
ABSTRACT
Ferroptosis is an iron-dependent, non-apoptotic form of regulated cell death caused by lipid peroxidation, which is controlled by integrated oxidation and antioxidant systems. The iron-containing enzyme lipoxygenase is the main promoter of ferroptosis by producing lipid hydroperoxides, and its function relies on the activation of ACSL4-dependent lipid biosynthesis. In contrast, the selenium-containing enzyme GPX4 is currently recognized as a central repressor of ferroptosis, and its activity depends on glutathione produced from the activation of the cystine-glutamate antiporter SLC7A11. Many metabolic (especially involving iron, lipids, and amino acids) and degradation pathways (macroautophagy/autophagy and the ubiquitin-proteasome system) orchestrate the complex ferroptotic response through direct or indirect regulation of iron accumulation or lipid peroxidation. Although the detailed mechanism of membrane injury during ferroptosis remains a mystery, ESCRT III-mediated plasma membrane repair can make cells resistant to ferroptosis. Here, we review the recent rapid progress in understanding the molecular mechanisms of ferroptosis and focus on the epigenetic, transcriptional, and posttranslational regulation of this process.
Abbreviations: 2ME: beta-mercaptoethanol; α-KG: α-ketoglutarate; ccRCC: clear cell renal cell carcinoma; EMT: epithelial-mesenchymal transition; FAO: fatty acid beta-oxidation; GSH: glutathione; MEFs: mouse embryonic fibroblasts; MUFAs: monounsaturated fatty acids; NO: nitric oxide; NOX: NADPH oxidase; PPP: pentose phosphate pathway; PUFA: polyunsaturated fatty acid; RCD: regulated cell death; RNS: reactive nitrogen species; ROS: reactive oxygen species; RTAs: radical-trapping antioxidants; UPS: ubiquitin-proteasome system; UTR: untranslated region.
KEYWORDS:
Introduction
Cell death is a common process in all organisms, with different classifications being used over time. Early classifications based on cell morphology divided cell death into apoptosis (type I), autophagy (type II), and necrosis (type III) in the 1970s. According to the latest recommendations of the Cell Death Nomenclature Committee in 2018, there are currently two types of cell death, namely accidental cell death (ACD) and regulated cell death (RCD) [Citation1]. Accidental cell death is an uncontrolled and unavoidable cell death process resulting from chemical, physical, or mechanical severe stress, whereas RCD can be regulated by pharmacological or genetic interventions. RCD is further divided into apoptotic and non-apoptotic forms (e.g., ferroptosis [Citation2], necroptosis [Citation3], pyroptosis [Citation4], and alkaliptosis [Citation5,Citation6]), which have different characteristics of signal induction and molecular modulation as well as disease implications [Citation7]. Morphologically, ferroptotic cells have typical necrosis-like changes, such as cell swelling and plasma membrane rupture, which is different from apoptotic cells that are characterized by membrane blebbing and shrinkage. Biochemically, ferroptosis is characterized by the production of lethal levels of iron-dependent lipid peroxidation [Citation8,Citation9]. However, the classical biochemical features of apoptosis, such as chromatin fragmentation, caspase activation, and the release of mitochondrial CYCS (cytochrome c, somatic), are rarely observed during ferroptosis [Citation2]. Although ferroptosis was initially described as an autophagy-independent type of cell death [Citation2], there is growing evidence that autophagy, especially selective autophagy, plays a context-dependent role in promoting ferroptotic cell death [Citation10–12]. Moreover, the interaction between ferroptosis and other types of RCD seems to be common in diseases and pathological conditions, indicating an intricate complex feedback mechanism between RCDs.
The concept of ferroptosis comes from efforts in precision oncology to develop compounds that selectively kill cells with an oncogenic RAS mutation. The first ferroptosis-inducing agent, erastin, was identified in 2003 by screening small-molecule drug libraries with selective lethality in an engineered cell line expressing an HRAS (HRas proto-oncogene, GTPase) mutation [Citation13]. In 2012, it was demonstrated that the anticancer activity of erastin relies on the induction of a new type of cell death, which can be completely prevented by iron chelators and lipophilic antioxidants, but not by apoptotic inhibitors (e.g., Z-VAD-FMK) [Citation2]. Therefore, the term “ferroptosis” was coined to describe this iron-dependent, non-apoptotic form of cell death [Citation2]. Subsequent screening studies showed that another small-molecule compound, RSL3, also induced ferroptosis, and further identified the antioxidant defense enzyme GPX4 (glutathione peroxidase 4) as a direct drug target of RSL3 by using a chemoproteomic approach [Citation14]. Erastin and RSL3 are currently the most commonly used classic reagents to induce ferroptosis. Although ferroptosis is caused by oxidative damage, it should be noted that not all sources of reactive oxygen species (ROS) contribute equally to ferroptotic cell death, and iron-dependent ROS production seems to be the main driver of ferroptosis by lipid peroxidation, indicating that ferroptosis requires a unique molecular machinery for its initiation and effector.
Mounting evidence has connected malfunctions in ferroptotic processes to iron- or ROS-related diseases, such as cancer, neurodegenerative disorders, infection, and inflammatory diseases [Citation8,Citation9]. Of note, ferroptosis may play a dual role in tumorigenesis and tumor therapy, depending on the tumor types and stages. The increase of iron can promote the occurrence and growth of tumors because iron is an important nutrient for cell proliferation and a co-factor for metabolic enzymes. In addition, ferroptotic cell death may trigger tumor initiation by increasing the inflammation response at an early stage [Citation15]. In contrast, increasing iron utilization by inducing ferroptosis appears to be an attractive method of killing various cancers at a later stage [Citation16]. In neuronal cells, the process of ferroptosis is similar to oxytosis, a type of oxidative cell death caused by glutamate toxicity [Citation17]. The inhibition of ferroptosis and oxytosis can help reduce brain damage in Alzheimer, Parkinson, and Huntington diseases. In addition to regulating infection responses by ferroptotic immune cells (e.g., T and B cells), ferroptosis-mediated sterile inflammation also plays a pathological role in the progression of ischemia/reperfusion (IR) injury of various tissues, such as those of the heart, liver, kidney, brain, intestines, and testis [Citation8,Citation9]. Therefore, pharmacological regulation of ferroptosis is a potential therapeutic approach for infection and tissue injury.
With the rapid expansion of studies on ferroptosis, new molecular mechanisms for orchestrating ferroptosis are being actively investigated. In this review, we summarize the current understanding of the process and basis of ferroptosis. In particular, we discuss how oxidant- and antioxidant-dependent signaling can modulate the complex ferroptotic response.
The antioxidant basis of ferroptosis
Ferroptosis is mainly caused by the inactivation of cellular antioxidant system, especially the system xc−-glutathione (GSH)-GPX4-dependent antioxidant defense, which leads to the accumulation of lipid hydroperoxides. The system xc− antiporter is responsible for the transmembrane import of extracellular cystine, which is reduced back to intracellular cysteine (a precursor amino acid for GSH synthesis). GSH acts as a necessary cofactor for the normal function of GPX4, which is an antioxidant enzyme that quenches phospholipid hydroperoxide. Selenium is an important component of selenocysteine-containing proteins (including but not limited to GPX4), which can increase the antioxidant capacity of cells during ferroptotic damage. In addition, the lipophilic antioxidant CoQ (CoQ) is converted to a reduced form, thereby protecting cells from ferroptosis in a GSH-independent manner. In this section, we summarize the present understanding of antioxidant system (including GSH, selenium, and CoQ system) in ferroptotic cell death.
GSH system
The antioxidant GSH is a tripeptide composed of glutamic acid, cysteine, and glycine. It is synthesized from its constituent amino acids by the consecutive actions of GCL (glutamate-cysteine ligase). GCL is composed of a catalytic subunit, GCLC (glutamate-cysteine ligase catalytic subunit), and a modifier subunit, GCLM (glutamate-cysteine ligase modifier subunit), that function as the rate-limiting enzymes in the de novo synthesis of GSH. Due to the limited concentration of cysteine in cells, cysteine is considered to be the rate-limiting precursor for GSH synthesis. Cysteine is imported into cells by system xc− in its oxidized form cystine, and cystine is immediately reduced to cysteine within the cells . As a cystine and glutamate antiporter, system xc− in the plasma membrane is a heterodimeric protein complex comprised of SLC7A11/xCT (solute carrier family 7 member 11) and SLC3A2 (solute carrier family 3 member 2). Mice lacking SLC7A11 are healthy in appearance and fertile, whereas fibroblasts isolated from these mice undergo cell death, which can be rescued by the presence of beta-mercaptoethanol (2ME), N-acetyl cysteine (NAC), or vitamin E [Citation18]. A plausible explanation for the phenotype difference between in vivo and in vitro slc7a11 deletion is that other transport systems may bypass the dependence on system xc− in vivo. In the case of iron overload, the absence of SLC7A11 promotes liver damage associated with ferroptosis [Citation19]. slc7a11 deletion suppresses KRAS (KRAS proto-oncogene, GTPase)-driven development of pancreatic duct adenocarcinoma in mice, indicating a role for cysteine depletion in the induction of ferroptosis during tumor suppression [Citation20].
Figure 1. Core mechanism of ferroptosis. System xc−-mediated cystine uptake and subsequent GSH production and GPX4 activation play a central role in protecting cells from ferroptosis. Alternatively, AIFM2 inhibits ferroptosis by catalyzing the production of CoQ10H2 from CoQ10 or promoting ESCRT-III–dependent membrane repair. Ferritinophagy and lipophagy provide the substrates Fe2+ and FA, respectively, for the execution of lipid peroxidation during ferroptosis. This process is accompanied by ACSL4-catalyzed arachidonic acid-CoA formation, followed by LPCAT3-mediated arachidonic acid-CoA esterification to phospholipids (PL). Several ROS-producing enzymes (e.g., ALOXs, POR, and NOXs) are also involved in the induction of lipid peroxidation. The red text and blue text indicate ferroptosis inducers and inhibitors, respectively. Abbreviations: 2ME, beta-mercaptoethanol; AA, arachidonic acid; ACLS4, acyl-CoA synthetase long chain family member 4; Ada, adrenic acid; AIFM2, apoptosis inducing factor mitochondria associated 2; ALOXs, lipoxygenases; ANGPTL4, angiopoietin like 4; CoA, coenzyme A; CoQ10, coenzyme Q10; CoQ10H2, ubiquinol; DPP4, dipeptidyl peptidase 4; EMP1, epithelial membrane protein 1; ESCRT-III, endosomal sorting complex required for transport-III; FA, fatty acid; G6PD, glucose-6-phosphate dehydrogenase; GCL, glutamate-cysteine ligase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSR, glutathione-disulfide reductase; GSS, glutathione synthetase; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; HDDC3, HD domain containing 3; LPCAT3, lysophosphatidylcholine acyltransferase 3; NAC, N-acetyl cysteine; NADK, NAD kinase; NCOA4, nuclear receptor coactivator 4; NOXs, NADPH oxidases; PGD, phosphogluconate dehydrogenase; PL, phospholipid; PLOOH, phospholipid hydroperoxides; POR, cytochrome p450 oxidoreductase; PPP, pentose phosphate pathway; RAB7A, RAB7A, member RAS oncogene family; RTAs, radical-trapping antioxidants; Se, selenium; t-BuOOH, tert-butyl hydroperoxide
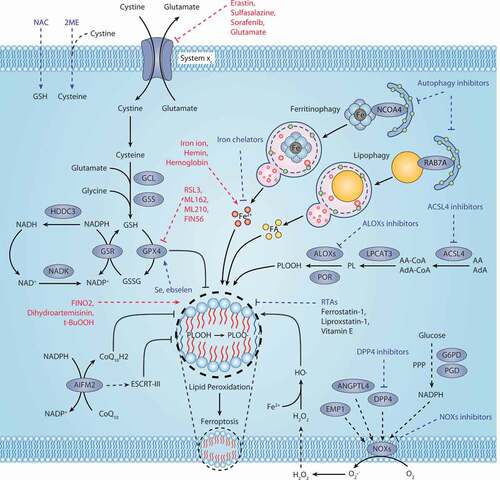
The GPXs family consists of multiple isozymes expressed in different subcellular locations and tissues. Compared to other GPXs members, GPX4 directly reduces lipid hydroperoxide to nontoxic lipid alcohol in the membrane . The availability of cellular GSH closely determines the proper GPX4 function. GPX4 is inactivated after GSH depletion, which can be triggered by system xc− inhibitors (e.g., erastin) or GCL inhibitors (e.g., butylthionine sulfoximine [BSO]) [Citation14]. GPX4 has an active site, namely selenocysteine, which forms a catalytic quadruplex with tryptophan, glutamine, and asparagine. Although this quadruplex is conserved in the GPXs family, only GPX4 inhibits ferroptosis. GPX4 catalyzes the reduction of lipid hydroperoxides involving a ping-pong mechanism, in which selenocysteine shuttles between reduced selenol (SeH) and oxidized selenic acid (SeOH) states. This dynamic process includes the following: (1) The selenol form of selenocysteine (GPX4-SeH) is oxidized by hydroperoxide into selenic acid intermediate (GPX4-SeOH), while hydroperoxide (LOOH) is reduced to alcohol (LOH); (2) This intermediate reacts with GSH to produce water and selenium-glutathione adduct (GPX4-Se-SG); (3) Subsequently, by reacting with the second equivalent of GSH, GPX4-Se-SG is converted back to selenol (GPX4-SeH) to produce oxidized glutathione (GSSG), resulting in the regeneration of the enzyme’s initial configuration.
GPX4 is a recognized ferroptosis gatekeeper and plays a central role in limiting lipid peroxidation. Unlike slc7a11 knockout mice, gpx4 knockout mice show early embryo lethality [Citation21,Citation22], indicating that SLC7A11 and GPX4 may have different functions in ferroptosis. Mice with a targeted mutation of the active site selenocysteine of GPX4 to serine (U46S) or alanine (U46A) also display embryonic lethality [Citation23,Citation24], indicating that the catalytic activity of GPX4 is essential for normal embryonic development. GPX4 has three isoforms: mitochondrial, cytosolic, and nuclear, but it is still unclear which isoform is the main regulator of anti-ferroptosis effects. It remains possible that these organelle-specific forms of GPX4 may act as independent regulators of local lipid hydroperoxides. In addition to ferroptosis, GPX4 also plays a role in restricting apoptosis [Citation25], necroptosis [Citation26] and pyroptosis [Citation27], indicating that lipid peroxidation may be a common signal for the induction of various types of RCD.
Selenium system
The essential trace element selenium plays a significant role in regulating cell redox during oxidative stress. Selenium is a component of selenocysteine at the catalytic site of antioxidant enzymes, such as GPXs, TXNRDs (thioredoxin reductases), and SELENOP (selenoprotein P). Selenium deficiency causes lipid ROS-dependent cell death in serum-free medium, and vitamin E may inactivate this process. Notably, the codon for selenocysteine is UGA, which is normally recognized as a translational termination signal. Selenoprotein synthesis is mediated by selenocysteine tRNA (tRNASec), which inserts selenocysteine in the UGA codons through a complex process. n-TUtca2/trsp/TRU-TCA1-1 (TRNA-SeC [nuclear-encoded tRNA selenocysteine 2 (anticodon TCA)]) knockout mice lacking all selenoproteins exhibit an embryonic-lethal phenotype [Citation28], indicating that selenoproteins are essential for mammalian life. Unlike the early embryonic lethality observed in gpx4 knockout mice, GPX4U46C/U46C (referred to as GPX4Cys/Cys) mice with a replacement of the active site selenocysteine to the cysteine variant have unexpectedly normal embryogenesis, although they show severe spontaneous seizures after birth [Citation29]. GPX4Cys/Cys mouse embryonic fibroblasts (MEFs) are highly sensitive to low concentrations of hydrogen peroxide (H2O2)-induced ferroptotic cell death [Citation29], further supporting a pro-survival role of GPX4 in response to oxidative damage. The knockout of n-TUtca2/TRU-TCA1-1 in GPX4Cys/Cys MEFs is feasible, but not in wild-type GXP4 MEFs, implying that selenocysteine incorporation is responsible for blocking ferroptosis when GPX4 function is maintained [Citation29]. However, it is still unknown whether GPX4Cys/Cys might rescue the development of mice lacking n-TUtca2/TRU-TCA1-1. In addition to GPX4, it remains to be clarified whether other selenoproteins are also involved in the control of ferroptosis.
Similar to ferrostatin-1, selenium strongly protects intracerebral hemorrhage by reducing ferroptosis in experimental animals. In particular, the addition of selenium (sodium selenite) induces the transcriptional upregulation of selenoprotein genes (including Gpx4, Selenop, Txnrd1 [thioredoxin reductase 1], and Gpx3 [glutathione peroxidase 3]), and inhibits homocysteine (glutamate analog) or heme-induced ferroptosis during intracerebral hemorrhage [Citation30]. Selenium treatment also abrogates erastin-induced ferroptosis in mouse primary cortical neurons and HT1080 cells, but it fails to block RSL3- and FIN56-induced ferroptosis in mouse primary cortical neurons [Citation30]. One possible explanation is that GPX4 is responsible for the anti-injury activity of selenium. However, the overexpression of GPX4 reverses RSL3 lethality in HT1080 cells, promoting the argument that GPX4 is not the only target of RSL3 [Citation14]. Indeed, RSL3 can also bind to other selenoproteins, such as TXNRD1, SELENOK/SELK (selenoprotein K), and SELENOT/SELT (selenoprotein T) [Citation14,Citation31], further raising the likelihood of other selenoproteins regulating ferroptosis. Consistent with this hypothesis, a TXN (thioredoxin) inhibitor ferroptocide robustly induces ferroptotic cell death in ES-2 ovarian cancer cells [Citation32]. Therefore, selenoproteins in an integrated regulatory network may cooperate and complement each other to inhibit ferroptosis.
CoQ system
CoQ, also known as ubiquinone, is an endogenously produced isoprenyl benzoquinone compound that is ubiquitous in nature. CoQ10, where 10 refers to the number of isoprene units in its side chain tail, is the most common form of CoQ as a dietary supplement. CoQ10 plays a fundamental role in the mitochondrial electron transport chain (also known as respiratory chain) through carrying electrons from complex I and II to III. In addition, the reduced form of CoQ10, namely ubiquinol (CoQ10H2), is used as an effective lipophilic antioxidant involved in the recovery of other antioxidants, such as tocopherol and ascorbate. The isoprenoid side chain of CoQ10 is synthesized through the mevalonate pathway in eukaryotes. Supplementation with farnesyl pyrophosphate (an upstream product for CoQ10 synthesis) or idebenone (a hydrophilic analog of CoQ10) suppresses the lethality of FIN56 [Citation33], suggesting that CoQ10 is an endogenous suppressor of ferroptosis. AIFM2/FSP1 (apoptosis inducing factor mitochondria associated 2) is identified as a repressor of ferroptosis through the production of CoQ10 [Citation34,Citation35], which is parallel to the GSH-dependent GPX4 pathway . AIFM2 has NADH:ubiquinone oxidoreductase activity, which can reduce CoQ10 to ubiquinol. The mutation of the E156 site in AIFM2 impairs the oxidoreductase activity of AIFM2 and fails to reverse RSL3-induced ferroptotic cell death, whereas the overexpression of AIFM2 mediates resistance to RSL3 [Citation35]. The depletion of COQ2 (coenzyme Q2, polyprenyltransferase), a CoQ10 biosynthesis enzyme, abolishes the anti-ferroptotic function of AIFM2 [Citation34,Citation35]. In addition, the metabolic derivatives tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) synthesized by GCH1 (GTP cyclohydrolase 1) may antagonize ferroptotic cell death by controlling the production of CoQ10 [Citation36]. These findings indicate that multiple pathways contribute to CoQ10 production in order to neutralize free radicals produced during ferroptosis.
In addition to mediating reduced CoQ10 production, AIFM2 is also implicated in ferroptosis resistance through activating the endosomal sorting complexes required for transport (ESCRT)-III–dependent membrane repair in the plasma membrane, supporting an anti-injury role of membrane AIFM2 [Citation37]. Some studies have also shown that, similar to AIFM1 (apoptosis inducing factor mitochondria associated 1), AIFM2 promotes caspase-independent apoptosis during oxidative stress [Citation38]. AIFM2 is adducted by 4-hydroxy-2-nonenal (4HNE, a major aldehyde product of lipid peroxidation), resulting in the inactivation of its NADH oxidoreductase activity and translocation from mitochondria to the nucleus to induce apoptosis [Citation39]. Although it is unclear how AIFM2 regulates the balance between apoptosis and ferroptosis, one possibility is that some unknown signals determine its subcellular location and subsequent enzymatic and non-enzymatic functions. In addition to synthesizing GSH, cysteine is used to synthesize coenzyme A (CoA, a potential substrate for CoQ10 synthesis), thereby reducing the sensitivity of cells to ferroptosis [Citation20]. Together, CoQ10 and GSH may work in separate pathways to inhibit ferroptosis, although there are still many links between these antioxidants.
The metabolic basis of ferroptosis
Iron metabolism
Iron is an indispensable element in human beings, and physiological iron concentration plays multiple roles in metabolic processes, such as oxygen transport, electron transport, and DNA synthesis. Due to its ability to accept and donate electrons, pathological iron accumulation can cause oxidative damage and even death in cells. In mammalian cells, non-heme and heme iron absorption pathways involve various transporters or receptors that provide iron for subsequent lipid peroxidation. In addition to mediating the production of ROS through the Fenton reaction, iron is also transported to several iron-containing enzymes involved in lipid peroxidation. In contrast, increasing iron storage through ferritin or exporting iron through SLC40A1/ferroportin-1 (solute carrier family 40 member 1) limits intracellular iron utilization, thereby limiting ferroptosis. Thus, the coordinated changes of regulators of iron homeostasis influence the sensitivity of cells to ferroptosis. In this section, we discuss the link between ferroptosis and iron metabolism, which includes at least four parts: uptake, storage, utilization, and efflux .
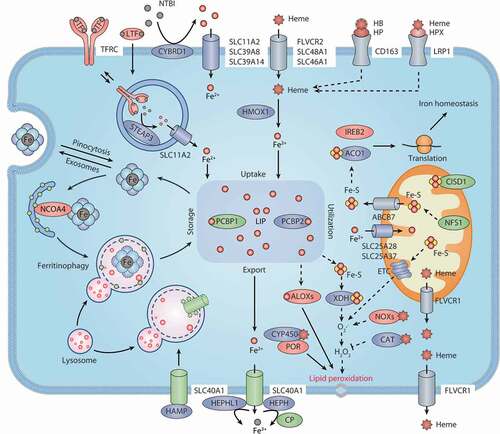
Figure 2. Iron metabolism and ferroptosis. Extracellular iron enters cells, mainly through endosome-mediated internalization of the TF-TFRC complex and receptor-mediated NTBI or heme uptake. Free iron in the cells is stored in ferritin, and iron is released again by ferritinophagy-mediated ferritin degradation. The iron chaperones PCBP1 and PCBP2 may play an important role in cellular iron transmission. SLC40A1 is an iron exporter, cooperating with iron oxidases (e.g., CP, HEPH, and HEPHL1) to transport iron to the extracellular space. Iron, heme, or Fe-S are all incorporated into ROS-generating enzymes (e.g., ALOXs, NOXs, XDH, CYP/CYP450, and ETC complexes) or antioxidant enzymes (e.g., CAT). ACO1 and IREB2 regulate iron homeostasis by controlling the translation of mRNA related to iron metabolism. Abbreviations: ABCB7, ATP binding cassette subfamily B member 7; ACO1, aconitase 1; ALOXs, lipoxygenases; CAT, catalase; CISD1, CDGSH iron sulfur domain 1; CP, ceruloplasmin; CYBRD1, cytochrome B reductase 1; CYP450, cytochrome P450; ETC, electron transport chain; Fe-S, iron-sulfur cluster; FLVCR1, FLVCR heme transporter 1; FLVCR2, FLVCR heme transporter 2; HAMP, hepcidin antimicrobial peptide; HB, hemoglobin; HEPH, hephaestin; HEPHL1, hephaestin like 1; HMOX1, heme oxygenase 1; HP, haptoglobin; HPX, hemopexin; IREB2, iron responsive element binding protein 2; LIP, labile iron pool; LRP1, LDL receptor related protein 1; LTF, lactotransferrin; NCOA4, nuclear receptor coactivator 4; NFS1, NFS1 cysteine desulfurase; NOXs, NADPH oxidases; NTBI, nontransferrin-bound iron; PCBP1, poly(RC) binding protein 1; PCBP2, poly(rC) binding protein 2; POR, cytochrome p450 oxidoreductase; SLC11A2, solute carrier family 11 member 2; SLC25A28, solute carrier family 25 member 28; SLC25A37, solute carrier family 25 member 37; SLC39A8, solute carrier family 39 member 8; SLC39A14, solute carrier family 39 member 14; SLC40A1, solute carrier family 40 member 1; SLC46A1, solute carrier family 46 member 1; STEAP3, STEAP3 metalloreductase; TF, transferrin; TFRC, transferrin receptor; XDH, xanthine dehydrogenase
Iron uptake
Most cells acquire nonheme iron mainly through two ways: TF (transferrin)-bound iron uptake and non-TF-bound iron (NTBI) uptake. Physiologically, almost all circulating iron binds to TF/TRF, which is a plasma glycoprotein that binds tightly to iron but is reversible. Each molecule of TF has two specific ferric iron (Fe3+) binding sites. Iron-containing TF can further bind to TFRC (transferrin receptor), thus causing membrane invagination and the formation of specialized endosomes. After internalization, TF is delivered to the endosome, where the pH drops rapidly, resulting in the release of Fe3+ from TF. Fe3+ is then reduced to Fe2+ by an endosomal ferrireductase, namely STEAP3 (STEAP3 metalloreductase). Fe2+ can traverse the endosomal membrane to enter the cytoplasm via SLC11A2/DMT1 (solute carrier family 11 member 2). Iron-free TF is then returned to the cell surface and dissociates from TFRC to enable further iron uptake. The knockdown of TF decreases siramesine- and lapatinib-induced ferroptosis in MDA-MB-231 and SKBR3 cancer cell lines [Citation40]. Similarly, the loss of TFRC suppresses cystine starvation- or erastin-induced ferroptotic cell death [Citation41,Citation42]. These findings indicate that TF and TFRC are required for ferroptotic cell death.
In the case of iron overload, such as hemochromatosis, excess iron may overwhelm the carrying capacity of TF and circulate in the form of NTBI. Although the exact mechanism of NTBI uptake is still elusive, it is generally accepted that the presence of one or more ferrireductases (e.g., CYBRD1 [cytochrome B reductase 1]) on the cell surface or the release of cellular reductants (e.g., ascorbate) [Citation43] reduce NTBI iron to the ferrous state and then import it through transmembrane transporters, such as SLC11A2, SLC39A8/ZIP8 (solute carrier family 39 member 8), or SLC39A14/ZIP14 (solute carrier family 39 member 14) [Citation44]. The mRNA expression of both TFRC and SLC11A2 is upregulated in HT1080 cells following erastin treatment [Citation45], suggesting that both TF-bound iron and NTBI might contribute to the induction of ferroptosis. Indeed, the knockout of Slc39a14 suppresses ferroptosis-related liver fibrosis in hepatocyte-specific trf knockout mice [Citation46].
Free HB and heme in plasma can be captured by HP (haptoglobin) and HPX (hemopexin), respectively. After binding to the macrophage scavenger receptor CD163 (CD163 molecule) and LRP1/CD91 (LDL receptor related protein 1) receptor, HB-HP and heme-HPX complexes are removed from circulation and internalized into endosomes [Citation47]. The uptake of extracellular nonprotein-bound heme or albumin-bound heme is mediated by various transporters, such as FLVCR2 (FLVCR heme transporter 2), SLC48A1/HRG-1 (solute carrier family 48 member 1), and SLC46A1/HCP1 (solute carrier family 46 member 1). In cells, heme is processed by the cytosolic HMOX1 (heme oxygenase 1), result in the release of Fe2+. Hemin is a stimulator of HMOX1 synthesis and induces ferroptosis in vitro and in vivo [Citation48,Citation49]. However, HMOX1 also shows anti-ferroptotic effects in hepatocellular carcinoma cells due to its antioxidant activity [Citation50]. The dual role of HMOX1 in ferroptosis may rely on its expression and the cell type.
Iron utilization
The iron in a cell enters a metabolically active pool called the “labile iron pool” (LIP). The LIP is exchangeable and can be stored, exported, or utilized. Most (>80%-90%) of the cytoplasmic LIP is in the reduced state (Fe2+) [Citation51,Citation52], which can be partially explained by the presence of excess iron-reducing agents (e.g., GSH and NADPH) in the cytosol. The exact form of this cytoplasmic iron pool is unclear, but at least some iron is bound by iron chaperones, such as poly(rC)-binding proteins (PCBPs). PCBP1 (poly(rC) binding protein 1) and PCBP2 appear to be important regulators for the fate of LIP by regulating the metalation of iron-containing proteins and the storage and export of Fe2+ [Citation53]. For instance, in the cytoplasm, PCBP1 and PCBP2 can deliver iron to HIF (hypoxia inducible factor) prolyl hydroxlases [Citation54]. Importantly, PCBP1 plays a role in limiting the toxicity of cytosolic iron and inhibiting ferroptotic cell death [Citation55]. The depletion of PCBP1 in mouse hepatocytes leads to the accumulation of lipid peroxidation and steatosis, which can be improved by supplementing with vitamin E [Citation55].
Generally, most of the iron in the LIP is transferred to mitochondria, where it is used to synthesize heme or iron-sulfur (Fe-S) clusters. NFS1 (NFS1 cysteine desulfurase) functions as an Fe-S cluster biosynthetic enzyme, and protects cancer cells from ferroptosis by iron regulatory protein (IRP)-dependent translational mechanisms [Citation56]. SLC25A37/mitoferrin-1 (solute carrier family 25 member 37) and SLC25A28/mitoferrin-2 (solute carrier family 25 member 28) are the main mitochondrial iron importers in the outer mitochondrial membrane, although they have other locations [Citation57,Citation58]. Moreover, a mitochondrial quality control pathway that depends on PINK1 (PTEN induced kinase 1)-PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase) limits mitochondrial iron accumulation through promoting the degradation of SLC25A37 and SLC25A28, indicating a potential role of mitophagy in the modulation of mitochondrial iron metabolism [Citation59,Citation60]. In contrast, heme and Fe-S clusters can be exported from mitochondria to the cytoplasm through the mitochondrial receptors FLVCR1 (FLVCR heme transporter 1) and ABCB7 (ATP binding cassette subfamily B member 7), respectively [Citation61,Citation62]. CISD1/mitoNEET (CDGSH iron sulfur domain 1) is an iron-containing outer mitochondrial outer membrane protein that limits the uptake of mitochondrial iron, thereby inhibiting ferroptotic cell death [Citation63]. Therefore, the level of mitochondrial iron is fine-tuned by different mitochondrial membrane proteins, which is important for maintaining mitochondrial homeostasis during ferroptosis.
Iron, heme, or Fe-S clusters can be incorporated into ROS-producing enzymes, such as ALOX (arachidonate lipoxygenase), NOX (NADPH oxidase), XDH (xanthine dehydrogenase), CYP/CYP450s, and mitochondrial electron transport chain complexes, but also are co-factors of antioxidant enzymes, such as CAT (catalase). These ROS generators or scavengers are involved in the regulation of lipid peroxidation-dependent ferroptosis in a context-dependent manner.
Iron storage
Ferritin is the main iron storage protein and was discovered in 1937. It stores 70%-80% of newly imported iron. Although mitochondrial and nuclear forms have been proposed, ferritin is mainly located in the cytoplasm. Ferritin also appears in human serum and is used to diagnose and monitor diseases with iron overload or iron deficiency. In vertebrates, ferritin is composed of 24 similar subunits of two types, namely H and L, forming a hollow spherical macro-inorganic complex. The H subtype, also known as FTH1 (ferritin heavy chain 1), has ferroxidase activity that is responsible for the oxidation of Fe2+ to Fe3+. The L subtype, also known as FTL (ferritin light chain), assists in iron nucleation and mineralization. Iron enters the lumen of ferritin through pores in the shell, reaches the center of catalytic ferroxidase, and is deposited inside in the form of ferrihydrite. One ferritin sphere may store up to 4000 iron atoms. Although iron can be excreted through the pores on ferritin shells, the most important mechanism of iron release involves ferritin degradation by NCOA4 (nuclear receptor coactivator 4)-mediated ferritinophagy [Citation64]. In addition to selective autophagy, ferritin is also degraded by the activation of the ubiquitin-proteasome system (UPS) [Citation65]. Thus, the UPS and autophagy are two complementary proteolytic pathways required for maintaining intracellular ferritin levels.
Obviously, ferritin plays a significant role in preventing iron-mediated oxidative damage. In addition to cytoplasmic ferritin, the overexpression of mitochondrial ferritin diminishes erastin-induced ferroptosis in neuronal cells [Citation66]. In contrast, the downregulation of FTH1 and FTL is associated with ferroptotic sensitivity in oncogenic RAS-harboring cancer cells [Citation42]. The mutation of FTH1 produces a sharp growth defect in the larval discs and adult wings of Drosophila, which is partly due to the induction of ferroptosis, indicating that model organisms can be used to study the mechanism of ferroptosis. As expected, autophagic degradation of ferritin by ferritinophagy promotes ferroptosis through increasing LIP [Citation67,Citation68] (discussed in the section “Ferritinophagy”). Additionally, ferritin and its stored iron are released from cells through exosomes, and this process is mediated by PROM2 (prominin 2), which facilitates ferroptosis resistance [Citation69]. Overall, the level of intracellular ferritin is not only regulated by the degradation pathway, but also by the secretory pathway, resulting in changes in LIP.
Iron export
SLC40A1 is currently the only known iron exporter in mammalian cells. SLC40A1 has twelve transmembrane domains with intracellularly located N and C termini. It is generally thought that SLC40A1 only transports Fe2+, and this process requires the extracellular oxidation of Fe2+ to Fe3+, resulting in Fe3+ binding to TF immediately. There are three ferroxidases known to oxidize iron from SLC40A1, namely CP (ceruloplasmin), HEPH (hephaestin), and HEPHL1 (hephaestin like 1). Among them, CP is a negative regulator of erastin- and RSL3-induced ferroptosis in live cancer cells [Citation70]. The depletion of CP increases the accumulation of intracellular Fe2+ and lipid peroxidation, depending on SLC40A1 [Citation70]. In contrast, parenteral CP treatment may prevent ferroptotic damage after ischemic stroke in mice [Citation71]. These findings suggest that CP is an important regulator of ferroptosis through SLC40A1-dependent iron export.
SLC40A1 is negatively regulated by HAMP (hepcidin antimicrobial peptide), a peptide hormone secreted by the liver. After interacting with HAMP, SLC40A1 is internalized and degraded in lysosomes, resulting in reduced iron export. Mutations in the SLC40A1 gene cause defects in SLC40A1 transport activity and subsequent iron overload-mediated ferroportin disease (also known as type 4 hereditary hemochromatosis) [Citation72]. An abnormal HAMP-SLC40A1 axis is implicated in tumor growth and development. SLC40A1 inhibits tumor growth by restricting the supply of iron, and the upregulation of HAMP may accelerate tumor progression by promoting the degradation of SLC40A1 and subsequent iron accumulation [Citation73]. There is some evidence that the overexpression of SLC40A1 ameliorates ferroptosis, whereas the knockdown of SLC40A1 promotes ferroptosis [Citation40,Citation74]. Therefore, iron output mediated by SLC40A1 plays a different role in tumorigenesis and therapy, which also affects ferroptosis by lipid peroxidation.
Lipid metabolism
Lipid peroxidation is a hallmark of ferroptosis and is caused by a complex process of lipid metabolism, involving non-enzymatic Fenton reaction and enzymatic reaction pathways. Polyunsaturated fatty acids (PUFAs) are one of the main targets of lipid peroxidation. Therefore, the production of PUFA mediated by lipid synthesis leads to increased sensitivity to ferroptosis. In contrast, the fatty acid beta-oxidation (FAO) in mitochondria usually consumes most of the fatty acids, thus leading to a reduction in the rate of lipid peroxidation. Lipid droplets form the main lipid in eukaryotic cells, thereby keeping PUFA away from lipid oxidative damage during ferroptosis. Incorporation of PUFA into phospholipids requires ACSL4 (acyl-CoA synthetase long-chain family member 4), which is an important event of ferroptosis. In this section, we discuss the mechanisms of lipid synthesis, storage, utilization, and peroxidation during the modulation of ferroptosis .
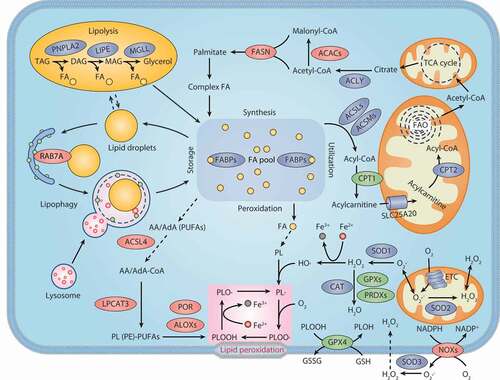
Figure 3. Lipid metabolism and ferroptosis. Both lipid catabolism and anabolic changes affect lipid peroxidation during ferroptosis. De novo lipogenesis is the synthesis of fatty acids from acetyl-CoA produced by the mitochondrial tricarboxylic acid (TCA) cycle. FA is transported into mitochondria for β-oxidation to release acetyl-CoA. Lipid droplets are intracellular sites for neutral lipid storage, and lipophagy induces autophagic degradation of lipid droplets and promotes the release of free FA. Lipolysis is defined as the catabolism of TAG stored in cell lipid droplets, which is regulated by PNPLA2/ATGL, LIPE/HSL, and MGLL. Oxygenases, such as ALOX and POR, catalyze the oxidation of PUFA to activate lipid peroxidation. ACSL4 and LPCAT act as positive regulators of lipid peroxidation by shaping cellular phospholipids. The NOX protein family reduces oxygen to produce free radicals, thereby attacking PUFA-containing lipids. Abbreviations: AA, arachidonic acid; AdA, adrenic acid; ACACs, acetyl-CoA carboxylases; ACLY, ATP citrate lyase; ACSL4, acyl-CoA synthetase long chain family member 4; ACSLs, acyl-CoA synthetase long chain family member proteins; ACSMs, acyl-CoA synthetase medium chain family member proteins; ALOXs, lipoxygenases; CAT, catalase; CoA, coenzyme A; CPT1, carnitine palmitoyltransferase 1; CPT2, carnitine palmitoyltransferase 2; DAG, diacylglycerol; DAGL, diacylglycerol lipase; ETC, electron transport chain; FA, fatty acid; FABPs, fatty acid binding proteins; FASN, fatty acid synthase; GPX4, glutathione peroxidase 4; GPXs, glutathione peroxidases; GSH, glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; LIPE, lipase E, hormone sensitive type; LPCAT3, lysophosphatidylcholine acyltransferase 3; MAG, monoacylglycerol; MGLL, monoglyceride lipase; NOXs, NADPH oxidases; PE, phosphatidylethanolamine; PL, phospholipid; PNPLA2, patatin like phospholipase domain containing 2; POR, cytochrome p450 oxidoreductase; PRDXs, peroxiredoxins; PUFAs, polyunsaturated fatty acids; RAB7A, RAB7A, member RAS oncogene family; SLC25A20, solute carrier family 25 member 20; SOD1, superoxide dismutase 1; SOD2, superoxide dismutase 2; SOD3, superoxide dismutase 3; TAG, triacylglycerol; TCA cycle, tricarboxylic acid cycle
Lipid synthesis
Fatty acids are important precursors for all biofilm lipids and are substrates for energy metabolism. There are two essential fatty acids, linoleic acid and α-linolenic acid, which cannot be synthesized de novo. Fatty acid synthesis occurs in the cytoplasm. The key metabolic substrate for fatty acid synthesis is cytoplasmic acetyl-CoA, which is mainly converted from mitochondrial-derived citric acid through the action of ACLY (ATP citrate lyase). ACAC (acetyl-CoA carboxylase) catalyzes the synthesis of malonyl-CoA from acetyl-CoA, which is the rate-limiting step in fatty acid synthesis. Then FASN (fatty acid synthase) catalyzes the condensation of malonyl-CoA and acetyl-CoA to produce the 16-carbon fatty acid palmitate (C16:0). The initial product of fatty acid synthesis is then extended by ELOVL (ELOVL fatty acid elongase) and desaturated by fatty acid desaturase. SCD/SCD1 (stearoyl-CoA desaturase) catalyzes the synthesis of monounsaturated fatty acids (MUFAs) from saturated fatty acids and inhibits ferroptosis in ovarian and lung cancer cells [Citation75,Citation76]. This is consistent with the observation that exogenous MUFAs protect cells from ferroptotic cell death [Citation77]. However, it is not clear how MUFA competitively affects the oxidation of PUFA.
Both ACSF2 (acyl-CoA synthetase family member 2) and CS (citrate synthase) are required for mitochondrial fatty acid metabolism. The knockdown of ACSF2 or CS reverses erastin-induced ferroptosis [Citation2], indicating a potential role of mitochondrial fatty acid metabolism in promoting ferroptosis. 5-(tetradecyloxy)-2-furoic acid (TOFA) is an allosteric inhibitor of ACACA/ACC1 (acetyl-CoA carboxylase alpha) and ACACB/ACC2 (acetyl-CoA carboxylase beta). TOFA inhibits ferroptosis caused by various stimuli (e.g., erastin, cystine depletion, RSL3, and FIN56) [Citation33,Citation78], further supporting the key role of PUFA as a substrate for lipid peroxidation in ferroptosis. Consistently, enhanced ACACA phosphorylation limits ACACA activity, thus protecting cells from erastin-induced cell death [Citation78]. Together, increased fatty acid biogenesis contributes to the induction of ferroptosis.
Lipid storage
Lipid droplets are ubiquitous in cells and can buffer and store excess lipid. They are highly dynamic organelles composed of triglycerides and cholesteryl esters, whose hydrophobic core is surrounded by a monolayer of phospholipids and various related proteins. It should be noted that lipid droplets interact with various organelles such as the endoplasmic reticulum (ER), peroxisomes, mitochondria, and lysosomes, thus making their functions more complicated. Generally, lipid droplets are formed from the cytoplasmic leaflets of the ER membrane, which is coupled to the synthesis of neutral lipids, mainly triacylglycerols and sterols. Lipid droplet formation prevents palmitic acid-induced lipotoxicity by isolating damaged membranes [Citation79]. Thus, this physical barrier function of lipid droplets may defend against various types of cell death.
However, the decomposition of lipid droplets by lipolysis not only provides a substrate for ATP production in mitochondrial fatty acid beta-oxidation but also increases the chance of lipid peroxidation during ferroptosis. Although many proteins are involved in the regulation of lipolysis, PNPLA2/ATGL (patatin like phospholipase domain containing 2) and LIPE/HSL (lipase E, hormone sensitive type) are the two main regulatory enzymes. Autophagy also selectively regulates the degradation of lipid droplets, a process named lipophagy. Increased lipophagy promotes the sensitivity of liver cancer cells to ferroptosis, further confirming the effect of selective autophagy in promoting the latter process [Citation80] (discussed in the section “Lipophagy”). Therefore, increased lipid storage through the formation of lipid droplets limits ferroptosis, whereas increased degradation of lipid droplets promotes ferroptosis. Monitoring the dynamic balance of lipid droplet formation and degradation is thus important for evaluating the progress of ferroptosis.
Lipid utilization
Fatty acids are catabolized by fatty acid FAO, which occurs mainly in the mitochondria, and involves a series of reactions that lead to the shortening of fatty acids. These reactions generate acetyl-CoA, NADH, and FADH2 in each oxidation cycle. Acetyl-CoA enters the Krebs cycle (TCA cycle), while NADH and FADH2 enter the electron transport chain to allow the production of ATP. The first step in FAO is fatty acid activation; that is, ACS (acyl-CoA synthetase) catalyzes the conversion of long-chain fatty acids to long-chain acyl-CoA. However, long-chain acyl-CoA itself fails to penetrate the inner mitochondrial membrane and requires L-carnitine as a co-factor to complete the transport process. CPT1 (carnitine palmitoyltransferase 1) is present in the outer mitochondrial membrane and catalyzes the conversion of acyl-CoA to the corresponding carnitine ester (acylcarnitine), which is the rate-limiting step of FAO. Acylcarnitine is then imported into the mitochondria through SLC25A20/CACT (solute carrier family 25 member 20) and finally converted back to acyl-CoA by the internal mitochondrial membrane enzyme CPT2 (carnitine palmitoyltransferase 2). Overexpressed CPT1 is tightly associated with tumor progression, whereas inhibiting CPT1 suppresses cancer cell growth. Etomoxir is an inhibitor of CPT1, which enhances RSL3-induced ferroptosis, suggesting that preserving PUFA for the oxidation reaction may promote ferroptosis [Citation81].
Consistently, the restoration of functional VHL (von Hippel-Lindau tumor suppressor) renders clear cell renal cell carcinoma (ccRCC) cells insensitive to ferroptosis due to the upregulation of genes involved in beta-oxidation (e.g., CPT1A [carnitine palmitoyltransferase 1A]) [Citation82]. Additionally, the knockdown of DECR1 (2,4-dienoyl-CoA reductase 1), a mitochondrial enzyme required for the beta-oxidation of PUFAs, causes cellular accumulation of polyunsaturated lipids and enhances ferroptosis in prostate cancer cells [Citation83]. Although these findings usually indicate that increased FAO may make cancer cells more resistant to ferroptotic cell death, the production of acetyl-CoA by FAO may promote ferroptosis through the increased synthesis of fatty acids. The enzymatic stage of mammalian mitochondrial beta-oxidation may be different, which determines its final role in regulating ferroptosis.
Lipid peroxidation
Lipid peroxidation is the process by which oxidants (e.g., free radicals or non-free radical substances) attack the carbon-carbon double bonds of lipids (especially PUFAs). Phospholipids, glycolipids, and cholesterol are common targets for this peroxidation modification. ROS is formed due to the incomplete reduction of oxygen and includes superoxide anion (O2·−), H2O2, and hydroxyl radical (HO·). The mitochondria electron transport chain and NOXs, and many other enzymes, produce O2·−, which is converted to H2O2 by SOD (superoxide dismutase) enzymes. Lipid peroxidation also occurs in a non-enzymatic manner through the Fenton reaction, which is a catalytic process in which Fe2+ reacts with H2O2 to generate Fe3+, HO·, and OH-. Moreover, the O2·− reacts with Fe3+ to regenerate Fe2+, a process that is called the Haber-Weiss cycle. The free radical ions generated from iron-catalyzed reactions cause oxidative damage to various components, including membrane lipids, proteins, and nucleic acids.
The process of lipid peroxidation can be divided into three stages: initiation, propagation, and termination. In the initiation stage, a co-oxidant (such as OH∙) extracts hydrogen atoms from the methylene carbon, which bridges the two double bonds of multiple unsaturated lipids, forming a lipid radical centered on carbon (L∙). The lipid free radical (L∙) then reacts with oxygen to produce a lipid peroxy radical (LOO∙), which extracts hydrogen atoms from another lipid to generate a new L∙ (propagating phase) and lipid hydroperoxide (LOOH). These propagation reactions can be terminated by antioxidant molecules, such as vitamin E, which provide hydrogen atoms to lipid peroxy radicals (LOO∙), thereby forming vitamin E free radicals (TOC∙). Finally, TOC∙ reacts with another LOO∙ to form nonradical products.
Several studies have identified ACSL4 as a key determinant of ferroptosis sensitivity [Citation81,Citation84,Citation85]. As mentioned earlier, ACSL4 catalyzes the addition of CoA to the long-chain polyunsaturated bonds of arachidonic acid, thereby promoting the esterification of PUFA to phospholipids. After ACSL4 activation, LPCAT3 (lysophosphatidylcholine acyltransferase 3) participates in ferroptotic lipid signaling through inserting acyl groups into lysophospholipids, specifically toward the phospholipids phosphatidylcholine and phosphatidylethanolamine (PE) [Citation81]. Notably, ferroptosis can also occur in an ACSL4-independent manner [Citation86]. Further investigation is needed to check whether other ACSL family members are involved in ACSL4-independent ferroptosis.
Finally, lipids are directly oxidized by oxygenases, such as ALOXs, CYP/CYP450, and PTGS/COX (prostaglandin-endoperoxide synthase). ALOXs are nonheme iron dioxygenases and have 6 subtypes in humans, namely ALOX5 (arachidonate 5-lipoxygenase), ALOX12 (arachidonate 12-lipoxygenase, 12S type), ALOX12B (arachidonate 12-lipoxygenase, 12 R type), ALOX15 (arachidonate 15-lipoxygenase), ALOX15B (arachidonate 15-lipoxygenase type B), and ALOXE3 (arachidonate lipoxygenase 3) [Citation87]. GPX4 limits the activity of ALOX15 by reducing hydroperoxylipid, which is the activator of the ALOX reaction [Citation88]. Thus, silencing or inactivating GPX4 tends to enhance ALOX-dependent lipid peroxide formation. In contrast, the genetic or pharmacological inhibition of ALOX enzymes prevents ferroptosis to a certain extent [Citation89,Citation90]. ALOXs initiate ferroptosis by oxidizing PUFA-PE [Citation81]. This process is further regulated by PEBP1/RKIP (phosphatidylethanolamine binding protein 1) because it interacts with ALOX15 and enables its catalytic ability for PUFA-PE, thereby promoting ferroptosis [Citation91]. Interestingly, the knockdown of ALOXs does not suppress RSL3-induced ferroptosis in G-401 cells [Citation90], although ALOX inhibitors block RSL3-induced ferroptosis [Citation81]. The knockout of alox15 does not reverse the phenotype of GPX4-deficient mice [Citation92,Citation93], further suggesting that ALOXs may not be the only oxygenase of lipid peroxidation in ferroptosis. Alternatively, POR (cytochrome p450 oxidoreductase) drives lipid peroxidation in an ALOX-independent manner during ferroptosis [Citation94]. PTGSs are the key enzymes in the synthesis of prostaglandins from arachidonic acid (C20:4). PTGS2/COX2 (prostaglandin-endoperoxide synthase 2) is markedly upregulated during ferroptosis [Citation14]. However, it seems that PTGS2 is not involved in the production of lipid peroxidation during ferroptosis, because the PTGS inhibitor indomethacin has little effect on the lethality of elastin or RSL3 [Citation14,Citation90]. It remains to be seen whether the knockdown of PTGS2 affects the sensitivity to ferroptosis.
Amino acid metabolism
The transsulfuration pathway
When the availability of cysteine is limited, certain cells use the transsulfuration pathway to biosynthesize cysteine from methionine. CBS (cystathionine beta-synthase) uses homocysteine derived from methionine to produce cystathionine, which is then converted to cysteine by CTH (cystathionine gamma-lyase). Inhibitors of CBS (e.g., CH004) or CTH (e.g., propargylglycine) trigger or enhance ferroptosis in hepatocellular carcinoma cells [Citation95] or motor neuron-like NSC-34 cells [Citation96], respectively. CBS expression is negatively regulated by MIR6852 (microRNA 6852) [Citation97], which provides a mechanism for fine-tuning ferroptosis through modulating the transsulfuration pathway.
CARS1/CARS (cysteinyl-tRNA synthetase 1) is identified as a positive regulator of erastin (but not BSO, RSL3, and FIN56)-induced ferroptosis through inhibiting the transsulfuration pathway [Citation98]. In contrast, the activation of ATF4 (activating transcription factor 4) through the phosphorylation of EIF2 (eukaryotic translation initiation factor 2) is a possible mechanism for the upregulation of the transsulfuration pathway [Citation98]. AHCY/SAHH (adenosylhomocysteinase) is the only known enzyme that catalyzes the hydrolysis of S-adenosyl-L-homocysteine/SAH to homocysteine. AHCY acts as a downstream effector of PARK7/DJ-1 (Parkinsonism associated deglycase) to suppress ferroptosis [Citation99]. The activation of the transsulfuration pathway inhibits ferroptosis by the generation of cysteine or GSH in parallel with a system xc−-dependent pathway.
The glutaminolysis pathway
Glutamine is produced by glutamate and ammonia catalyzed by GLUL/glutamine synthetase (glutamate-ammonia ligase) in cells. This enzyme is required for cystine starvation- and erastin-induced ferroptosis via the activation of glutaminolysis, a process of catabolizing glutamine to glutamate [Citation41]. The initial deamination of glutamine requires GLS/GLS1 (glutaminase) and GLS2 (glutaminase 2). Glutamate is then converted to α-ketoglutarate (α-KG, an important TCA cycle intermediate) in mitochondria. The mitochondrial glutaminase GLS2, but not cytosolic glutaminase GLS, is required for ferroptotic cell death, indicating that mitochondria are involved in ferroptosis induction [Citation41,Citation68,Citation100,Citation101]. GLS2 is negatively regulated by MIR103A1 (microRNA 103a-1), and is required for ferroptosis induced by physcion 8-O-β-glucopyranoside (a chemical component of Rumex japonicus Houtt) [Citation102], providing an example of the posttranscriptional regulation of ferroptosis by miRNA.
As expected, the knockdown of two glutamine transporters (namely SLC38A1 [solute carrier family 38 member 1] and SLC1A5 [solute carrier family 1 member 5]) or the pharmacological inhibition of SLC1A5 reverse cystine starvation-induced ferroptosis [Citation41,Citation68,Citation100]. Consistently, MIR137 (microRNA 137) negatively regulates SLC1A5, thus decreasing glutamine uptake and the sensitivity of melanoma cells to erastin- or RSL3-induced ferroptosis [Citation103]. Exogenous α-KG mimics the death-promoting activity of glutamine, supporting the role of glutaminolysis in promoting ferroptosis [Citation41,Citation100]. In addition to being used by DLD (dihydrolipoamide dehydrogenase) to produce ROS in mitochondria, α-KG can be further converted into acetyl-CoA in the cytosol for fatty acid synthesis and subsequent ferroptosis [Citation100]. Thus, glutaminolysis, a traditional pro-survival pathway, acts as a positive regulator of ferroptosis. What checkpoints distinguish between the survival and death functions of glutaminolysis remains to be determined.
Glucose metabolism
Glucose is the main source of acetyl-CoA used to synthesize fatty acids. Glucose starvation largely inhibits various types of ferroptosis in MEFs through the activation of the energy sensor AMP-activated protein kinase (AMPK) [Citation78]. Although AMPK-mediated phosphorylation of ACACA inhibits PUFA biosynthesis and subsequent ferroptosis [Citation78], AMPK-mediated BECN1 (beclin 1) phosphorylation promotes ferroptosis [Citation45]. These findings indicate that AMPK plays a dual role in ferroptosis, depending on its substrate. The inhibition of HK (hexokinase; an upstream rate-limiting enzyme in glycolysis) by 2-deoxy-d-glucose not only reduces erastin-, cystine depletion- or RSL3-induced ferroptosis in MEFs [Citation78], but also protects against ferroptosis-related renal IR injury. Notably, the activation of glucose-mediated oxidative phosphorylation shares the initial pathway of glycolysis, including HK. Under anaerobic conditions, glycolysis eventually leads to the conversion of pyruvate to lactate. Whether lactate plays a similar role to 2-deoxy-d-glucose in suppressing ferroptosis is unclear. The well-known function of PHKG2 (phosphorylase kinase catalytic subunit gamma 2) is to activate PYG (glycogen phosphorylase), which breaks down glycogen into glucose-1-phosphate. However, PHKG2 is essential for erastin-induced ferroptosis in a glycogen-independent manner [Citation90]. Alternatively, PHKG2 plays a previously unknown role in promoting iron accumulation, although the exact mechanism is still unclear [Citation90].
The pentose phosphate pathway (PPP) is a glucose-related metabolic pathway parallel to glycolysis. One of the main functions of PPP activation is the production of NADPH, which is an essential electron donor in all organisms. NADPH seems to be a double-edged sword for ferroptosis. On the one hand, NADPH is used for the reduction of GSSG to GSH by GSR (glutathione-disulfide reductase), which indicates that NADPH has a suppressive role in ferroptosis. The decreased level of NADPH may be a biomarker predicting the sensitivity to ferroptosis [Citation104]. Erastin, RSL3, and FIN56 all reduce both NAD(H) and NADP(H) levels during ferroptosis [Citation104]. Inhibiting IDH2 (isocitrate dehydrogenase [NADP{+}] 2), a NADPH-producing metabolic enzyme, sensitizes cancer cells to erastin-induced ferroptosis [Citation105]. Moreover, the IDH1 (isocitrate dehydrogenase [NADP{+}] 1) mutation enhances erastin-induced ferroptosis by converting α-KG to (D)-2-hydroxyglutarate [Citation106], further supporting a role of the IDH family in ferroptosis. Other NADPH-associated kinases also play a context-dependent role in ferroptosis. For example, the knockdown of NADK (NAD kinase), a regulator that catalyzes the phosphorylation of NAD+ to NADP+, increases the sensitivity to ferroptosis by downregulating NADP(H) levels [Citation104,Citation107]. In contrast, the knockdown of HDDC3/MESH1 (HD domain containing 3), a regulator that hydrolyzes the phosphate group of NADPH to produce NADH, inhibits the sensitivity to ferroptosis by sustaining NADP(H) levels [Citation107]. These findings shed new light on the role of NADPH in protection against ferroptosis.
On the other hand, NADPH acts as an electron donor for NOXs, which may contribute to ferroptosis. NOXs are transmembrane enzymes that promote ferroptosis by catalyzing electron transfer from NADPH to O2 to produce O2- in a context-dependent manner [Citation2]. For instance, NOX1 (NADPH oxidase 1), CYBB/NOX2 (cytochrome b-245 beta chain), and NOX4 (NADPH oxidase 4) have been implicated in the initiation of lipid ROS during ferroptosis [Citation108–110]. Consistently, the PPP inhibitor 6-aminonicotinamide attenuates erastin-induced ferroptosis in HT1080 and Calu-1 cells [Citation2]. The knockdown of two PPP enzymes, G6PD (glucose-6-phosphate dehydrogenase) and PGD (phosphogluconate dehydrogenase), also prevents erastin- or cystine starvation-induced ferroptosis [Citation2,Citation68]. These findings also suggest that the metabolic products of PPP, such as NADPH, may contribute to NOX-dependent ferroptosis. However, it is still difficult to distinguish what proportion of NADPH is used by different anti-ferroptosis or pro-ferroptosis enzymes.
The membranous basis of ferroptosis
A lipid bilayer forms the basis of the membrane barrier that separates the cell from the extracellular space and the organelle from the cytoplasm. Membrane repair is a stress response to cell membrane damage. The balance between damage and repair of the cell membrane affects the consequences of ferroptosis.
Membrane injury
Where lipid peroxidation occurs during ferroptosis remains a mystery. Based on different morphological or functional assays, plasma membranes and multiple intracellular organelle membranes are candidates. The uptake of propidium iodide (PI) fluorescent dye indicates that cells undergoing ferroptosis exhibit a loss of plasma membrane integrity [Citation68]. In contrast, ferroptotic cells still maintain the structural integrity of the nucleus, lacking chromatin condensation and marginalization [Citation2]. Mitochondria in ferroptotic cells have reduced size, increased membrane density, and rupture of the outer membrane [Citation2,Citation92]. Although mitochondria are the center of cell death, their role in ferroptosis remains controversial. Early studies show that cells lacking mitochondria are still sensitive to ferroptosis caused by erastin, RSL3, and FIN56, indicating that ferroptosis is a mitochondria-independent type of cell death [Citation2,Citation111]. Recent studies indicate that mitochondria-depleted cells are resistant to cystine starvation- or erastin-induced ferroptosis, supporting the idea that mitochondria are required for ferroptosis [Citation112]. Using a bio-orthogonal imaging technique, the accumulation of ferrostatins has been observed in the ER, indicating that the ER may be critical to ferroptosis initiation [Citation111]. Consistently, using the fluorescent lipid peroxidation probe LiperFluo, lipid hydroperoxide has been found mainly in the ER [Citation81]. After treatment with erastin, ALOX5 translocates to the nuclear membrane, indicating that lipid peroxidation also occurs in the nuclear membrane [Citation90]. The above differences suggest that membrane damage may be a dynamic process involving various membrane organelles.
Understanding how lipid peroxidation causes cell death is still a huge challenge. Although the exact mechanism of membrane damage during ferroptosis is still unclear, lipid peroxidation has been shown to alter the assembly, structure, and dynamics of biological membrane. X-ray diffraction analysis and kinetic studies of lipid bilayers provide direct evidence that oxidized lipids reduce membrane thickness and increase membrane permeability. Lipid peroxidation also inhibits membrane fluidity. In experiments using giant monolayer vesicles, membrane changes, such as membrane thinning and increased curvature after peroxidation, have been observed [Citation113], providing evidence that lipid peroxidation impairs membrane integrity during ferroptosis. Lipid hydroperoxides degrade into several toxic aldehydes, such as 4HNE and malondialdehyde (MDA). These aldehydes can be used to covalently modify biomolecules, including proteins and amino lipids, to generate corresponding covalent adducts, thereby aggravating membrane damage [Citation114]. Erastin-resistant cells show a significant upregulation of aldehyde detoxification enzymes, especially AKR1C1 (aldo-keto reductase family 1 member C1), AKR1C2, and AKR1C3 [Citation115], indicating that lipid aldehydes may directly cause cell death. Pore-forming proteins, such as GSDMD (gasdermin D) and MLKL (mixed lineage kinase domain like pseudokinase), mediate the membrane damage of pyroptosis and necroptosis, respectively [Citation7]. Whether there is a similar pore-forming protein-mediated membrane rupture of ferroptotic cells remains to be clarified.
Membrane repair
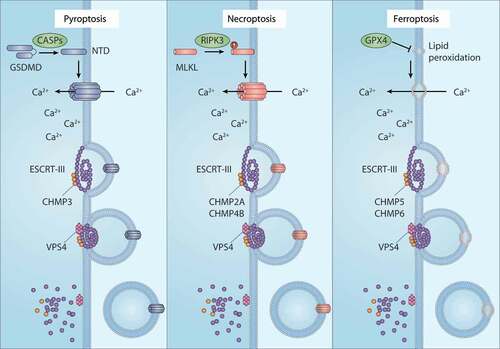
Several plasma membrane repair mechanisms have been proposed, such as exocytosis of lysosomes, endocytosis of caveolar vesicles, and membrane budding mediated by the ESCRT complex. The ESCRT machinery is composed of the ESCRT-0, -I, -II, and -III complexes, VPS4 (vacuolar protein sorting 4), and other related proteins. Among them, ESCRT-III is the main membrane remodeling mechanism, involving repair of plasma membrane and nuclear membrane [Citation116]. The ESCRT complex consists of polymerized or oligomeric small alpha-helix charged multivesicular body proteins (CHMPs), which are recruited and assembled at the damage site. The AAA-ATPase VPS4 is essential for the disassembly of the ESCRT-III complex after membrane rupture. Ferroptosis activators, such as erastin and RSL3, induce the accumulation of certain ESCRT-III subunits, especially CHMP5 (charged multivesicular body protein 5) and CHMP6 (charged multivesicular body protein 6), in the plasma membrane of pancreatic cancer cells (PANC1) [Citation117]. Importantly, the knockdown of CHMP5 or CHMP6 makes cells sensitive to ferroptosis induced by erastin or RSL3 in vitro or in animal models [Citation117]. In addition to CoQ10, ESCRT-III-dependent membrane repair contributes to AIFM2-mediated ferroptosis resistance [Citation117]. The loss of AIFM2 prevents the accumulation of CHMP5 and CHMP6 in the plasma membrane during ferroptosis [Citation117]. An overexpression of CHMP5 blocks ferroptosis induced by erastin, sorafenib, or RSL3 in both wild-type and AIFM2-knockdown HepG2 cells [Citation37]. These findings demonstrate a role of ESCRT-III machinery in the inhibition of ferroptosis.
In addition to ferroptosis, other early studies have shown that CHMP2A (charged multivesicular body protein 2A), CHMP4B (charged multivesicular body protein 4B), and CHMP3 (charged multivesicular body protein 3) play selective roles in ESCRT-III–mediated membrane repair to limit necroptosis and pyroptosis [Citation118–]. The influx of Ca2+ ions is especially important in initiating the recruitment of ESCRT-III machinery to damaged membrane regions during various types of RCD, including ferroptosis [Citation117], necroptosis [Citation118], and pyroptosis [Citation119]. Overall, the ESCRT-III complex seems to play a universal role in the inhibition of various kinds of RCD through repairing damaged membranes [Citation120] .
Figure 4. The membrane repair function of ESCRT-III in RCD. ESCRT-III plays a key role in plasma membrane repair in three forms of regulated cell death (RCD), including pyroptosis, necroptosis, and ferroptosis. In pyroptosis and necroptosis, GSDMD and MLKL are transported from the cytoplasm to the cell membrane and form oligomers and large permeability pores, resulting in cell death. In the case of ferroptosis, the accumulation of lethal lipid peroxidation production or the translocation of unknown pore-forming protein in the cell membrane leads to cell death. ESCRT-III machines, including the CHMP family and VPS4, can promote membrane sprouting and shedding of injured plasma membranes. Abbreviations: CASPs, caspases; CHMP2A, charged multivesicular body protein 2A; CHMP3, charged multivesicular body protein 3; CHMP4B, charged multivesicular body protein 4; CHMP5, charged multivesicular body protein 5; CHMP6, charged multivesicular body protein 6; ESCRT-III, endosomal sorting complex required for transport-III; GPX4, glutathione peroxidase 4; GSDMD, gasdermin D; MLKL, mixed lineage kinase domain like pseudokinase; RIPK3, receptor interacting serine/threonine kinase 3; VPS4, vacuolar protein sorting 4
The chemical basis of ferroptosis
Ferroptosis inducers
Ferroptosis can be induced by small-molecule compounds or drugs targeting transporters or enzymes in the following ways:
Table 1. Ferroptosis inducers
(1) The depletion of GSH. GSH, the most abundant intracellular antioxidant, is required for the activity of various antioxidant enzymes (e.g., GPX4). The synthesis of intracellular GSH depends on system xc−-mediated cystine uptake. Erastin induces ferroptosis mainly by inhibiting the activity of system xc− [Citation2]. Notably, erastin analogs, such as piperazine erastin and imidazole ketone erastin, have better metabolic stability and water solubility than erastin in vivo [Citation14,Citation121]. Some drugs (e.g., sulfasalazine and sorafenib) as well as exogenous glutamate also trigger ferroptosis by inhibiting system xc− [Citation2,Citation115]. In addition, the inhibition of GCL, the first rate-limiting enzyme of GSH synthesis, by BSO, increases the sensitivity to ferroptosis.
(2) The inhibition of GPX4. GPX4 is a phospholipid hydroperoxidase that plays a universal role in preventing membrane lipid peroxidation. Several reagents containing electrophilic chloroacetamide, such as RSL3 and ML162/DPI7, induce ferroptosis by covalently interacting with the active site of GPX4 (namely selenocysteine) and inhibiting its activity [Citation14]. Nitrile oxide electrophiles, such as ML210/DPI10, JKE-1674, and JKE-1716, also covalently target the selenocysteine residue of GPX4 to trigger ferroptosis [Citation122,Citation123]. In contrast, the ferroptosis activator FIN56 acts by promoting the degradation of GPX4 protein in an ACAC-dependent manner [Citation33].
(3) Organic peroxides. Organic peroxides are compounds containing one or more oxygen-oxygen bonds (ROOR). The O-O linkage can be broken down easily, producing free radicals in the form of RO· (alkoxy anions). Organic peroxides are often exploited in models to produce oxidative damage in cells. If one of the H atoms is replaced by an organic group R, these compounds are called organic hydroperoxides (ROOH). Tert-butyl hydroperoxide (t-BuOOH) is such a lipid peroxide analog and is widely regarded as a stimulus of lipid peroxidation-dependent ferroptosis [Citation124]. Artemisinin and its derivatives (e.g., dihydroartemisinin and artesunate) are 1,2,4-trioxane-based organic peroxides, and also effectively cause ferroptotic cancer cell death. FINO2 is an organic peroxide containing a 1,2-dioxolane skeleton and has a dual induction mechanism for ferroptosis, involving direct iron oxide or indirect inhibition of GPX4 activity [Citation125].
(4) Iron overload. Excess nonheme iron (Fe2+ and Fe3+) accumulation induces ferroptosis in mouse cardiomyocytes, hepatocytes, bone marrow-derived macrophages, and organotypic hippocampal slice cultures. Exogenous hemin or HB (hemoglobin) triggers ferroptosis in vitro and intracerebral hemorrhage in vivo [Citation19,Citation49,Citation126,Citation127]. In addition, the pharmacological activation of ferritinophagy-mediated ferritin degradation increases the intracellular free iron contents and subsequent ferroptosis, supporting a role of selective autophagy in mediating ferroptosis [Citation128–130].
(5) Other ferroptosis inducers. A variety of nanomaterials have the capacity to cause ferroptosis through inducing lipid peroxidation. Drugs that damage mitochondrial DNA, such as zalcitabine, also induce autophagy-dependent ferroptosis in human pancreatic cancer cells [Citation131], indicating a connection between mitochondrial dysfunction, autophagy activation, and DNA sensor pathways.
Ferroptosis inhibitors
The most common strategy for preventing ferroptosis is to inhibit the formation of lipid peroxides through either an enzyme-dependent or -independent manner by the following means:
Table 2. Ferroptosis inhibitors
(1) Radical-trapping antioxidants (RTAs). RTAs (also known as chain-breaking antioxidants) scavenge chain-carrying radicals, thereby terminating the self-oxidizing chain reaction. The most common chain-breaking RTAs are phenols and aromatic amines, which have relatively weak O-H and N-H bonds, respectively [Citation132]. For example, the natural phenolic substance α-tocopherol is the most active form of vitamin E. Although it works at a relatively high concentration (approximately 100 μM) in an in vitro culture model, α-tocopherol effectively suppresses ferroptosis [Citation33]. As synthetic phenolic compounds, butylated hydroxytoluene and butylated hydroxyanisole are currently used as food additives and can inhibit ferroptosis [Citation14,Citation33]. Other synthetic phenolic compounds, such as the tetrahydronaphthyridinols, also exhibit promising anti-ferroptotic activities [Citation133]. Two aromatic amines, ferrostatin-1 and liproxstatin-1, are identified as classical ferroptosis inhibitors used in vitro and in vivo [Citation2,Citation92]. Surprisingly, ferrostatin-1 is not consumed when inhibiting the iron-induced peroxidation reaction, and it appears to eliminate alkoxy radicals generated by ferrous iron from lipid hydroperoxides [Citation134], supporting the idea that the aromatic amines ferrostatin-1 and liproxstain-1 act as potent ferroptosis inhibitors. Diarylamine compounds (e.g., phenoxazines and phenothiazines) [Citation135] and certain nitroxides [Citation136,Citation137] are also effective RTA inhibitors, preventing lipid peroxidation-dependent ferroptosis. Notably, it is still unclear how these RTAs selectively prevent ferroptosis but not other types of oxidative death.
(2) Iron chelators. The important role of iron in ferroptosis is related to the production of lipid peroxidation, which is through the induction of a non-enzymatic iron-mediated Fenton reaction or the activation of iron-containing lipid oxygenases (e.g., ALOX [Citation81,Citation90,Citation138] and CYP/CYP450/cytochrome P450 oxygenases [Citation94]). Accordingly, ferroptotic cell death is blocked by iron chelators, such as deferoxamine, cyclipirox, and deferiprone [Citation2]. It should be noted that H2O2-induced necrosis is inhibited by iron chelators, but not by ferrostatin-1 or liproxstatin-1 [Citation92,Citation139], indicating that iron may participate in other types of cell death through different mechanisms.
(3) Enzyme inhibitors. ACSL4 adds CoA to long-chain fatty acids, especially arachidonic acid, which seems to be an important early event for subsequent ALOX-dependent lipid peroxidation. ACSL4 inhibitors (e.g., thiazolidinediones [TZNs] and triacsin C) [Citation81,Citation84] and ALOX inhibitors (e.g., cinnamyl-3,4-dihydroxya-cyanocinnamate [CDC], baicalein, PD146176, zileuton, AA-861, ML351, and NCTT-956) [Citation81,Citation90] prevent ferroptotic cell death. Some ALOX inhibitors (e.g., CDC, baicalein, PD146176, and zileuton) may inhibit ferroptosis through their off-target antioxidant activity [Citation132]. The NOXs on the cell membranes are a resource of cellular ROS for ferroptosis. Consequently, erastin-induced ferroptosis is suppressed by NOX inhibitors, such as diphenylene iodonium, GKT137831, and 2-acetylphenothiazine [Citation2,Citation108]. It is recommended to use a combination of iron chelator, RTA, and related enzyme inhibitors to clarify the different activation pathways of ferroptosis.
(4) Protein degradation inhibitors. Many ferroptosis activators have the ability to induce the degradation of GPX4, causing lipid peroxidation [Citation33,Citation140–143]. In contrast, the ACAC inhibitor TOFA, the neurotransmitter dopamine, and the HSP90 (heat shock protein 90) inhibitor 2-amino-5-chloro-N,3-dimethylbenzamide (CDDO) block FIN56- or erastin-induced GPX4 degradation [Citation33,Citation140,Citation143]. Therefore, pharmacologically blocking the degradation of GPX4 may enhance antioxidant capacity in the course of ferroptosis.
(5) Other ferroptosis inhibitors. N-acetyl cysteine (NAC, a GSH precursor), ebselen (a GPX mimic), and CoQ10 are reported to protect against various oxidative cell deaths, including ferroptosis [Citation2,Citation34]. The reducing agent 2ME reacts with cystine to generate mixed disulfides, which are taken up by system xc−-independent transporters (e.g., the L system in cell membranes) to generate intracellular cysteine [Citation144]. Unlike NAC, 2ME suppresses the lethality of erastin, but not that of RSL3, indicating that the oxidant activity between erastin and RSL3 is different [Citation115]. Of note, the MAP2K/MEK (mitogen-activated protein kinase kinase) inhibitor U0162 is widely used to suppress ferroptosis through its nonspecific antioxidant activity [Citation41]. Exogenous MUFAs or deuterated PUFAs display potent protective effects against ferroptosis, which may be associated with displacing PUFA from phospholipids, thus reducing the accumulation of lipid peroxidation [Citation77,Citation90]. However, the mechanism involved in cell protection mediated by these fatty acids is still unclear.
The transcriptional regulation of ferroptosis
Increasing evidence has emerged for the role of transcription factors in the regulation of ferroptosis, and these proteins act as both promoters and blockers through regulating the expression of target genes involved in metabolism and antioxidant pathways [Citation145] . The complex transcriptional regulatory network affects the sensitivity to ferroptosis. Some ferroptosis-related genes may also be simultaneously regulated by multiple transcription factors. For example, SLC7A11 is upregulated by transcription factors NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2), ATF4, and ARNTL (aryl hydrocarbon receptor nuclear translocator like), but is downregulated by transcription factors TP53 (tumor protein p53), ATF3 (activating transcription factor 3), BACH1 (BTB domain and CNC homolog 1), and STAT1 (signal transducer and activator of transcription 1). In this section, we mainly introduce the complex functions of several ferroptosis-related transcription factors, such as TP53, NFE2L2, YAP1/YAP (Yes 1 associated transcriptional regulator), and WWTR1/TAZ (WW domain containing transcription regulator 1).
Table 3. Transcriptional regulators in ferroptosis
TP53
TP53 was originally identified as a cellular protein complex with TEAD1/SV40 (TEA domain transcription factor 1) T antigen in mouse cells infected and transformed with TEAD1. TP53 is currently a recognized tumor suppressor gene that regulates various cellular stresses, including DNA damage, hypoxia, oncogene activation, and ribosomal stress. TP53 activation induces cell cycle arrest, DNA damage repair, aging, cell death, and metabolic reprogramming. In particular, TP53 regulates the sensitivity to ferroptosis bidirectionally through transcription-dependent and transcription-independent mechanisms [Citation146] .
Figure 5. Dual role of TP53 in ferroptosis. 1) Pro-ferroptosis role of TP53. TP53 inhibits the expression of SLC7A11 or promotes the expression of SAT1, thus regulating ALOX12- or ALOX15-dependent lipid peroxidation reactions, respectively. 2) Anti-ferroptosis role of TP53. TP53 binds with DDP4 in the nucleus, which limits DPP4 binding to NOX1 to mediate ROS production in the cell membrane. TP53 also promotes the expression of CDKN1A, thereby inducing the production of GSH to inhibit lipid peroxidation. Abbreviations: ALOX12, arachidonate 12-lipoxygenase; ALOX15, arachidonate 15-lipoxygenase; CDKN1A, cyclin dependent kinase inhibitor 1A; DPP4, dipeptidyl peptidase 4; GSH, glutathione; NOX1, NADPH oxidase 1; SAT1, spermidine/spermine N1-acetyltransferase 1; SLC7A11, solute carrier family 7 member 11; TP53, tumor protein P53
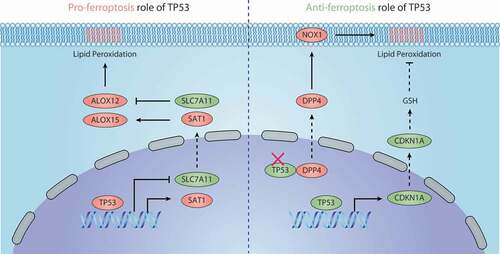
The SLC7A11 gene was identified as a repression target of TP53 through microarray screening in tetracycline-controlled TP53-inducible H1299 cell lines [Citation147]. The knock-in of TP53[3KR], an acetylation-deficient mutant at 3 lysine residues (at positions 117, 161, and 162), transcriptionally represses SLC711A expression in H1299 cell lines [Citation147]. However, the forced expression of TP53[3KR] fails to activate pro-apoptotic genes (e.g., BBC3/PUMA [BCL2 binding component 3]) and cell cycle arrest (e.g., CDKN1A/p21 [cyclin dependent kinase inhibitor 1A]) [Citation147]. Both Trp53+/+ and Trp533KR/3KR MEFs are more sensitive to erastin-induced ferroptosis than trp53−/- MEFs [Citation147], indicating a pro-ferroptotic role of TP53. Surprisingly, although TP53 activation induces the downregulation of SLC7A11 and partially inhibits cystine uptake, it cannot affect GSH levels and GPX4 function [Citation86]. This may be due to the activation of other TP53 targets, such as TIGAR (TP53 induced glycolysis regulatory phosphatase) [Citation148], GLS2 [Citation149], and CDKN1A [Citation150], which increase the production of GSH. It is worth noting that TP53-mediated ferroptosis requires ALOX12, whereas ACSL4 is optional [Citation86].
SAT1 (spermidine/spermidine N1-acetyltransferase 1) encodes the rate-limiting enzyme in the metabolism of polyamines and is the transcription target of TP53. SAT1 expression also contributes to TP53-mediated ferroptosis in MCF7, U2OS, A375, and H1299 cells following ROS treatment [Citation151]. ALOX15, but not ALOX5 or ALOX12, is critical for SAT1-induced ferroptosis during TP53 activation [Citation151]. These findings suggest that different ALOXs may mediate different types of TP53 target gene-induced ferroptosis, although the SAT1-mediated ALOX15 expression mechanism is still unknown. In addition, a variant of TP53 (P47S) shows a decreased ability to regulate specific TP53 target genes (e.g., GLS2 and SAT1), which may lead to ferroptosis tolerance [Citation101,Citation152].
TP53 also has an anti-ferroptosis effect. For example, TP53 inhibits ferroptosis in human colon cancer cells (e.g., HCT116 and SW48) in a transcription-independent manner [Citation108]. Mechanistically, TP53 binds to DPP4/CD26 (dipeptidyl peptidase 4) in the nucleus and retains DPP4 in an inactive form, whereas the loss of TP53 promotes DDP4 localization in the plasma membrane. Then, membrane DDP4 promotes ferroptosis by binding to NOX1, thereby increasing NOX1 activity to promote lipid peroxidation [Citation108]. Remarkably, the knockdown of DPP4 or administration of DPP4 inhibitors (e.g., vildagliptin, alogliptin, and linagliptin) blocks ferroptosis in TP53-deficient colon cancer cells [Citation108]. TP53 also suppresses erastin2 (an erastin analog)-induced ferroptosis through transcriptional induction of CDKN1A expression in HT1080 cells [Citation150]. The activation of the TP53-CDKN1A axis seems to be less dependent on SLC7A11-mediated cystine uptake and subsequent GSH synthesis, although its underlying mechanism remains unclear [Citation150]. Overall, the role of TP53 in ferroptosis may depend on the environment and cell type, as well as on its modification, binding protein, and target gene.
NFE2L2
Transcription factor NFE2L2 is a member of the cap “n” collar (CNC) family of basic region leucine zipper (bZIP) transcription factors, which was first cloned in 1994. NFE2L2 maintains cell redox homeostasis by combining with an antioxidant response element in target genes. Under normal conditions, NFE2L2 is an unstable protein due to KEAP1 (kelch like ECH associated protein 1)-mediated NFE2L2 degradation. Upon ferroptosis, the stability and activity of NFE2L2 increases, which plays a key role in preventing lipid peroxidation and cell death [Citation50,Citation153–158]. Ferroptosis caused by sorafenib in hepatocellular carcinoma cell lines reveals the initial link between NFE2L2 and an anti-ferroptosis effect [Citation50]. Certain ferroptosis activators (e.g., sorafenib, erastin, and BSO) induce the interaction between SQSTM1/p62 (sequestosome 1) and KEAP1 in hepatocellular carcinoma cell lines, resulting in the stabilization of NFE2L2 and a subsequent NFE2L2 activation as well as anti-ferroptosis gene expression [Citation50,Citation153]. In addition to its impact on various cancer cells, the activation of NFE2L2 mediates ferroptosis resistance in normal cells and tissues, highlighting the widespread role of NFE2L2 in preventing lipid peroxidation [Citation159].
Several ferroptosis-related genes are transcriptionally regulated by NFE2L2. First, NFE2L2 inhibits ferroptosis through controlling genes involved in iron metabolism, including FTH1, SLC40A1, HMOX1, and MT1G (metallothionein 1 G). Second, NFE2L2 activation upregulates genes related to GSH synthesis, metabolism, and release, such as SLC7A11, GCLM, CBS, CHAC1 (ChaC glutathione specific gamma-glutamylcyclotransferase 1), ABCC1/MRP1 (ATP binding cassette subfamily C member 1), GCLC, and GSS (glutathione synthetase). Third, some NFE2L2 target genes are involved in detoxification or antioxidant responses, which may inhibit the sensitivity to ferroptosis. These genes include NQO1 (NAD(P)H quinone dehydrogenase 1), TXNRD1, AKR1C1/2/3, SESN2 (sestrin 2), GSTP1 (glutathione S-transferases pi 1), GPXs, GSR, SRXN1 (sulfiredoxin 1), ALDH (aldehyde dehydrogenase), and PRDXs (peroxiredoxins). Thus, NFE2L2 plays a central role in the transcriptional regulation of ferroptosis mainly through expressing anti-injury genes [Citation159].
YAP1 and WWTR1
YAP1 and WWTR1 are two key downstream transcription coactivators of the Hippo pathway, involving the regulation of organ size, tissue homeostasis, and tumorigenesis. The nuclear translocation of YAP1 and WWTR1 is negatively regulated by phosphorylation, which is mediated by the activation of Hippo pathway kinase LATS1 (large tumor suppressor kinase 1)-LATS2 (large tumor suppressor kinase 2). The Hippo LATS1-LATS2-YAP1-WWTR1 pathway plays a major role in promoting cell density-induced resistance to ferroptosis [Citation109,Citation110,Citation160,Citation161]. The knockdown of the key Hippo pathway effectors, such as NF2/merlin (neurofibromin 2), LATS1, and LATS2, restores the sensitivity of cancer cells to ferroptosis under high cell density conditions [Citation160]. Similarly, the overexpression of mutant YAP1S127A that cannot be phosphorylated by LATS1 or LATS2 also increases the sensitivity to ferroptosis by inducing nuclear retention of YAP1 under high cell density conditions [Citation160]. The expression of the constitutively active form of WWTR1 (S89A mutant) similarly increases the sensitivity to ferroptosis in renal cell carcinoma [Citation109], indicating that WWTR1 may coordinate with YAP1 in the regulation of ferroptosis. Unlike the transcriptional upregulation of TFRC and ACSL4 that may lead to YAP1-dependent ferroptosis, the WWTR1-mediated expression of EMP1 (epithelial membrane protein 1) and ANGPTL4 (angiopoietin like 4) promotes ferroptosis in a NOX4- or CYBB-dependent manner, respectively [Citation109,Citation110]. Overall, YAP1 and WWTR1 are transcriptional regulators in cell density-mediated ferroptosis resistance, but further studies are still needed to clarify the effect of this pathway in vivo.
The epigenetic regulation of ferroptosis
DNA methylation regulates ferroptosis
HELLS/LSH (helicase, lymphoid specific), a member of the SNF2 family of chromatin remodeling proteins, cooperates with WDR76 (WD repeat domain 76) to inhibit ferroptosis by activating metabolic genes, including SCD and FADS2 (fatty acid desaturase 2) [Citation76]. The upregulation of SCD and FADS2 prevents ferroptosis by lowering lipid ROS and iron levels [Citation76]. HELLS also induces epigenetic silencing of cytosolic lncRNA LINC00472/P53RRA (long intergenic non-protein coding RNA 472), which is downregulated in cancers and functions as a tumor suppressor. LINC00472 may promote ferroptosis by displacing TP53 from G3BP1 (G3BP stress granule assembly factor 1), causing TP53 to remain in the nucleus, which finally affects TP53-dependent metabolic gene expression [Citation162]. In addition to inhibiting LINC00472, HELLS promotes nuclear lncRNA LINC00336 (long intergenic non-protein coding RNA 336) expression. LINC00336, competing for MIR6852, upregulates the expression of CBS, which mediates ferroptosis inhibition [Citation97]. Therefore, the expression of HELLS is important for integrated epigenetic and transcriptional regulation of ferroptosis.
Histone modification regulates ferroptosis
Histone ubiquitination is an important form of ferroptosis involving epigenetic regulation. Histone 2A ubiquitination (H2Aub) epigenetically activates the expression of SLC7A11 to inhibit ferroptosis. The tumor suppressor BAP1 (BRCA1 associated protein 1) is a member of the ubiquitin C-terminal hydrolase (UCH) subfamily of deubiquitinases that negatively regulates H2Aub, leading to SLC7A11 inhibition and the accumulation of lipid peroxidation for ferroptosis [Citation163]. Histone 2B ubiquitination (H2Bub) also epigenetically induces SLC7A11 expression during ferroptosis [Citation164]. This process requires USP7 (ubiquitin specific peptidase 7), which is a deubiquitinase responsible for the deubiquitination of histone 2B. TP53 decreases H2Bub by interacting with USP7 and inducing the nuclear translocation of USP7 through a transcription‐independent mechanism, thus reducing the level of H2Bub [Citation164]. TP53 reduces the occupation of H2Bub in the regulatory region of the SLC7A11 gene and inhibits the expression of SLC7A11 during erastin-induced ferroptosis [Citation164]. These findings indicate that through epigenetic control of H2Bub-mediated gene expression, TP53 has an atypical role in ferroptosis.
Histone methylation also regulates ferroptosis. KDM3B (lysine demethylase 3B), a histone H3 lysine 9 demethylase, protects against erastin-induced ferroptosis through activating the expression of SLC7A11 [Citation165]. NFE2L2 is acetylated by EP300 (E1A binding protein p300)- CREBBP/CBP (CREB binding protein), leading to its enhanced DNA binding and gene transactivation activities [Citation166]. Although CDKN2A/ARF (cyclin dependent kinase inhibitor 2A) stabilizes and activates TP53 by promoting the degradation of MDM2 (MDM2 proto-oncogene) [Citation167], ARF promotes ferroptosis in a TP53-independent manner. Alternatively, ARF impairs the interaction between CREBBP and NFE2L2, thereby weakening the acetylation of NFE2L2 and inhibiting NFE2L2-mediated SLC7A11 expression [Citation158]. The bromodomain-containing (BRD) family plays a role in epigenetic regulation by recognizing acetylated lysine residues on histones or other nuclear proteins. The BRD4 (bromodomain containing 4) inhibitor JQ1 induces ferroptosis via downregulating the expression of GPX4, SLC7A11, and SLC3A2 in breast and lung cancer cells [Citation130], indicating that BRD4 is required for the expression of anti-ferroptosis genes.
The posttranslational regulation of ferroptosis by degradation
Autophagy
Autophagy is an evolutionarily conserved degradation process used to remove dysfunctional proteins, damaged organelles, and invading pathogens. Based on the different degradation mechanisms and mediators, autophagy can be divided into three subtypes: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [Citation168,Citation169]. Macroautophagy (hereafter referred to as autophagy) is the most studied type and its abnormality is associated with various diseases. In the process of autophagy, a small cup-shaped membrane precursor (namely a phagophore) forms a double-membrane structure (namely an autophagosome), which is then fused with a lysosome to degrade the engulfed contents. The proteins encoded by the autophagy-related (ATG) genes are essential for the activation and execution of the autophagy process through the formation of various protein complexes, which is initiated by the inhibition of MTOR (mechanistic target of rapamycin kinase) or the activation of AMPK [Citation170]. The formation of autophagic membrane structures is further regulated by lipid metabolism [Citation171]. In addition to nonselective autophagy, selective autophagy recognizes specific targets and recruits corresponding autophagy receptors for subsequent lysosomal degradation. Various ferroptosis activators can inhibit MTOR or activate AMPK, leading to the formation of autophagosomes [Citation45,Citation172]. Several stress-related proteins, such as STING1/TMEM173 (stimulator of interferon response CGAMP interactor 1) and DUSP1 (dual-specificity phosphatase 1), are important regulators of autophagy-dependent ferroptosis in cancer therapy [Citation131,Citation173]. The excessive activation of several selective types of autophagy (e.g., ferritinophagy, lipophagy, and clockophagy) as well as CMA mediate ferroptotic cell death, which is discussed below ().
Figure 6. Mechanisms of autophagy-dependent ferroptosis. Several types of selective autophagy, including ferritinophagy, lipophagy, clockophagy, and CMA, promote ferroptotic cell death by inducing the degradation of ferritin, lipid droplets, ARNTL, and GPX4, respectively. Abbreviations: ARNTL, aryl hydrocarbon receptor nuclear translocator like; CMA, chaperone-mediated autophagy; GPX4, glutathione peroxidase 4; HSP90, heat shock protein 90; NCOA4, nuclear receptor coactivator 4; RAB7A, RAB7A, member RAS oncogene family; SQSTM1, sequestosome 1
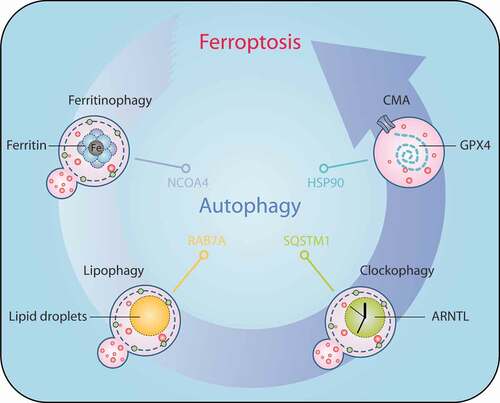
Ferritinophagy
Ferritinophagy is a process of selective degradation of ferritin and was discovered in 2014 [Citation64]. Quantitative proteomics was used to identify NCOA4 as an autophagy receptor responsible for ferritinophagy. The direct binding of the key surface arginine in FTH1 to the C-terminal element in NCOA4 is observed during ferritinophagy [Citation64]. Gene disruption of mouse Ncoa4 leads to an accumulation of ferritin in tissues and causes anemia, suggesting the potential role of NCOA4 in iron homeostasis [Citation174]. As free iron increases after ferritin degradation, ferritinophagy is an important regulator of hypertrophy. The deletion of Atg5 (autophagy related 5) or Atg7 limits erastin-induced ferroptosis, indicating that the general autophagy machinery contributes to ferroptosis [Citation67]. Notably, the knockdown of NCOA4 increases ferritin levels and inhibits ferroptosis, whereas overexpression of NCOA4 increases ferritin degradation and promotes ferroptosis in PANC-1 and HT1080 cells, indicating a specific role of ferritinophagy-mediated ferritin degradation in promoting ferroptosis [Citation67]. Similarly, RNAi screening and subsequent genetic analysis confirm that ATG13 and ATG3 are positive regulators of cystine starvation-induced ferroptosis and ferritinophagy in MEFs [Citation68,Citation175]. The knockdown of BECN1 or MAP1LC3B (microtubule associated protein 1 light chain 3 beta) also prevents ferritin degradation and ferroptosis following erastin treatment [Citation175], further indicating that ferroptosis is a type of autophagy-dependent cell death.
Lipophagy
Lipophagy is defined as the autophagic degradation of intracellular lipid droplets, which was first described in mouse hepatocytes [Citation176]. PLIN (perilipin)-family proteins not only cover the surface of lipid droplets but also absorb other regulatory proteins onto the surface of lipid droplets. During the ferroptosis caused by RSL3 in mouse or human hepatocytes, the accumulation of lipid droplets and protein levels of PLIN2 increases at an early stage, but decreases at a later stage [Citation80]. The suppression of TPD52 (tumor protein D52), a key regulator of lipid droplet formation, enhances RSL3-induced ferroptotic cell death. In contrast, the overexpression of TPD52 increases the accumulation of lipid droplets, which leads to the inhibition of RSL3-induced ferroptosis, supporting the anti-injury effect of lipid droplets during ferroptosis. In contrast, the induction of lipophagy promotes RSL3-induced ferroptosis [Citation80]. RAB7A (RAB7A, member RAS oncogene family) is a key regulator in the trafficking of multivesicular bodies and lysosomes to the lipid droplet surface during lipophagy. The knockdown of RAB7A prevents RSL3-induced lipid peroxidation and subsequent ferroptosis in vitro and in vivo [Citation80], indicating the potential role of lipophagy in promoting ferroptotic cell death. Given that a deficiency in lipophagy is associated with obesity, steatosis, and atherosclerosis, it will be interesting to determine the role of ferroptosis in these metabolic diseases.
Clockophagy
The mammalian circadian clock is regulated by a self-regulating transcriptional feedback mechanism that oscillates on a 24-h cycle. The key component of circadian clock is a heterodimeric transcriptional activator, which contains two basic helix-loop-helix PER-ARNT-SIM (bHLH-PAS) proteins (namely CLOCK and ARNTL) that transcriptionally upregulate a series of genes. The autophagic degradation of ARNTL, a process called clockophagy, was recently shown to promote ferroptosis induced by type II ferroptosis activators (e.g., RSL3 and FIN56), but not by type I ferroptosis activators (e.g., erastin, sulfasalazine, and sorafenib) [Citation177,Citation178]. Mass spectrometric analysis of immunoprecipitated ARNTL complexes indicates that SQSTM1 is an autophagy receptor for clockophagy-dependent ARNTL degradation [Citation177]. ATG5 and ATG7, but not ATG9A, contribute to autophagic degradation of ARNTL during ferroptosis [Citation177]. ARNTL regulates gene expression by recognizing the E-box motif (CAGCTG or CACGTG) in the promoter. The E-box-containing gene EGLN2 (egl-9 family hypoxia inducible factor 2) is a transcriptional target of ARNTL, which mediates HIF1A degradation and subsequent lipid peroxidation [Citation177], indicating a molecular connection between circadian rhythm and the hypoxic pathway during ferroptosis.
Despite the lack of direct evidence of dysregulated clockophagy in acute pancreatitis, mice lacking ARNTL still show a worsening of the disease induced by L-arginine, which is related to ferroptotic damage [Citation179]. Mechanistically, ARNTL coordinates the expression of genes encoding antioxidants (e.g., SLC7A11, GPX4, SOD1, TXN, and NFE2L2) or membrane repair (e.g., CHMP5) to suppress pancreatic ferroptosis during inflammation [Citation179]. Melatonin is a pineal hormone that regulates the circadian rhythm of vertebrates. Interestingly, melatonin reduces hemoglobin-induced ferroptosis in platelets [Citation180], indicating that the circadian clock may be an important regulator of ferroptotic response in the coagulation system. Circadian rhythm dysfunction is observed in neurodegenerative disorders (e.g., Alzheimer, Parkinson, and Huntington diseases) and cancer. Further research is necessary to determine the specific function of the circadian rhythm in ferroptosis-related diseases.
CMA
CMA is a kind of selective autophagy, which involves molecular chaperone HSPA8/Hsc70 (heat shock protein family A [Hsp70] member 8) binding to a selected protein substrate with a KFERQ-like motif, which is then targeted to the lysosome for degradation [Citation181]. LAMP2A (lysosomal associated membrane protein type 2A) is a receptor in the lysosomal membrane and mediates the transport of substrates into the lysosome in CMA. CMA is activated by oxidative stress and helps to effectively remove oxidized proteins. The chemical inactivation of GPX4 resulting from treatment with the ferroptosis inducer RSL3 also causes significant degradation of GPX4, indicating that the activity of GPX4 is regulated by its stability [Citation81,Citation141]. HSPA5/GRP78/BIP is a chaperone expressed primarily in the ER. HSPA5 stabilizes GPX4 by forming an HSPA5-GPX4 complex, which causes resistance to ferroptosis [Citation142]. In contrast, HSP90 promotes CMA-mediated degradation of GPX4 by improving the stability of LAMP2A, thereby promoting ferroptosis [Citation140]. However, it is still unknown whether CMA-mediated degradation of GPX4 is a common event in response to various ferroptosis activators.
UPS
The UPS degrades most intracellular proteins through a multistep dynamic process, including the polyubiquitination of substrate proteins and subsequent destruction via the 26S proteasome. A deubiquitinase is a protease that removes a single ubiquitin or multiple ubiquitin chains from a target protein. OTUB1 (OTU deubiquitinase, ubiquitin aldehyde binding 1) directly interacts with and stabilizes SLC7A11 by removing ubiquitin moieties [Citation182]. Therefore, inhibiting OTUB1 enhances the proteasome-dependent degradation of SLC7A11, which leads to the sensitivity of cancer cells to ferroptosis [Citation182]. The UPS also regulates the protein stability of ferroptosis promoters such as ACSL4 [Citation183], VDAC2 (voltage dependent anion channel 2) [Citation184], or VDAC3 [Citation184]. The expression of ACSL4 is downregulated in liver tissues of mice fed a high-fat diet [Citation183]. Arachidonic acid as the preferred substrate for ACSL4 is the main factor that selectively induces ACSL4 degradation by the UPS [Citation183]. Earlier studies indicate that VDAC2 and VDAC3 are direct targets of erastin responsible for ferroptosis through unknown mechanisms [Citation184]. Recent studies have shown that erastin can induce rapid degradation of VDAC2 and VDAC3, thus forming a negative feedback mechanism to inhibit ferroptosis [Citation184]. Mechanistically, NEDD4 (NEDD4 E3 ubiquitin protein ligase) mediates VDAC2 and VDAC3 degradation in response to erastin [Citation185]. Unlike NEDD4, NEDD4L (NEDD4-like E3 ubiquitin protein ligase)-mediated lactotransferrin (LTF) protein degradation inhibits intracellular iron accumulation and subsequent lipid peroxidation-dependent ferroptosis in cancer cells [Citation186]. As mentioned earlier, the UPS strictly controls the activation of transcription factors (e.g., NFE2L2, TP53, IREB2, and HIF) associated with ferroptosis, although autophagy can act synergistically. Therefore, targeting specific protein degradation may represent a potential strategy for treating diseases associated with impaired ferroptosis.
Conclusions and perspectives
The sensitivity of ferroptosis seems to be highly dependent on the state of cellular metabolism (especially the metabolism of lipids, iron, and amino acids) and degradation systems (autophagy and the UPS), which involve a complex network to shape oxidative stress. Although their activities and effects are not equal, ferroptotic processes can be regulated by a variety of reagents, which is related to disease treatment. Pharmacologically inducing ferroptosis represents a promising anticancer strategy, although its role in tumor prevention is currently unclear. In particular, some traditional drug-resistant cancer cells are vulnerable to GPX4 inhibitors in vitro [Citation187]. Because slc7a11 knockout mice are feasible to generate [Citation18] and gpx4 knockout mice are embryonically lethal [Citation21,Citation22], it is speculated that SLC7A11 inhibitors may be clinically safer than GPX4 inhibitors. Several iron chelators (e.g., deferoxamine, deferiprone, and deferasirox) and antioxidants (e.g., vitamin E and CoQ10) have been used in the clinic. It will be interesting to see if these drugs can protect against ferroptosis-mediated tissue injury in patients. In addition, certain synthetic small-molecule compounds (e.g., ferrostatin-1 and liproxstatin-1) are effective inhibitors of ferroptosis in preclinical animal models, and further clinical studies are needed to determine their safety and effectiveness.
Given the fundamental role of inflammation in various human diseases, further research is still needed to clarify the interaction between ferroptotic cell death and the immune system. It is generally thought that damage-associated molecular patterns (DAMPs) are immune mediators of various types of RCD. DAMPs are endogenous molecules that can be released by dead or dying cells and eventually stimulate inflammation and immune responses through binding their receptors in various immune cells (e.g., macrophages and monocytes) [Citation188]. Recent research highlights that HMGB1 (high mobility group box 1), a typical DAMP, is released by ferroptotic cells and drives inflammation by activating macrophages to produce pro-inflammatory cytokines [Citation189,Citation190]. Furthermore, intermediate or final products of lipid peroxidation (e.g., 4HNE) may be other resources that modulate the immune response during ferroptotic cell death [Citation191]. The specific immune signals associated with ferroptosis in the early or late stages must be further determined.
In summary, although there are a large number of regulators that directly or indirectly affect iron accumulation and lipid peroxidation to regulate ferroptosis, there are still many questions that have not been answered: (1) What is the executor of ferroptotic cell death? (2) What is the key molecular marker of ferroptosis? (3) What is the physiological and pathological function of ferroptosis in human health and disease? (4) How do we define the crosstalk between ferroptosis, autophagy, and other types of RCD? Further functional investigations into the complicated machinery and regulation of ferroptosis will provide a new way to effectively treat diseases related to iron overload or lipid peroxidation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072.
- Weinlich R, Oberst A, Beere HM, et al. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–136.
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
- Song X, Zhu S, Xie Y, et al. JTC801 Induces pH-dependent Death Specifically in Cancer Cells and Slows Growth of Tumors in Mice. Gastroenterology. 2018;154(5):1480–1493.
- Liu J, Kuang F, Kang R, et al. Alkaliptosis: a new weapon for cancer therapy. Cancer Gene Ther. 2020;27(5):267–269.
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364.
- Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379.
- Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285.
- Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435.
- Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2019. DOI:https://doi.org/10.1016/j.semcancer.2019.03.002
- Kang R, Tang D. Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep. 2017;5(2):153–159.
- Dolma S, Lessnick SL, Hahn WC, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296.
- Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331.
- Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;1–15. DOI:https://doi.org/10.1080/15548627.2020.1714209
- Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–849.
- Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1(6):497–506.
- Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280(45):37423–37429.
- Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–465.
- Badgley MA, Kremer DM, Maurer HC, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–89.
- Imai H, Hirao F, Sakamoto T, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305(2):278–286.
- Yant LJ, Ran Q, Rao L, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34(4):496–502.
- Ingold I, Aichler M, Yefremova E, et al. Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J Biol Chem. 2015;290(23):14668–14678.
- Brutsch SH, Wang CC, Li L, et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal. 2015;22(4):281–293.
- Ran Q, Van Remmen H, Gu M, et al. Embryonic fibroblasts from Gpx4± mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic Biol Med. 2003;35(9):1101–1109.
- Canli O, Alankus YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–148.
- Kang R, Zeng L, Zhu S, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe. 2018;24(1):97–108 e4.
- Bosl MR, Takaku K, Oshima M, et al. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc Natl Acad Sci U S A. 1997;94(11):5531–5534.
- Ingold I, Berndt C, Schmitt S, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2018;172(3):409–22 e21.
- Alim I, Caulfield JT, Chen Y, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262–79 e25.
- Gao J, Yang F, Che J, et al. Selenium-encoded isotopic signature targeted profiling. ACS Cent Sci. 2018;4(8):960–970.
- Llabani E, Hicklin RW, Lee HY, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. 2019;11(6):521–532.
- Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503.
- Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698.
- Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692.
- Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53.
- Dai E, Zhang W, Cong D, et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523(4):966–971.
- Wu M, Xu LG, Li X, et al. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem. 2002;277(28):25617–25623.
- Miriyala S, Thippakorn C, Chaiswing L, et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic Biol Med. 2016;91:68–80.
- Ma S, Henson ES, Chen Y, et al. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307.
- Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
- Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–245.
- Lane DJ, Merlot AM, Huang ML, et al. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta. 2015;1853(5):1130–1144.
- Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med. 2019;133:101–111.
- Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28(15):2388–99 e5.
- Yu Y, Jiang L, Wang H, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739.
- Gozzelino R, Soares MP. Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal. 2014;20(11):1754–1769.
- Imoto S, Kono M, Suzuki T, et al. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus Apher Sci. 2018;57(4):524–531.
- Hassannia B, Wiernicki B, Ingold I, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341–3355.
- Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184.
- Egyed A, Saltman P. Iron is maintained as Fe(II) under aerobic conditions in erythroid cells. Biol Trace Elem Res. 1984;6(4):357–364.
- Breuer W, Epsztejn S, Cabantchik ZI. Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II). J Biol Chem. 1995;270(41):24209–24215.
- Philpott CC, Jadhav S. The ins and outs of iron: escorting iron through the mammalian cytosol. Free Radic Biol Med. 2019;133:112–117.
- Nandal A, Ruiz JC, Subramanian P, et al. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011;14(5):647–657.
- Protchenko O, Baratz E, Jadhav S, et al. Iron chaperone PCBP1 protects murine liver from lipid peroxidation and steatosis. Hepatology. 2020. DOI:https://doi.org/10.1002/hep.31328.
- Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639–643.
- Shaw GC, Cope JJ, Li L, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96–100.
- Paradkar PN, Zumbrennen KB, Paw BH, et al. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol. 2009;29(4):1007–1016.
- Li C, Zhang Y, Cheng X, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46(4):441–55 e8.
- Kang R, Xie Y, Zeh HJ, et al. Mitochondrial quality control mediated by PINK1 and PRKN: links to iron metabolism and tumor immunity. Autophagy. 2019;15(1):172–173.
- Chiabrando D, Marro S, Mercurio S, et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest. 2012;122(12):4569–4579.
- Muckenthaler MU, Rivella S, Hentze MW, et al. A red carpet for iron metabolism. Cell. 2017;168(3):344–361.
- Yuan H, Li X, Zhang X, et al. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478(2):838–844.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109.
- De Domenico I, Vaughn MB, Li L, et al. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. Embo J. 2006;25(22):5396–5404.
- Wang YQ, Chang SY, Wu Q, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308.
- Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428.
- Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032.
- Brown CW, Amante JJ, Chhoy P, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575–86 e4.
- Shang Y, Luo M, Yao F, et al. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020;72:109633.
- Tuo QZ, Lei P, Jackman KA, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22(11):1520–1530.
- Detivaud L, Island ML, Jouanolle AM, et al. Ferroportin diseases: functional studies, a link between genetic and clinical phenotype. Hum Mutat. 2013;34(11):1529–1536.
- Bogdan AR, Miyazawa M, Hashimoto K, et al. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41(3):274–286.
- Geng N, Shi BJ, Li SL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826–3836.
- Tesfay L, Paul BT, Konstorum A, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79(20):5355–5366.
- Jiang Y, Mao C, Yang R, et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7:3293–3305.
- Magtanong L, Ko PJ, To M, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420–32 e9.
- Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234.
- Plotz T, Hartmann M, Lenzen S, et al. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr Metab (Lond). 2016;13(1):16.
- Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003.
- Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90.
- Miess H, Dankworth B, Gouw AM, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37(40):5435–5450.
- Blomme A, Ford CA, Mui E, et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat Commun. 2020;11(1):2508.
- Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98.
- Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343.
- Chu B, Kon N, Chen D, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–591.
- Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10):5866–5898.
- Schnurr K, Belkner J, Ursini F, et al. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J Biol Chem. 1996;271(9):4653–4658.
- Conrad M, Sandin A, Forster H, et al. 12/15-lipoxygenase-derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2010;107(36):15774–15779.
- Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–75.
- Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628–41 e26.
- Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191.
- Matsushita M, Freigang S, Schneider C, et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555–568.
- Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309.
- Wang L, Cai H, Hu Y, et al. A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 2018;9(10):1005.
- Martinez AM, Mirkovic J, Stanisz ZA, et al. NSC-34 motor neuron-like cells are sensitized to ferroptosis upon differentiation. FEBS Open Bio. 2019;9(4):582–593.
- Wang M, Mao C, Ouyang L, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019;26(11):2329–2343.
- Hayano M, Yang WS, Corn CK, et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278.
- Cao J, Chen X, Jiang L, et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun. 2020;11(1):1251.
- Shin D, Lee J, You JH, et al. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020;30:101418.
- Jennis M, Kung CP, Basu S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30(8):918–930.
- Niu Y, Zhang J, Tong Y, et al. Physcion 8-O-beta-glucopyranoside induced ferroptosis via regulating miR-103a-3p/GLS2 axis in gastric cancer. Life Sci. 2019;237:116893.
- Luo M, Wu L, Zhang K, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25(8):1457–1472.
- Shimada K, Hayano M, Pagano NC, et al. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. 2016;23(2):225–235.
- Kim H, Lee JH, Park JW. Down-regulation of IDH2 sensitizes cancer cells to erastin-induced ferroptosis. Biochem Biophys Res Commun. 2020;525(2):366–371.
- Wang TX, Liang JY, Zhang C, et al. The oncometabolite 2-hydroxyglutarate produced by mutant IDH1 sensitizes cells to ferroptosis. Cell Death Dis. 2019;10(10):755.
- Ding CC, Rose J, Sun T, et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat Metab. 2020;2(3):270–277.
- Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–1704.
- Yang WH, Ding CC, Sun T, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28(10):2501–8 e4.
- Yang WH, Huang Z, Wu J, et al. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. 2020;18(1):79–90.
- Gaschler MM, Hu F, Feng H, et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13(4):1013–1020.
- Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–63 e3.
- Agmon E, Solon J, Bassereau P, et al. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8(1):5155.
- Catala A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids. 2009;157(1):1–11.
- Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, et al. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136.
- Dai E, Meng L, Kang R, et al. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522(2):415–421.
- Gong YN, Guy C, Olauson H, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169(2):286–300 e16.
- Ruhl S, Shkarina K, Demarco B, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–960.
- Liu J, Kang R, Tang D. ESCRT-III-mediated membrane repair in cell death and tumor resistance. Cancer Gene Ther. 2020.
- Zhang Y, Tan H, Daniels JD, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26(5):623–33 e9.
- Eaton JK, Furst L, Ruberto RA, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16(5):497–506.
- Eaton JK, Ruberto RA, Kramm A, et al. Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J Am Chem Soc. 2019;141(51):20407–20415.
- Wenz C, Faust D, Linz B, et al. t-BuOOH induces ferroptosis in human and murine cell lines. Arch Toxicol. 2018;92(2):759–775.
- Gaschler MM, Andia AA, Liu H, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14(5):507–515.
- Li Q, Han X, Lan X, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2(7):e90777.
- Baba Y, Higa JK, Shimada BK, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314(3):H659–H68.
- Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145.
- Mai TT, Hamai A, Hienzsch A, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9(10):1025–1033.
- Sui S, Zhang J, Xu S, et al. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019;10(5):331.
- Li C, Zhang Y, Liu J, et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2020 Mar 18;1-13. doi: https://doi.org/10.1080/15548627.2020.1739447.
- Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15(12):1137–1147.
- Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3(3):232–243.
- Miotto G, Rossetto M, Di Paolo ML, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328.
- Shah R, Margison K, Pratt DA. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem Biol. 2017;12(10):2538–2545.
- Griesser M, Shah R, Van Kessel AT, et al. The catalytic reaction of nitroxides with peroxyl radicals and its relevance to their cytoprotective properties. J Am Chem Soc. 2018;140(10):3798–3808.
- Krainz T, Gaschler MM, Lim C, et al. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent Sci. 2016;2(9):653–659.
- Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4(3):387–396.
- Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–4556.
- Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996–3005.
- Muller T, Dewitz C, Schmitz J, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74(19):3631–3645.
- Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064–2077.
- Wang D, Peng Y, Xie Y, et al. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. 2016;480(4):602–607.
- Ishii T, Bannai S, Sugita Y. Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. Role of the mixed disulfide of 2-mercaptoethanol and cysteine. J Biol Chem. 1981;256(23):12387–12392.
- Dai C, Chen X, Li J, et al. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020. DOI:https://doi.org/10.1038/s41417-020-0170-2
- Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–168.
- Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62.
- Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120.
- Hu W, Zhang C, Wu R, et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107(16):7455–7460.
- Tarangelo A, Magtanong L, Bieging-Rolett KT, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–575.
- Ou Y, Wang SJ, Li D, et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–E12.
- Leu JI, Murphy ME, George DL. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A. 2019;116(17):8390–8396.
- Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500.
- Shin D, Kim EH, Lee J, et al. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med. 2018;129:454–462.
- Roh JL, Kim EH, Jang H, et al. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–262.
- Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer. 2020;122(2):279–292.
- Fan Z, Wirth AK, Chen D, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6(8):e371.
- Chen D, Tavana O, Chu B, et al. NRF2 Is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68(1):224–32 e4.
- Anandhan A, Dodson M, Schmidlin CJ, et al. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27(4):436–447.
- Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–406.
- Wenz C, Faust D, Linz B, et al. Cell-cell contacts protect against t-BuOOH-induced cellular damage and ferroptosis in vitro. Arch Toxicol. 2019;93(5):1265–1279.
- Mao C, Wang X, Liu Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78(13):3484–3496.
- Zhang Y, Shi J, Liu X, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–1192.
- Wang Y, Yang L, Zhang X, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20(7):e47563.
- Wang Y, Zhao Y, Wang H, et al. Histone demethylase KDM3B protects against ferroptosis by upregulating SLC7A11. FEBS Open Bio. 2020;10(4):637–643.
- Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29(10):2658–2672.
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92(6):725–734.
- Klionsky DJ. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45.
- Xie Y, Li J, Kang R, et al. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431.
- Liu Y, Wang Y, Liu J, et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2020. DOI:https://doi.org/10.1038/s41417-020-0182-y
- Xie Y, Kuang F, Liu J, et al. DUSP1 blocks autophagy-dependent ferroptosis in pancreatic cancer. J Pancreatol. 2020;Publish Ahead of Print. DOI:https://doi.org/10.1097/JP9.0000000000000054
- Bellelli R, Federico G, Matte A, et al. NCOA4 deficiency impairs systemic iron homeostasis. Cell Rep. 2016;14(3):411–421.
- Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135.
- Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238.
- Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035.
- Liu Y, Wang Y, Liu J, et al. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun. 2020;525(3):620–625.
- NaveenKumar SK, Hemshekhar M, Kemparaju K, et al. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: protection by Melatonin. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2303–2316.
- Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3(4):295–299.
- Liu T, Jiang L, Tavana O, et al. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019;79(8):1913–1924.
- Kan CF, Singh AB, Stafforini DM, et al. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J Lipid Res. 2014;55(8):1657–1667.
- Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868.
- Yang Y, Luo M, Zhang K, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020;11(1):433.
- Wang Y, Liua Y, Liua J, et al. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020. DOI:https://doi.org/10.1016/j.bbrc.2020.07.032
- Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247–250.
- Tang D, Kang R, Coyne CB, et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–175.
- Wen Q, Liu J, Kang R, et al. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278–283.
- Yu Y, Xie Y, Cao L, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2(4):e1054549.
- Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405–414.
- Zhang L, Liu W, Liu F, et al. IMCA Induces Ferroptosis Mediated by SLC7A11 through the AMPK/mTOR Pathway in Colorectal Cancer. Oxid Med Cell Longev. 2020;2020:1675613.
- Lorincz T, Jemnitz K, Kardon T, et al. Ferroptosis is involved in acetaminophen induced cell death. Pathol Oncol Res. 2015;21(4):1115–1121.
- Woo JH, Shimoni Y, Yang WS, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162(2):441–451.
- Meng P, Zhang S, Jiang X, et al. Arsenite induces testicular oxidative stress in vivo and in vitro leading to ferroptosis. Ecotoxicol Environ Saf. 2020;194:110360.
- Ooko E, Saeed ME, Kadioglu O, et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22(11):1045–1054.
- Eling N, Reuter L, Hazin J, et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532.
- Chang LC, Chiang SK, Chen SE, et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–137.
- Basit F, van Oppen LM, Schockel L, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3):e2716.
- Alborzinia H, Ignashkova TI, Dejure FR, et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol. 2018;1(1):210.
- Deng G, Li Y, Ma S, et al. Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic Biol Med. 2020;148:151–161.
- Zeng T, Deng G, Zhong W, et al. Indoleamine 2, 3-dioxygenase 1enhanceshepatocytes ferroptosis in acute immune hepatitis associated with excess nitrative stress. Free Radic Biol Med. 2020;152:668–679.
- Chen GQ, Benthani FA, Wu J, et al. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242–254.
- Du J, Wang T, Li Y, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–369.
- Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680.
- Chen P, Wu Q, Feng J, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020;5(1):51.
- Fang S, Yu X, Ding H, et al. Effects of intracellular iron overload on cell death and identification of potent cell death inhibitors. Biochem Biophys Res Commun. 2018;503(1):297–303.
- Wu J, Yang JJ, Cao Y, et al. Iron overload contributes to general anaesthesia-induced neurotoxicity and cognitive deficits. J Neuroinflammation. 2020;17(1):110.
- Jiang T, Chu J, Chen H, et al. Gastrodin inhibits H2O2-induced ferroptosis through its antioxidative effect in rat glioma cell line C6. Biol Pharm Bull. 2020;43(3):480–487.
- Wang W, Green M, Choi JE, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274.
- Lang X, Green MD, Wang W, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9(12):1673–1685.
- Lei G, Zhang Y, Koppula P, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–162.
- Ye LF, Chaudhary KR, Zandkarimi F, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15(2):469–484.
- Liang C, Zhang X, Yang M, et al. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51):e1904197.
- Shen Z, Song J, Yung BC, et al. Emerging strategies of cancer therapy based on ferroptosis. Adv Mater. 2018;30(12):e1704007.
- Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52(3):1011–1022.
- Wang Y, Tang M. PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ Pollut. 2019;254:112937.
- Wang Z, Ding Y, Wang X, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21–33.
- Wu C, Zhao W, Yu J, et al. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci Rep. 2018;8(1):574.
- Palmer LD, Jordan AT, Maloney KN, et al. Zinc intoxication induces ferroptosis in A549 human lung cells. Metallomics. 2019;11(5):982–993.
- Torii S, Shintoku R, Kubota C, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473(6):769–777.
- Gao H, Bai Y, Jia Y, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. 2018;503(3):1550–1556.
- Xie Y, Song X, Sun X, et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016;473(4):775–780.
- Neitemeier S, Jelinek A, Laino V, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–570.
- Jelinek A, Heyder L, Daude M, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med. 2018;117:45–57.
- Mishima E, Sato E, Ito J, et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J Am Soc Nephrol. 2020;31(2):280–296.
- Do Van B, Gouel F, Jonneaux A, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178.
- Li X, Zou Y, Xing J, et al. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3beta/Nrf2 pathway. Oxid Med Cell Longev. 2020;2020:6286984.
- Liu B, Zhao C, Li H, et al. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun. 2018;497(1):233–240.
- Wang L, Liu Y, Du T, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. 2020;27(2):662–675.
- Chen Y, Mi Y, Zhang X, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38(1):402.
- Chen D, Fan Z, Rauh M, et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36(40):5593–5608.
- Wang N, Zeng GZ, Yin JL, et al. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem Biophys Res Commun. 2019;519(3):533–539.
- Chen MS, Wang SF, Hsu CY, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget. 2017;8(70):114588–114602.
- Nishizawa H, Matsumoto M, Shindo T, et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J Biol Chem. 2020;295(1):69–82.
- Wang GX, Tu HC, Dong Y, et al. DeltaNp63 inhibits oxidative stress-induced cell death, including ferroptosis, and cooperates with the BCL-2 family to promote clonogenic survival. Cell Rep. 2017;21(10):2926–2939.
- Zhang X, Du L, Qiao Y, et al. Ferroptosis is governed by differential regulation of transcription in liver cancer. Redox Biol. 2019;24:101211.
- Zou Y, Palte MJ, Deik AA, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617.
- Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625.
- Chen Y, Zhu G, Liu Y, et al. O-GlcNAcylated c-Jun antagonizes ferroptosis via inhibiting GSH synthesis in liver cancer. Cell Signal. 2019;63:109384.
- Chen PH, Wu J, Ding CC, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27(3):1008–1022.
- Cao JY, Poddar A, Magtanong L, et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 2019;26(6):1544–56 e8.
- Gagliardi M, Cotella D, Santoro C, et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019;10(12):902.
- Park SJ, Cho SS, Kim KM, et al. Protective effect of sestrin2 against iron overload and ferroptosis-induced liver injury. Toxicol Appl Pharmacol. 2019;379:114665.
- Li Y, Feng D, Wang Z, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26(11):2284–2299.
- Brown CW, Amante JJ, Goel HL, et al. The alpha6beta4 integrin promotes resistance to ferroptosis. J Cell Biol. 2017;216(12):4287–4297.
- Li L, Sun S, Tan L, et al. Polystyrene nanoparticles reduced ros and inhibited ferroptosis by triggering lysosome stress and TFEB nucleus translocation in a size-dependent manner. Nano Lett. 2019;19(11):7781–7792.