-treated (B) Hsc70-4Δ16 homozygous mutant cells (lacking GFP expression; outlined in yellow in A’, B’) show a partially reduced eMI response (red) compared to neighboring cells. Nuclei are in blue in merged images. Monochrome images show indicated channel. Scale bar: 20 µm. (C, D) Quantification of eMI response in fat body cells treated with Eto (C) or paraquat (D) expressing indicated knockdown constructs. One-way ANOVA (Dunnett corrections) p < 0.001; ***, P < 0.001. (E) Compared to the WT sensor, a sensor in which the KFERQ motif was mutated to KFEAA (AA) does not show a significant puncta formation. One-way ANOVA (Tukey corrections) p < 0.05; *, P < 0.05. ns: not significant
eMI response is enhanced in the absence of macroautophagy
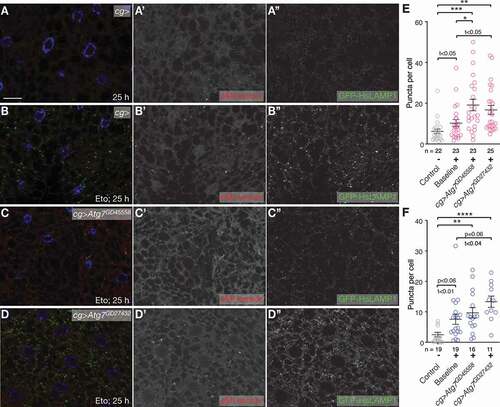
Various proteostasis pathways communicate with one another. It is, however, unknown if eMI shows crosstalk with other forms of autophagy. We, therefore, knocked down Atg7, a key MA gene in the fat body [Citation32] using RNAi lines that have previously been shown to prevent the induction of starvation-induced MA [Citation26]. Compared to Eto treatment alone, we found an increased eMI response in the absence of MA (; quantified in ). A similar tendency was found upon eMI induction by oxidative stress (quantified in ). Our data thus suggest that the eMI response is enhanced to compensate for the absence of MA and that there is a crosstalk between those two types of autophagy in the Drosophila fat body. Additionally, this demonstrates that similar to starvation-induced eMI [Citation26], the eMI sensor puncta due to exposure to DNA damage or ROS are distinct from of MA.
Figure 5. eMI compensates for lack of MA. (A-D) Compared to untreated fat body cells (A) and Eto treatment alone (B), RNAi mediated knockdown of Atg7 with indicated RNAi lines (C, D) leads to an enhanced eMI response (red; (endo)lysosomes marked by GFP-HsLAMP1 in green, nuclei in blue). Monochrome images show indicated channels. (E, F) Quantification of the eMI response in indicated genotypes upon treatment with Eto (E) and paraquat (F). One-way ANOVAs (Tukey correction; (E) p < 0.001; (F) p < 0.0001). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Unpaired T-test results are indicated as “t-values”
Mechanism of eMI induction
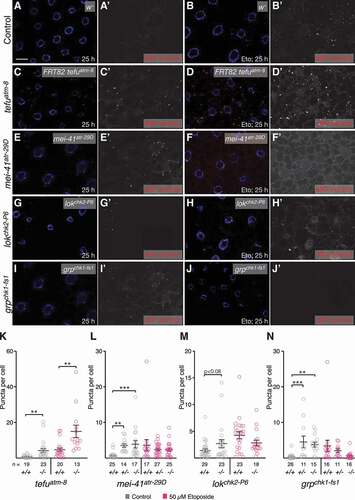
eMI triggered by nutrient deprivation in Drosophila is controlled by inhibition of Tor and requires Atg1 and Atg13 [Citation26], but it is unknown how ROS or DNA damage induced by Eto activate eMI. Key to DNA repair are the kinases ATM (ATM serine/threonine kinase) and ATR (ATR serine/threonine kinase), both members of the phosphatidylinositol 3-kinase-related kinases (reviewed in [Citation37,Citation52]). DNA double-strand breaks such as the ones created by TOP2A/Topo II inhibitors are recognized by the MRN complex (MRE11, RAD50, NBN/Nbs1), which leads to the recruitment and activation of ATM (tefu in Drosophila [Citation53]). Single-strand breaks of base adducts lead to the activation of ATR (mei-41 in Drosophila [Citation54]). Although there is a lot of crosstalk between the ATM-ATR pathways, ATM generally activates CHEK2/CHK2 (lok [loki] in flies [Citation55]) and TP53, leading to cell cycle arrest, DNA repair and induction of apoptosis and MA. ATR phosphorylates CHEK1/CHK1 (grp [grapes] in flies [Citation56]), triggering cell cycle arrest in G2 and DNA repair. We thus tested whether mutation of these genes affected eMI in the fat body.
In the absence of externally induced DNA damage, homozygous tefuatm−8, mei-41atr−29D, and grpchk1-fs−1 mutants showed an increase in eMI (; quantified in ; lokchk2-P6 shows a similar tendency). Interestingly, heterozygosity for mei-41/atr and grp/chk1 was sufficient for eMI induction (see discussion). eMI induction under these conditions is likely due to accumulating DNA damage due to repair defects, supporting our data showing that Eto-induced sensor puncta are indeed due to DNA damage. In the case of mei-41/atr and grp/chk1, similar results were obtained with RNAi-mediated knockdown (Fig. S3). When DNA damage was induced by Eto, tefuatm−8 showed a dramatic increase of eMI (, quantified in ), while no further stimulation of eMI was seen in mei-41atr−29D, lokchk2-P6, or grpchk1-fs−1 mutants (; quantified in ). Taken together, these data show that neither the ATM-CHEK1 nor the ATR-CHEK2 branch of DNA damage repair are uniquely required for the eMI response due to DNA damage.
Figure 6. Loss of components of the ATM and ATR pathways induces eMI. (A, C, E, G, I) Untreated fat body. Compared to w- control (A), tefuatm−8 (C), mei-41atr−29D (E), and grpchk1-fs1 (I) mutants show increased eMI activity (red). lokchk2-P6 (G) may show a tendency toward increased sensor signal. (B, D, F, H, J) Eto-treated larvae. Compared to w− control-treated larvae (B), atm/tefu-8 (D) larvae show an enhanced eMI response, while mei-41atr−29D (F), lokchk2-P6 (H), and grpchk1-fs1 (J) mutants show no significant change. Nuclei are in blue, monochrome images show eMI. Scale bar: 20 µm. (K-N) Quantification of eMI response in untreated (gray) and Eto-treated larvae (magenta) of indicated genotypes. Note that mei-41atr−29D and grpchk1-fs1 heterozygotes also show a phenotype in the absence of drug. (K, M) t-tests. (L, N) One-way ANOVAs (Dunnett corrections): p < 0.0001 (L); p < 0.001 (N). *, P < 0.05; **, P < 0.01; ***, P < 0.001
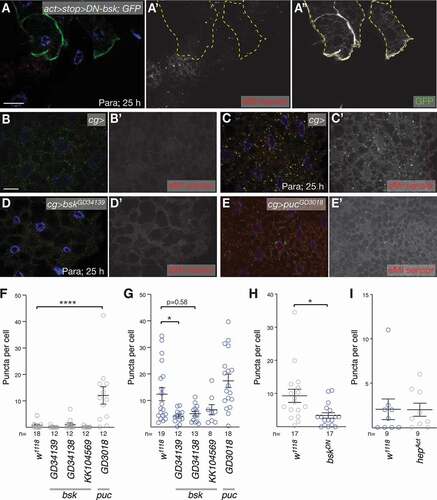
Mild oxidative stress activates MA in several ways (reviewed in [Citation40,Citation45]). AMP-activated protein kinase (AMPK) itself is redox-sensitive and can activate the ULK1/Atg1 complex leading to activation of MA. Alternatively, MAPK/JNK can phosphorylate BECN1 (Beclin 1), reducing its affinity to BCL2, thus triggering MA. We thus tested whether Bsk/MAPK/JNK (Basket) affects the eMI response to ROS. Dominant-negative bsk efficiently reduces MAPK/JNK signaling in the Drosophila eye [Citation57]. Its expression in a mosaic pattern in the fat body reduced the eMI response cell-autonomously (; quantified in ). Similarly, RNAi-mediated knockdown of bsk reduced the eMI response upon paraquat exposure (; quantified in ). In contrast, expression of an activated form of the Drosophila hep/MAPKK/JNKK (hemipterous) was not sufficient to induce eMI (). Importantly, knockdown of puc (puckered), a known feedback inhibitor of Bsk, was sufficient to induce eMI in the absence of oxidative stress (), suggesting that activation of Bsk is sufficient for eMI induction. Together, our data thus show that ROS-mediated eMI induction in the fly fat body is mediated by MAPK/JNK signaling.
Figure 7. JNK signaling regulates ROS induced eMI. (A) Expression of DN-Bsk marked by co-expression of GFP (A”) cell-autonomously blocks eMI (A’) induced by paraquat. (B-D) Compared to paraquat-treated control larvae that show an eMI response (C; untreated control in B), knockdown of bsk leads to a reduction of eMI (D). (E) In contrast, knockdown of the JNK feedback inhibitor puc with RNAi lines GD3018 is sufficient to induce an eMI response in the absence of drug. (F-G) Quantification of the eMI response of controls and indicated RNAi knockdowns for untreated (F), paraquat-treated (G) larvae. (H) Quantification of eMI response comparing control with neighboring DN-Bsk expressing fat body cells. (I) Quantification of eMI response upon expression of hepAct. Single-channel images show grayscale image of indicated channels. In composite images, nuclei are in blue. Scale bars: 20 µm. (F,G) One-way ANOVAs (Dunnett corrections) with p < 0.0001 (F) and p < 0.001 (G), respectively. (H,I) t-tests. *, P < 0.05; ****, P < 0.0001
Discussion
Autophagy is an evolutionarily conserved catabolic process that reduces the burden of damaged proteins that otherwise could contribute to aging and diverse pathologies including neurodegenerative diseases and cancer [Citation3,Citation4,Citation21]. Only very limited information is available about the physiological role and the regulation of eMI. It has been suggested that in aging mice, failure to remove oxidized proteins in MVBs could contribute to reduced antigen processing and presentation [Citation21,Citation28]. In flies, the negative control of eMI by TOR signaling in the fat body suggests that eMI contributes to the recycling of nutrients under adverse growth conditions [Citation26], while at larval neuromuscular junctions, basal eMI is required for synaptic fitness [Citation27]. To shed light on possible physiological roles of eMI, we tested whether other common forms of cellular stress elicited an eMI response, as this would suggest a role for eMI in cellular damage control. Indeed, we found that paraquat and menadione, which cause increased ROS levels in vivo [Citation46,Citation48], and dsDNA breaks induced by Eto and doxorubicin elicit a robust eMI response in the fat body of feeding Drosophila larvae (). Importantly, mutations in genes required for the DDR also lead to an eMI response (), as does genetic manipulation of JNK signaling (), suggesting that the chemically induced eMI response is indeed due to DNA damage and ROS, rather than indirectly due to larvae avoiding toxic food. Significantly, we also found an enhanced eMI response upon exposure to Eto and ROS in the absence of MA. The existence of compensatory mechanisms between eMI and catabolic pathways have not been identified yet. However, in most mammalian tissues in the absence of CMA, MA is upregulated and can compensate for some of the degradative functions, and vice versa [Citation58,Citation59]. The existence of compensation between MA and eMI thus supports the idea that in flies, eMI could take over some functions of CMA that they lack due to the absence of LAMP2A [Citation26].
Characteristic for eMI, the stress-induced activation is strictly dependent on the ESCRT machinery for MVB formation (). In contrast, we find that it is only partially dependent on Hsc70-4 and a KFERQ motif in case of DNA damage (). As in mammals, eMI thus also occurs in bulk [Citation24]. Hsc70-4 has a membrane-binding activity for MVB formation [Citation27] and, in mammals, mediates the KFERQ-dependence of CMA [Citation18,Citation20]. Similarly, it was shown that regulation of the synaptic protein Comt (comatose), an endogenous eMI substrate in larval neuromuscular junctions, by Hsc70-4 was KFERQ-dependent [Citation27] and our lab originally reported a strong dependence of starvation-induced eMI on the presence of a KFERQ motif [Citation26]. We note though, that for unknown reasons, we more recently also found a strong bulk eMI component upon starvation (not shown).
Interestingly, however, not all forms of stress tested caused the activation of eMI. Despite a robust induction of ER stress measured by the Xbp1 reporter (), neither tunicamycin nor thapsigargin turn on eMI, suggesting a selective regulation of eMI in vivo as a reaction to distinct cellular requirements. Most misfolded proteins due to ER stress are degraded by the proteasome as part of the ER-associated degradation response (ERAD; reviewed in [Citation50,Citation60]). However, in mammals, ER sub-structures and some proteins including collagen aggregates, are also disposed of via ER-to-lysosome associated degradation [Citation60,Citation61]. This term encompasses different routes of degradation including ER-phagy, a selective form of MA, microautophagy, and transport of single-membrane, ER-derived vesicles to the endosomal system. It is possible that the MI (or eMI) branch of this degradative pathway is not conserved in Drosophila. Alternatively, the specific substrates that would be targets of such an (e)MI based process may not be relevant for cellular homeostasis in the larval fat body.
Clearly though, eMI is induced upon ROS exposure and DNA damage. MA and, as has been suggested, eMI are conserved means of degradation of oxidized proteins in mammals [Citation28,Citation40,Citation45] and in flies ([Citation47] and this work). Although JNK signaling is not required for starvation-induced MA, JNK signaling is required and sufficient for survival of flies and MA induction upon paraquat exposure [Citation62], likely by upregulating MA genes [Citation47,Citation62,Citation63] or altering Atg6 (the BECN1 homolog) availability, as it does in mammalian cells [Citation47,Citation62,Citation63]. Interestingly, however, expression of an activated form of Hep, the Drosophila MKK7 homolog, is unable to induce eMI () despite being sufficient to activate MA upon oxidative stress [Citation47]. Still, enhancing JNK activity by blocking the JNK feedback inhibitor Puc () was sufficient to induce eMI in the absence of oxidative stress, opening the possibility that eMI may be activated in a Hep-independent manner. It is also worth noting that despite ER stress not triggering eMI in the fly fat body, it can induce JNK signaling in mammalian cells [Citation64]. Our data thus suggest that, at least in this tissue, such a connection may not be conserved.
Autophagy and DNA damage are interlinked. On the one hand, autophagy helps to clear specific substrates to regulate genotoxic stress [Citation37,Citation65,Citation66], and loss of MA causes replication stress [Citation67]. On the other hand, the DDR can induce MA through lysosomal translocation of the MTOR regulator TSC2 [Citation68]. Similarly, CMA is induced upon DNA damage and ATM-mediated phosphorylation of CHEK1 kinase is not only required for CMA activation upon DNA damage but is also a signal for its degradation via CMA [Citation41]. Conversely, in the absence of CMA, active CHEK1 remains in the nucleus, preventing cell-cycle reentry after completion of DNA repair. Consistent with accumulation of spontaneous DNA lesions due to lack of DNA repair, mutation of either of the four major DDR kinases ATM, ATR, CHEK2, and CHEK1 is sufficient to elicit an eMI response. This response is even higher in atmtefu−8 mutants in the presence of Eto, consistent with ATM being key to repairing double-stranded DNA breaks (reviewed in [Citation69–71]). Heterozygosity for mei-41/atr and lok/chk2 was sufficient for eMI induction, the former consistent with previous reports that mei-41/atr mutants are semi-dominant [Citation72]. To our knowledge, no such characteristic has been reported for chk1grp mutants, but CHEK1 in mammalian cells is haploinsufficient [Citation73]. Interestingly, in the absence of Eto, none of the four genes is uniquely required for eMI induction, suggesting that there could be redundant functions between the ATM and ATR branches of the DDR machinery. Indeed, extensive crosstalk between these branches has been reported [Citation37,Citation69,Citation71]. Alternatively, other proteins could serve as damage sensors for eMI. In this respect, it is important to note that the genome of Drosophila lacks DNA-PKcs (DNA-dependent protein kinase catalytic subunit, aka PRKDC), an additional phosphatidylinositol 3-kinase-related kinase family member, which ATM and ATR are members of.
Taken together, our in vivo data show that selective forms of cellular stress induce eMI in response to distinct signals in larval fat body cells that function in a similar manner to the mammalian liver [Citation74,Citation75], indicating that the physiological function of eMI in flies includes adaptation to cytotoxic stress.
Materials and Methods
Fly strains and genetics
eMI sensor and UAS-GFP-HsLAMP1 [Citation43] strains were described in [Citation26]. tubGal4> UAS-Xbp1(fl)-EGFP flies were a kind gift of Dr. H. D. Ryoo (NYU) [Citation76]. hsFLP; r4-Gal4 FRT82B UAS-GFPnls, hsFLP; cgGal4 FRT42D UAS-GFPnls [Citation77], FRT42 Vps25A3, FRT42 shrb/Vps32 G5 (aka. shrub) [Citation78], FRT82 Hsc70-4Δ16 [Citation79] were gifts of Drs. T. Neufeld (University of Minnesota), T. Vaccari (University of Milan, Italy), and H. Chang (Purdue University), respectively.
Df(3 R)BSC471/TM6 C, Df(3 R)BSC750/TM6 C (both uncovering Hsc70-4 and atmtefu), Df(2 R)ED1742/SM6a, Df(2 R) Exel8047/CyO (both uncovering Vps25) were from BDSC. RNAi lines KK101734, GD26465, GD50222 (all Hsc70-4), GD45558, GD27432 (both Atg7), KK108074 (tefu/atm), KK103624, GD11251 (both mei-41/atr); KK110342 (lok/chk2), KK110076 (grp/chk1), GD34138, GD34139, KK104569 (all bsk), GD3018 (puc) were from VDRC and RNAi line JF01422 (BL#31,635; tefu/atm) was from the BDSC. hsFlp act>CD2> Gal4 UAS-GFP, UAS-hep-ACT and UAS-DN-Bsk [Citation57] were kind gifts of Dr. G. Júhasz (Eotvos Lorand University, Hungary) and M. Mlodzik (Mount Sinai, New York). tefu8 (atm) [Citation80,Citation81], mei4129D (atr; a semi dominant allele) [Citation72], lokP6 (chk2) [Citation82], grpfs−1 (chk1), both protein null alleles [Citation82,Citation83], were donated by Drs. K. McKim, J. Sekelsky, and W. Theurkauf, respectively. For verification of key alleles, see Table S1.
Flies were kept on standard food at 25°C unless noted. To induce mitotic clones with hsFLP [Citation84] 6–8 h old embryos of appropriate genotype were incubated for 1.5 h at 38°C. For clonal overexpression using hsFlp act>CD2> Gal4 UAS-GFP, we took advantage of the spontaneous activity of hsFLP of this line in the fat body (no heat-shock required).
See Table S2 for a list of exact genotypes for each experiment.
Molecular biology
To construct an eMI sensor directly driven by the FB-specific r4 promoter, the r4 repeats with the transcription start site of Gal4 were PCR amplified from r4-Gal4 fly DNA [Citation85] with primers pCasp-rev (GCGCTGACTTTGAGTGGAAT) and Gal4-Nrev (AGCGGAGACCTTTTGGTTTT) and cloned into pSca (Agilent, #240,205). The internal EcoRI site was destroyed by re-cloning the EcoRI (blunt)/BbsI fragment into the HpaI/BbsI sites of the original construct. The r4 promoter including the transcription start site (of Gal4; StuI/HindIII [blunt]) were then cloned into the BamHI (blunt) site of pCasper3 to give pCasp3-r4. The sensor was then inserted from pUAS-KFERQ-PAmCherry [Citation26] as StuI/EcoRI fragment. Transgenics were made by Rainbow Transgenic Flies (Camarillo, CA, USA).
eMI assay and drug treatments
eMI assays were performed as described [Citation26]. Briefly, 10 virgin females were crossed with 5 males of appropriate genotypes on standard fly food at 25°C until late 2nd/early 3rd instar. Larvae were washed 3 times in H2O, and the sensor photoactivated by exposure to 405-nm light for 10 min on ice in 800 µl Graces medium (Invitrogen, 11,605–094) containing 10% heat-inactivated FBS (Atlanta Biochemicals, S11050). Larvae were then washed 3 times in H2O and transferred to 35 mm cell culture dishes with 3 filter papers containing 1 ml of 20% sucrose with heat-inactivated yeast and the indicated concentrations of drugs (paraquat [Sigma, 856,177–1 G] dissolved in H2O; menadione Na-bisulfite [Sigma, M5750-25 G] in H2O; Etoposide [Sigma E1383-25 MG; dissolved in DMSO; Fisher, MT25-950-CQC]; 2 mg/ml doxorubicin HCl isotonic solution [Pfizer, 00069–3032-20]; tunicamycin [Fisher Scientific, ICN15002805; 2.5 mg/ml in H2O with 2 µl 10 M NaOH]; Thapsigargin [Callbiochem, 586,005] dissolved in DMSO) for 25 h in the dark (dying larvae were removed after a few hours). For each experiment (repeated at east 3 times), approximately 20 3rd instar larvae were processed together. Larvae were inverted, fixed in 4% freshly made paraformaldehyde (Fisher ICN15014601) in 1x phosphate-buffered saline (PBS; Corning Cellgro, 55–031-PC) at 4°C overnight. After 3 washes in 1x PBS, fat body lobes of approximately 5 larvae were mounted in 20 µl DAPI fluoromount-G (Southern Biotech 0100–20) per slide (up to 3 slides per sample). Images were taken on an ApoTome.2 system using an Axiovert 200 equipped with a 63 × 1.4 NA oil lens (Carl-Zeiss, Oberkochen, Germany) and quantified using the SParQ plugin of Fiji/ImageJ [Citation86,Citation87].
Note that the solvent DMSO at final concentrations of 0.1% and 0.2% does not affect eMI (Fig. S1A). Graphs represent means and standard error of means (SEM).
Immunohistochemistry
After treatment and fixation as above, larvae were washed 3 times in PBX (PBS with 0.1% Triton X-100 [Fisher, BP151-500]) and blocked in blocking buffer (PBX with 5% normal goat serum [Fisher, 50–588-35]) for 1 h. Samples were then incubated with primary antibody overnight in blocking buffer at 4°C in a humidified box. After 3 washes in PBX, larvae were incubated in secondary antibody for 2 h on a rotating platform at room temperature. Mounting was as above. Anti-Dcp-1 (Cell Signaling Technology, 9578) was used at a dilution of 1:100.
Statistical analysis
Statistical tests were done with GraphPad Prism using one-way ANOVAs with indicated corrections or, where appropriate, unpaired T-tests. Each larva is bilaterally symmetric and has two main fat body lobes that can fragment during processing. Although only one picture per tissue fragment was taken, we cannot exclude that two fragments may have originally belonged to the same lobe in the preparation. As each larva has two major lobes, there is a good chance that pictures of each side were included for some of the larvae. Because usually 3 slides were prepared with independent animals, it is thus a reasonable estimate that the number of animals is minimally about n/2 and maximally equal to n. Thus, “n = ” indicates fields of view unless indicated differently.
Supplemental Material
Download Zip (14.8 MB)Acknowledgments
We thank Drs. N. Baker, H. Chang, G. Juhasz, K. McKim, M. Mlodzik, T. Neufeld, H. D. Ryoo, J. Sekelsky, W. Theurkauf, T. Vaccari, U. Weber, and the Vienna Drosophila Resource and Bloomington Drosophila Stock centers for kindly sharing fly strains. We thank Drs. Mimi Kim (Division of Biostatistics, Einstein) and Ana Maria Cuervo for advice and Drs. J. Secombe and H. Bülow for their comments on this manuscript. We thank the Einstein Analytical Imaging Facility (Grant # P30CA013330). This work was supported by AHA postdoctoral fellowship 18POST34030231 (to A.M) and NIH/NIGMS grant GM119160 (to A.J.).
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review]. Exp Gerontol. 2005 Aug-Sep;40(8–9):622–633.
- Vellai T, Takacs-Vellai K. Regulation of protein turnover by longevity pathways [Research Support, Non-U.S. Gov’t Review]. Adv Exp Med Biol. 2010;694:69–80.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):349–364.
- Wollert T. Autophagy. Curr Biol. 2019 Jul 22;29(14):R671–R677.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011 Nov 11;147(4):728–741.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Nature. 2008 Feb 28;451(7182):1069–1075.
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging [Research Support, Non-U.S. Gov’t Review]. Cell. 2011 Sep 2;146(5):682–695.
- Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol. 2015 Jan;11(1):34–45.
- Gelino S, Hansen M. Autophagy - An emerging anti-aging mechanism. J Clin Exp Pathol. 2012 Jul;12(Suppl 4).
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015 May 20;16(6):345–357.
- Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Nat Rev Drug Discov. 2007 Apr;6(4):304–312.
- Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016 Jun;2(85):685–713.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014 Jan;24(1):24–41.
- Juhasz G, Neufeld TP. Experimental control and characterization of autophagy in Drosophila. Methods Mol Biol. 2008;445:125–133.
- Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015 Apr 7;25(7):376–387.
- Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017 Apr 10;41(1):10–22.
- Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018 Feb 19;28(4):R170–R185.
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis [Research Support, U.S. Gov’t, P.H.S. Review]. Trends Biochem Sci. 1990 Aug;15(8):305–309.
- Chiang HL, Terlecky SR, Plant CP, et al. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins [Research Support, U.S. Gov’t, P.H.S.]. Science. 1989 Oct 20;246(4928):382–385.
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Science. 1996 Jul 26;273(5274):501–503.
- Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2018 Apr 13;293(15):5414–5424.
- Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011 Jul;7(7):673–682.
- Santambrogio L, Cuervo AM. Chasing the elusive mammalian microautophagy. Autophagy. 2011 Jun;7(6):652–654.
- Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes [Research Support, N.I.H., Extramural]. Dev Cell. 2011 Jan 18;20(1):131–139.
- Kirchner P, Bourdenx M, Madrigal-Matute J, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019 May;17(5):e3000301.
- Mukherjee A, Patel B, Koga H, et al. Selective endosomal microautophagy is starvation-inducible in Drosophila. Autophagy. 2016;3:1984–1999.
- Uytterhoeven V, Lauwers E, Maes I, et al. Hsc70-4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron. 2015 Nov 18;88(4):735–748.
- Cannizzo ES, Clement CC, Morozova K, et al. Age-related oxidative stress compromises endosomal proteostasis. Cell Rep. 2012 Jul 26;2(1):136–149.
- Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997 Feb 28;272(9):5606–5615.
- Cuervo AM, Knecht E, Terlecky SR, et al. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1995 Nov;269(5 Pt 1):C1200–8.
- Wing SS, Chiang HL, Goldberg AL, et al. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J. 1991 Apr 1;275(Pt 1):165–169.
- Juhasz G, Erdi B, Sass M, et al. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Genes Dev. 2007 Dec 1;21(23):3061–3066.
- Kang KB, Zhu C, Yong SK, et al. Enhanced sensitivity of celecoxib in human glioblastoma cells: induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol Cancer. 2009 Aug 25;8:66.
- Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006 Jan-Feb;8(1–2):152–162.
- Kiffin R, Christian C, Knecht E, et al. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004 Nov;15(11):4829–4840.
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998 Feb 13;273(7):3963–3966.
- Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, et al. DNA damage and autophagy. Mutat Res. 2011 Jun 3;711(1–2):158–166.
- Simonsen A, Cumming RC, Brech A, et al. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila [Research Support, Non-U.S. Gov’t]. Autophagy. 2008 Feb;4(2):176–184.
- Xiong Y, Contento AL, Nguyen PQ, et al. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007 Jan;143(1):291–299.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015 Mar;22(3):377–388.
- Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015;6:6823.
- Chelouah S, Monod-Wissler C, Bailly C, et al. An integrated Drosophila model system reveals unique properties for F14512, a novel polyamine-containing anticancer drug that targets topoisomerase II. PLoS One. 2011;6(8):e23597.
- Pulipparacharuvil S, Akbar MA, Ray S, et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J Cell Sci. 2005 Aug 15;118(Pt 16):3663–3673.
- Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013 Feb;65(2):157–170.
- Navarro-Yepes J, Burns M, Anandhan A, et al. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014 Jul 1;21(1):66–85.
- Rzezniczak TZ, Douglas LA, Watterson JH, et al. Paraquat administration in Drosophila for use in metabolic studies of oxidative stress. Anal Biochem. 2011 Dec 15;419(2):345–347.
- Wu H, Wang MC, Bohmann D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech Dev. 2009 Aug-Sep;126(8–9):624–637.
- Lushchak OV, Gospodaryov DV, Rovenko BM, et al. Specific dietary carbohydrates differentially influence the life span and fecundity of Drosophila melanogaster. J Gerontol A Biol Sci Med Sci. 2014 Jan;69(1):3–12.
- Wang Y, Singh R, Massey AC, et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2008 Feb 22;283(8):4766–4777.
- Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015 Mar;40(3):141–148.
- Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490:71–92.
- Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015 May;149:124–138.
- Oikemus SR, McGinnis N, Queiroz-Machado J, et al. Drosophila atm/telomere fusion is required for telomeric localization of HP1 and telomere position effect. Genes Dev. 2004 Aug 1;18(15):1850–1861.
- Hari KL, Santerre A, Sekelsky JJ, et al. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell. 1995 Sep 8;82(5):815–821.
- Xu J, Xin S, Du W. Drosophila Chk2 is required for DNA damage-mediated cell cycle arrest and apoptosis. FEBS Lett. 2001 Nov 23;508(3):394–398.
- Fogarty P, Campbell SD, Abu-Shumays R, et al. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr Biol. 1997 Jun 1;7(6):418–426.
- Weber U, Paricio N, Mlodzik M. Jun mediates Frizzled-induced R3/R4 cell fate distinction and planar polarity determination in the Drosophila eye. Development. 2000 Aug;127(16):3619–3629.
- Massey AC, Kaushik S, Sovak G, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006 Apr 11;103(15):5805–5810.
- Kaushik S, Massey AC, Mizushima N, et al. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008 May;19(5):2179–2192.
- De Leonibus C, Cinque L, Settembre C. Emerging lysosomal pathways for quality control at the endoplasmic reticulum. FEBS Lett. 2019 Sep;593(17):2319–2329.
- Forrester A, De Leonibus C, Grumati P, et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. Embo J. 2019 Jan 15;38(2). DOI:10.15252/embj.201899847.
- Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008 Jun 20;30(6):678–688.
- Pattingre S, Bauvy C, Carpentier S, et al. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009 Jan 30;284(5):2719–2728.
- Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000 Jan 28;287(5453):664–666.
- Anand SK, Sharma A, Singh N, et al. Entrenching role of cell cycle checkpoints and autophagy for maintenance of genomic integrity. DNA Repair (Amst). 2019 Nov 13;86:102748.
- Siggens L, Figg N, Bennett M, et al. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. Faseb J. 2012 May;26(5):2117–2124.
- Vanzo R, Bartkova J, Merchut-Maya JM, et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019 Aug 14:1134–1153.
- Liu M, Zeng T, Zhang X, et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat Commun. 2018 Oct 8;9(1):4139.
- Lanz MC, Dibitetto D, Smolka MB. DNA damage kinase signaling: checkpoint and repair at 30 years. Embo J. 2019 Sep 16;38(18):e101801.
- Sekelsky J. DNA repair in Drosophila: mutagens, models, and missing genes. Genetics. 2017 Feb;205(2):471–490.
- Song YH. Drosophila melanogaster: a model for the study of DNA damage checkpoint response. Mol Cells. 2005 Apr 30;19(2):167–179.
- Laurencon A, Purdy A, Sekelsky J, et al. Phenotypic analysis of separation-of-function alleles of MEI-41, Drosophila ATM/ATR. Genetics. 2003 Jun;164(2):589–601.
- Lam MH, Liu Q, Elledge SJ, et al. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004 Jul;6(1):45–59.
- Arquier N, Leopold P. Fly foie gras: modeling fatty liver in Drosophila. Cell Metab. 2007 Feb;5(2):83–85.
- Deutsch J, Laval M, Lepesant JA, et al. Larval fat body-specific gene expression in D. melanogaster. Dev Genet. 1989;10(3):220–231.
- Sone M, Zeng X, Larese J, et al. A modified UPR stress sensing system reveals a novel tissue distribution of IRE1/XBP1 activity during normal Drosophila development. Cell Stress Chaperones. 2013 May;18(3):307–319.
- Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation [Research Support, N.I.H., Extramural]. Mol Biol Cell. 2009 Apr;20(7):2004–2014.
- Vaccari T, Rusten TE, Menut L, et al. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J Cell Sci. 2009 Jul 15;122(Pt 14):2413–2423.
- Bronk P, Wenniger JJ, Dawson-Scully K, et al. Drosophila Hsc70-4 is critical for neurotransmitter exocytosis in vivo. Neuron. 2001 May;30(2):475–488.
- Pedersen M, Tiong S, Campbell SD. Molecular genetic characterization of Drosophila ATM conserved functional domains. Genome. 2010 Oct;53(10):778–786.
- Silva E, Tiong S, Pedersen M, et al. ATM is required for telomere maintenance and chromosome stability during Drosophila development. Curr Biol. 2004 Aug 10;14(15):1341–1347.
- Brodsky MH, Weinert BT, Tsang G, et al. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004 Feb;24(3):1219–1231.
- Sibon OC, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997 Jul 3;388(6637):93–97.
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993 Apr;117(4):1223–1237.
- Lee G, Park JH. Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics. 2004 May;167(1):311–323.
- Mesquita A, Pereira J, Jenny A. Streamlined particle quantification (SParQ) plug-in is an automated fluorescent vesicle quantification plug-in for particle quantification in Fiji/ImageJ. Autophagy. 2019 Nov;27:1–7.
- Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012 Jun 28;9(7):676–682.