
ABSTRACT
Macroautophagy/autophagy refers to the engulfment of cellular contents selected for lysosomal degradation. The final step in autophagy is the fusion of autophagosome with the lysosome, which is mediated by SNARE proteins. Of the SNAREs, autophagosome-localized Q-SNAREs, such as STX17 and SNAP29, and lysosome-localized R-SNAREs, such as VAMP8 or VAMP7, have been reported to be involved. Recent studies also reveal participation of the R-SNARE, YKT6, in autophagosome-lysosome fusion. These SNAREs, with the help of other regulatory factors, act coordinately to spatiotemporally control the fusion process. Besides regulating autophagosome-lysosome fusion, some SNAREs, such as STX17, also function in other autophagic processes, including autophagosome formation and mitophagy. A better understanding of the functions of SNAREs will shed light on the molecular mechanisms of autophagosome-lysosome fusion as well as on the mechanisms by which autophagy is globally regulated.
Abbreviations: ATG: autophagy related; DNM1L: dynamin 1 like; ER: endoplasmic reticulum; GABARAP: GABA type A receptor-associated protein; GABARAPL1: GABA type A receptor associated protein like 1; IRGM: immunity related GTPase M; LAMP2: lysosomal associated membrane protein 2; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; PIK3R4: phosphoinositide-3-kinase regulatory subunit 4; PLEKHM1: pleckstrin homology and RUN domain containing M1; PRKN: PRKN RBR E3 ubiquitin protein ligase; RAB2A: RAB2A, member RAS oncogene family; RAB33B: RAB33B, member RAS oncogene family; RAB7A: RAB7A, member RAS oncogene family; RB1CC1: RB1 inducible coiled-coil 1; RTN3: reticulon 3; RUBCNL: rubicon like autophagy enhancer; SNARE: soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SNAP29: synaptosomal associated protein 29; STX17: syntaxin 17; ULK1: unc-51 like autophagy activating kinase 1; VAMP7: vesicle associated membrane protein 7; VAMP8: vesicle associated membrane protein 8; YKT6: YKT6 v-SNARE homolog
Introduction
Macroautophagy/autophagy is a conserved catabolic process in which selected cellular components are delivered to and degraded within lysosomes to maintain cellular homeostasis [Citation1–3]. Upon starvation, the autophagic negative regulator, MTOR (mechanistic target of rapamycin), is inactivated and dissociates from dephosphorylated ULK1, which phosphorylates and forms a complex with ATG13 and RB1CC1/FIP200 (RB1 inducible coiled-coil 1) to initiate nucleation of the phagophore [Citation1–3]. Another complex comprised of ATG14 (autophagy related 14), BECN1/Beclin 1 (an ortholog of yeast Vps30/Atg6), PIK3R4/Vps15, and PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) serves to phosphorylate phosphatidylinositol to phosphatidylinositol-3-phosphate (PtdIns3P) to expand the phagophore during the nucleation phase. Elongation of the phagophore during the expansion phase is facilitated by two ubiquitin-like conjugation systems, the ATG12 conjugation system and the Atg8-family protein conjugation system. Finally, the completely closed autophagosome fuses with a lysosome, which is mediated by SNARE proteins, to form the autolysosome [Citation1–3].
In this review, we will primarily discuss the roles of SNARE proteins in the autophagosome-lysosome fusion process and provide perspectives for future studies.
Autophagosome-lysosome fusion
The fusion of autophagosomes and lysosomes is the final step of autophagy and requires the coordination of SNAREs, small GTPases, tethering factors, and other proteins [Citation4]. In yeast, dissociation of Atg proteins from the autophagosomes, which is sensitive to the level of PtdIns3P, is a prerequisite for the initiation of autophagosome-lysosome fusion [Citation5]. Depletion of Ymr1 (yeast myotubularin related), the sole yeast ortholog of the mammalian MTM/MTMR (myotubularin/myotubularin related) family of PtdIns3P phosphatases, causes accumulation of autophagosomes as a result of the Atg proteins’ dissociation defects [Citation6]. However, a different role is determined in mammalian cells, as MTMR14 (myotubularin related protein 14) negatively regulates autophagosome formation [Citation7]. Formation of PtdIns4P on the autophagosomes [Citation8] and the proper ratio and amount of PtdIns(3,5)P2 and PtdIns3P on the lysosomes [Citation9] are reported to be required for autophagosome-lysosome fusion. The exact role of lipid composition in autophagy regulation merits further exploration.
Normally, interaction between the scattered autophagosomes in the cytoplasm and the perinuclear-enriched lysosomes depends on their bidirectional movement on microtubules, especially dynein-mediated autophagosome perinuclear movement [Citation10]. Loss of dynein results in increased levels of LC3-II, which is associated with impaired autophagosome-lysosome fusion [Citation11]. Depletion of KIF5B, the heavy chain of kinesin-1, triggers peripheral aggregation of lysosomes and perinuclear accumulation of autophagosomes, indicating an important role for kinesin-mediated transport in the correct positioning of these organelles [Citation12]. Consistently, disrupting anterograde transport of lysosomes promotes their interactions with autophagosomes, and forced dispersion of lysosomes to the periphery blocks fusion [Citation13]. Actin filaments have also been shown to be involved in regulation of autophagosome-lysosome fusion [Citation14].
A major event during autophagosome-lysosome fusion is the SNARE complex-mediated fusion process. Autophagosome-localized STX17, SNAP29, and lysosome-localized VAMP8 [Citation15] or VAMP7 [Citation16] are believed to be the key factors involved in this process. A recent report shows that YKT6 also regulates autophagosome-lysosome fusion with the help of SNAP29, independent of the STX17 pathway [Citation17]. However, the Drosophila Ykt6 homolog forms a temporary complex with Syx17/STX17 until the intervention of Vamp7, which has a higher binding capacity for Syx17 than Ykt6 [Citation18]. The differences between these studies’ results may be due to differences in the model organisms used. Nevertheless, the significance of SNARE proteins’ involvement in autophagosome-lysosome fusion is well established.
To promote a systematic fusion process, other regulators, such as small GTPases and tethering factors, are also involved. RAB GTPases, such as RAB7A [Citation19], RAB33B [Citation20], and RAB2A [Citation21], have all been reported to regulate autophagosome-lysosome fusion. PLEKHM1 (pleckstrin homology and RUN domain containing M1), a RAB7A effector, is able to recruit a tethering factor, the homotypic fusion and protein sorting (HOPS) complex, to lysosome membranes to control the fusion step [Citation22]. The HOPS complex is reported to interact with STX17 and regulate assembly of the SNARE complex [Citation23,Citation24]. Another RAB7A effector, EPG5 (ectopic P-granules autophagy protein 5 homolog), binds to LC3 on autophagosomes and stabilizes the STX17-SNAP29-VAMP7/8 complex, facilitating autophagosome-lysosome fusion [Citation25]. In addition, ATG14, which is well-known for its role in autophagosome initiation, also directly binds to the STX17-SNAP29 binary t-SNARE complex to promote its fusion with lysosomal VAMP8 [Citation26]. Considering the complicated and delicate regulation of the SNARE-mediated fusion process, more regulators are likely involved and remain to be identified.
Assembly of SNARE complexes during autophagosome-lysosome fusion
In SNARE-mediated membrane fusion, four types of SNARE motifs, Qa, Qb, Qc and R, provided by each of the SNAREs form a parallel four alpha-helix bundle [Citation27]. In the case of autophagosome-lysosome fusion, the Q-SNAREs, STX17 and SNAP29, preform the Qabc bundle on the autophagosome followed by its forming a complex with a lysosome-localized R-SNARE, such as VAMP8 [Citation15]. The crystal structure shows that the Q196 of STX17, Q84 and Q230 of SNAP29, and R37 of VAMP8 constitute the ionic layer of the fusion-competent SNARE complex [Citation26]. A recent study shows that SNARE priming mediated by NAPA/αSNAP, the adaptor for NSF (N-ethylmaleimide sensitive factor, vesicle fusing ATPase), also plays important roles in producing autophagosomes capable of fusing with the lysosomes, without affecting autophagosome formation or autophagosome localization of STX17 [Citation28].
SNAP29, which lacks any transmembrane domain or membrane targeting motifs, is recruited to autophagosomes by interacting with STX17 after STX17 is transferred to autophagosomes [Citation15]. STX17 has two tandem transmembrane domains containing glycine zipper-liker motifs, both of which are required for the autophagosome localization of STX17 [Citation15]. However, the two transmembrane domains have low hydrophobicity, leading to a relatively large amount of cytosol-localized STX17. Combined with the observation that STX17 only presents on completely closed autophagosomes, autophagosomal STX17 likely originates from the cytosolic pool, but the detailed mechanism of its translocation remains undescribed [Citation15]. Autophagosomal recruitment of STX17 seems to occur immediately before or after autophagosome closure when the elliptic elongating phagophore becomes spherical in shape [Citation29]. It is reported that the autophagosome targeting of STX17 is affected in the absence of the lysosomal protein, LAMP2 [Citation30], as well as autophagosomal IRGM (immunity related GTPase M) with the help of LC3 [Citation31]. However, the role of Atg8-family members in recruiting STX17 to the autophagosome is controversial, as STX17 still presents on autophagosomal structures in cells deficient in all eight members of the Atg8 protein family during mitophagy [Citation32]. Consistently, STX17-positive autophagosome-like structures are still formed in the absence of ATG conjugation systems, but at a much lower rate [Citation29]. One plausible explanation is that LC3 alone is not sufficient for STX17 autophagosomal transfer but assists the recruitment and occludes premature binding of other fusion machineries to the SNARE domain of STX17 [Citation31].
SNARE bundle formation is also regulated by post-transcriptional modifications, which either alter ionic charges critical for protein interaction or result in steric hindrance within the SNARE complex [Citation33,Citation34]. For example, a recent study shows that K219 and K223, two sites in the SNARE domain of STX17 that face the outside of the alpha-helical bundle, are acetylated by CREBBP (CREB binding protein) under normal conditions and are deacetylated by HDAC2 (histone deacetylase 2) upon autophagy stimulation [Citation35]. SNAP29 and VAMP8 display a binding preference for deacetylated STX17 over acetylated STX17, indicating that the capacity for recruiting SNAP29, and thus subsequent SNARE complex formation, is the mechanism underlying the requirement of STX17 deacetylation during autophagosome-lysosome fusion [Citation35]. The O-GlcNAc-modification on SNAP29 also hinders the formation of the STX17-SNAP29-VAMP8 SNARE complex, possibly due to steric hindrance caused by this modification [Citation36].
Key players of the SNARE systems in autophagosome-lysosome fusion
STX17
STX17 was the first Qa-SNARE found to be involved in autophagosome-lysosome fusion [Citation15]. The conformation of STX17 is unique, as it contains two transmembrane domains in its C terminus; these form a hairpin structure that results in both the N terminus and C terminus localizing to the cytosol (). STX17 is broadly expressed in multiple tissues and is primarily considered to be an endoplasmic reticulum (ER)-localized SNARE [Citation37]. The C terminal tail of STX17 is required for its ER localization, while the N terminal domain, especially the conserved tyrosine residue Y156, regulates its exit from the ER into the ER-Golgi intermediate compartment (ERGIC) [Citation38]. The Y156 residue can be phosphorylated by ABL1/c-Abl and dephosphorylated by TC48, an isoform of PTPN2 (protein tyrosine phosphatase non-receptor type 2), to modulate the interaction of STX17 with COPI vesicles [Citation39]. Thus, STX17 is thought to cycle between the ER and ERGIC, which maintains the architecture of ERGIC and the Golgi apparatus [Citation38]. Overexpression of STX17 redistributes COPB1 (COPI coat complex subunit beta 1) and blocks anterograde transport of VSVG (vesicular stomatitis virus G-protein) in the ERGIC [Citation38]. Nuclear localization of STX17 is reportedly observed in several cancer cell lines, suggesting the existence of non-SNARE functions of STX17 [Citation40]. STX17 has also been reported to present on the mitochondria, mitochondria-associated membranes (MAM), and in the cytosol [Citation15,Citation41]. Therefore, STX17 exhibits a diversity of subcellular localizations, indicating the complexity of its functions and mechanisms of regulation ().
Figure 1. Schematic illustration of autophagosome-lysosome fusion-related SNARE proteins. The schematic drawing of the protein structures for STX17, SNAP29, VAMP7, VAMP8, and YKT6. TM, transmembrane domain; NPF, the Asp-Pro-Phe (NPF) motif; CCAIM, the C terminal prenylation consensus site; N, the N terminus
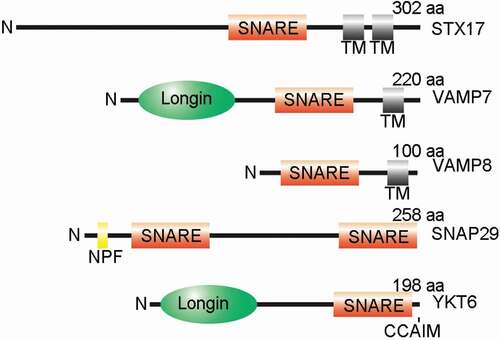
STX17 is thought to be transferred to autophagosomes upon nutrient starvation, and a large number of autophagosomes accumulate upon STX17 depletion [Citation15]. Mechanistically, STX17 and the Qbc-SNARE SNAP29 interact on the autophagosomes and mediate autophagosome-lysosome fusion by further coupling with lysosome-localized VAMP8 [Citation15] (). Essential for autophagosome localization, the two transmembrane domains of STX17 are also required for autophagic regulatory function, as quadruple mutations in G244, G248, G264 and G268, which disrupt the organization of the transmembrane domains, fail to rescue defects in LC3-II accumulation caused by STX17 knockdown [Citation15].
Figure 2. The diverse functions of STX17 in autophagy-related processes. Under normal conditions, STX17 recruits DNM1L at the mitochondria-ER-microtubule contact site to drive mitochondrial fission [Citation49]. Mitochondria-localized STX17 also controls mitochondria quality by MDV [Citation51], by PRKN-dependent mitophagy through regulation of the PGAM5-FUNDC1 interaction [Citation52], or by PRKN-independent mitophagy in the absence of FIS1 [Citation53]. Upon starvation, dephosphorylated MAP1B-LC1 releases STX17 from the microtubule [Citation55]. STX17 binds to ATG14 at MAM and initiates formation of the phagophore [Citation41]. Golgi apparatus-localized STX17, which is phosphorylated by TBK1, transfers to the phagophore assembly site to promote assembly of the RB1CC1-ATG13-ULK1 complex [Citation47]. On the completely closed autophagosome, STX17 is recruited from the cytosol by LC3 and IRGM [Citation31], followed by SNARE-mediated fusion with the help of various regulators, such as the HOPS complex (including VPS33A) [Citation23,Citation24], ATG14 [Citation26], DIPK2A [Citation67] and ULK1 [Citation48]. Upon ULK1 phosphorylation by PRKCA, ULK1 is degraded [Citation48]
![Figure 2. The diverse functions of STX17 in autophagy-related processes. Under normal conditions, STX17 recruits DNM1L at the mitochondria-ER-microtubule contact site to drive mitochondrial fission [Citation49]. Mitochondria-localized STX17 also controls mitochondria quality by MDV [Citation51], by PRKN-dependent mitophagy through regulation of the PGAM5-FUNDC1 interaction [Citation52], or by PRKN-independent mitophagy in the absence of FIS1 [Citation53]. Upon starvation, dephosphorylated MAP1B-LC1 releases STX17 from the microtubule [Citation55]. STX17 binds to ATG14 at MAM and initiates formation of the phagophore [Citation41]. Golgi apparatus-localized STX17, which is phosphorylated by TBK1, transfers to the phagophore assembly site to promote assembly of the RB1CC1-ATG13-ULK1 complex [Citation47]. On the completely closed autophagosome, STX17 is recruited from the cytosol by LC3 and IRGM [Citation31], followed by SNARE-mediated fusion with the help of various regulators, such as the HOPS complex (including VPS33A) [Citation23,Citation24], ATG14 [Citation26], DIPK2A [Citation67] and ULK1 [Citation48]. Upon ULK1 phosphorylation by PRKCA, ULK1 is degraded [Citation48]](/cms/asset/c9b40ec5-6b76-4688-8d97-2e7091d560f7/kaup_a_1823124_f0002_c.jpg)
During STX17-mediated autophagosome-lysosome fusion, the fusion process is precisely regulated. The HOPS complex has been reported to interact with STX17 and translocate to STX17-positive autophagosomes [Citation23,Citation24]. The recruitment of HOPS to autophagosomes is precisely regulated by RUBCNL/PACER (rubicon like autophagy enhancer), whose autophagosome targeting is also regulated by STX17 [Citation42]. Knockdown of the HOPS components, VPS33A, VPS16, or VPS39, blocks autophagic flux and leads to autophagosome accumulation [Citation24]. The N terminal domain of STX17 is required for its interaction with VPS33A [Citation43]. Overexpression of an STX17 mutant lacking the N terminal domain causes a dominant-negative effect in HeLa cells, with accumulation of a large number of undigested autophagosomes occurring as a result of a defect in HOPS binding [Citation43]. HOPS may act as a scaffold for organizing the fusion machinery [Citation24]. PLEKHM1 interacts with HOPS through the RUN domain and is colocalized at vesicle contact sites with RAB7A and HOPS [Citation22]. PLEKHM1 is able to contact the autophagosomes as it binds to autophagosome-localized LC3 through the LC3-interacting region (LIR). Similar to PLEKHM1, BIRC6/BRUCE, a lysosome-localized inhibitor of apoptosis protein (IAP), is recently shown to promote autophagosome-lysosome fusion by bridging STX17 and LAMP2-labeled lysosomes [Citation44]. Unlike PLEKHM1, which nonspecifically interacts with all six Atg8-family members, BIRC6 interacts with autophagosomes through a binding preference for GABARAP and GABARAPL1. Notably, VPS33A belongs to the Sec1/Munc18 (SM) protein family, the well-known SNARE assembly regulators, and thus may promote the fusion process by stabilizing the SNARE bundle [Citation45,Citation46].
Our understanding of STX17 in autophagy regulation was significantly expanded by the identification of MAM-localized STX17 [Citation41], pointing to a role for STX17 in autophagosome formation. MAM is a newly identified hot spot for origination of autophagosomes. STX17 binds to ATG14 and recruits it to the MAM after starvation. In STX17 knockdown cells, recruitment of ATG14 to MAM is hindered, and ATG5-positive dots are accumulated [Citation41]. Interestingly, ATG14 is also involved in autophagosome-lysosome fusion through binding to the SNARE domain of STX17, stabilizing the STX17-SNAP29 binary t-SNARE complex on autophagosomes [Citation26]. The autophagosome initiation function of STX17 is further supported by the recent observation that STX17 is phosphorylated by TBK1 at S202 [Citation47]. Upon starvation, phosphorylated STX17 (S202) translocates from the Golgi apparatus to the phagophore assembly site to orchestrate the assembly of the ATG13-RB1CC1-ULK1 complex [Citation47]. In addition, the ULK1-STX17 interaction is also necessary for the later step of autophagosome-lysosome fusion by enhancing STX17-SNAP29 coupling [Citation48]. PRKCA/PKCα phosphorylates ULK1 at S423, which decreases ULK1-STX17 interactions. Phosphorylated ULK1 has an attenuated ability to regulate autophagosome-lysosome fusion and is degraded via chaperone-mediated autophagy.
STX17 is able to present on the mitochondria; this presentation is dependent on both its transmembrane domains and C terminal domain [Citation49]. Replacement of the K254 residue of STX17 is sufficient to disrupt its mitochondrial localization [Citation49]. Consistently, the transmembrane domains and the C terminal domain are also responsible for STX17’s regulation of mitochondrial dynamics, as a mutant with their depletion fails to rescue the mitochondria elongation caused by STX17 knockdown, which also disrupts DNM1L/DRP1 (dynamin 1 like) puncta formation [Citation49]. However, there is disagreement regarding whether STX17 is involved in mitophagy regulation. In HeLa cells, STX17 knockdown blocks hypoxia- or deferiprone (DFP)-induced mitophagy as a result of autophagosome-lysosome fusion defects [Citation50]; however, STX17 depletion does not affect the levels of mitochondrial proteins in oligomycin/antimycin A (OA)-induced mitophagy under mCherry-PRKN (parkin RBR E3 ubiquitin protein ligase)-expressing conditions [Citation32]. Another study that uses GFP-PRKN-expressing U2OS cells induced by carbonyl cyanide m-chlorophenylhydrazone (CCCP) shows that STX17 is dispensable for depolarization-induced mitochondrial degradation as well as PRKN translocation [Citation51]. Only recently is the role of STX17 in PRKN-mediated mitophagy verified; the detachment of PGAM5 from STX17 facilitates the interaction between PGAM5 and FUNDC1 [Citation52]. In this case, mitophagy is inhibited upon depletion of STX17 or PGAM5. A recent study further shows that STX17 is also involved in PRKN-independent mitophagy. STX17 interacts with the TPR2 domain of FIS1 (fission, mitochondrial 1) through its N terminal domain and transmembrane domain, and STX17 overexpression induces mitophagy upon FIS1 depletion in a PINK1/PRKN-independent manner [Citation53]. STX17 recruits ATG14 and, subsequently, the core autophagy proteins, to the mitochondria and MAM to form mitophagosomes, which finally fuse with lysosomes through RAB7A and TBC1D15 (TBC1 domain family member 15). STX17 is found to present on mitochondrial-derived vesicles (MDVs), which are shed from the mitochondria to protect cells against oxidative stress [Citation51,Citation54]. STX17, together with SNAP29 and VAMP7, mediate fusion between MDVs and lysosomes. In summary, STX17 clearly plays important roles in mitochondrial quality control ().
Considering the diverse distribution of STX17 in the cell, questions remain regarding how the protein senses upstream signals and then transfers to autophagosomes. Autophagosome-localized STX17 primed for autophagosome-lysosome fusion is believed to come from the cytosolic pool following autophagosome closure [Citation15]. Surprisingly, STX17 is able to bind to Atg8-family members, including LC3B and GABARAP, through two LIR motifs (LIR1172–175 and LIR2189–192), mutations to which reduce the autophagosomal localization of STX17 [Citation31]. Another LC3-interacting protein, IRGM, binds to the transmembrane domains of STX17, the regions required for its insertion into autophagosome membranes, to facilitate the recruitment of STX17 to autophagosomes by forming a STX17-LC3B-IRGM holocomplex [Citation31].
How STX17 serves to precisely coordinate a diversity of subcellular functions remains undefined. Likely, under non-starvation conditions, STX17 associates with the microtubules, the ER, and the mitochondria to regulate mitochondrial division by recruiting DNM1L [Citation49]. MAP1B-LC1 (microtubule associated protein 1B-light chain 1), a proteolytic product of the polyprotein MAP1B, is able to link STX17 to microtubules by binding both [Citation55]. Starvation causes the dephosphorylation of MAP1B-LC1T217, which decreases its interaction with STX17. Subsequently, released STX17 further regulates autophagy initiation by binding to ATG14 [Citation55]. Thus, STX17 performs different functions by binding to diverse partners ().
SNAP29
SNAP29, the Qab-SNARE, belongs to the SNAP25-protein family, members of which possess two SNARE domains separated by a linker region () [Citation37,Citation56]. SNAP29 lacks a transmembrane domain and thus transiently associates with the membrane of organelles, including the Golgi apparatus, endosomes, and lysosomes, by interacting with other endocytic factors, such as syntaxins [Citation37,Citation56]. The wide distribution of SNAP29 allows it to function in a variety of membrane trafficking processes, from endolysosomal trafficking to secretion [Citation57]. SNAP29 was also found to localize to the outer kinetochore and to function in organizing kinetochore formation [Citation58].
In Drosophila fat bodies, Snap29 depletion results in complete inhibition of starvation-induced autolysosome formation as well as in accumulation of Atg8-positive autophagosomes, similar to Syx17 or Vamp7 depletion, indicating a role for Snap29 in autophagosome-lysosome fusion [Citation16]. In starved mammalian cells, SNAP29 colocalizes with STX17-positive LC3 structures, and SNAP29 knockdown leads to LC3-II accumulation and inhibition of autophagic flux [Citation15].
As discussed above, the autophagic activity of SNAP29 is regulated by O-GlcNAc modification [Citation36]. OGT (O-linked N-acetylglucosamine (GlcNAc) transferase) mediates O-GlcNAcylation of SNAP29 at multiple sites (S2, S61, T130, and S153); these modifications are removed by OGA (O-GlcNAcase). Upon starvation, the levels of O-GlcNAcylation on SNAP29 are reduced, and unmodified SNAP29 forms a more stable complex with STX17 and VAMP8 to mediate autophagosome-lysosome fusion. Expression of the O-GlcNAc-defective SNAP29 mutant, SNAP29S2A/S61G/T130A/S153G, enhances autophagic activity and facilitates autophagic degradation of protein aggregates even under nutrient-rich conditions [Citation36]. Further work in type I diabetic rats confirms the role of O-GlcNAc modification of SNAP29 in vivo, highlighting its functions and molecular mechanisms in disease [Citation59]. A recent report demonstrates that DAPK3 (death associated protein kinase 3), a member of the DAPK family of proteins involved in gestational diabetes mellitus (GDM), enhances the STX17-SNAP29 interaction by binding to SNAP29 [Citation60]. Interestingly, silencing DAPK3 modulates autophagy only in high-glucose conditions [Citation60]. It remains undescribed if there is a link between the O-GlcNAc modification of SNAP29 and DAPK3 function. Thus, fine tuning of SNAP29 modifications may represent a potential target for clinical application.
VAMP8 and VAMP7
Lysosome-localized R-SNAREs function as couplers for autophagosomal STX17-SNAP29; these include VAMP8 in mammals and Vamp7 in Drosophila [Citation15,Citation16]. The trafficking of VAMP8 (or VAMP7) to lysosomes is regulated by RAB21 and MTMR13, depletion of which reduces VAMP8 (or VAMP7) lysosomal distribution and attenuates autophagosome-lysosome fusion [Citation61].
A recent study using high-resolution fluorescence lifetime imaging (FLIM)-FRET reveals that STX17 heterodimerizes with VAMP7 in situ, highlighting a functional role for VAMP7 in autophagosome clearance in mammalian cells [Citation45]. VAMP7 appears to be more reliable than VAMP8 in coupling with the STX17-SNAP29 complex due to its greater proximity to STX17 [Citation45]. If not the dominant R-SNARE in autophagosome-lysosome fusion, VAMP7 is at least an alternative SNARE in this process.
VAMP7 is a member of the longin family of SNARE proteins. Longin SNAREs, including VAMP7, YKT6, and SEC22B, all contain a coiled-coil longin domain in their N terminus (); this domain is an important regulatory domain for trans-SNARE complex fusion and thus membrane fusion [Citation62]. The longin domain of VAMP7 is able to interact with its own SNARE domain to autoinhibit the protein itself [Citation63]. However, the longin domain also regulates the distribution and function of VAMP7 by binding to various partners [Citation64–66]. Interestingly, an isoform of VAMP7, VAMP7B, lacks the longin domain of the predominant form of VAMP7 (VAMP7A), and is able to inhibit autophagosome-lysosome fusion by ameliorating the interaction of VAMP7A and STX17 [Citation67]. VAMP7B binds to the SNARE domain of STX17 competitively with VAMP7A through its C terminal domain, while DIPK2A (divergent protein kinase domain 2A), a lysosome-localized protein, disrupts their interaction. Thus, DIPK2A promotes autophagosome-lysosome fusion by enhancing the STX17-VAMP7A interaction [Citation67].
Apart from its role in mediating autophagosome-lysosome fusion, VAMP7 is also involved in early autophagic process regulation. Knockdown of VAMP7 results in decreased LC3-II levels both in the presence and absence of the autophagy inhibitor bafilomycin A1 (Baf A1), indicating an inhibition of autophagosome formation [Citation68]. Similarly, VAMP7 knockout attenuates autophagosome formation, leading to accumulation of dysfunctional mitochondria in pancreatic β-cells [Citation69]. Moreover, VAMP7 colocalizes and interacts with ATG9A on recycling endosomes [Citation70]. AGFG1/Hrb, which binds to the longin domain of VAMP7, recruits VAMP7 and ATG9A to recycling endosomes from the plasma membrane, followed by VAMP7-, STX16 (syntaxin 16)-, and SNAP47-mediated fusion of ATG9A-resident vesicles, which finally contributes to autophagosome formation [Citation70]. VAMP7, ATG16L1, and ATG5 are colocalized on LC3-negative autophagic precursors as well as on LC3-positive phagophores [Citation68]. VAMP7 promotes the homotypic fusion of ATG16L1 precursors and regulates the size of ATG16L1 vesicles, which are important for autophagic membrane expansion. The ATG16L1-positive pool of VAMP7 also originates from the plasma membrane in a AGFG1-dependent manner [Citation68], perhaps partially from the ATG9A pathway as well [Citation70]. VAMP7 has also been reported to recruit LC3 to Yersinia pseudotuberculosis-containing vacuoles (YCVs) in infected macrophages, paving the road for this pathogen during its autophagic journey [Citation71]. All of these studies expand our understanding of VAMP7 in autophagy regulation.
YKT6
YKT6, an R-SNARE protein, has recently received increased attention in the field of autophagosome-lysosome fusion [Citation17,Citation18]. YKT6 contains a longin domain in its N terminus, similar to VAMP7, but does not contain a transmembrane domain () [Citation72]. Thus, membrane targeting of YKT6 depends on its C terminal prenylation consensus site (CCAIM), which is reversibly modulated by palmitoylation [Citation72]. YKT6 exhibits variable functions, and yeast YKT6 is reported to be required for autophagosome formation [Citation73].
In Drosophila, large amounts of autophagosomes accumulate in Ykt6-depleted cells as a result of failed autophagosome-lysosome fusion [Citation18]. Lysosome-localized Ykt6 is able to form a SNARE complex with Syx17-Snap29, until participation of Vamp7, which outcompetes Ykt6 from the temporary SNARE complex [Citation18]. Mutation in the SNARE domain does not affect the autophagic ability of Ykt6, indicating that Ykt6 acts as a regulator rather than as the fusion machinery in this process [Citation18]. The role of YKT6 in mammalian cells differs from that in Drosophila, where it mediates the autophagosome-lysosome fusion process by coupling with SNAP29 and STX7 in a STX17-independent manner [Citation17]. In this case, YKT6 is recruited to the autophagosomes, but not to LC3-negative lysosomes, through its longin domain. Thus, YKT6-SNAP29-STX7 likely functions as a second functional SNARE complex in addition to the STX17-SNAP29-VAMP8 complex in mammals [Citation17]. Studies in yeast further demonstrate the functional conservation of YKT6, which is localized on the autophagosomes, in autophagosome-vacuole fusion using in vitro autophagosome-vacuole fusion assays [Citation74,Citation75]. Though differences remain between species, the involvement of YKT6 in regulating the autophagosome-lysosome fusion process is unequivocal.
Perspectives
Along with the discovery of an increasing number of regulators in autophagosome-lysosome fusion, the mechanism of the process is becoming clearer. However, questions still remain regarding how different SNARE couplers and regulatory factors cooperate to ensure the process proceeds efficiently and precisely and how the fusion activity of SNARE proteins is spatiotemporally regulated.
Owing to its diverse localization in the cell, STX17 is a “Jack of all trades” in numerous cellular functions. How STX17 balances its variable roles in different cellular pathways remains unclear. One possibility is that STX17 exerts these functions through different domains. For example, the roles of STX17 in autophagy and mitochondria fission regulation diverge from each other, as elongated mitochondria can be rescued by a STX17 mutant defective in autophagosome-lysosome fusion [Citation76]. However, how these distinct functions are precisely regulated remains unclear.
It is well-known that organelle interactions play important roles in cellular functions [Citation77]. The function of STX17 is likely regulated by signaling from different organelles. For example, SIGMAR1 (sigma non-opioid intracellular receptor 1), an ER-resident chaperone protein, interacts with STX17, along with ATG14 and VAMP8, and promotes autophagosome-lysosome fusion during mitophagy [Citation78]. The STX17 and ATG14 interaction is disrupted by BCAP31 (B cell receptor associated protein 31), an ER membrane protein with an ER stress-modulating function [Citation79]. Dysfunctional mitochondrial respiration can lead to transfer of STX17 from the ER to the mitochondria [Citation53]. STX17 has also been reported to interact with ACSL3 (acyl-CoA synthetase long chain family member 3), a key enzyme in lipid droplet biogenesis, at the ER-mitochondria interface through its SNARE domain [Citation80]. STX17 knockdown disrupts ACSL3 localization from the ER to the lipid droplet, attenuating lipid droplet maturation [Citation80]. In summary, a better understanding of its functions will provide more insights into how STX17 is coordinated in various cellular processes.
A recent study reveals an ER-to-lysosome-associated degradation (ERLAD) pathway, in which some ER-localized proteasome-resistant cargos, such as the Z variant of SERPINA1/alpha1-antitrypsin Z/ATZ, are segregated into ER-derived single membrane vesicles with the help of a reticulophagy receptor, RETREG1/FAM134B, followed by direct fusing with lysosomes through STX17-VAMP8-mediated fusion [Citation81]. ERLAD differs from reticulophagy in its lack of dependence on autophagosome biogenesis machinery. Additional reticulophagy receptors, such as RTN3 (reticulon 3) [Citation82], ATL3 [Citation83], and TEX264 [Citation84], have been identified recently. It will be interesting to investigate whether STX17, as well as other SNARE proteins, coordinate with these factors and also regulate ER quality control.
Our current understanding of the mechanism of autophagosome-lysosome fusion largely benefits from traditional biochemistry, molecular, and cell biology techniques, such as analyzing autophagic protein turnover, localization, and pH alteration [Citation85]. In vitro-purified proteoliposome fusion assay, which is based on Förster/fluorescence resonance energy transfer/FRET, also represents a reliable means for analyzing SNARE-mediated vesicle fusion during autophagosome-lysosme fusion [Citation26,Citation86]. However, the need to reveal the spatiotemporal dynamics and regulatory mechanisms of autophagosome-lysosome fusion necessitates the development of new tools to describe the fusion process more precisely and quantitively. With the improvements in the resolution of optical microscopes, biocellular processes, such as organelle interactions, can be captured and quantified accurately by super-resolution microscopy methods. For example, a recent study analyzes mitophagy events by quantifying mitochondria-lysosome contacts with structured illumination microscopy/SIM [Citation87]. Combined with single synapse imaging, time-lapse imaging or fluorescence recovery after photobleaching/FRAP, the analyses of SNARE protein assembly dynamics during synaptic vesicle exocytosis have been well-established by stochastic optical reconstruction microscopy/STORM [Citation88], stimulated emission depletion/STED fluorescence microscopy [Citation89], and total internal reflection fluorescence/TIRF microscopy at single-molecule levels [Citation90]. Application of these techniques to autophagosome-lysosome fusion may explain the mechanisms underlying the accurate regulation of SNARE assembly in this process.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
References
- Hale AN, Ledbetter DJ, Gawriluk TR, et al. Autophagy: regulation and role in development. Autophagy. 2013 July;9(7):951–972.
- Ge L, Baskaran S, Schekman R, et al. The protein-vesicle network of autophagy. Curr Opin Cell Biol. 2014 Aug;29:18–24.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132.
- Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. 2019 Mar 4;218(3):757–770.
- Yu ZQ, Ni T, Hong B, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012 June;8(6):883–892.
- Cebollero E, van der Vaart A, Zhao M, et al. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol. 2012 Sept 11;22(17):1545–1553.
- Vergne I, Roberts E, Elmaoued RA, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. Embo J. 2009 Aug 5;28(15):2244–2258.
- Wang H, Sun HQ, Zhu X, et al. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A. 2015 June 2;112(22):7015–7020.
- Hasegawa J, Iwamoto R, Otomo T, et al. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. Embo J. 2016 Sept 1;35(17):1853–1867.
- Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33(1):109–122.
- Ravikumar B, Acevedo-Arozena A, Imarisio S, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nature Genet. 2005 July;37(7):771–776.
- Cardoso CM, Groth-Pedersen L, Hoyer-Hansen M, et al. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS One. 2009;4(2):e4424.
- Korolchuk VI, Saiki S, Lichtenberg M, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011 Apr;13(4):453–460.
- Tumbarello DA, Waxse BJ, Arden SD, et al. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012 Oct;14(10):1024–1035.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 7;151(6):1256–1269.
- Takats S, Nagy P, Varga A, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013 May 13;201(4):531–539.
- Matsui T, Jiang P, Nakano S, et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol. 2018 Aug 6;217(8):2633–2645.
- Takats S, Glatz G, Szenci G, et al. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet. 2018 Apr;14(4):e1007359.
- Gutierrez MG, Munafo DB, Beron W, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004 June 1;117(Pt 13):2687–2697.
- Itoh T, Kanno E, Uemura T, et al. OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol. 2011 Mar 7;192(5):839–853.
- Ding X, Jiang X, Tian R, et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy. 2019 Oct;15(10):1774–1786.
- McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015 Jan 8;57(1):39–54.
- Takats S, Pircs K, Nagy P, et al. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. 2014 Apr;25(8):1338–1354.
- Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014 Apr;25(8):1327–1337.
- Wang Z, Miao G, Xue X, et al. The vici syndrome protein EPG5 is a rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016 Sept 1;63(5):781–795.
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015 Apr 23;520(7548):563–566.
- Lou X, Shin YK. SNARE zippering. Biosci Rep. 2016 June;36(3). DOI:https://doi.org/10.1042/BSR20160004
- Abada A, Levin-Zaidman S, Porat Z, et al. SNARE priming is essential for maturation of autophagosomes but not for their formation. Proc Natl Acad Sci U S A. 2017 Nov 28;114(48):12749–12754.
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016 Nov 25;354(6315):1036–1041.
- Hubert V, Peschel A, Langer B, et al. LAMP-2 is required for incorporating syntaxin-17 into autophagosomes and for their fusion with lysosomes. Biol Open. 2016 Oct 15;5(10):1516–1529.
- Kumar S, Jain A, Farzam F, et al. Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J Cell Biol. 2018 Mar 5;217(3):997–1013.
- Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016 Dec 19;215(6):857–874.
- Xu NJ, Yu YX, Zhu JM, et al. Inhibition of SNAP-25 phosphorylation at Ser187 is involved in chronic morphine-induced down-regulation of SNARE complex formation. J Biol Chem. 2004 Sept 24;279(39):40601–40608.
- Malmersjo S, Di Palma S, Diao J, et al. Phosphorylation of residues inside the SNARE complex suppresses secretory vesicle fusion. Embo J. 2016 Aug 15;35(16):1810–1821.
- Shen Q, Shi Y, Liu J, et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy. 2020 Apr;15:1–13.
- Guo B, Liang Q, Li L, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014 Dec;16(12):1215–1226.
- Steegmaier M, Yang B, Yoo JS, et al. Three novel proteins of the syntaxin/SNAP-25 family. J Biol Chem. 1998 Dec 18;273(51):34171–34179.
- Muppirala M, Gupta V, Swarup G. Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol Cell. 2011 July;103(7):333–350.
- Muppirala M, Gupta V, Swarup G. Tyrosine phosphorylation of a SNARE protein, syntaxin 17: implications for membrane trafficking in the early secretory pathway. Biochim Biophys Acta. 2012 Dec;1823(12):2109–2119.
- Zhang Q, Li J, Deavers M, et al. The subcellular localization of syntaxin 17 varies among different cell types and is altered in some malignant cells. J Histochem Cytochem. 2005 Nov;53(11):1371–1382.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 Mar 21;495(7441):389–393.
- Cheng X, Ma X, Ding X, et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol Cell. 2017 Mar 16;65(6):1029–1043 e5.
- Uematsu M, Nishimura T, Sakamaki Y, et al. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy. 2017 Aug 3;13(8):1452–1464.
- Ebner P, Poetsch I, Deszcz L, et al. The IAP family member BRUCE regulates autophagosome-lysosome fusion. Nat Commun. 2018 Feb 9;9(1):599.
- Saleeb RS, Kavanagh DM, Dun AR, et al. A VPS33A-binding motif on syntaxin 17 controls autophagy completion in mammalian cells. J Biol Chem. 2019 Mar 15;294(11):4188–4201.
- Lobingier BT, Nickerson DP, Lo SY, et al. SM proteins Sly1 and Vps33 co-assemble with Sec17 and SNARE complexes to oppose SNARE disassembly by Sec18. eLife. 2014 May 16;3:e02272.
- Kumar S, Gu Y, Abudu YP, et al. Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev Cell. 2019 Apr 8;49(1):130–144 e6.
- Wang C, Wang H, Zhang D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018 Aug 28;9(1):3492.
- Arasaki K, Shimizu H, Mogari H, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. 2015 Feb 9;32(3):304–317.
- Yamashita SI, Jin X, Furukawa K, et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol. 2016 Dec 5;215(5):649–665.
- McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016 Aug 1;214(3):275–291.
- Sugo M, Kimura H, Arasaki K, et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. Embo J. 2018 Nov 2;37(21). DOI:https://doi.org/10.15252/embj.201798899
- Xian H, Yang Q, Xiao L, et al. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun. 2019 May 3;10(1):2059.
- Soubannier V, Rippstein P, Kaufman BA, et al. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. 2012;7(12):e52830.
- Arasaki K, Nagashima H, Kurosawa Y, et al. MAP1B-LC1 prevents autophagosome formation by linking syntaxin 17 to microtubules. EMBO Rep. 2018 June 19;19. DOI:https://doi.org/10.15252/embr.201745584
- Hohenstein AC, Roche PA. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem Biophys Res Commun. 2001 July 13;285(2):167–171.
- Morelli E, Ginefra P, Mastrodonato V, et al. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy. 2014;10(12):2251–2268.
- Morelli E, Mastrodonato V, Beznoussenko GV, et al. An essential step of kinetochore formation controlled by the SNARE protein Snap29. Embo J. 2016 Oct 17;35(20):2223–2237.
- Huang L, Yuan P, Yu P, et al. O-GlcNAc-modified SNAP29 inhibits autophagy-mediated degradation via the disturbed SNAP29-STX17-VAMP8 complex and exacerbates myocardial injury in type I diabetic rats. Int J Mol Med. 2018 Dec;42(6):3278–3290.
- Wang Y, Ji L, Peng Z, et al. Silencing DAPK3 blocks the autophagosome-lysosome fusion by mediating SNAP29 in trophoblast cells under high glucose treatment. Mol Cell Endocrinol. 2020 Feb 15;502:110674.
- Jean S, Cox S, Nassari S, et al. Starvation-induced MTMR13 and RAB21 activity regulates VAMP8 to promote autophagosome-lysosome fusion. EMBO Rep. 2015 Mar;16(3):297–311.
- Daste F, Galli T, Tareste D. Structure and function of longin SNAREs. J Cell Sci. 2015 Dec 1;128(23):4263–4272.
- Vivona S, Liu CW, Strop P, et al. The longin SNARE VAMP7/TI-VAMP adopts a closed conformation. J Biol Chem. 2010 June 4;285(23):17965–17973.
- Schafer IB, Hesketh GG, Bright NA, et al. The binding of Varp to VAMP7 traps VAMP7 in a closed, fusogenically inactive conformation. Nat Struct Mol Biol. 2012 Dec;19(12):1300–1309.
- Pryor PR, Jackson L, Gray SR, et al. Molecular basis for the sorting of the SNARE VAMP7 into endocytic clathrin-coated vesicles by the ArfGAP Hrb. Cell. 2008 Sept 5;134(5):817–827.
- Kent HM, Evans PR, Schafer IB, et al. Structural basis of the intracellular sorting of the SNARE VAMP7 by the AP3 adaptor complex. Dev Cell. 2012 May 15;22(5):979–988.
- Tian X, Zheng P, Zhou C, et al. DIPK2A promotes STX17- and VAMP7-mediated autophagosome-lysosome fusion by binding to VAMP7B. Autophagy. 2020 May;16(5):797–810.
- Moreau K, Ravikumar B, Renna M, et al. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011 July 22;146(2):303–317.
- Aoyagi K, Ohara-Imaizumi M, Itakura M, et al. VAMP7 regulates autophagy to maintain mitochondrial homeostasis and to control insulin secretion in pancreatic beta-cells. Diabetes. 2016 June;65(6):1648–1659.
- Aoyagi K, Itakura M, Fukutomi T, et al. VAMP7 regulates autophagosome formation by supporting Atg9a functions in pancreatic beta-cells from male mice. Endocrinology. 2018 Nov 1;159(11):3674–3688.
- Ligeon LA, Moreau K, Barois N, et al. Role of VAMP3 and VAMP7 in the commitment of Yersinia pseudotuberculosis to LC3-associated pathways involving single- or double-membrane vacuoles. Autophagy. 2014 Sept;10(9):1588–1602.
- Fukasawa M, Varlamov O, Eng WS, et al. Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc Natl Acad Sci U S A. 2004 Apr 6;101(14):4815–4820.
- Nair U, Jotwani A, Geng J, et al. SNARE proteins are required for macroautophagy. Cell. 2011 July 22;146(2):290–302.
- Gao J, Reggiori F, Ungermann C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J Cell Biol. 2018 Oct 1;217(10):3670–3682.
- Bas L, Papinski D, Licheva M, et al. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion. J Cell Biol. 2018 Aug 10;217:3656–3669.
- Wang B, Xiao X, Huang F, et al. Syntaxin-17-dependent mitochondrial dynamics is essential for protection against oxidative-stress-induced apoptosis. Antioxidants. 2019 Oct 30;8(11):522.
- Zheng P, Chen Q, Tian X, et al. DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling. Cell Res. 2018 Aug 28;28(8):833–854.
- Yang H, Shen H, Li J, et al. SIGMAR1/Sigma-1 receptor ablation impairs autophagosome clearance. Autophagy. 2019 Sept;15(9):1539–1557.
- Machihara K, Namba T. BAP31 inhibits cell adaptation to er stress conditions, negatively regulating autophagy induction by interaction with STX17. Cells. 2019 Oct 30;8(11):1350.
- Kimura H, Arasaki K, Ohsaki Y, et al. Syntaxin 17 promotes lipid droplet formation by regulating the distribution of acyl-CoA synthetase 3. J Lipid Res. 2018 May;59(5):805–819.
- Fregno I, Fasana E, Bergmann TJ, et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. Embo J. 2018 Sept 3;37(17). DOI:https://doi.org/10.15252/embj.201899259
- Grumati P, Morozzi G, Holper S, et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife. 2017 June;15:6.
- Chen Q, Xiao Y, Chai P, et al. ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr Biol. 2019 Mar 4;29(5):846–855 e6.
- An H, Ordureau A, Paulo JA, et al. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for er remodeling during nutrient stress. Mol Cell. 2019 June 6;74(5):891–908 e10.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
- Diao J, Li L, Lai Y, et al. In vitro reconstitution of autophagosome-lysosome fusion. Methods Enzymol. 2017;587:365–376.
- Chen Q, Shao X, Hao M, et al. Quantitative analysis of interactive behavior of mitochondria and lysosomes using structured illumination microscopy. Biomaterials. 2020 Aug;250:120059.
- Sakamoto H, Ariyoshi T, Kimpara N, et al. Synaptic weight set by Munc13-1 supramolecular assemblies. Nat Neurosci. 2018 Jan;21(1):41–49.
- Sieber JJ, Willig KI, Kutzner C, et al. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007 Aug 24;317(5841):1072–1076.
- Barg S, Knowles MK, Chen X, et al. Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc Natl Acad Sci U S A. 2010 Nov 30;107(48):20804–20809.