
ABSTRACT
Macrophage derived foam cells in atherosclerotic plaques are the major factor responsible for the pathogenesis of atherosclerosis (AS). During advanced AS, macrophage-specific macroautophagy/autophagy is dysfunctional. 1, 25-dihydroxy vitamin D3 (VitD3) and its receptor VDR (vitamin D receptor) are reported to inhibit foam cell formation and induce autophagy; however, the role of VitD3-VDR-induced autophagy and foam cell formation in AS has not been explored. Here we find that VitD3 significantly recovered oxidized low-density lipoprotein-impaired autophagy, as well as increased autophagy-mediated lipid breakdown in mouse bone marrow-derived macrophages and human monocyte-derived macrophages, thus inhibiting the conversion of macrophages into foam cells. Importantly, VitD3 functions through its receptor VDR to upregulate autophagy and attenuate the accumulation of lipids in macrophages. Moreover, this study is the first occasion to report the interesting link between VitD3 signaling and PTPN6/SHP-1 (protein tyrosine phosphatase non-receptor type 6) in macrophages. VitD3-induced autophagy was abrogated in the presence of the PTPN6/Ptpn6 shRNA or inhibitor. VDR along with RXRA (retinoid X receptor alpha), and NCOA1 (nuclear receptor coactivator 1), are recruited to a specific response element located on the gene promoter and induce PTPN6 expression. PTPN6 contributes to VitD3-mediated autophagy by regulating autophagy-related genes via activation of MAPK1 (mitogen-activated protein kinase 1) and CEBPB (CCAAT enhancer binding protein beta). Furthermore, expression of PTPN6 is also crucial for VitD3-mediated inhibition of macrophage foam cell formation through autophagy. Thus, VitD3-VDR-PTPN6 axis-regulated autophagy attenuates foam cell formation in macrophages.
Introduction
Atherosclerosis (AS) is a chronic inflammatory disease exhibiting lipoprotein-driven metabolic dysfunction, characterized by the progressive build-up of plaques in the arterial vasculature [Citation1,Citation2]. It is the major cause of cardiovascular diseases (CVDs) arising from myocardial infarction, stroke, or ischemic heart failure [Citation3,Citation4]. Plaque development initiates through modification of low-density lipoprotein (LDL) molecules into oxidized LDL (Ox-LDL) by free radicals [Citation5]. Accumulation of these toxic lipid molecules leads to damage to the arteries. This initiates a repair process leading to a series of reactions, including the recruitment of monocytes to the growing plaque and their subsequent differentiation into macrophages. Ox-LDL uptake by macrophages leads to foam cell formation, thereby leading to intracellular lipid accumulation in the form of lipid droplets (LDs) and development of AS [Citation6]. Therefore, degrading toxic lipids and inhibiting the conversion of macrophages into foam cells would be an important protective strategy against AS.
Macroautophagy/autophagy is a lysosomal-dependent catabolic process, which involves the sequestration of self-components in double-membrane organelles called autophagosomes, and their degradation through autolysosome formation [Citation7]. Apart from the classical function of autophagy as a self-renewing mechanism under different internal or external stimuli, the immunological function of autophagy has also been researched in recent years [Citation8-10]. Autophagy, especially macrophage autophagy, plays a very decisive role in inflammation and infection [Citation9,Citation11]. Lipophagy is a type of selective autophagy where lipids are selectively delivered to autophagosomes for lysosome-mediated lipid breakdown [Citation12,Citation13]. Therefore, lipophagy helps in the regulation of lipid metabolism and cholesterol homeostasis, hence playing an important role in the pathogenesis of AS [Citation14-17]. In advanced AS, macrophage autophagy becomes dysfunctional due to atherogenic factors, such as toxic lipids (e.g., Ox-LDL), inflammation, and reactive oxygen species, which promote plaque development and disease progression [Citation18,Citation19]. Recent data show that macrophage-specific deletion of ATG5 (autophagy related 5), an important protein in the autophagy process, leads to the development of more atherosclerotic plaques in mice fed with a high fat diet [Citation15,Citation20,Citation21]. In addition, stent based delivery of MTOR (mechanistic target of rapamycin kinase) inhibitor, everolimus (an autophagy inducer), in cholesterol-fed rabbits demonstrates an atheroprotective role [Citation22,Citation23]. Moreover, the atheroprotective role of rapamycin in AS has also been attributed to up-regulation of autophagy [Citation24,Citation25]. These reports suggest that induction of macrophage autophagy would be a possible target for inhibiting AS development.
Vitamin D deficiency is a global health problem affecting almost 50% of the population worldwide [Citation26,Citation27]. Epidemiological and clinical evidence show a link between vitamin D deficiency and an increased risk of CVD, including coronary artery calcification, coronary stenosis, and carotid AS [Citation28]. VDR (vitamin D receptor) is a member of the nuclear receptor super-family and mediates the major known functions of 1, 25-dihydroxy vitamin D3 (VitD3), a biologically active form of vitamin D [Citation29,Citation30]. Vitamin D and VDR appear to be important immunological regulators of AS [Citation31-34]. vdr knockout (KO) mice develop more atherosclerotic plaques, while VDR signaling inhibits foam cell formation by decreasing macrophage cholesterol uptake [Citation35,Citation36]. LDL receptor and VDR double-knockout mice display accelerated atherosclerotic lesion development, increase in cholesterol influx in macrophages, and proinflammatory cytokines in the aorta [Citation37]. Hence, these studies suggest a critical role of VDR in controlling lipid homeostasis and regulation of atherogenesis. Upon ligand binding, VDR binds to the promoter region of various target genes with specific DNA sequences of direct repeat 3 type of steroid response element [Citation38-41]. Another study has shown that VitD3-mediated inhibition of mycobacterial growth is dependent on induction of autophagy by upregulation of autophagy related genes such as BECN1 (beclin 1) and ATG5 as well as by phagosome maturation [Citation42,Citation43]. Although autophagy, VDR, and vitamin D, are all involved in the pathogenesis of AS, it is still unclear whether they are related or these processes function independently. Various reports have identified vitamin D as a potent stimulator of autophagy in inflammation and infection [Citation44-46]; however, no study has directly addressed the role of vitamin D-mediated autophagy in AS.
Our earlier studies have demonstrated the cardinal role of nuclear receptors in autophagy modulation and macrophage biology [Citation47-50]. In this current study, we present evidence to suggest that vitamin D in macrophages inhibits AS by inducing autophagy. We found that VitD3, through VDR signaling, rescues Ox-LDL-impaired autophagy in both mouse bone marrow-derived macrophages (mBMDMs) and human monocyte-derived macrophages (hMDMs), and inhibited macrophage foam cell conversion in autophagy-dependent manner. To the best of our knowledge, we are reporting for the first time that VitD3 promotes the expression and function of PTPN6/SHP-1 (protein tyrosine phosphatase non-receptor type 6), which is a novel target gene of VDR. Ligand bound VDR binds to the promoter region of PTPN6/Ptpn6 and regulates its expression. Furthermore, we have shown that PTPN6 plays an important role in VitD3-mediated autophagy and that the VitD3-VDR-PTPN6 pathway inhibits the conversion of macrophages into foam cells. Hence, our study completes an existing knowledge gap by describing a defined role for macrophage VDR signaling in atheroprotection, through the process of autophagy.
Results
VitD3 rescues Ox-LDL-impaired autophagic flux in murine macrophages
To investigate whether vitamin D could rescue the impairment in autophagic flux induced via Ox-LDL, mBMDMs were stimulated with Ox-LDL and subsequently treated with VitD3, in the presence or absence of bafilomycin A1 (BafA) an inhibitor of late stage autophagy. BafA blocks the maturation of autophagosome by inhibiting autophagosome lysosome fusion and therefore blocks the degradation of two commonly used autophagy markers: MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta)-II, which is involved in autophagosome formation, and SQSTM1/p62 (sequestosome 1) which is an autophagy receptor required for removing protein aggregates via autolysosomes [Citation51]. Hence, the accumulation of MAP1LC3B and SQSTM1 is observed upon BafA treatment. Furthermore, we investigated Ox-LDL-mediated disruption of autophagy in mBMDMs for which 50 μg/ml Ox-LDL was used for 48 h (arrived at by concentration series and time kinetics of Ox-LDL treatment) (Fig. S1A-C). Treatment with Ox-LDL led to a slight increase in the level of MAP1LC3B-II by western blotting ( and S2A; increase in MAP1LC3B-II:MAP1LC3B-I is not significant), as well as the number of MAP1LC3B puncta (p<0.05) by immunofluorescence staining () as compared to the controls. However, the level of SQSTM1 was significantly enhanced in comparison to the control group (p<0.001). In addition, while BafA treatment increased the level of MAP1LC3B-II and SQSTM1 compared to control cells (p<0.01), no significant difference was observed with BafA treatment along with Ox-LDL, as compared to treatment with Ox-LDL alone ().
Figure 1. VitD3 improved Ox-LDL-impaired autophagy in murine macrophages. (A) Immunoblot analysis of MAP1LC3B-II and SQSTM1 protein levels in mBMDMs treated with Ox-LDL (50 μg/ml) for 48 h in the presence or absence of VitD3 (20 nM) for 18 h and/or BafA (200 nM) treatment for 2 h. ACTB/beta-actin (actin, beta) was used as a loading control. (B) Changes in MAP1LC3B-II and SQSTM1 protein levels are demonstrated by densitometric analysis of MAP1LC3B-II:ACTB and SQSTM1:ACTB ratios in bar histograms. (C) Treated mBMDMs were immunostained for MAP1LC3B or SQSTM1. (D) Number of MAP1LC3B and SQSTM1 puncta per cell were counted for 50 cells in each group. Images are representative of three independent experiments. Data are representative and ± standard deviation (SD) from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
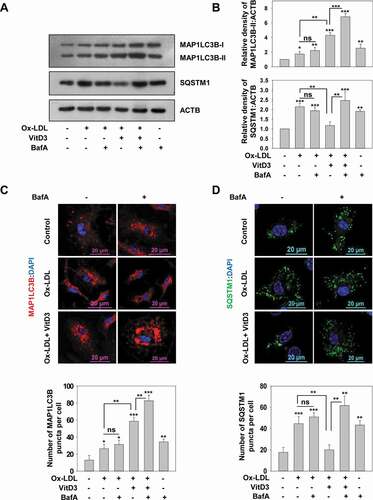
It is known that accumulation of SQSTM1 protein levels correlates with dysfunctional autophagy and is a feature of impaired autophagic flux due to toxic lipid accumulation [Citation21,Citation52,Citation53]; therefore, these results indicate that Ox-LDL cause dysfunctional autophagic flux in mBMDMs. However, treatment with VitD3 markedly increased the level of MAP1LC3B-II (p<0.01), as well as the number of MAP1LC3B puncta in Ox-LDL-stimulated macrophages (p<0.01); and decreased the accumulation of SQSTM1 (p<0.01) as compared to treatment with Ox-LDL alone ( and S2A). Moreover, treatment of BafA in Ox-LDL-stimulated macrophages in combination with VitD3 resulted in a significant increase in the levels of MAP1LC3B-II (p<0.001) and SQSTM1 (p<0.01) in comparison to Ox-LDL and VitD3 treatment. Therefore, these data suggest that VitD3 treatment of macrophages may improve the Ox-LDL-induced impairment of autophagic flux. To investigate the rescue effect of VitD3 on prolonged Ox-LDL treatment, the mBMDMs were treated with Ox-LDL for longer time (72 h) and autophagic flux was determined. VitD3 was equally efficacious in rescuing Ox-LDL-impaired autophagy at 48 h and 72 h (Fig. S1C).
VitD3 functions through VDR to regulate lipophagy in Ox-LDL-stimulated macrophages
VitD3 is a biologically active form of vitamin D and mediates its effects through transcriptional regulation via its receptor VDR [Citation29,Citation30]. We used BMDMs from wild-type (WT) and vdr KO mice to monitor the effect of VDR deficiency on VitD3 rescue of impairment in autophagy induced via Ox-LDL. VitD3 treatment significantly increased MAP1LC3B-II levels in Ox-LDL stimulated WT mBMDMs compared to control and Ox-LDL treatment alone (p<0.01). However, no such difference was found in the case of vdr KO mBMDMs. Absence of VDR led to abrogation of VitD3-induced MAP1LC3B-II levels in Ox-LDL-stimulated mBMDMs (p<0.01) ( and S2B). Furthermore, the protein levels of SQSTM1 were significantly reduced upon VitD3 treatment of Ox-LDL-stimulated WT mBMDMs compared to Ox-LDL treatment alone (p<0.001); however, this effect was abrogated in the case of vdr KO mBMDMs (p<0.01) () indicating the role of VDR signaling in VitD3 rescue of impairment in autophagy induced via Ox-LDL.
Figure 2. VDR deficiency leads to defective autophagic flux and lipophagy dysfunction in macrophages. (A) Immunoblot analysis of autophagy proteins in BMDMs from wild-type (WT) and vdr KO mice treated with Ox-LDL (50 μg/ml) for 48 h in the presence or absence of VitD3 (20 nM) for 18 h. (B) Densitometric analysis represents the fold-change after normalizing the protein band intensity to ACTB. (C) Fluorescence confocal microscopy images of Ox-LDL-stimulated BMDMs from WT and vdr KO mice treated with or without VitD3 for 18 h, and stained with Nile Red, to demonstrate neutral lipid, and anti-MAP1LC3B, to show autophagsosomes. Images are representative of three independent experiments. (D) The numbers of lipid droplets (LDs), MAP1LC3B puncta and percent colocalization of lipid with MAP1LC3B per cell were counted for 50 cells in each group. Data are representative and mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
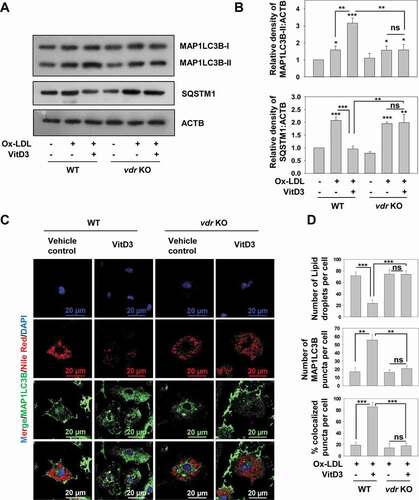
Ox-LDL is primarily stored as LDs in macrophage foam cells and can be selectively delivered to autophagosomes for its degradation via autophagy [Citation12]. Therefore, to further examine whether VitD3 and VDR can improve the breakdown of lipids inside macrophages via autophagy, autophagosomes were labeled with an anti-MAP1LC3B antibody and LDs were stained with Nile Red (a fluorescent dye generally used to stain intracellular lipids) and the colocalization of MAP1LC3B with LDs was observed by immunofluorescence. We found that cells with more MAP1LC3B puncta had fewer LDs, whereas cells with fewer MAP1LC3B puncta had more LDs (), suggesting that the breakdown of lipids was mediated via autophagy. Importantly, there was a significant increase in the percentage colocalization of MAP1LC3B with LDs in mBMDMs treated with VitD3 and Ox-LDL, compared with Ox-LDL alone (p<0.001); however, this increase was abrogated in the absence of VDR (p<0.001) (). Therefore, these data indicate that VitD3, via VDR, elevates autophagy and promotes the degradation of lipids accumulated in macrophage foam cells.
PTPN6 is essential for VitD3 to restore Ox-LDL-impaired autophagy
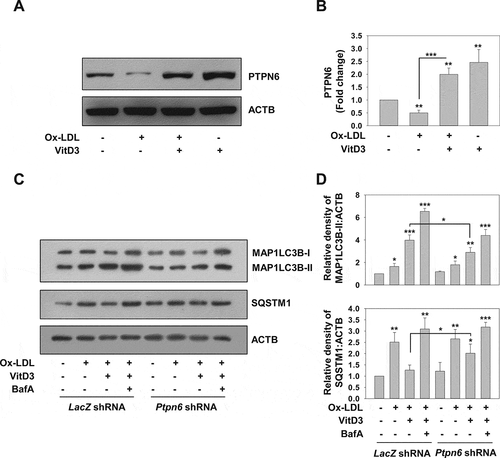
PTPN6 is a non-receptor tyrosine phosphatase that expresses at the highest level in hematopoietic cells, and plays a crucial role in the differentiation and proliferation of macrophages, T and B lymphocytes, and other erythroid cells [Citation54]. It has been shown to play a crucial role in vascular homeostasis by preventing excessive inflammation and thrombus formation [Citation55,Citation56]. PTPN6 negatively regulates superoxide formation, and decrease in the activity of PTPN6 contributes to the progression of atherosclerotic disease [Citation55-58]. Recently, it has been reported that treatment with modified lipids (Ox-LDL) suppresses PTPN6 expression in mouse vascular smooth muscle cells [Citation59]. We found that PTPN6 expression was also reduced in mBMDMs treated with Ox-LDL (p<0.01). Intriguingly, VitD3 treatment increased PTPN6 expression (p<0.001), thereby inhibiting the Ox-LDL-mediated reduction of PTPN6 (). PTPN6 has also been shown to induce autophagy by inhibiting STAT3 (signal transducer and activator of transcription 3) [Citation60,Citation61]. Therefore, we tested the role of PTPN6 in VitD3-induced autophagy using Ptpn6 shRNA in mBMDMs. mBMDMs were transduced with adenovirus expressing LacZ or Ptpn6 shRNA. We observed that knockdown of PTPN6 curtailed VitD3-induced autophagy in mBMDMs stimulated with Ox-LDL as evident by a decrease in MAP1LC3B-II (p<0.01) and increase in SQSTM1 (p<0.05) ( and S2C). Moreover, the increase in the levels of MAP1LC3B-II upon treatment of BafA in Ox-LDL-stimulated macrophages in combination with VitD3 was abrogated in PTPN6 knockdown background. The knockdown efficiency was confirmed by western blot analysis with PTPN6 antibody (Fig. S2D). These results suggest that PTPN6 is required for VitD3 to rescue Ox-LDL-impaired autophagy, and thus it plays an essential role in VitD3 mediated autophagy induction.
Figure 3. PTPN6 facilitates the VitD3-mediated restoration of Ox-LDL-impaired autophagy in macrophages. (A and B) Immunoblot for PTPN6 protein in mBMDMs treated with or without Ox-LDL for 48 h in the presence or absence of VitD3 for 18 h. (C) Immunoblot analysis of MAP1LC3B-II and SQSTM1 protein levels in control (LacZ shRNA) and PTPN6 knockdown (Ptpn6 shRNA) mBMDMs treated with or without Ox-LDL and VitD3 in the presence or absence of BafA. (D) Fold change in MAP1LC3B-II and SQSTM1 protein levels is demonstrated in bar histograms after normalizing protein band intensity to ACTB. Data are representative and mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
The role of PTPN6 in VitD3-VDR autophagy
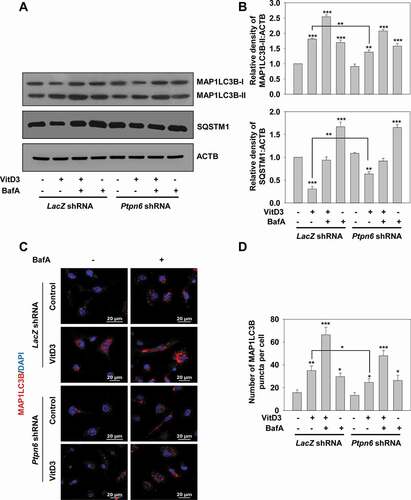
Next, we investigated in detail the role of PTPN6 in VitD3-induced autophagy using either Ptpn6 shRNA or a pharmacological inhibitor of PTPN6, sodium stibogluconate (SSG), in mBMDMs. mBMDMs transduced with LacZ or Ptpn6 shRNA were incubated in the presence and absence of VitD3 and/or BafA. MAP1LC3B-II and SQSTM1 protein levels were measured by western blotting () and quantification was represented by bar histograms (). The results suggest that PTPN6 knockdown inhibited VitD3-induced autophagy as evident by the levels of MAP1LC3B-II and SQSTM1 (p<0.01) ( and S3A). Additionally, we observed that SSG also inhibited VitD3-mediated autophagy by comparing mBMDMs cultured in the presence of VitD3 and SSG, or VitD3 alone (p<0.05) (Fig. S3B). VitD3-induced MAP1LC3B puncta formation was still observed in the presence of Ptpn6 shRNA; however, the number of MAP1LC3B puncta were decreased as compared to LacZ shRNA (p<0.05) (). The MAP1LC3B puncta were observed by confocal microscopy, quantified, and then represented in insets as MAP1LC3B puncta per cell ().
Figure 4. PTPN6 modulates VitD3-induced autophagy in macrophages. (A) Immunoblot analysis of MAP1LC3B-II and SQSTM1 protein levels in control (LacZ shRNA) and PTPN6 knockdown (Ptpn6 shRNA) mBMDMs treated with or without VitD3 and/or BafA. (B) Changes in protein levels are demonstrated by MAP1LC3B-II:ACTB and SQSTM1:ACTB ratios in bar histograms as in insets. (C) Enumeration of MAP1LC3B puncta by confocal microscopy in control and PTPN6 knockdown mBMDMs treated with or without VitD3 and/or BafA. Images are representative of three independent experiments. (D) The number of MAP1LC3B puncta were counted for 50 cells in each group and represent the MAP1LC3B puncta per cell. Data are representative and mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
VitD3-VDR induces PTPN6 expression and function
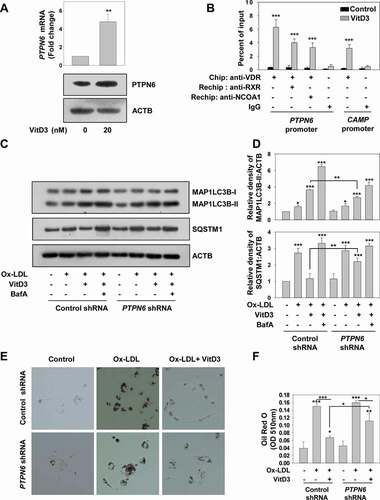
Previously, VitD3 has been shown to inhibit STAT3 phosphorylation; however, the mechanism was not identified [Citation62,Citation63]. STAT3 is a well-known target of PTPN6 and is also known to inhibit autophagy [Citation60,Citation61]. Therefore, to investigate the effect of VitD3 on PTPN6 activity, we verified the phosphorylation state of STAT3. The status of phosphorylated STAT3 (p-STAT3) and STAT3 upon VitD3 treatment was examined in mBMDMs transduced with LacZ or Ptpn6 shRNA. VitD3 significantly inhibited STAT3 phosphorylation and this inhibition was remarkably reduced in the presence of Ptpn6 shRNA indicating that VitD3 induces the dephosphorylation of STAT3 via PTPN6 (). Next, we performed the expression analysis of PTPN6/Ptpn6 in the presence of different concentrations of VitD3. Elevated expression of PTPN6/Ptpn6 was observed under VitD3 stimulation in mBMDMs, and this induction was most significant at 20 nM concentration (p<0.001) (). Additionally, we examined the time kinetics of PTPN6/Ptpn6 expression under VitD3 stimulation and we observed maximum expression at 18-24 h (p<0.001) (). To determine the possible role of VDR in VitD3-induced expression of PTPN6, we analyzed PTPN6/Ptpn6 expression in VitD3 treated WT and vdr KO mBMDMs. Treatment of WT mBMDMs with VitD3 induced the expression of PTPN6/Ptpn6 both at mRNA and protein levels (p<0.001). However, VitD3 failed to induce PTPN6/Ptpn6 expression in vdr KO mBMDMs (). Thus, VitD3-VDR plays a crucial role in modulating PTPN6 expression and function.
Figure 5. The VitD3-VDR pathway modulates PTPN6 activity and expression. (A and B) Immunoblot analysis of STAT3 and phosphorylated STAT3 (p-STAT3) levels in control (LacZ shRNA) and PTPN6 loss of function (Ptpn6 shRNA) mBMDMs treated with or without VitD3. (C and D) PTPN6/Ptpn6 expression in mBMDMs treated with different concentrations of VitD3 analyzed by qRT-PCR (C) and immunoblotting (D). (E and F) Time kinetics of PTPN6/Ptpn6 expression in mBMDMs treated with 20 nM of VitD3 analyzed by qRT-PCR (E) and immunoblotting (F). (G and H) PTPN6/Ptpn6 expression in control or vdr KO mBMDMs treated with or without VitD3 analyzed by qRT-PCR (G) and immunoblotting (H). Data are representative and mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
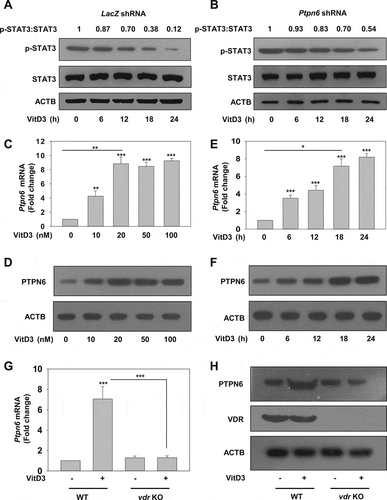
Ptpn6 is a direct target gene of VDR
To investigate whether VDR modulates PTPN6 expression by genomic mechanism, we performed a bioinformatics search to identify if there is a putative vitamin D response element (VDRE) on the Ptpn6 promoter. Interestingly, we identified a potential VDRE on the Ptpn6 promoter at position −357, which suggests possible direct gene regulation of Ptpn6 via VDR (). Chromatin immunoprecipitation (ChIP) was performed in mBMDM cells treated with or without VitD3. Immunoprecipitation was performed using VDR and RXRA (retinoid X receptor alpha) antibodies. We identified that VDR-RXRA heterodimeric complex was recruited to the Ptpn6 promoter (p<0.001) (). We also identified NCOA1 (nuclear receptor coactivator 1) as a coactivator binding partner to this transcriptional complex by re-ChIP experiments (). Atg16l1 (autophagy related 16 like 1) was used as a positive control for ChIP experiments [Citation64]. To check the binding of VDR and RXRA to the putative VDRE on the Ptpn6 promoter in-vitro, EMSA was performed. The VDR-RXRA heterodimer formed a strong complex with the oligonucleotides pertaining to the VDRE of the Ptpn6 promoter (). Osteopontin oligonucleotides were used as a positive control for EMSA experiments. Moreover, mutant Ptpn6 oligonucleotide VDRE sequence and cold competition assays were performed to further define the specificity of VDR-RXRA binding to the putative response element of Ptpn6 promoter (). Additionally, we also performed a luciferase reporter assay in COS1 cells, which further confirmed the positive regulation of PTPN6 expression by VitD3 (). The above experimental evidence and the underlying mechanism of VDR-mediated induction of Ptpn6 expression are depicted in .
Figure 6. VitD3 regulates the expression of PTPN6 by a genomic mechanism. (A) Pictorial representation of a VDRE on the mouse Ptpn6 promoter. (B) ChIP assays were performed with mBMDM cells cultured in the presence or absence of VitD3. Immunoprecipitation was performed with antibodies to VDR, RXRA, and NCOA1. Eluted DNA after the immunoprecipitation was amplified using Atg16l1 and Ptpn6 promoter-specific primers and as a control, Gapdh promoter-specific primers were used. VDR binding to the Ptpn6 promoter was analyzed by qRT-PCR and represented as percent of input. (C) EMSA was performed by incubating end-labeled Ptpn6, and end labeled osteopontin oligonucleotides, with in vitro translated VDR and RXRA proteins, and complex formation was observed. (D) EMSA was performed with WT and mutant probe. (E) Competition experiments were carried out in the absence or presence of 5-, 10-, and 50- fold excess of the cold (unlabeled) putative Ptpn6 oligonucleotide. (F) Luciferase assays in COS-1 cells co-transfected with pFLAG-Vdr, pFLAG-Rxra, and pGL3-Ptpn6 promoter plasmids, cultured in the absence or presence of 20 nM VitD3. (G) Schematic representation of the proposed mechanism underlying the modulation of PTPN6 expression by VitD3. VitD3 activated VDR forms a complex with RXRA. This VDR-RXRA complex then binds to the positive VDRE on the mouse Ptpn6 gene promoter, and recruitment of the co-activator NCOA1 results in upregulation of Ptpn6 gene expression. Data are representative and mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 are considered significant over control
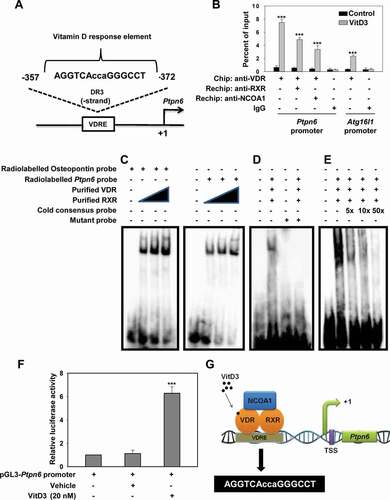
VitD3 modulates the expression of autophagy-related genes through PTPN6
It has previously been reported that VitD3 modulates autophagy through the induction of two core autophagy proteins, BECN1 and ATG5 [Citation42]. Therefore, we examined whether VitD3-PTPN6 modulated BECN1 or ATG5 in LacZ or Ptpn6 shRNA-transduced mBMDMs. Our qRT-PCR analysis clearly revealed that VitD3-induced expression of Becn1 and Atg5 (p<0.001) was significantly inhibited by PTPN6 knockdown (Becn1 (p<0.001) and Atg5 (p<0.01)) (). This VitD3-mediated induction was previously attributed to the MAPK1/ERK2 (mitogen-activated protein kinase 1) kinases and CEBPB/C/EBPβ (CCAAT enhancer binding protein beta) [Citation42]. Hence, we studied the expression, as well as the activation status, of CEBPB and MAPK1 (), in VitD3 treated mBMDMs (control or PTPN6 knockdown). It was observed that VitD3 treatment induced the expression of CEBPB in mBMDMs but PTPN6 knockdown had no effect on VitD3-induced CEBPB expression. However, a decrease in phosphorylated CEBPB (p-CEBPB) levels was observed in PTPN6 knockdown cells treated with VitD3 compared to control cells (p<0.001) (). Furthermore, we also monitored the levels of MAPK1 and phosphorylated MAPK1 (p-MAPK1) in control and PTPN6 knockdown mBMDMs treated with or without VitD3. We observed an increase in p-MAPK1 upon treatment with VitD3 (p<0.01) and this increase in p-MAPK1 was abrogated in PTPN6 knockdown cells (p<0.001) (). These results lend support to the previously reported role for PTPN6 in the regulation of MAPK1 activation, which contributes to CEBPB activity [Citation65].
Figure 7. PTPN6 is required for VitD3-mediated expression of autophagy-related genes. (A and B) qRT-PCR analysis of autophagy-related genes (Becn1 and Atg5) in control (LacZ shRNA) and PTPN6 knockdown (Ptpn6 shRNA) mBMDMs treated with or without VitD3. (C and E) Immunoblot analysis of CEBPB, p-CEBPB (C) and MAPK1, p-MAPK1 (E) in control and PTPN6 knockdown mBMDMs treated with or without VitD3. (D and F) p-CEBPB:CEBPB (D) and p-MAPK1:MAPK1 (F) blots were quantified and represented as bar histograms. Data are representative and mean ± SD from three independent experiments.*p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
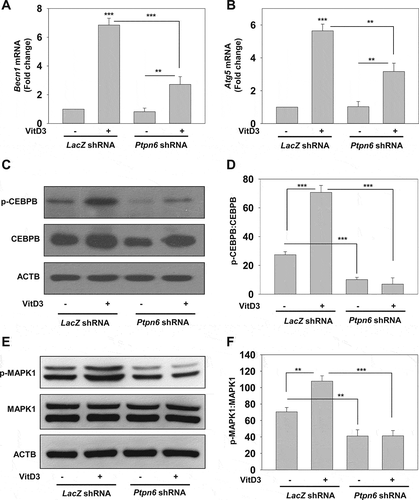
VitD3-mediated autophagy reduces lipid accumulation and macrophage conversion into foam cells through PTPN6
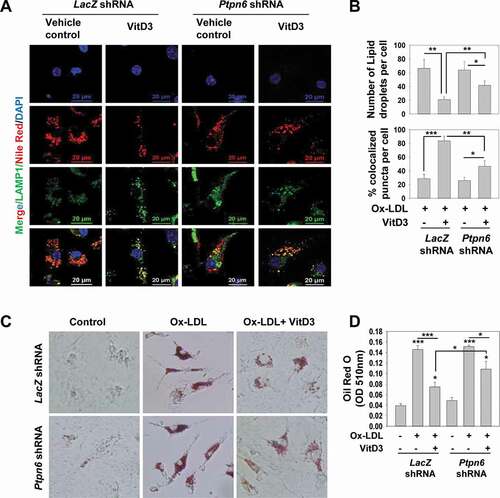
Accumulated lipids stored in the form of LDs in macrophages are degraded by lysosomes via autophagy, which is seriously impaired in advanced AS with macrophages being transformed into foam cells [Citation12,Citation18,Citation19]. To investigate the influence of VitD3 and PTPN6 on autolysosome fusion, Ox-LDL stimulated macrophages were fluorescently labeled with Nile Red to stain LDs, as well as with LAMP1 (lysosomal associated membrane protein 1) antibody. The colocalization of LAMP1 with LDs was determined by confocal microscopy; VitD3 treatment reduced the number of LDs (p<0.01) and increased the percentage colocalization of LAMP1 (p<0.001) with LDs compared to the vehicle control group (). There was a significant decrease in VitD3-mediated colocalization of LDs with LAMP1 in PTPN6 knockdown mBMDMs (p<0.01) (), suggesting a role of PTPN6 in VitD3-mediated lipid breakdown via autophagy. We further analyzed whether PTPN6 activation by VitD3 inhibits foam cell formation in mBMDMs. VitD3 treatment led to decrease in foam cell formation in Ox-LDL stimulated macrophages (p<0.001) (). However, knockdown of PTPN6 significantly curtailed this decrease in foam cell formation (p<0.05) (). Taken together, our data have shown that VitD3 inhibited the conversion of macrophages into foam cells through PTPN6 in an autophagy dependent manner ().
Figure 8. PTPN6 is required for VitD3-mediated inhibition of macrophages foam cell formation by modulating lipophagy. (A) Immunostained microscopy images of Ox-LDL stimulated control (LacZ shRNA) and PTPN6 knockdown (Ptpn6 shRNA) mBMDMs, treated with or without VitD3 for 18 h, and stained with Nile Red and anti-LAMP1. (B) The numbers of LDs and percentage colocalization of lipid with LAMP1 per cell were counted for 50 cells in each group. (C) Comparison of in vitro foam cells formation in control and PTPN6 knockdown mBMDMs stimulated with Ox-LDL and treated with or without VitD3 for 18 h, and stained with Oil Red O solution (D) Quantification of ORO staining. Images are representative of three independent experiments. Data are representative and mean ± SD from three independent experiments.*p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
VitD3-VDR-PTPN6 axis rescues Ox-LDL-impaired autophagic flux in human macrophages
To investigate whether VitD3-VDR modulates the expression of PTPN6 in human macrophages, hMDMs were treated with VitD3 and PTPN6/PTPN6 expression was checked by qRT-PCR and western blot. Upon VitD3 treatment, there was increased expression of PTPN6 both at mRNA (p<0.01) and protein level (). We then looked for the putative vitamin D response element (VDRE) on the human PTPN6 promoter, and identified a potential VDRE on the PTPN6 promoter at position −769 (Fig. S4A). Therefore, to confirm the direct gene regulation, hMDMs were treated with or without VitD3 and ChIP was performed. We observed the recruitment of VDR-RXRA to the human PTPN6 promoter (p<0.001) (). Re-ChIP experiment confirmed the binding of coactivator NCOA1 to transcriptional complex on the human PTPN6 promoter. CAMP/Cathelicidin (cathelicidin antimicrobial peptide) was used as positive control. Furthermore, we elucidated whether PTPN6 is essential for VitD3 to rescue Ox-LDL-impaired autophagy in control or PTPN6 shRNA transduced hMDMs. We observed that in hMDMs stimulated with Ox-LDL, VitD3-induced autophagy was inhibited in PTPN6 knockdown background (p<0.01) ( and S4B). PTPN6 knockdown efficiency was validated by western blot (Fig. S4C). To further explore the role of PTPN6 in VitD3-VDR-induced autophagy, number of MAP1LC3B puncta were calculated in the presence or absence of PTPN6 shRNA. As expected, the number of VitD3-induced MAP1LC3B puncta were significantly decreased in cells incubated with PTPN6 shRNA as compared to cells treated with control shRNA (p<0.05) (Fig. S4D,E). We also examined whether VitD3-PTPN6 modulated autophagy related genes in hMDMs and found that VitD3-induced expression of BECN1 and ATG5 was abrogated in PTPN6 knockdown conditions (Fig. S4F). Next, we analyzed the effect of VitD3-induced PTPN6 on foam cell formation. Expectedly, VitD3 treatment led to decrease in foam cell formation in Ox-LDL-stimulated macrophages (p<0.001). However, this decrease was curtailed in PTPN6 knockdown condition (p<0.05) (). Thus, VitD3-VDR-PTPN6 axis recues Ox-LDL-impaired autophagy, which inhibits foam cell formation in human macrophages as well.
Figure 9. VitD3-VDR modulates PTPN6 expression and rescues Ox-LDL-impaired autophagy in hMDMs. (A) qRT-PCR and immunoblot analysis of PTPN6/PTPN6 in presence or absence of VitD3 in hMDMs. (B) ChIP assay with hMDMs cultured in the presence or absence of VitD3. (C) Immunoblot analysis of MAP1LC3B-II and SQSTM1 protein levels in control (control shRNA) and PTPN6 knockdown (PTPN6 shRNA) hMDMs treated with or without Ox-LDL and VitD3 in the presence or absence of BafA. (D) Densitometric analysis represents the fold-change after normalizing the protein band intensity to ACTB. (E) Foam cell formation in control and PTPN6 knockdown hMDMs stimulated with Ox-LDL and treated with or without VitD3 for 18 h, and stained with ORO solution. Images are representative of three independent experiments. (F) Quantification of ORO staining. Data are representative and mean ± SD from three independent experiments.*p < 0.05, **p < 0.01, ***p < 0.001 compared to control or as indicated
Discussion
Lipophagy, a specific type of autophagy, has critical roles in lipid metabolism and in the pathogenesis of AS [Citation12,Citation18,Citation21,Citation53]. In this study, we have provided evidence supporting the protective role of VitD3-mediated autophagy in AS. By determining the level of the autophagy marker MAP1LC3B-II and chaperone SQSTM1 [Citation51], we have shown that VitD3 recovers the Ox-LDL-impaired autophagic flux in both mBMDMs and hMDMs (). VitD3 functions mainly through its receptor VDR [Citation29,Citation30,Citation66], and the absence of VDR led to attenuation of macrophage lipophagy and resulted in the accumulation of lipids inside the macrophages ().
VitD3 has been shown to modulate the activity of many serine-threonine and tyrosine kinases [Citation67-70]. PTPN6 is a non-receptor tyrosine phosphatase and its significance in the phagolysosomal fusion and localization on phagolysosomes has been characterized [Citation71]. Recently, PTPN6 has been shown to induce autophagy by inhibiting the phosphorylation of STAT3 [Citation60,Citation61]. In this study, we have found a novel signaling pathway where VitD3-VDR induces autophagy by modulating the protein tyrosine phosphatase PTPN6. We have provided evidence that PTPN6 positively modulates VitD3-mediated autophagy in Ox-LDL-stimulated macrophages (). VitD3-mediated autophagy was significantly reduced upon inhibition of PTPN6 either through knockdown or treatment with SSG (, S3B and S4D,E). Moreover, the expression and function of PTPN6 was found to be induced by VitD3 (). Further, we have also identified a putative VDRE on the PTPN6/Ptpn6 gene promoter and demonstrated that VDR, its coactivator NCOA1, and its heterodimeric partner, RXRA, are recruited to the PTPN6/Ptpn6 promoter in a VitD3-dependent manner ( and S4A). Our results also demonstrated that the VitD3-VDR-PTPN6 pathway induced autophagy by elevating the expression of autophagy-related genes such as BECN1/Becn1 and ATG5/Atg5 in macrophages ( and S4F).
Vitamin D deficiency is associated with increased incidence of CVD, thereby suggesting its atheroprotective role [Citation28,Citation33,Citation35]. Several reports have shown that vitamin D promotes autophagy and also inhibits foam cell formation in macrophages [Citation35,Citation37,Citation40,Citation42,Citation72-75]. PTPN6 function in AS and autophagy has only been reported recently [Citation55,Citation56,Citation60,Citation61]. This study provides evidence that PTPN6 is crucial for VitD3-mediated decrease in LDs, and their colocalization with autolysosomes indicates that PTPN6 can induce macrophage lipophagy (). Accumulation of LDs inside macrophages leads to foam cell formation which is a hallmark feature of atherosclerotic lesions. PTPN6 also contributed to VitD3-mediated inhibition of foam cell formation in both mBMDMs and hMDMs (). Hence, this study reports a novel pathway of VitD3-VDR-PTPN6 in macrophages, which inhibited the conversion of macrophage to foam cells through modulation of the cellular autophagy process ().
Figure 10. A schematic representation of a novel signaling pathway VitD3-VDR-PTPN6 leading to autophagy induction and inhibition of macrophage foam cell formation. Pictorial representation depicts the recruitment of VDR-RXRA complex along with NCOA1 to the positive VDRE on the Ptpn6 promoter, leading to the increase in the expression of PTPN6. Upon induction, PTPN6 modulates the cellular autophagy process by increasing the expression of two core autophagy proteins: BECN1 and ATG5, through activation of MAPK1 and CEBPB. Additionally, PTPN6 also inhibits the activation of STAT3, which is a negative regulator of autophagy by its tyrosine phosphatase activity. This induction in autophagy subsequently leads to degradation of accumulated lipids inside the macrophages
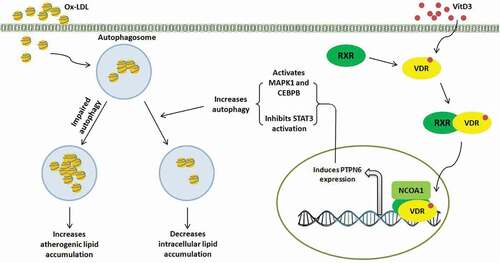
Vitamin D is reported to activate or inhibit several protein kinases, thereby, modulates the phosphorylation status within the cells, and thus the cellular function [Citation67,Citation68,Citation70]. The non-receptor tyrosine phosphatase PTPN6 has been shown to play a pivotal role in immune cells where it functions downstream to many of the cell surface receptors and transmit signals by dephosphorylating its substrates [Citation76,Citation77]. In the process of investigating signaling pathways downstream of VitD3, we have identified a very interesting association between VitD3 and PTPN6. VitD3 treatment led to dephosphorylation of STAT3 via PTPN6 in macrophages (). Dephosphorylation of STAT3 is known to inhibit dimer formation, nuclear localization and transcriptional repression of autophagy genes [Citation78]. VitD3 via PTPN6 also activated MAPK1 and CEBPB which are known to modulate autophagy related genes [Citation42]. Hence, VitD3-VDR-PTPN6 axis elevated the expression of autophagy related genes such as BECN1/Becn1 and ATG5/Atg5 in macrophages ( and S4F). VitD3-induced PTPN6 inhibited STAT3 phosphorylation and elevated the expression of Becn1 and Atg5 by activating MAPK1 and CEBPB, thereby regulating autophagy in atherosclerotic macrophages. This novel signaling pathway partially explains the ability of VitD3 to induce autophagy and inhibit macrophage foam cell formation through PTPN6.
Our study demonstrates a novel role of PTPN6 in VitD3-mediated suppression of macrophage foam cell formation. Furthermore, we have shown that PTPN6 plays a decisive role in AS by promoting VitD3-mediated autophagy in both human and mice macrophages. Thus, our study has provided a new pathway which will significantly aid in the research for AS and would provide novel targets for pharmaceutical intervention.
Materials and methods
Cell culture and plasmids
COS1 cells were procured from the National Centre for Cell Science, India, and were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Invitrogen, 10566-016) supplemented with 1% penicillin streptomycin (Gibco, Invitrogen, 15140-122), and 10% fetal bovine serum (FBS; Gibco, Invitrogen, 26140079). mBMDMs were cultured, as previously described [Citation49]. Briefly, bone marrow precursors were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Invitrogen, 11875), 1% penicillin-streptomycin, 10% newborn calf serum (NBCS; Gibco, Invitrogen, 16010159) and 50 ng/ml CSF2/GM-CSF (colony stimulating factor 2; eBioscience, 14-8331-80). PBMCs were isolated from fresh blood of healthy individuals using a Ficoll-Paque (Sigma-Aldrich, GE17-5442-03) gradient centrifugation (400 × g, 30 min, 20ºC). After 2-3 h of incubation at 37ºC, cells were washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4) to remove non-adherent cells. Cells were grown in RPMI supplemented with 10% FBS and 50 ng/ml CSF2/GM-CSF (eBioscience, BMS324) at 37ºC and 5% CO2 for 7 d. Mouse Rxra (8861822, GeneBankBC138800) and Vdr (3710866, GeneBankBC006716) cDNA were procured from Open Biosystems and sub-cloned into the mammalian expression vector pFLAG (Sigma-Aldrich, E7033). The mouse Ptpn6 promoter was cloned into the pGL3 vector, which was procured from Promega (E1751).
Human ethics statement
The project was approved by the Ethics Committee of the Government Medical College and Hospital, Sector 32 (Chandigarh, India), and the Ethics and Biosafety Committee of IMTECH, Sector 39A (Chandigarh, India). The study was conducted strictly in accordance with the Ethical Guidelines for Biomedical Research on Human Subjects by the Central Ethics Committee on Human Research, Indian Council of Medical Research-2000 and those as contained in the Declaration of Helsinki. Each subject was provided with written information about the study, and written consent on the consent form was obtained from each healthy volunteer before his or her induction in the study in the language (English, Hindi, and Punjabi) familiar to them.
Mice and animal ethics statement
vdr KO (B6.129S4-vdr1tm1Mbd/J) mice were procured from the Jackson Laboratory (006133; Heterozygous males and females were bred to generate WT and KO mice for the experiment). C57BL/6 mice (Charles River Laboratories, 027) were procured from the animal facility of the Institute of Microbial Technology (IMTECH). All animals were housed in the pathogen-free animal facility at IMTECH. All animal experiments were approved by the Institutional Animal Ethics Committee at the IMTECH and were performed as per the guidelines issued by the Committee for the Purpose of Supervision of Experiments on Animals (No.55/1999/CPCSEA), Ministry of Environment and Forest, Government of India.
Gene silencing, Adenovirus production and infection
For the loss of gene function experiments in macrophages, adenovirus-expressing short hairpin RNA (shRNA) for LacZ (LacZ shRNA) and mouse Ptpn6 (Ptpn6 shRNA) were used. Adenovirus-expressing shRNAs were generated using pAd/BLOCK-iT-DEST (Invitrogen, V49220) according to the manufacturer’s instructions. For transduction, macrophages were incubated with adenovirus in RPMI 1640 medium supplemented with 1% penicillin-streptomycin and 10% NBCS for 24 h, followed by the replacement of culture supernatant. Cells were further incubated for an additional 24 h before the experimental assays. Control shRNA and human PTPN6 shRNA were purchased from Sigma-Aldrich (SHC002V, SHCLNV). The silencing efficiency was monitored by qRT-PCR as well as immunoblot analysis.
Chromatin immunoprecipitation (ChIP)
ChIP was carried out using the protocol described previously [Citation67]. In brief, 1×107 cells were fixed in 1% (w:v) formaldehyde and the reaction was stopped by adding 150 mM glycine (Sigma-Aldrich, 50046). The cells were then washed with ice-cold 1X PBS and lysis was performed in sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0) followed by sonication to obtain DNA fragments of suitable length. Immunoprecipitation was carried out with ChIP grade VDR (Santa Cruz Biotechnology, sc-13133), RXRA (Santa Cruz Biotechnology, sc-553), and NCOA1 (Santa Cruz Biotechnology, sc-6096) antibodies. The immune complexes were collected by centrifugation and sequentially washed with a set of buffers. The immunoprecipitated DNA was eluted in ChIP elution buffer (0.1% SDS, 0.1 M NaHCO3) and proceeded for purification by ethanol precipitation. The purified DNA was used for amplification with Ptpn6 promoter-specific primers. The primers used in the ChIP assay were mouse Ptpn6 promoters forward 5ʹ- TGGCAAGCAGAATTCAAAAG’ and reverse 5ʹ-GTCATTTAATGGAGTGTCGCA-3ʹ, mouse Atg16l1 promoters forward 5ʹ-GGTTCCGTTCTTGTTTCT-3ʹ and reverse 5ʹ-TCAAGTTGTCTCCAAGATTAT-3ʹ, human CAMP promoters forward 5ʹ-ACCGTGCCCTGCCTCATTC-3ʹ and reverse 5ʹ-TGGTCCCCATGTCTGCCTC-3ʹ and human PTPN6 promoters forward 5ʹ-AGATCAACCAGCCCTGAGAAA-3ʹand reverse 5ʹ-CTTGCCAATGCTTGCAAGAGA-3ʹ.
Confocal microscopy
For imaging experiments, macrophages were cultured on cover slips and subjected to relevant treatments. At the end of the treatments, cells were fixed in 4% paraformaldehyde and processed for immunostaining. To enumerate the MAP1LC3B and SQSTM1 puncta, cells were incubated overnight at 4ºC with rabbit anti-MAP1LC3B (Sigma-Aldrich, L8918) and anti-SQSTM1 primary antibodies (MBL International, PM045), and washed three times with 1X PBS followed by incubation with a secondary goat anti-rabbit antibody conjugated with Texas Red (TR; Santa Cruz Biotechnology, sc-2780) or fluorescein isothiocyanate (FITC; Santa Cruz Biotechnology, sc-2012). For colocalization studies, intracellular lipids were stained with Nile Red (3.3 mg/ml; Molecular Probes, N1142) for 15 min. MAP1LC3B was stained using rabbit anti-MAP1LC3B and goat anti-rabbit secondary antibody conjugated with Alexa Flour 633 (Invitrogen, A-21072); LAMP1 (lysosomal associated membrane protein 1) was stained using mouse anti-LAMP1 (Santa Cruz Biotechnology, sc-17768) followed by anti-mouse secondary antibody conjugated with Alexa Flour 633 (Invitrogen, A-21063). 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, D8417) was used to stain the nuclei. The percentage colocalization of MAP1LC3B or LAMP1 puncta with Nile Red puncta was calculated. Images were acquired using a Nikon AIR confocal microscope (Nikon, Yokohama, Japan) at the CSIR-IMTECH, India.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed with 5′-end labeled oligonucleotides, using T4-polynucleotide kinase (New England Biolabs, M0201L) and [γ-32P]-ATP. Recombinant mouse VDR and RXRA proteins were obtained by in vitro transcription and translation of pFLAG-Vdr and pFLAG-Rxra plasmids using the 1-Step Human Coupled IVT Kit (Thermo Scientific, 88882) according to the manufacturer’s instructions. DNA binding to recombinant proteins was determined as described previously [Citation79]. For competition experiments, 5-, 10-, and 50- fold excess of the unlabeled cold probe was added to compete out the DNA binding. The oligonucleotide sequence corresponding to the site −357 to −372 on Ptpn6 promoter (5ʹ-TGAGGAATGAGGCCCTGGTGACCTGTAACTCA-3ʹ and 5ʹ-TGAGTTACAGGTCAC CAGGGCCTCATTCCTCA-3ʹ) were used as test. Mouse osteopontin oligonucleotides (5ʹ-GATCCACAAGGTTCACGAGGTTCACGTCTG-3ʹ and 5ʹ-CAGACGTGAACCTCGTGAACCTTGTGGATC-3ʹ) were used as positive control. Mutant Ptpn6 oligonucleotides (5ʹ-TGAGGAATGAGGCCCTGGTGATTTGTAACTCA-3ʹ and 5ʹ-TGAGTTACAAATCACCAGGGCCTCATTCCTCA-3ʹ) were generated whereby the mutations were done in the core motif.
Immunoblot analysis
For immunoblot analysis, cells were subjected to lysis using mammalian cell lysis buffer (50 mM NaCl, 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% Triton X-100 [Sigma-Aldrich, T8787], 1× protease inhibitor cocktail [Merck Millipore, 539134], 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich, P7626]) for 15 min on ice, and protein concentrations in the samples were quantified by Bradford reagent (Sigma-Aldrich, B6916). Furthermore, 12% SDS-polyacrylamide gel electrophoresis was used to resolve the protein followed by transfer to polyvinylidene difluoride membrane (Immobilon-P; Millipore, IPVH00010). Membranes were blocked with 5% bovine serum albumin (Merck Millipore, 12659) and incubated overnight at 4ºC with primary antibodies. The following primary antibodies were used: ACTB/β-actin (sc-47778), VDR (sc-9164), PTPN6 (sc-7289), p-STAT3 (sc-8059), STAT3 (sc-483), CEBPB (sc-150), MAPK1 (sc-135900), p-MAPK1 (sc-7383) (all from Santa Cruz Biotechnology), MAP1LC3B (Sigma-Aldrich, L8918) and SQSTM1 (MBL International, PM045). Membranes were then washed three times with Tris-HCl-buffered saline (TBS; 20 mM Tris-HCl, pH 7.4, 150 mM NaCl) with 0.1% Tween 20 (Sigma-Aldrich, P9416; TBST, pH 7.4), followed by incubation with appropriate HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, sc-2313, sc-2314) for 60 min at room temperature and visualized by chemiluminescent HRP substrate Luminata Forte (Millipore, WBLUF0500).
Luciferase assay
COS1 cells were grown in 24-well dishes and allowed to proliferate to 70% confluency in DMEM supplemented with 10% FBS. Cells were then transiently transfected with pFLAG-Vdr, and pFLAG-Rxra plasmids along with the pGL3-Ptpn6 promoter plasmid using turbofect transfection reagent (Thermo Scientific, R0531). pBIND vector (Promega, E245A) was used as a transfection control. Dual luciferase activity was monitored, as per the manufacturer’s instructions (Promega, E1960) using a GloMax 20/20 luminometer (Promega, USA). Normalized luciferase activities (relative light units) were plotted as an average of triplicate samples.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from macrophages using the TRIzol reagent (Ambion, Invitrogen, 15596026) of which 1 µg RNA was reverse transcribed with the Verso cDNA Synthesis Kit (Thermo Scientific, AB-1453/B) as per the manufacturer instructions. The cDNA was then subjected to SYBR green (Thermo Scientific, F-416L)-based qRT-PCR with gene-specific primers and Rn18s rRNA as a control. The relative abundance of the genes was calculated by using the formula 2−ΔΔCt.
Oil red O (ORO) staining
Cells were fixed with 10% formalin for 1 h, rinsed with 60% isopropanol, and stained with freshly filtered 0.5% ORO for 30 min at RT. Cells were then rinsed with 60% isopropanol and washed with PBS. The cells were then mounted on glass slides, observed, and imaged by Olympus IX71 inverted microscope (Olympus, Waltham, USA). To quantify lipid accumulation, ORO (Sigma-Aldrich, O0625) stain was eluted with 100% isopropanol and the optical density (OD) of the elutes was measured at 520 nm using a spectrophotometer.
Statistical analysis
Results are expressed as the mean ± standard deviation (SD) unless otherwise mentioned. Sigma Plot was used for statistical analysis. Two tailed student’s t-tests were performed to obtain p-values. Statistical significance was established at *p < 0.05, **p < 0.01, ***p < 0.001.
Disclosures statement
The authors have no financial conflicts of interest.
Supplemental Material
Download MS Word (5 MB)Acknowledgments
We thank IMTECH (a constituent laboratory of the Council of Scientific and Industrial Research) for facilities and financial support.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011 Nov 7;17(11):1410–1422.
- Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013 Oct;13(10):709–721.
- Lusis AJ. Atherosclerosis. Nature. 2000 Sep 14;407(6801):233–241.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005 Apr 21;352(16):1685–1695.
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006 Jul;6(7):508–519.
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001 Feb 23;104(4):503–516.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014 Jan;24(1):24–41. DOI:https://doi.org/10.1038/cr.2013.168.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008 Feb 28;451(7182):1069–1075.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737.
- Chandra V, Bhagyaraj E, Parkesh R, et al. Transcription factors and cognate signalling cascades in the regulation of autophagy. Biol Rev Camb Philos Soc. 2016 May;91(2):429–451.
- Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. 2012 Nov 13;109(46):E3168–76.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009 Apr 30;458(7242):1131–1135.
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018 Mar;20(3):233–242.
- Chen K, Yuan R, Zhang Y, et al. Tollip Deficiency Alters Atherosclerosis and Steatosis by Disrupting Lipophagy. J Am Heart Assoc. 2017 Apr 10;6(4). DOI:https://doi.org/10.1161/JAHA.116.004078.
- Ouimet M, Franklin V, Mak E, et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011 Jun 8;13(6):655–667.
- Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013 Jan;20(1):3–11.
- Yang M, Zhang Y, Ren J. Autophagic Regulation of Lipid Homeostasis in Cardiometabolic Syndrome. Front Cardiovasc Med. 2018;5:38.
- Sergin I, Razani B. Self-eating in the plaque: what macrophage autophagy reveals about atherosclerosis. Trends Endocrinol Metab. 2014 May;25(5):225–234.
- Emanuel R, Sergin I, Bhattacharya S, et al. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol. 2014 Sep;34(9):1942–1952. DOI:https://doi.org/10.1161/ATVBAHA.114.303342.
- Liao X, Sluimer JC, Wang Y, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012 Apr 4;15(4):545–553.
- Razani B, Feng C, Coleman T, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012 Apr 4;15(4):534–544.
- Martinet W, Verheye S, De Meyer GR. Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy. 2007 May-Jun;3(3):241–244.
- Verheye S, Martinet W, Kockx MM, et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol. 2007 Feb 13;49(6):706–715.
- Sun RZ, Fan Y, Liang X, et al. Rapamycin and FTY720 Alleviate Atherosclerosis by Cross Talk of Macrophage Polarization and Autophagy. Biomed Res Int. 2018;2018:1010248.
- Liu Y, Yang F, Zou S, et al. Rapamycin: A Bacteria-Derived Immunosuppressant That Has Anti-atherosclerotic Effects and Its Clinical Application. Front Pharmacol. 2018;9:1520.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007 Jul 19;357(3):266–281.
- Roth DE, Abrams SA, Aloia J, et al. Global prevalence and disease burden of vitamin D deficiency: a roadmap for action in low- and middle-income countries. Ann N Y Acad Sci. 2018 Oct;1430(1):44–79. DOI:https://doi.org/10.1111/nyas.13968.
- Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res. 2014 Jan 17;114(2):379–393.
- Pike JW, Meyer MB, Lee SM, et al. The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J Clin Invest. 2017 Apr 3;127(4):1146–1154.
- Demay MB. Mechanism of vitamin D receptor action. Ann N Y Acad Sci. 2006 Apr;1068:204–213.
- Gardner DG, Chen S, Glenn DJ. Vitamin D and the heart. Am J Physiol Regul Integr Comp Physiol. 2013 Nov 1;305(9):R969–77.
- Kassi E, Adamopoulos C, Basdra EK, et al. Role of vitamin D in atherosclerosis. Circulation. 2013 Dec 3;128(23):2517–2531.
- Yin K, You Y, Swier V, et al. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler Thromb Vasc Biol. 2015 Nov;35(11):2432–2442. DOI:https://doi.org/10.1161/ATVBAHA.115.306132.
- Bozic M, Alvarez A, de Pablo C, et al. Impaired Vitamin D Signaling in Endothelial Cell Leads to an Enhanced Leukocyte-Endothelium Interplay: Implications for Atherosclerosis Development. PLoS One. 2015;10(8):e0136863. DOI:https://doi.org/10.1371/journal.pone.0136863.
- Oh J, Weng S, Felton SK, et al. 1,25(OH)2 vitamin d inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation. 2009 Aug 25;120(8):687–698.
- Oh J, Riek AE, Darwech I, et al. Deletion of macrophage Vitamin D receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell Rep. 2015 Mar 24;10(11):1872–1886.
- Szeto FL, Reardon CA, Yoon D, et al. Vitamin D receptor signaling inhibits atherosclerosis in mice. Mol Endocrinol. 2012 Jul;26(7):1091–1101. DOI:https://doi.org/10.1210/me.2011-1329.
- Dwivedi PP, Omdahl JL, Kola I, et al. Regulation of rat cytochrome P450C24 (CYP24) gene expression. Evidence for functional cooperation of Ras-activated Ets transcription factors with the vitamin D receptor in 1,25-dihydroxyvitamin D(3)-mediated induction. J Biol Chem. 2000 Jan 7;275(1):47–55.
- Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005 Jul;19(9):1067–1077.
- Wang TT, Nestel FP, Bourdeau V, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004 Sep 1;173(5):2909–2912.
- Kato S. The function of vitamin D receptor in vitamin D action. J Biochem. 2000 May;127(5):717–722.
- Yuk JM, Shin DM, Lee HM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009 Sep 17;6(3):231–243.
- Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8(5):e1002689.
- Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015 Jul;64(7):1082–1094. DOI:https://doi.org/10.1136/gutjnl-2014-307436.
- Das LM, Binko AM, Traylor ZP, et al. Vitamin D improves sunburns by increasing autophagy in M2 macrophages. Autophagy. 2019 May;15(5):813–826. DOI:https://doi.org/10.1080/15548627.2019.1569298.
- Hoyer-Hansen M, Nordbrandt SP, Jaattela M. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol Med. 2010 Jul;16(7):295–302.
- Chandra V, Bhagyaraj E, Nanduri R, et al. NR1D1 ameliorates Mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy. 2015 Nov 2;11(11):1987–1997.
- Dkhar HK, Nanduri R, Mahajan S, et al. Mycobacterium tuberculosis keto-mycolic acid and macrophage nuclear receptor TR4 modulate foamy biogenesis in granulomas: a case of a heterologous and noncanonical ligand-receptor pair. J Immunol. 2014 Jul 1;193(1):295–305.
- Mahajan S, Saini A, Chandra V, et al. Nuclear receptor Nr4a2 promotes alternative polarization of macrophages and confers protection in sepsis. J Biol Chem. 2015 Jul 24;290(30):18304–18314.
- Nanduri R, Kalra R, Bhagyaraj E, et al. AutophagySMDB: a curated database of small molecules that modulate protein targets regulating autophagy. Autophagy. 2019 Jul;15(7):1280–1295. DOI:https://doi.org/10.1080/15548627.2019.1571717.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
- Sergin I, Bhattacharya S, Emanuel R, et al. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal. 2016 Jan 5;9(409):ra2.
- Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017 Jun 7;8:15750. DOI:https://doi.org/10.1038/ncomms15750.
- Pao LI, Badour K, Siminovitch KA, et al. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol. 2007;25:473–523.
- Koch E, Pircher J, Czermak T, et al. The endothelial tyrosine phosphatase SHP-1 plays an important role for vascular haemostasis in TNFalpha -induced inflammation in vivo. Mediators Inflamm. 2013;2013:279781.
- Krotz F, Engelbrecht B, Buerkle MA, et al. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. J Am Coll Cardiol. 2005 May 17;45(10):1700–1706.
- Jiang Z, Qin JJ, Zhang Y, et al. LILRB4 deficiency aggravates the development of atherosclerosis and plaque instability by increasing the macrophage inflammatory response via NF-kappaB signaling. Clin Sci (Lond). 2017 Jul 25;131:2275–2288. DOI:https://doi.org/10.1042/CS20170198.
- Yi X, Zhang J, Zhuang R, et al. Silencing LAIR-1 in human THP-1 macrophage increases foam cell formation by modulating PPARgamma and M2 polarization. Cytokine. 2018 Nov;111:194–205.
- Qi W, Li Q, Liew CW, et al. SHP-1 activation inhibits vascular smooth muscle cell proliferation and intimal hyperplasia in a rodent model of insulin resistance and diabetes. Diabetologia. 2017 Mar;60(3):585–596. DOI:https://doi.org/10.1007/s00125-016-4159-1.
- Tai WT, Shiau CW, Chen HL, et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485.
- Su JC, Tseng PH, Hsu CY, et al. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget. 2014 Jul 15;5(13):4909–4919.
- Muthian G, Raikwar HP, Rajasingh J, et al. 1,25 dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006 May 15;83(7):1299–1309.
- So JY, Smolarek AK, Salerno DM, et al. Targeting CD44-STAT3 signaling by Gemini vitamin D analog leads to inhibition of invasion in basal-like breast cancer. PLoS One. 2013;8(1):e54020. DOI:https://doi.org/10.1371/journal.pone.0054020.
- Huang FC. Vitamin D differentially regulates Salmonella-induced intestine epithelial autophagy and interleukin-1beta expression. World J Gastroenterol. 2016 Dec 21;22(47):10353–10363.
- Rego D, Kumar A, Nilchi L, et al. IL-6 production is positively regulated by two distinct Src homology domain 2-containing tyrosine phosphatase-1 (SHP-1)-dependent CCAAT/enhancer-binding protein beta and NF-kappaB pathways and an SHP-1-independent NF-kappaB pathway in lipopolysaccharide-stimulated bone marrow-derived macrophages. J Immunol. 2011 May 1;186(9):5443–5456.
- Haussler MR, Jurutka PW, Mizwicki M, et al. Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab. 2011 Aug;25(4):543–559. DOI:https://doi.org/10.1016/j.beem.2011.05.010.
- Nanduri R, Mahajan S, Bhagyaraj E, et al. The active form of vitamin D transcriptionally represses Smad7 signaling and activates Extracellular Signal-regulated Kinase (ERK) to inhibit the differentiation of a inflammatory T helper cell subset and suppress experimental autoimmune encephalomyelitis. J Biol Chem. 2015 May 8;290(19):12222–12236.
- Ravid A, Rubinstein E, Gamady A, et al. Vitamin D inhibits the activation of stress-activated protein kinases by physiological and environmental stresses in keratinocytes. J Endocrinol. 2002 Jun;173(3):525–532. DOI:https://doi.org/10.1677/joe.0.1730525.
- Buitrago C, Boland R, de Boland AR. The tyrosine kinase c-Src is required for 1,25(OH)2-vitamin D3 signalling to the nucleus in muscle cells. Biochim Biophys Acta. 2001 Dec 19;1541(3):179–187.
- Morelli S, Buitrago C, Vazquez G, et al. Involvement of tyrosine kinase activity in 1alpha,25(OH)2-vitamin D3 signal transduction in skeletal muscle cells. J Biol Chem. 2000 Nov 17;275(46):36021–36028.
- Gómez CP, Tiemi Shio M, Duplay P, et al. The protein tyrosine phosphatase SHP-1 regulates phagolysosome biogenesis. J Immunol. 2012;189(5):2203–2210. DOI:https://doi.org/10.4049/jimmunol.1103021.
- Salamon H, Bruiners N, Lakehal K, et al. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J Immunol. 2014 Jul 01;193(1):30–34.
- Hmama Z, Sendide K, Talal A, et al. Quantitative analysis of phagolysosome fusion in intact cells: inhibition by mycobacterial lipoarabinomannan and rescue by an 1alpha,25-dihydroxyvitamin D3-phosphoinositide 3-kinase pathway. J Cell Sci. 2004 Apr 15;117(Pt 10):2131–2140.
- Wu S, Sun J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov Med. 2011 Apr;11(59):325–335.
- Tavera-Mendoza LE, Westerling T, Libby E, et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc Natl Acad Sci USA. 2017 Mar 14;114(11):E2186–E2194.
- Frank C, Burkhardt C, Imhof D, et al. Effective dephosphorylation of Src substrates by SHP-1. J Biol Chem. 2004 Mar 19;279(12):11375–11383.
- Keilhack H, Tenev T, Nyakatura E, et al. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J Biol Chem. 1998;273(38):24839–24846. DOI:https://doi.org/10.1074/jbc.273.38.24839.
- You L, Wang Z, Li H, et al. The role of STAT3 in autophagy. Autophagy. 2015;11(5):729–739. DOI:https://doi.org/10.1080/15548627.2015.1017192.
- Chandra V, Mahajan S, Saini A, et al. Human IL10 gene repression by Rev-erbalpha ameliorates Mycobacterium tuberculosis clearance. J Biol Chem. 2013 Apr 12;288(15):10692–10702.