
ABSTRACT
Iron metabolism is involved in numerous physiological processes such as erythropoiesis, oxidative metabolism. However, the in vivo physiological functions of the iron metabolism-related gene Hfe in immune response during viral infection remain poorly understood. Here, we identified 5 iron metabolism-associated genes specifically affected during RNA virus infection by a high-throughput assay and further found that HFE was a key negative regulator of RIG-I-like receptors (RLR)-mediated type I interferons (IFNs) signaling. RNA virus infection inhibited the binding of HFE to MAVS (mitochondrial antiviral signaling protein) and blocked MAVS degradation via selective autophagy. HFE mediated MAVS autophagic degradation by binding to SQSTM1/p62. Depletion of Hfe abrogated the autophagic degradation of MAVS, leading to the stronger antiviral immune response. These findings established a novel regulatory role of selective autophagy in innate antiviral immune response by the iron metabolism-related gene Hfe. These data further provided insights into the crosstalk among iron metabolism, autophagy, and innate immune response.
Abbreviations: ATG: autophagy-related; BAL: bronchoalveolar lavage fluid; BMDMs: bone marrow-derived macrophages; CGAS: cyclic GMP-AMP synthase; CQ: chloroquine; Dpi: days post-infection; ELISA: enzyme-linked immunosorbent assay; GFP: green fluorescent protein; HAMP: hepcidin antimicrobial peptide; Hpi: hours post-infection; HJV: hemojuvelin BMP co-receptor; IFNs: interferons; IL6: interleukin 6; IRF3: interferon regulatory factor 3; ISRE: interferon-stimulated response element; Lipo: clodronate liposomes; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MAVS: mitochondrial antiviral signaling protein; MEFs: mouse embryonic fibroblasts; SLC40A1/FPN1: solute carrier family 40 (iron-regulated transporter), member 1; flatiron; SQSTM1/p62: sequestosome 1; STAT1: signal transducer and activator of transcription 1; STING1/STING: stimulator of interferon response cGAMP interactor 1; TBK1: TANK-binding kinase 1; TFRC/TfR1: transferrin receptor; TNF/TNFα: tumor necrosis factor; WT: wild type.
Introduction
Iron is an essential micronutrient and is required for cellular respiration, oxygen transport, erythropoiesis and immune responses [Citation1]. The role of the hereditary hemochromatosis protein HFE in the regulation of iron metabolism has been widely established [Citation2–4], and it binds to TFRC/TFR1 (transferrin receptor) and reduces cellular iron uptake. In hepatocytes, HFE modifies the formation of HAMP (hepcidin antimicrobial peptide), a critical iron-regulatory hormone, and thus affects systemic iron balance [Citation5–7]. In contrast, HFE in monocytes and intestinal cells promotes iron accumulation by suppressing iron release, and this activity is independent of its interaction with TFRC [Citation8]. Host iron metabolism is also correlated with various virus infections [Citation9–16]. HFE is a common target when various viral infections attempt to escape the host immune defenses. The human immunodeficiency virus-1 Nef protein downregulates HFE expression and causes a 90% HFE reduction by rerouting HFE to the trans-Golgi network [Citation17,Citation18]. In human cytomegalovirus-infected cells, the viral protein US2 induces HFE degradation via proteolysis [Citation19]. Despite considerable efforts to elucidate its physiologic function, the specific role of HFE in the innate immune response has remained largely unknown.
As the first line of defense against viral infections, the type I interferons (IFNs) play a pivotal role in innate immunity, and numerous studies have shown that MAVS/IPS-1/Cardif/VISA (mitochondrial antiviral signaling protein) is essential for the innate immune response against RNA virus infections [Citation20–23]. Upon binding to viral RNA, RIG I-like receptors (RLRs) interact with MAVS for antiviral signaling transduction. MAVS further triggers the TBK1 (TANK binding kinase 1)-mediated activation of interferon regulatory factor IRF3 and IRF7, which results in the production of type I IFNs [Citation24]. Previous studies have indicated that the activity and stability of MAVS are regulated by multiple posttranslational modifications [Citation25], and the proteasome-dependent degradation of MAVS that is triggered by viral infection is mediated by several K48-linked ubiquitin-associated proteins such as SMURF2, TRIM25, and ITCH/AIP4 [Citation26–28]. Additionally, autolysosome also plays a pivotal role in the stability of MAVS to maintain mitochondrial homeostasis. MAVS binding to LC3 maintains mitochondrial homeostasis via mitochondria-associated autophagic signaling [Citation29]. BST2/TETHERIN and RNF34 target MAVS for CALCOCO2/NDP52 -mediated selective autophagic degradation [Citation30,Citation31]. Hantan virus glycoprotein triggers the mitophagy-dependent degradation of MAVS by interacting with TUFM [Citation32]. However, whether lysosomes are also involved in regulating the stability of MAVS, and the potential role of HFE in this process, remains poorly investigated.
In the present study, we identified HFE as a negative regulator of antiviral innate immunity by using influenza A virus (H7N9) as model virus. HFE was significantly downregulated in mice by H7N9 virus infection both in vitro and in vivo, and hfe knockout caused mice to become more resistant to H7N9 virus infection compared to wild-type (WT) mice. Our studies had led to the discovery of a mechanism for HFE targeting MAVS for SQSTM1/p62 (sequestosome 1)-mediated MAVS degradation through a selective autophagy-lysosome pathway. Hfe deficiency potentiated the activation of TBK1 and IRF3, the production of IFNB1/IFN-beta, and innate antiviral responses to RNA viruses. These findings established a pivotal role for HFE in controlling viral infection by regulating the induction of type I IFNs in mouse embryonic fibroblasts (MEFs) and innate immune cells.
Results
RNA virus infection alters the expression of iron metabolism-associated genes
To investigate the effect of virus infection on the phenotype of iron metabolism-associated genes, we analyzed the gene expression profile in mouse bone marrow-derived macrophages (BMDMs) infected in vitro with H7N9 virus as a model. As shown in and Table S1, we evaluated 35 differentially expressed genes that might be correlated with iron metabolism. Among these genes, H7N9 virus infection specifically affected five iron metabolism-associated genes, including Hfe, Hjv (hemojuvelin BMP co-receptor), Hamp, Slc40a1/Fpn1 (solute carrier family 40 [iron-regulated transporter], member 1; flatiron) and Tfrc (transferrin receptor). We next investigated the expression of these genes in various tissues from mice inoculated with H7N9 virus. As shown in , the mRNA levels of Hfe and Hjv were significantly downregulated in the liver, spleen, and lung at 5 dpi (days post-infection). Compared with Hfe reduction, Hamp expression was greatly enhanced in these three tissues after virus infection. Interestingly, the mRNA transcriptional dynamics of Tfrc was similar with that of Hamp in liver and Hfe in the lung; however, the transcriptional dynamics of Slc40a1 was in contrast to that of Tfrc among different tissues. Similarly, the protein levels of HFE, HJV, SLC40A1, TFRC in the liver, spleen, and lung at 5 d post-infection were consistent with the mRNA levels of these genes ().
Figure 1. Reduced expression of Hfe is associated with H7N9 virus infection. (A) Microarray analysis of differentially expressed genes at 12 hpi in mouse BMDMs infected with H7N9 virus. The heatmap showed the abundance of iron homeostasis-regulating genes following H7N9 virus infection. Untreated macrophages were used as controls. Data are representative of three independent experiments. (B and D) qRT-PCR was performed at the indicated time points to measure the mRNA levels of Hfe, Slc40a1, Hamp, Hjv and Tfrc in various tissues in mice infected with H7N9 virus (B) and in mouse BMDMs and MEFs infected with H7N9 virus (D). (C) Immunoblot analysis of the indicated proteins in lysates of various tissues in mice infected with H7N9 virus. (E) Mice alveolar epithelial cells were infected with VSV, SeV for the indicated times, and qRT-PCR was used to measure the Hfe, Hjv, Slc40a1, Hamp, Tfrc mRNA level. All qRT-PCR data are normalized to the Actb gene and are representative of three independent experiments. Control group is used as a reference. The error bars represent the standard deviation, and differences between the experimental and control groups were determined by one-way ANOVA (*p < 0.05, **p < 0.01, and ***p < 0.001).
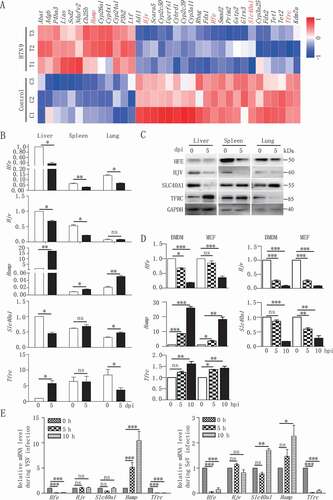
To further characterize the direct effect of viral infection on the transcription of these genes, we inoculated BMDMs and MEFs with H7N9 virus for 5 and 10 h. The kinetics of Hfe, Hjv, Hamp, Slc40a1 and Tfrc induction in vitro were consistent with the in vivo observations (). We further measured the transcription levels of these genes after infection with other RNA viruses. The infection with the RNA viruses including vesicular stomatitis virus (VSV) and Sendai virus (SeV) suppressed the mRNA level of Hfe in mice alveolar epithelial cells (). Together, these data suggested that RNA virus triggered changes in iron metabolism-associated genes both in vivo and in vitro.
Hfe-deficient mice strengthen resistance to virus infection
We employed the hfe knockout (−/-) mice to study the in vivo function of HFE. The frequency of dendritic cells (DCs) in the spleen was comparable between WT and hfe−/- mice (Figure S1A). The hfe−/- mice also did not show any significant abnormalities in thymocyte development or homeostasis of peripheral T and B cells (Figures S1B and S1C). These data suggested that hfe knockout did not affect the development or homeostasis of major subpopulations of immune cells.
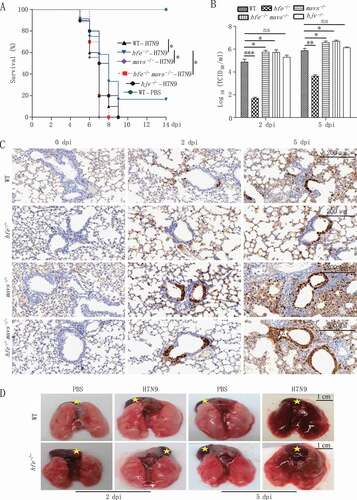
To determine the in vivo role of HFE in viral infection, we evaluated the antiviral immune responses using hfe−/- mice. WT and hfe−/- mice were intranasally infected with H7N9 virus and then monitored for pathologic symptoms for 2 weeks. Compared to their WT littermates, hfe−/- mice exhibited 16.7% survival rate (). We further determined virus titer in the lungs from H7N9 virus-infected mice. As shown in , the viral titers in the lungs from hfe−/- mice were significantly reduced compared to WT controls. Immunohistochemistry staining of the viral proteins NP indicated that WT mice developed severe fulminant viral pneumonia that was associated with extensive edema and red blood cell extravasation (). In addition, H7N9 virus infection in WT mice also resulted in profound immune cell infiltration and severe lung injury (including increased alveolar wall thickness, formation of hyaline membranes, and proteinaceous debris filling the airspaces) ( and S2A-C). Moreover, we also checked the in vivo role of HJV in H7N9 virus infection by hjv−/- mice. Interestingly, hjv−/- mice had 100% mortality to H7N9 virus infection (). These results indicated that Hfe deficiency but not Hjv deficiency protected partially mice from H7N9 virus infection by restricting viral replication and influenza A virus -driven inflammation during the acute phase.
Figure 2. Hfe deficiency protects mice from influenza virus H7N9 infection. (A) The survival of H7N9 virus-infected WT, hfe−/-, mavs−/-, hfe−/- mavs−/- and hjv−/- mice were monitored for 14 d (n = 12 per group). (B) Viral titers in the lung were quantified at 2 or 5 dpi by TCID50 assay (n = 3 per group). (C) Immunohistochemical analysis of the influenza virus proteins NP at 2 or 5 dpi in the lungs of WT, hfe−/-, mavs−/-, hfe−/- mavs−/- mice infected with H7N9 virus. Scale bars: 200 μm. (D) WT and hfe−/- mice were infected with H7N9 for the indicated number of days, and their lung pathology was observed by the indicate of stars (scale bars: 1 cm). The error bars represent standard deviations, and differences between the experimental and control groups are determined by 2-way ANOVA and the mouse survival study, Kaplan-Meier survival curves are generated and analyzed by Log-Rank test (*p < 0.05, **p < 0.01 and ***p < 0.001).
Hfe deficiency promotes the induction of type I interferons by MAVS signaling pathway during virus infection
The type I IFNs play a critical role in innate immunity against influenza virus infection [Citation33], and influenza viruses can suppress Ifnb1 gene induction and its receptor signaling in order to escape the host immune defense. To evaluate the role of HFE in innate immune responses, we compared the differences in the expression of various cytokines between WT and hfe−/- mice. Compared to WT controls, at 2 dpi the mRNA and protein levels of Ifnb1 were significantly increased in lung, serum and bronchoalveolar lavage fluid (BAL) of hfe−/- mice when infected with H7N9 virus (), suggesting that HFE specifically suppressed the production of type I IFNs and inhibited the antiviral innate response.
Figure 3. Hfe deficiency enhances type I IFNs induction in response to H7N9 virus infection. (A) The mRNA transcription of the genes Ifnb1, Il6 and Tnf in the lungs of H7N9 virus-infected WT and hfe−/- mice were analyzed by qRT-PCR at 2 dpi. The concentrations of IFNB1, IL6 and TNF were evaluated in the serum and BAL at 2 dpi (n = 5). (B and C) The heatmaps showed 30 genes that were differentially expressed in the BMDMs of H7N9 virus-infected WT and hfe−/- mice and that were involved in the innate response (B) and inflammation (C). (D and E) qRT-PCR and ELISA analysis of the mRNA and expression of the genes Ifnb1, Il6 and Tnf in BMDMs infected with H7N9 virus at the indicated time points. (F) TCID50 was used to measure the H7N9 virus titers in the supernatants of WT and hfe−/- BMDMs infected with H7N9 virus. (G and H) qRT-PCR and ELISA analyzed the mRNA and expression of the genes Ifnb1, Il6 and Tnf in BMDMs of WT or hfe−/- mice transfected with poly (I:C) (1 μg/ml) or HSV60 (1 μg/ml) for 4 h. (I) Immunoblot analysis of the indicated proteins in lysates of WT and hfe−/- BMDMs infected with VSV or H7N9 virus. (J) Immunoblot analysis of the indicated proteins in lysates of WT and hfe−/- BMDMs transfected with poly (I:C) (1 μg/ml) or HSV60 (1 μg/ml) for 4 h. All qRT-PCR data are normalized to the Actb gene and used the control group as a reference. Data are representative of at least three independent experiments, and differences between the experimental and control groups are determined by 2-way ANOVA (*p < 0.05, **p < 0.01, and ***p < 0.001).
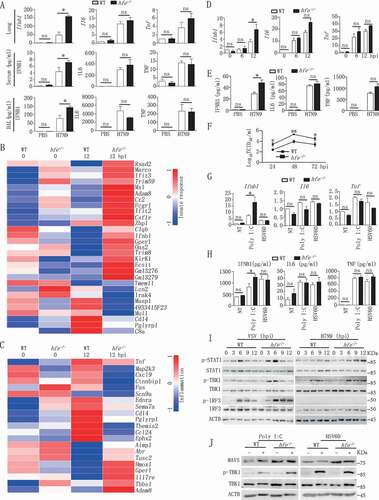
To further confirm the role of HFE in the influenza A virus -induced expression of type I IFNs, we analyzed the changes in global gene expression between WT and hfe−/- BMDMs induced by H7N9 virus. Microarray analysis revealed that the greatest changes (fold-change >1.5, p < 0.05) in expression were in genes that regulate the innate immune responses (). We also measured gene expression at 6 and 12 hpi (hours post-infection) by qRT-PCR in WT and hfe−/- BMDMs infected with H7N9 virus (). We observed increased Ifnb1 expression in hfe−/- BMDMs and was consistent with observations in vivo, the mRNA levels of Il6 (interleukin 6) and Tnf (tumor necrosis factor) in hfe−/- BMDMs were comparable with WT controls. Enzyme linked immunosorbent assay (ELISA) also gave similar results for secreted cytokines in the supernatant (). Viral titer revealed a significant reduction in the supernatant of BMDMs from hfe−/- mice (). Similar to H7N9 virus infection, increased expression of Ifnb1, Il6 and Tnf were exhibited in hfe−/- BMDMs transfected with polyinosine-polycytidylic acid (poly I:C) but not HSV60 (CDS agonist HSV-60) compared to WT BMDMs (). These results suggested that HFE selectively suppressed the production of type I IFNs induced by RNA virus but not DNA virus.
To further investigate the potential mechanism by which HFE suppressed type I IFNs expression, we checked the activation of MAVS-mediated signaling pathway by H7N9 virus and VSV infections. Immunoblot analysis indicated that Hfe deficiency promoted the phosphorylation of TBK1, and was consistent with the phosphorylation of STAT1 (signal transducer and activator of transcription), a downstream signal of type I IFNs receptors, was also significantly enhanced in hfe−/- BMDMs (). Similar results displayed in hfe−/- BMDMs transfected with poly I:C but not HSV60 (). In order to deeply validate the relation between MAVS pathway and HFE, we evaluated the antiviral immune responses by generating hfe−/- mavs−/- mice. hfe−/- mavs−/- and mavs−/- mice were intranasally infected with H7N9 virus. Compared to their hfe−/- littermates, hfe−/- mavs−/- and mavs−/- mice exhibited 100% mortality (p < 0.05), the average death time of hfe−/- mavs−/- mice were quicker than mavs−/- mice (), and the transcription and expression of Ifnb1 gene were very low in serum, BAL and lung of hfe−/- mavs−/- and mavs−/- mice as well as in hfe−/- mavs−/- and mavs−/- BMDMs (Figure S3). These data demonstrated that HFE inhibited specifically the antiviral innate immune response by MAVS pathway but not CGAS (cyclic GMP-AMP synthase)-STING1/STING (stimulator of interferon response cGAMP interactor 1) pathways.
Targeting of MAVS protein stability by HFE
To identify the target of HFE in the regulation of type I IFNs expression, we examined HFE’s effect on IFNB1 promoter activity using luciferase reporter assays in human embryonic kidney (HEK) 293 T cells co-transfected with HFE plus various inducers. As shown in , HFE selectively suppressed MAVS and DDX58/RIG-I (DExD/H-box helicase 58)-mediated IFNB1 and interferon-stimulated response element (ISRE) activation but had no effect on TBK1-, CGAS-, STING1-, or IRF3-stimulated IFNB1 or ISRE activation. Thus, we hypothesized that HFE might target MAVS to inhibit the antiviral signaling pathway, and our reporter assay indicated that HFE inhibited MAVS-induced IFNB1 and ISRE activation in a dose-dependent manner (). Previous studies demonstrate that catalytically inactive HFE mutants (C282Y and H63D) lose their function in iron metabolism [Citation34], but surprisingly we found that neither HFEC282Y nor HFEH63D lost their capacity to inhibit MAVS-induced IFNB1 and ISRE reporter activation (). These results suggested that HFE negatively regulated the antiviral host defense by targeting MAVS independently of its role in regulating iron homeostasis.
Figure 4. HFE inhibits type I IFNs signaling by promoting the degradation of MAVS. (A) Luciferase assay of HEK293T cells transfected with the IFNB1 or ISRE reporter plasmids together with DDX58, MAVS, CGAS, STING1, TBK1, or IRF3 and with or without the HFE plasmid. (B) The promoter activity of IFNB1 and ISRE was evaluated in HEK293T cells co-transfected with plasmids expressing MAVS and different doses of HFE. (C) The effect of HFE-WT, HFEH63D, and HFEC282Y on the activity of MAVS was measured by luciferase assay in HEK293T cells. (D) Immunoblot analysis of MAVS or MAVS-Pex (indicating MAVS on peroxisomes) or VDAC1 or SLC25A6 probed with anti-MAVS or GFP or FLAG antibody in lysates of HEK293T cells transfected with different doses of HFE. (E) Immunoblot analysis of MAVS in lysates of WT and hfe−/- BMDMs, BMDCs, and MEF and HFE knockdown THP-1 cells. THP-1 cells were transfected with small interfering RNA (siRNA) targeting HFE for 36 h. These tested cells were infected with H7N9 virus. (F) Immunoblot analysis of MAVS in lysates of lung from WT and hfe−/- mice. (G) Confocal microscopy of HEK293T cells transfected for 24 h with FLAG-MAVS and MYC-HFE. Immunofluorescence was performed using anti-MYC (green) and anti-FLAG (red) antibodies and DAPI (blue). Scale bars: 10 μm. (H) Immunoblot analysis of lysates of HEK293T cells transfected with plasmids encoding FLAG-MAVS, FLAG-TBK1, FLAG-STING1 and MYC-HFE for 24 h followed by immunoprecipitation with anti-FLAG or anti-MYC antibodies. The asterisk indicated the heavy chains. (I) Immunoprecipitation analysis of the interaction between endogenous MAVS and HFE in H7N9-infected BMDMs for the indicated times. Data are representative of three independent experiments, and differences between the experimental and control groups are determined by 2-way ANOVA in (A) and One-way ANOVA in (B and C) (***p < 0.001).
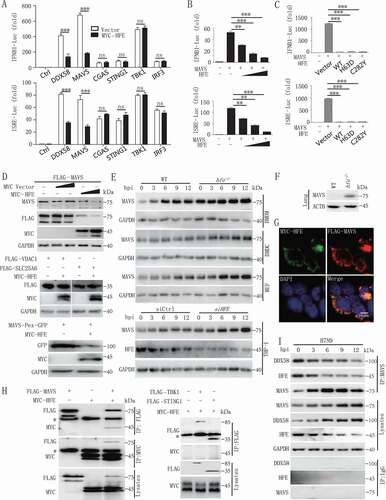
We next evaluated the function of HFE on the stability of MAVS by co-expressing these two molecules in HEK293T cells, and we found that HFE significantly induced only MAVS degradation in the mitochondria and peroxisomes; however, HFE did not degrade VDAC1 (voltage dependent anion channel protein 1) and SLC25A6/ANT3 (solute carrier family 25 member 6) on the mitochondrial membrane (), suggesting that HFE directly degraded MAVS but not the whole mitochondria. Consistently, Hfe deficiency in BMDMs, BMDCs (mouse bone marrow-derived dendritic cells) and MEFs of mice and in human THP-1 cells significantly increased endogenous levels of MAVS compared to WT (), and MAVS expression also increased in lung of hfe−/- mice in comparison with WT mice (). This increase in MAVS was not due to increase transcription, and similar Mavs mRNA levels were seen in WT and hfe−/- BMDMs (Figure S4A). In addition, the truncation of HFE, which lost its function in iron metabolism, retained its ability to induce the degradation of MAVS (Figure S4B), indicating that the iron metabolism-associated residues HFE (H63) and HFE (C282) of were not the functional domains that degraded MAVS.
To explore the underlying mechanism by which HFE reduced the level of the MAVS protein, we first determined the physical interaction between HFE and MAVS. Confocal microscopy and co-immunoprecipitation assay indicated that HFE interacted with MAVS but not TBK1, STING1 and IRF3 in HEK293T cells (, S4C and S4D). Interestingly, H7N9 virus infection disrupted this association, which might contribute to the stabilization of MAVS during antiviral signal transduction (). Together, these results suggested that HFE promoted MAVS degradation dependent of their physical association.
HFE promotes MAVS degradation via the autophagy-lysosome pathway
To clarify the mechanism of HFE-mediated MAVS degradation, we evaluated the protein level of MAVS after incubating with proteasome or lysosome inhibitors, respectively. HFE-mediated degradation of MAVS was completely inhibited by the lysosomal inhibitor chloroquine (CQ) but not MG132, which disrupted proteasome activity (). Confocal microscopy further indicated that MAVS did not co-localize with lysosomes in uninfected WT or hfe−/- BMDMs. While H7N9 virus infection increased the amount of MAVS localized at lysosomes in WT BMDMs, Hfe deficiency disrupted this co-localization, as indicated in and S4E. We next transfected hfe−/- BMDMs with mCherry-HFE and found that HFE clearly promoted the association between MAVS and lysosomes (). Collectively, these results showed that HFE promoted the translocation of MAVS to the lysosomes and thus reduced the MAVS protein level in WT BMDMs.
Figure 5. HFE acts as a novel suppressor of MAVS stability through the autophagy-lysosome pathway. (A and B) HEK293T cells were transfected with FLAG-MAVS and HA-HFE plasmids for 24 h and then treated with 20 μM MG132 (M) (A) or 50 µM CQ (B) for the indicated times. The total cell lysates were immunoblotted with the indicated antibodies. (C) Confocal microscopy of WT and hfe−/- BMDMs transfected with GFP-Mavs (green) and stained with LysoTracker Red DND-99 (red) after H7N9 virus infection for 18 h. Scale bars, 10 μm. (D) Confocal microscopyof hfe−/- BMDMs transfected with mCherry-Hfe (red) and GFP-Mavs (green) for 24 h and then stained with LysoTracker Deep Red (blue) for 2 h. Scale bars: 10 μm (1 μm in the inset). (E) MYC-HFE and GFP-LC3B were co-transfected into HEK293T cells. Immunoprecipitation and immunoblotting analysis of the lysates were performed with various combinations of anti-MYC anti-FLAG and anti-GFP antibodies. (F) hfe−/- BMDMs transfected with the mCherry-Hfe (red) expression vector and GFP-Lc3b (green). These BMDMs were stained with LysoTracker Deep Red (blue) for 2 h after treated with 50 μM CQ for another 12 h. Scale bars: 10 μm (1 μm in the inset). (G) Immunoblotting analysis of FLAG-MAVS in lysates of WT, atg5−/- and atg7 −/- cells transfected with different doses of MYC-Hfe. The total cell lysates were immunoblotted with the indicated antibodies. (H) WT, atg5−/- and atg7 −/- MEFs were co-transfected with GFP-Mavs (green) and mCherry-Hfe (red). Cells were stained 24 h later with LysoTracker Deep Red (blue) for 2 h and subjected to confocal microscopy. Scale bars: 10 μm (1 μm in inset). Data are representative of at least three independent experiments, and differences between the experimental and control groups are determined by 2-way ANOVA (***p < 0.001).
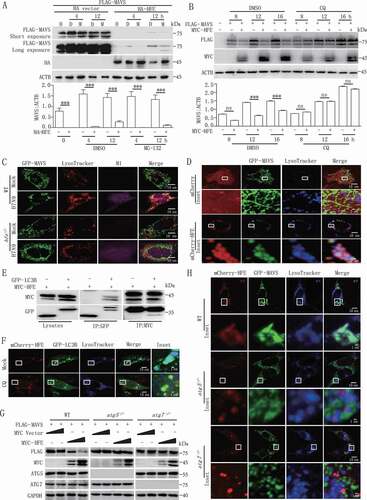
To determine the underlying mechanism of lysosome-dependent MAVS degradation by HFE, we identified a conserved region at positions 101 to 105 (Ac-F-x-x-V/I/L) in HFE that interacted with MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) (Figure S5A). Immunoprecipitation assays also confirmed that HFE physically interacted with MAP1LC3B (). In hfe−/- BMDMs reconstituted with mCherry-HFE, we also found co-localization of HFE with GFP (green fluorescent protein)-LC3B autophagosomes and lysosomes, and this co-localization was impaired by incubation with CQ in WT BMDMs ().
We next determined the function of HFE in autophagosome maturation. We infected WT and hfe−/- BMDMs with adenovirus that expressed mCherry-GFP-LC3B, which was used to discriminate autophagosomes (expressing both mCherry and GFP fluorescent) from acidified autolysosomes (expressing red fluorescence only). We found that Hfe deletion did not affect autophagosome maturation in Earle’s Balanced Salt Solution (EBSS)-starved or CQ-treated cells (Figure S5B). To further confirm the role of autophagosomes in HFE-mediated MAVS degradation, we co-transfected HFE and MAVS plasmids in WT, atg5−/- or atg7−/- (autophagy-related gene 5/7) MEFs. HFE strongly induced the degradation of MAVS in a dose-dependent manner in WT MFEs, but HFE lost this ability in atg5−/- and atg7−/- MEFs (). Consistent with this, confocal microcopy also indicated that HFE, MAVS, and lysosomes were co-localized in WT MEFs, but not in atg5−/- or atg7−/- MEFs (). Collectively, these results suggested that HFE functions as a novel adaptor for mediating lysosome-dependent MAVS degradation during RNA virus infection.
Hfe deficiency resistance to virus infection is not affected by dietary iron
To investigate whether iron was related to the resistance of hfe−/- mice to virus infection, we tested the iron status of hfe−/- mice. We observed that a high iron concentration presented in serum and liver of hfe−/- mice when compared to WT mice, however, the macrophages in hfe−/- mice were similar iron concentration with that in WT mice (), indicating that iron concentration was not affected in macrophages of hfe−/- mice. Subsequently, we explored if macrophages played a role in the resistance of hfe−/- mice to virus infection, macrophages in hfe−/- mice were cleared by clodronate liposomes (Lipo). As expected, after H7N9 virus infection, the mortality of hfe−/- mice without macrophages was similar with WT mice (), moreover, the level of mRNA transcript and proteins of Ifnb1, Il6 and Tnf decreased significantly in serum and BAL of hfe−/- mice without macrophages (), demonstrating that macrophages played an important role in virus infection to hfe−/- mice.
Figure 6. Hfe deficiency resistance to virus infection is macrophage-dependent and not affected by dietary iron. (A) The hfe−/- mice were fed low, medium and high dietary iron for 14 d, and infected with H7N9 virus. Iron levels in serum, liver and lung were measured with ELISA (n = 5 per group). (B) Flow cytometry analysis of the median fluorescence intensity of calcein in peritoneal macrophages from WT and hfe−/- mice. (C) The survival of H7N9 virus-infected WT and hfe−/- mice with or without Lipo treatment for 48 h was monitored for 14 d (n = 10 per group). (D) The mRNA levels of the indicated genes in the lungs of WT and hfe−/- mice with or without Lipo treatment for 48 h were analyzed by qRT-PCR at 2 dpi. ELISA was used for evaluating the concentrations of the indicated cytokines in the serum and BAL at 2 dpi (n = 5). (E) The survival of H7N9 virus-infected WT and hfe−/- mice fed low, medium and high dietary iron was monitored for 14 d (n = 10 per group). (F) Viral titers in lung were quantified at 5 dpi by TCID50 assay (n = 3 per group). (G) The concentrations of the IFNB1 in the serum of WT mice fed with low, medium and high iron when infected with H7N9 virus for 5 d (n = 3 per group). Data are representative of at least three independent experiments, and the error bars represent the standard deviation. Differences between the experimental and control groups are determined by 2-way ANOVA and the mouse survival study, Kaplan-Meier survival curves are generated and analyzed by Log-Rank test (*p < 0.05, **p < 0.01 and ***p < 0.001).
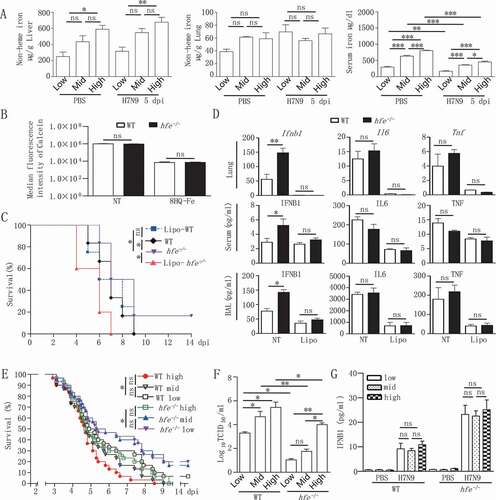
To further investigate whether iron supplementation affected the resistance of hfe−/- mice to virus infection, we subsequently fed hfe−/- mice diets with low, medium levels of iron for 2 weeks, or high levels of iron for 1 week, and then inoculated these mice with H7N9 virus. The results showed that hfe−/- mice fed with low-iron diet revealed the similar survival rate to hfe−/- mice fed with mid-iron diet (), hfe−/- mice fed with high-iron diet postponed death time when compared to WT mice fed with high-iron diet, suggesting that the concentration of diet iron was irrelevant to resistance of hfe−/- mice to virus infection. Surprisingly, as a parallel control, WT mice fed with low-iron diet had survivals when compared to WT mice fed with mid-iron diet, and viral loads in lung homogenate supernatants from mice fed the low-iron diet were significantly reduced compared to mice fed the high- and medium-iron diet (). Interestingly, there was no obvious difference in IFNB1 alteration in WT mice fed with different levels of dietary iron (). This implied that iron-deficient diet promoted host resistance to H7N9 virus infection. Collectively, these results indicated that the resistance of hfe−/- mice to RNA virus infection was irrelevant to diet iron, Hfe deficiency was critical to resist virus infection and HFE might be a potential drug target to improve the clinical outcome of H7N9 virus infection.
HFE binding to SQSTM1 mediates MAVS autophagic degradation
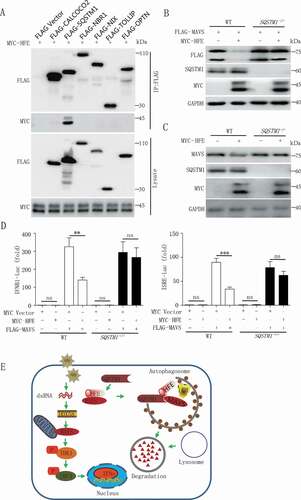
Substantial pieces of evidence suggest that cargo receptors play necessary roles in delivering substrates to the autophagosome for selective degradation [Citation35–37]. As we all know that HFE was not a cargo receptor, so we supposed that HFE might bridge MAVS to the cargo receptors for autophagic degradation. We found that although HFE interacted with LC3B (), it mainly associated with SQSTM1 rather than other cargo receptors CALCOCO2, NBR1, NIX, TOLLIP and OPTN (). We next generated SQSTM1 knockout cell lines and observed that MAVS degradation was abolished in SQSTM1 knockout cells (). In addition, the inhibition of type I IFNs activation by HFE was abolished in SQSTM1 knockout cells (). All together, these results indicated that SQSTM1 was the only cargo receptor for HFE mediated selective autophagic degradation of MAVS.
Figure 7. HFE interaction with SQSTM1 facilitates MAVS autophagic degradation. (A) HFE interaction with SQSTM1 but not CALCOCO2, NBR1, NIX, TOLLIP and OPTN. HEK293T cells were transfected with vectors encoding MYC-HFE and indicated Flag-tagged cargo receptors, followed by co-immunoprecipitation with anti-FLAG antibody and immunoblot analysis with anti-MYC antibody. (B and C) MAVS degradation via SQSTM1. WT and SQSTM1 KO (−/-) 293 T cells were transfected with only MYC-HFE or FLAG-MAVS and MYC-HFE plasmids. Cell lysates were subjected to immunoblot analysis. (D) Luciferase assay of IFNB1 or ISRE. HEK293T WT and SQSTM1 KO cells were co-transfected with the IFNB1 or ISRE reporter plasmid together with MAVS in the presence or absence of HFE plasmid. Data are representative of at least three independent experiments, and differences between the experimental and control groups are determined by 2-way ANOVA (**p < 0.01, ***p < 0.001). (E) A proposed working model to illustrate how HFE negatively regulates type I IFNs signaling during RNA virus infection.
Discussion
Iron is an essential micronutrient, and iron balance is involved in multiple physiological processes, including the transportation of oxygen, cellular respiration, and immune responses [Citation1]. Iron is also essential for pathogenic microbes, and thus one of the host defenses against parasite or bacterial growth is through restricting iron absorption by these pathogens [Citation38]. Current evidence suggests that microbes also regulate host iron metabolism in order to escape from host immune surveillance [Citation39]. Some reports show that iron status elevated has been associated with increased mortality in human immunodeficiency virus-1 [Citation9–12], human hepatitis B virus, and human hepatitis C virus infection [Citation39]. Although the link between iron uptake and viral infection has been established, the iron metabolism-associated genes reprogram immune responses to viral infection remain poorly investigated. In the present study, we firstly identified the iron metabolism-associated genes Hfe, Hjv, Hamp, Slc40a1 and Tfrc during H7N9 virus infection. Only hfe−/- mice exhibited less than 20% survival rate to H7N9 virus infection (). However, the survival of hfe−/- mice to H7N9 virus infection was irrelevant to the concentration of diet iron (). This suggested that iron intake was not related with the resistance of hfe−/- mice to H7N9 virus infection.
Hereditary hemochromatosis is a common genetic disorder with a C282Y mutation in the Hfe gene that leads to iron overload [Citation34], and the HFE protein has been shown to physically interact with TFRC and to impair the uptake of iron by cells [Citation40]. Here, we showed that HFE was downregulated by H7N9 virus infection both in vitro and in vivo and that hfe−/- mice were more resistant to H7N9 virus and produced higher IFNB1 levels during early phases of infection compared to their WT littermates. These results are consistent with previous findings that hfe−/- mice are resistant to systemic infection with Salmonella [Citation41]. Collectively, our findings revealed an unexpected function of HFE in antiviral innate immune regulation and type I IFNs induction. In addition to HFE, the peptide HAMP/hepcidin, which is encoded by the Hamp gene, is a pivotal regulator of iron homeostasis, also contributes to host defense [Citation42]. The hamp−/- mice are more susceptible to lethal infection, although similar iron levels and iron stores are seen in WT mice [Citation43]. These findings suggested that iron metabolism-associated genes played essential roles in host defense, but the underlying molecular mechanism had remained in need of further clarification.
MAVS has been identified as an essential adaptor protein for controlling RLR-independent cytosolic signaling and for promoting the production of type I IFNs [Citation22]. Influenza virus protein PB1-F2 is identified to interact with MAVS and to inhibit the induction of type I IFNs by decreasing mitochondrial membrane potential [Citation44]. In our study, we showed that H7N9 virus triggered type I IFNs and that interferon-induced genes, such as Ifnb1, were enhanced in macrophage cells from hfe−/- mice compared to such cells from WT mice. A reporter assay further indicated that HFE greatly impaired the activity of the IFNB1 promoter triggered by MAVS or the upstream component of DDX58. Thus, we concluded that HFE functions in the innate antiviral response by modulating the stability of MAVS. The hfe−/- mice were more resistant to H7N9 virus mortality than their WT counterparts, and the viral titers were significantly decreased in the hfe−/- mice. HFE bound to MAVS and promoted SQSTM1-mediated degradation of MAVS via the selective autophagy-lysosome pathway. This suggested that HFE recruited LC3 to form autophagosomes and delivers substrates to autophagic vesicles by cargo receptor SQSTM1, which fuse with lysosomes for MAVS degradation ().
In summary, we had identified an unexpected function of HFE in innate immunity. Together, our findings showed crosstalk between HFE regulation by iron homeostasis and its effect on innate immunity, and they suggested that HFE might be a potential therapeutic target for the treatment of iron metabolic imbalance and RNA virus infection.
Materials and methods
Ethics statement
All animal care and experimental procedures were in accordance with the Animal Research Committee guidelines of Zhejiang University (No. ZJU20170066). Experiments using human samples were approved by the Institutional Review Board of the Zhejiang University and conducted in accordance with the principles expressed in the Declaration of Zhejiang University. Written informed consent was obtained from all adult subjects.
Mice
WT and hfe−/- mice in the C57BL/6-129/Ola genetic background were a kind gift from Dr. Nancy C. Andrews of the Department of Medicine and Pediatrics, Harvard Medical School, Boston. mavs−/- mice in the C57BL/6 genetic background were a kind gift from Dr. Dahua Chen of the Institute of Zoology, Chinese Academy of Sciences, Beijing. We constructed hjv−/- and hfe−/- mavs−/- mice in the C57BL/6-129/Ola genetic background. Mice were maintained under specific-pathogen-free conditions in a controlled environment of 20–22°C with 50%-70% humidity and food and water provided ad libitum. H7N9 virus infection was conducted in an ABSL3 facility. Experiments were conducted with blinding, with age- and sex-matched mice. After mice were infected with the H7N9 virus, the viability and weight loss were monitored daily for 14 d.
Cell culture
Human lung epithelia A549 cells, HEK293T cells, Madin-Darby canine kidney (MDCK) cells (American Type Culture Collection [ATCC], CRM-CCL-185/CRL-3216/CCL-34) and THP1 cells (Cell Bank of Chinese Academy of Sciences, SCSP-567) were routinely cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS; Gibco, 16140071) and 1% penicillin-streptomycin. Primary MEFs were prepared from day 13.5 hfe−/- mice embryos in the C57BL/6-129/Ola genetic background and cultured in DMEM supplemented with 10% FBS. SQSTM1 knockout HEK293T cells were from our laboratory. For preparation of BMDMs, the bone marrow cells were isolated from tibia and femur and were cultured in DMEM supplemented with 30% (v/v) L929 cell-conditioned medium (ATCC, CCL-197) and 20% FBS for 4 d. For preparation of BMDCs, the bone marrow cells were cultured in medium containing 20% FBS, murine CSF2/GM-CSF (50 ng/ml; Gibco, PMC2013) and IL4 (10 ng/ml; Gibco, BMS338) for 7 d. WT, atg5−/-, and atg7−/- MEFs were the kind gift of Professor Wei Liu of Zhejiang University, and lung fibroblasts were isolated from rats, and these cells were cultured in DMEM supplemented with 10% FBS. For growth under iron-replete conditions, CQ (Sigma-Aldrich, PHR1258) was used at a final concentration of 25 μM.
Virus and infection
All in vivo and in vitro infections were performed using influenza virus strain A/HangZhou/1/2013 (H7N9 virus GenBank accession code KC853766, obtained from the Center for Disease Control and Prevention of Zhejiang province). For in vivo infection, 6- to 8-week-old mice were inoculated intranasally under anesthesia with H7N9 virus in 50 μl phosphate-buffered saline, PBS (Sangon Biotech, E607008) at a dose of 5 × 104 pfu (plaque-forming unit). Viability and weight loss were monitored daily until 14 dpi. For the pathogenesis assay, the mice were sacrificed at the indicated time post-infection and their organs (lung, spleen, and liver) were removed aseptically for further analysis. For in vitro virus infection, cells were infected with H7N9 virus at a multiplicity of infection (MOI) of 1 at the indicated times in figure legends. After adsorption for 1 h at 37°C, the cells were washed with PBS and cultured in DMEM containing 2 μg/ml L-1-tosylamido-2-phenylethyl chloromethyl ketone/TPCK-treated trypsin (Sigma-Aldrich, 4370285). Virus stocks were propagated in embryonated chicken eggs, and virus titer was measured by 50% tissue culture infectious dose (TCID50) in MDCK cells. For the in vivo and in vitro infections with VSV, SeV (MOI = 1), the method was depicted in the figure legend.
Iron diet treatment
WT and hfe−/- mice at 6 weeks of age were fed diets with different concentrations of iron (low iron, medium iron, and high iron). The low-iron diet was the egg white-based AIN-76A diet (Research Diets, V10001) and contained 0.9 mg iron (Research Diets, V10001) per kg chow, the medium-iron diet was the control diet and contained 45 mg iron per kg chow, and the high-iron diet contained 225 mg iron per kg chow as previously described [Citation45]. Mice were fed the different iron diets for 2 weeks after which they were euthanized to determine the local and systemic effects of dietary iron intervention alone. For the virus infection experiment, mice were fed the diets for 2 weeks and then challenged with H7N9 virus infection. The diets were continued during the infection. Body weights were monitored during the course of infection, and stool samples were taken at appropriate time points.
Serum and tissue iron measurement
All blood samples were taken from the mice by intracardiac puncture under anesthesia prior to being sacrificed. Serum iron was measured with the Iron/TIBC Reagent Set according to the manufacturer’s protocol (Pointe Scientific, 17504–60). For iron concentrations in tissues, mouse lung, spleen, and liver were dried overnight at 110°C, dissociated mechanically, weighed, and digested in 1 ml of 3 M HCl and 0.6 M trichloroacetic acid for 20 h at 65°C. The total non-heme iron content was then measured using the bathophenanthroline method as described previously [Citation46].
ELISA
Cell supernatants, mouse serum, and bronchoalveolar lavage fluid were collected at the indicated times post virus infection. Cytokine levels included IL6, IFNB1 and TNF were determined using a customized Bio-Plex Pro Assay according to the manufacturer’s instructions (Bio-Rad, MCA1490/PMP28Z/MCA1488).
Histology and immunohistochemistry
The tracheas and lungs from control and virus-infected mice were dissected at the indicated post-infection times, fixed in 10% formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin solution, and visualized by light microscopy for histologic changes. Pathological scores ranging from 0 (normal) to 4 (most severely inflamed) in the lung were assigned as previously reported [Citation47]. For immunohistochemistry assays, tissues sections were stained as described previously using an anti-influenza virus NP monoclonal antibody (stored in our laboratory) [Citation48]. Sections were examined by light microscopy (Olympus, Tokyo, Japan).
Microarray analysis
BMDMs were infected for 12 h with H7N9 virus (MOI = 1), and total RNA was prepared by TRIzol extraction (Invitrogen, 15596018). RNA integrity and concentration were evaluated with a 2100 Bioanalyzer and RNA 6000 Nano kit (Agilent Technologies Inc, California, USA). The preparation of cDNA and its hybridization to an Affymetrix Mouse Gene 1.0 ST Array were performed at the Guangzhou Ruibo Biological Technology Co. Ltd. Statistical analysis for comparison of replicate arrays was done with the local poor-error test. P-values were adjusted with the Bonferroni algorithm, and a threshold of p < 0.05 was used as the criterion for significance. Assay data that support the findings of this study were available from the authors and had been deposited in the Gene Expression Omnibus under accession number GSE112964.
Flow cytometry
The spleen, thymus, and peripheral lymph nodes were obtained from WT and hfe−/- mice, and the leukocytes were extracted from the lung tissue and BAL and prepared for analysis. After removal of red blood cells by ammonium chloride, the cells were subject to stain with the indicated antibodies for 30 min followed by flow cytometry analysis. The following fluorescence-labeled antibodies were from BD Biosciences: PE-conjugated anti-PTPRC/B220 (50–112-3546), anti-ITGAM/CD11B (50–140-13), and anti-CD8A/CD8 (MCD0817); FITC-conjugated anti-ITGAX/CD11 C (50–944-0) and CD4 (50–937-6); and APC-conjugated anti-CD3E (50–148-36). DAPI (4,6-diamidino-2-phenylindole) was from Life Technologies Corporation (P36931). Flow cytometry was performed on a CytoFlex (Beckman Coulter), and the data were processed with the FlowJo7.6.1 software (Tree Star).
Iron concentration in macrophages was determined by flow cytometry. Peritoneal macrophages from WT and hfe−/- mice were collected, incubated with APC-ADGRE1/F4/80) antibody (Invitrogen, 17–4801-80) at room temperature for 20 min, and centrifuged. The resultant cells were incubated with 0.25 μM Calcein-AM (Invitrogen, C1430) at 37°C for 10 min and were treated with or without 10 μM 8-hydroxyquinoline iron (8HQ-Fe; Sigma-Aldrich, 1070980250). The median fluorescence intensity of Calcein was recorded by flow cytometry.
Confocal microscopy
BMDMs, MEFs, and HEK293T cells were washed with PBS (Sangon Biotech, E607008) three times, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, 9002–93-1) in PBS/0.1% Tween 20 (Sigma-Aldrich, 9005–64–5) at 4°C for 20 min, and then washed again with PBS. The cells were then incubated with the appropriate antibodies. Nuclei were stained with DAPI (Life Technologies Corporation, P36931). For staining of acidic compartments, 50 nM LysoTracker Red DND-99 (Invitrogen, L7528) or 200 nM LysoTracker Deep Red (Invitrogen, L12492) was added to the medium. Following staining, the stained cells were examined under a Zeiss LSM 510 laser confocal microscopy (Zeiss, Oberkochen, Germany).
Quantitative real time PCR
RNA was isolated from the indicated cells or snap-frozen mouse tissues by using TRIzol reagent and purified with the RNeasy purification kit (QIAGEN, 74104). Total RNA was then reverse-transcribed with a Quantitec reverse transcription kit (QIAGEN, 205311). Quantitative real-time PCR (qRT-PCR) was carried out in triplicate using SYBR Green (Invitrogen, K0221) according to the manufacturer’s instructions, and fold-changes were calculated with the 2−ΔΔCt method, where ΔCt is the difference between the amplification fluorescence threshold of the mRNA of interest and the mRNA of Actb used as the internal reference.
Immunoprecipitation and immunoblotting
Whole-cell lysates were prepared by lysing the cells for 15 min at 4°C in NP-40 buffer (50 mM pH 7.4 Tris-HCl, 150 mM NaCl, 1% NP-40 [Beyotime, P0013 F], 0.5% sodium deoxycholate and 0.1% SDS) containing phenylmethylsulfonyl Fluoride (PMSF; Solarbio® Life Sciences, P8340) and a protease inhibitor cocktail (Beyotime Biotechnology, P0013 C). Lysates were centrifuged at 12,000 × g for 10 min at 4°C. Protein concentrations were determined using a bicinchoninic acid (BCA) assay kit (Novagen, Darmstadt, Germany) according to the manufacturer’s instructions. Immunoprecipitation was performed using mouse anti-FLAG (Millipore, MAB3118), anti-MYC (Millipore, 05–419), anti-GFP (HUABIO INC, EM30501), or anti-MAVS (Santa Cruz Biotechnology, sc-365333 or sc-166583) and anti-IgG (Beyotime Biotechnology, A7028) antibodies with protein A/G agarose beads (Santa Cruz Biotechnology, sc-2003). Western blotting of the cell lysates and immunoprecipitates was performed using mouse anti-FLAG, anti-GFP, anti-HA (HUABIO INC, 0906–1), anti-MYC, and other antibodies as indicated. Equal amounts of proteins were analyzed by immunoblot, and signals were developed with the SuperSignal West Femto Substrate Trial Kit (Thermo Scientific, 34096). ImageJ software (National Institutes of Health, Bethesda, USA) was used to quantify the intensities of the protein bands.
Dual luciferase reporter assay
Cells were transfected with plasmids encoding the IFNB1 or ISRE luciferase reporter gene together with the pRL-TK (Promega, E2241) and indicated plasmids in figure legends. 24 h after transfection, the cells were collected and lysed. Luciferase activity was measured with the dual luciferase reporter assay system (Promega, Madison, USA) according to the manufacturer’s protocols. Data were normalized by the ratio of firefly luciferase activity to renilla luciferase activity.
Reagents and constructs
Poly (I:C) (tlrl-piclv) and HSV60 (tlrl-hsv60n) were from InvivoGen. Rabbit antibodies against phospho-TBK1 (5483), TBK1 (3504), phospho-STAT1 (9177), STAT1 (14994), phospho-IRF3 (4947), IRF3 (4302), and DDX58 (3743) were from Cell Signaling Technology. Rabbit antibodies against ATG5 (ab108327), SQSTM1 (ab109012) and ATG7 (ab183188) were from Abcam. Rabbit antibody against HJV (AF3634) was from R&D Systems. Rabbit antibody against SLC40A1 (MTP-11A) was from Alpha Diagnostic International. Mouse antibodies against HFE (sc-514405) and TFRC (sc-393719) were from Santa Cruz Biotechnology. Mouse antibodies against ACTB (actin, beta; M1210-2) and GAPDH (EM1101) were from HUABIO INC. Goat anti-mouse IgG, IgM secondary antibody, FITC (A-10679) and donkey anti-mouse IgG secondary antibody, and Alexa Fluor® 647 (A-31571) and 546 (A10036) conjugates were obtained from Thermo Fisher Scientific. Mouse monoclonal antibodies against the NP and M1 proteins of the influenza virus were produced in our laboratory. EXFectTM Transfection Reagent (T101–01/02) was purchased from Vazyme Biotech. MAVS-Pex-GFP plasmid was kindly provided by Professor Chunfu Zheng of the Chinese Fujian Medical University. The IFNB1 and ISRE promoter luciferase reporter plasmids were kindly provided by Professor Zongping Xia of Zhejiang University, and mammalian expression plasmids for FLAG-tagged DDX58, MAVS, CGAS, STING1, TBK1, IRF3, CALCOCO2, SQSTM1, OPTN, NIX, TOLLIP, NBR1, VDAC1 and SLC25A6 had been stored in our laboratory. The mammalian expression plasmids for WT and mutant MYC-tagged HFE were constructed with standard molecular biology techniques, and these included the cytoplasmic (ΔCyto), transmembrane (ΔTM), and extracellular Δα1Δα2, Δα2, and Δα3 domain deletions of HFE protein and the HFE LC3-interacting region (LIR). HFE mutants were kindly provided by Professor Yin Xia of the Chinese University of Hong Kong.
Statistical analysis
Statistically significant differences between groups and treatments were determined by 2-way analysis of variance (ANOVA) and between groups were determined by one-way with the Tukey Multiple Comparison Test and using GraphPad Prism 5 software. For the mouse survival study, the Log-Rank test was used to analyze the Kaplan-Meier survival curves. A p-value < 0.05 was considered statistically significant.
Supplemental Material
Download Zip (707.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe. 2013;13(5):509–519.
- Ramalingam TS, West AP Jr., Lebron JA, et al. Binding to the transferrin receptor is required for endocytosis of HFE and regulation of iron homeostasis. Nat Cell Biol. 2000;2(12):953–957.
- Roy CN, Carlson EJ, Anderson EL, et al. Interactions of the ectodomain of HFE with the transferrin receptor are critical for iron homeostasis in cells. FEBS Lett. 2000;484(3):271–274.
- Salter-Cid L, Peterson PA, Yang Y. The major histocompatibility complex-encoded HFE in iron homeostasis and immune function. Immunol Res. 2000;22(1):43–59.
- Wu XG, Wang Y, Wu Q, et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood. 2014;124(8):1335–1343.
- Pantopoulos K. Iron regulation of hepcidin through Hfe and Hjv: common or distinct pathways? Hepatology. 2015;62(6):1922–1923.
- Wu Q, Wang H, An P, et al. HJV and HFE play distinct roles in regulating hepcidin. Antioxid Redox Signal. 2015;22(15):1325–1336.
- Drakesmith H, Sweetland E, Schimanski L, et al. The hemochromatosis protein HFE inhibits iron export from macrophages. Proc Natl Acad Sci U S A. 2002;99(24):15602–15607.
- Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol. 2003;38(1):61–88.
- Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352(10):1011–1023.
- Lundgren JD, Mocroft A. Anemia and survival in human immunodeficiency virus. Clin Infect Dis. 2003;37(Suppl 4S):297–303.
- O’Brien ME, Kupka R, Msamanga GI, et al. Anemia is an independent predictor of mortality and immunologic progression of disease among women with HIV in Tanzania. J Acquir Immune Defic Syndr. 2005;40(2):219–225.
- Poynard T, Yuen MF, Ratziu V, et al. Viral hepatitis C. Lancet. 2003;362(9401):2095–2100.
- Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. 2003;110(2):163–169.
- Chouteau P, Le Seyec J, Saulier-Le Drean B, et al. Inhibition of hepatitis B virus production associated with high levels of intracellular viral DNA intermediates in iron-depleted HepG2.2.15 cells. J Hepatol. 2001;34(1):108–113.
- Parrish CR. Pathogenesis of feline panleukopenia virus and canine parvovirus. Baillieres Clin Haematol. 1995;8(1):57–71.
- Schwartz O, Marechal V, Le Gall S, et al. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2(3):338–342.
- Chen N, McCarthy C, Drakesmith H, et al. HIV-1 down-regulates the expression of CD1d via Nef. Eur J Immunol. 2006;36(2):278–286.
- Ben-Arieh SV, Zimerman B, Smorodinsky NI, et al. Human cytomegalovirus protein US2 interferes with the expression of human HFE, a nonclassical class I major histocompatibility complex molecule that regulates iron homeostasis. J Virol. 2001;75(21):10557–10562.
- Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988.
- Xu LG, Wang YY, Han KJ, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19(6):727–740.
- Seth RB, Sun L, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682.
- Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172.
- Ding SW. RNA-based antiviral immunity. Nat Rev Immunol. 2010;10(9):632–644.
- Guan K, Wei C, Zheng Z, et al. MAVS promotes inflammasome activation by targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J Immunol. 2015;194(10):4880–4890.
- Pan Y, Li R, Meng JL, et al. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J Immunol. 2014;192(10):4758–4764.
- Castanier C, Zemirli N, Portier A, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;1044.
- You F, Sun H, Zhou X, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10(12):1300–1308.
- Sun X, Sun L, Zhao Y, et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2016;216024.
- Jin S, Tian S, Luo M, et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68(2):308–22 e4.
- He X, Zhu Y, Zhang Y, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. Embo J. 2019;38(14):e100978.
- Wang K, Ma H, Liu H, et al. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 2019;27(7):2075–91 e5.
- Liu Q, Ma J, Strayer DR, et al. Emergence of a novel drug resistant H7N9 influenza virus: evidence based clinical potential of a natural IFN-alpha for infection control and treatment. Expert Rev Anti Infect Ther. 2014;12(2):165–169.
- Ehrlich R, Lemonnier FA. HFE–a novel nonclassical class I molecule that is involved in iron metabolism. Immunity. 2000;13(5):585–588.
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662.
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75.
- Schaible UE, Kaufmann SH. Iron and microbial infection. Nat Rev Microbiol. 2004;2(12):946–953.
- Drakesmith H, Prentice A. Viral infection and iron metabolism. Nat Rev Microbiol. 2008;6(7):541–552.
- Lebron JA, West AP Jr., Bjorkman PJ. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J Mol Biol. 1999;294(1):239–245.
- Nairz M, Theurl I, Schroll A, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114(17):3642–3651.
- Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98(15):8780–8785.
- Michels K, Nemeth E, Ganz T, et al. Hepcidin and host defense against infectious diseases. PLoS Pathog. 2015;11(8):e1004998.
- Varga ZT, Grant A, Manicassamy B, et al. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. 2012;86(16):8359–8366.
- Zhang Z, Zhang F, Guo X, et al. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56(3):961–971.
- Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33(1):9–11.
- Han D, Hu Y, Li L, et al. Highly pathogenic porcine reproductive and respiratory syndrome virus infection results in acute lung injury of the infected pigs. Vet Microbiol. 2014;169(3–4):135–146.
- Yu Y, Wang X, Jin T, et al. Newly emergent highly pathogenic h5n9 subtype avian influenza A Virus. J Virol. 2015;89(17):8806–8815.