
ABSTRACT
The immunodeficiency observed in HIV-1-infected patients is mainly due to uninfected bystander CD4+ T lymphocyte cell death. The viral envelope glycoproteins (Env), expressed at the surface of infected cells, play a key role in this process. Env triggers macroautophagy/autophagy, a process necessary for subsequent apoptosis, and the production of reactive oxygen species (ROS) in bystander CD4+ T cells. Here, we demonstrate that Env-induced oxidative stress is responsible for their death by apoptosis. Moreover, we report that peroxisomes, organelles involved in the control of oxidative stress, are targeted by Env-mediated autophagy. Indeed, we observe a selective autophagy-dependent decrease in the expression of peroxisomal proteins, CAT and PEX14, upon Env exposure; the downregulation of either BECN1 or SQSTM1/p62 restores their expression levels. Fluorescence studies allowed us to conclude that Env-mediated autophagy degrades these entire organelles and specifically the mature ones. Together, our results on Env-induced pexophagy provide new clues on HIV-1-induced immunodeficiency.
Abbreviations: Ab: antibodies; AF: auranofin; AP: anti-proteases; ART: antiretroviral therapy; BafA1: bafilomycin A1; BECN1: beclin 1; CAT: catalase; CD4: CD4 molecule; CXCR4: C-X-C motif chemokine receptor 4; DHR123: dihydrorhodamine 123; Env: HIV-1 envelope glycoproteins; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; GFP-SKL: GFP-serine-lysine-leucine; HEK: human embryonic kidney; HIV-1: type 1 human immunodeficiency virus; HTRF: homogeneous time resolved fluorescence; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; NAC: N-acetyl-cysteine; PARP: poly(ADP-ribose) polymerase; PEX: peroxin; ROS: reactive oxygen species; siRNA: small interfering ribonucleic acid; SQSTM1/p62: sequestosome 1.
Introduction
HIV-1 infection is characterized by a progressive decline in the number of CD4+ T lymphocytes, ultimately leading to acquired immuno-deficiency syndrome/AIDS, in untreated patients [Citation1,Citation2]. Importantly, the majority of dying CD4+ T cells are not productively infected but are bystander uninfected cells [Citation3–6]. Among the several mechanisms identified to explain this depletion [Citation7–10], it has been demonstrated that HIV-1 envelope glycoproteins, composed of gp120 and gp41 associated in trimers (here called Env), play a major role. Indeed, Env expressed at the surface of infected cells is able to induce apoptosis in bystander CD4+ T cells after its interaction with the CD4 receptor and the co-receptor, either CCR5 or CXCR4 [Citation11,Citation12]. Our team has demonstrated that Env, and specifically the gp41 fusogenic function, triggers a massive induction of macroautophagy (hereafter referred to as autophagy) in bystander CD4+ T cells [Citation13,Citation14]. Importantly, this mechanism is necessary for the subsequent apoptosis, suggesting that Env-mediated autophagy could specifically degrade a component crucial for cellular survival [Citation14]. Interestingly, we and others, have also shown that Env triggers an oxidative stress in bystander CD4+ T cells, characterized by a strong production of ROS (Reactive Oxygen Species) [Citation15,Citation16].
Autophagy is a catabolic pathway involved in several cellular functions such as homeostasis, development, proliferation and immunity [Citation17]. It can be divided in three major steps: (i) the initiation step, with the formation of a double-membrane structure named a phagophore, (ii) the expansion step, where this structure expands engulfing portions of cytoplasm and then closes to form a double-membrane vesicle called an autophagosome, and (iii) the maturation step, where the autophagosomes fuse with lysosomes in order to degrade and recycle the sequestered material. More than 30 genes and several signaling platforms are involved in autophagosome formation, such as the BECN1 (beclin 1)-containing class III phosphatidylinositol 3-kinase complex, acting at the initiation step of the process, and two ubiquitination-like conjugation systems leading to the covalent conjugation of Atg8-family proteins to a lipid, phosphatidylethanolamine, involved in the expansion step of phagophore membranes [Citation18]. Autophagic degradation can be highly selective using proteins called autophagy receptors, such as SQSTM1/p62 (sequestosome 1) [Citation19]. These proteins bind to both the phosphatidylethanolamine-conjugated Atg8-family proteins and to substrates, allowing their targeting to phagophores [Citation20].
Autophagy and oxidative stress are intimately connected and the level of ROS production is tightly regulated. Low amounts of ROS are involved in the activation of survival signaling pathways [Citation21], whereas an excessive ROS production activates cell death [Citation22]. By degrading organelles involved in ROS production, such as damaged mitochondria, selective autophagy balances excessive ROS production [Citation23]. However, when the levels of ROS become excessive, cell death is initiated either by apoptosis or necrosis [Citation22]. Alternatively, autophagy controls the cellular oxidative status by its involvement in peroxisome homeostasis. Peroxisomes are essential single membrane-limited organelles containing multiple enzymes involved in metabolic and antioxidant reactions. CAT (catalase), one of the major peroxisomal enzyme, catalyzes the transformation of hydrogen peroxide into water, therefore detoxifying the cell from this harmful component [Citation24,Citation25]. PEX/peroxin (peroxisomal biogenesis factor) lumenal and membrane proteins control peroxisome assembly and division. Peroxisomes acquire their various components along their biogenesis and grow toward mature peroxisomes, reaching a size between 0.5 µm and 1 µm. Selective autophagy of peroxisomes, called pexophagy, is responsible for the number and quality of these organelles [Citation26] by maintaining an equilibrium between their biogenesis and breakdown [Citation27,Citation28].
The aim of the present study was to understand how Env-mediated autophagy could trigger apoptosis in bystander CD4+ T cells. Given that Env also induced an accumulation of ROS in target cells and considering the essential role of peroxisomes in the regulation of the oxidative status of the cell, we focused our research on the interplay between autophagy and peroxisomes upon Env exposure. Our results show that Env-mediated autophagy is responsible for ROS accumulation in bystander CD4+ T cells and demonstrate that Env-mediated autophagy selectively targets mature peroxisomes.
Results
Env-mediated autophagy and ROS production are responsible for bystander CD4+ T cells apoptosis
We previously demonstrated that Env, expressed at the cell surface, induces autophagy in target bystander CD4+ T cells, ultimately leading to apoptosis [Citation29]. These results were obtained with different types of effector cells and target cells also used in the present study (). These models, based on the co-culture between effector cells expressing Env and target cells that express CD4 and CXCR4, allow the analysis of the Env effect in the absence of viral replication. In the model 1, the target cells were either primary CD4+ T lymphocytes or the CEM-derived A201/CD4.403 T cell line. These target cells were co-cultured with HEK293 (human embryonic kidney cells) stably expressing Env (HEK.Env) during 48 h. The parental HEK293 cell line was used as a negative control. In the model 2, the target cells were HEK293 cells stably expressing CD4.403 and CXCR4 and are referred to as (HEK/CD4.403/CXCR4). These target cells were co-cultured for 48 h with 8.E5 cells expressing Env and harboring a single defective copy of replication incompetent HIV-1 proviral DNA. The parental CEM cell line was used as a negative control. Importantly, Env induces equivalent levels of autophagy (between 20 and 30%) and cell death (between 15 and 20%) in the two coculture models and, in both cases, Env-induced apoptosis depends on autophagy [Citation13,Citation14,Citation30,Citation31]. Moreover, it should be noted that the interaction between Env-expressing cells and target cells that express HIV-1 receptors, leads to a transient cell-to-cell fusion, also called hemifusion, or to a complete cell-to-cell fusion with the formation of syncytia, in both models. Importantly, Env-mediated autophagy and cell death are triggered in both single cells and syncytia [Citation12,Citation13,Citation15,Citation32].
Figure 1. Env-induced autophagy is a prerequisite for apoptosis in CD4+ T cells. (A) Description of the cellular models. The experiments are based on the co-culture between effector cells expressing Env at their surface and target cells expressing CD4 and CXCR4. In the model 1, target cells are either primary CD4+ T lymphocytes purified from healthy donor blood samples or a CD4+ T cell line expressing CXCR4. Effector cells are HEK293 stably expressing Env (HEK.Env). HEK293 cells are used as negative control. In the model 2, target cells are HEK293 cells stably expressing CD4 and CXCR4. Effector cells are 8.E5 cells, which are chronically infected cells and thus express Env at their surface. The parental CEM cell line is used as a negative control. (B-D) Env-induced apoptosis depends on autophagy. Target CD4+ T cells were co-cultured for 48 h with HEK293 or HEK.Env in the presence or absence of (B) AMD3100 (1 µg/ml), (C) spautin 1 (50 μM), (D) BafA1 (50 nM). Target cells were harvested and fold increase in Env-mediated apoptosis was studied by quantifying the cleavage of PARP using HTRF. Results are from at least 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. (E and F) Env-induced autophagy participated in ROS production that is responsible for uninfected CD4+ T cells apoptosis. Target CD4+ T cells (either T cell line, E or primary CD4+ T cells, F) were co-cultured for 48 h with HEK or HEK.Env in the presence or absence of NAC (10 mM). Target cells were harvested and fold increase in Env-mediated apoptosis was studied by quantifying the cleavage of PARP using HTRF. Data are representative of at least three independent experiments. **p < 0.01; ***p < 0.001. (G) Target CD4+ T cells were co-cultured for 48 h with HEK293 or HEK.Env in the presence or absence of spautin 1 (50 µM). Analysis of intracellular ROS production was measured by flow cytometry using DHR 123. Data are representative of at least three independent experiments. *p < 0.05; **p < 0.01
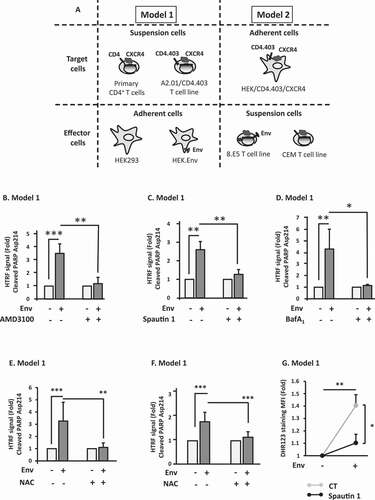
We first used model 1 to confirm the results obtained previously using a homogeneous time resolved fluorescence (HTRF) method. This method allows the quantitative detection of the cleaved PARP (poly(ADP-ribose) polymerase), which is a known substrate of CASP3 (caspase 3) when apoptosis is induced. As shown in , Env-induced cleavage of PARP is completely blocked in the presence of AMD3100, which is an antagonist of CXCR4 [Citation33], demonstrating that this cleavage is due to Env binding to this co-receptor. Moreover, Env-mediated cleavage of PARP is dependent on autophagy since it is abolished by addition of spautin 1 () and bafilomycin A1 (BafA1) (), which are inhibitors of the early and late steps of autophagy, respectively.
As mentioned above, we, and others, demonstrated that effector cells expressing Env induce an oxidative stress in target bystander CD4+ T cells characterized by a production of ROS [Citation15,Citation16]. Here, we evaluated the production of ROS in target cells after 48 h of coculture with effector cells (model 1) using both the dihydrorhodamine 123 (DHR123) probe and an oxyblot assay. The first assay allowed us to observe a significant increase in the production of free radicals (Figure S1A) and the second allowed us to observe a significant increase in the oxidation of total cellular proteins (Figure S1B). As oxidative stress induces apoptosis, we evaluated the involvement of the Env-induced production of ROS in this apoptosis. We thus co-cultured effector cells expressing Env with target CD4+ T cells, either a CD4+ T cell line () or blood-purified primary CD4+ T cells (), in the presence of N-acetyl-cysteine (NAC), which is a general antioxidant molecule. Addition of NAC completely abolished Env-induced apoptosis, demonstrating that Env-mediated accumulation of ROS is responsible for this cell death. NAC also abolished the total Env-mediated bystander CD4+ T cell death as shown by Propidium Iodide internalization (Figure S1C).
As the antioxidant properties of autophagy are well characterized, we then analyzed the effect of this process on the Env-induced ROS production. To this aim, using the model 1 of co-culture, we analyzed ROS production in target cells in conditions where autophagy is inhibited by addition of spautin 1. After 48 h, we added DHR123 to quantify ROS production. Surprisingly, we found that inhibition of autophagy leads to a decrease in ROS production (). These results strongly suggested that autophagy is involved in the Env-mediated accumulation of ROS, which is responsible for bystander CD4+ T cell apoptosis.
Autophagy selectively targets peroxisomal proteins
Peroxisomes are major cellular antioxidant organelles. Their number and integrity are regulated through selective degradation by autophagy in a process called pexophagy. We hypothesized that Env-induced autophagy could degrade peroxisomes in target cells, leading to the accumulation of ROS. To test this hypothesis, we analyzed the level of two peroxisomal proteins by western blot, in target cells after their co-culture during 48 h with Env-expressing cells in different conditions. We followed the level of expression of CAT, which is one of the major peroxisomal enzymes. As CAT is also present in mitochondria, we also analyzed PEX14, which is strictly located at the peroxisomes [Citation34]. As shown in and Figure S2A, the levels of PEX14 and CAT respectively decreased in target cells in contact with Env. AMD3100, which is known to block Env-mediated autophagy and cell death, rescued their level, confirming the involvement of Env in this effect. Next, to analyze the role of autophagy in Env-mediated PEX14 and CAT decreases, we added lysosomal protease inhibitors (AP for anti-proteases: E64d and pepstatin A) to the co-culture. We observed that the Env-mediated PEX14 and CAT decreases are mediated by lysosomal degradation ( and Figure S2B). No significant impact of AMD3100 or AP addition has been observed, at the basal level, on the PEX14:GAPDH ratio. Notably, we verified by RT-PCR that the observed diminution of PEX14 expression levels was not due to a difference in PEX14 mRNA production (Figure S2C).
Figure 2. PEX14 is degraded by Env-induced autophagy. (A) Env induces a decrease in PEX14 expression level. PEX14 expression level was analyzed in target CD4+ T cells after 48 h of co-culture with effector HEK293 cells expressing or not Env, in the presence or absence of AMD3100 (1 µg/ml). The expression level ratios of PEX14 were calculated between the conditions with or without Env and normalized to that obtained with anti-GAPDH Ab. Data are representative of at least 3 independent experiments. *p < 0.05. (B) PEX14 is degraded in lysosomes in response to Env. PEX14 expression level was analyzed in target CD4+ T cells after 48 h of co-culture with effector HEK293 cells expressing or not Env, in the presence or absence of AP (E64d + pepstatin A, 10 µM each). The expression level ratios of PEX14 were calculated between the conditions with or without Env and normalized to that obtained with anti-GAPDH antibody. Data are representative of at least 3 independent experiments. **p < 0.01. (C) Autophagy is involved in Env-mediated PEX14 degradation. HEK/CD4.403/CXCR4 cells were co-cultured during 48 h with effector cells expressing or not Env (8.E5 or CEM, respectively) after their transfection with BECN1 (siBECN1) or control (siCT) siRNAs. Reduction in BECN1 expression was analyzed by western blot using the specific Abs. The expression level ratios of PEX14 were calculated between the conditions with or without Env and normalized to that obtained with anti-GAPDH Ab. Data are representative of at least 3 independent experiments. **p < 0.01. (D) SQSTM1/p62 is involved in the Env-mediated PEX14 degradation. HEK/CD4.403/CXCR4 cells were co-cultured during 48 h with effector cells expressing or not Env (8.E5 or CEM, respectively) after their transfection with SQSTM11/p62 (siSQSTM1) or control (siCT) siRNAs. Reduction in SQSTM1/p62 expression was analyzed by western blot using the specific Abs. The expression level ratios of PEX14 were calculated between the conditions with or without Env and normalized to that obtained with anti-GAPDH Ab. Data are representative of at least 3 independent experiments. **p < 0.01
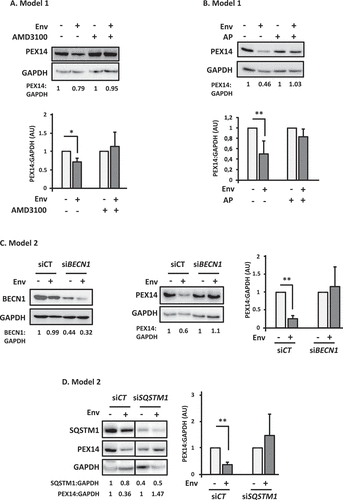
To definitively attest the role of Env-induced autophagy on PEX14 and CAT levels, we inhibited autophagy by transfecting siRNA targeting the expression of BECN1 in target cells before their co-culture with Env-expressing cells. We previously showed that Env-mediated cell death was completely inhibited in these conditions [Citation14]. We still observed a PEX14 decrease in target cells transfected with a control siRNA upon Env exposure, but PEX14 expression level remains unchanged when target cells, in which BECN1 was downregulated, are co-cultured with Env-expressing cells (). As the peroxisome number and quality are regulated by autophagy at the basal level in the cells, we checked if BECN1 siRNA could affect the expression level of PEX14 in the absence of Env. We found that siBECN1 had no significative effect on the PEX14:GAPDH ratio without Env (not shown). Of note, similar results were obtained for CAT (Figure S2D). Taken together, these results confirmed that Env induces the autophagic degradation of peroxisomal proteins.
To complete the analysis, we checked the involvement of the SQSTM1/p62 autophagy receptor, known for its involvement in pexophagy [Citation35]. We transfected siRNA targeting SQSTM1/p62 in target cells and co-cultured them with Env-expressing cells. The PEX14 protein is no longer degraded in target cells where the expression of SQSTM1/p62 has been downregulated (). Notably, the SQSTM1/p62 siRNA had no significant effect on the PEX14:GAPDH ratio without Env (not shown).
Together, these results showed that Env-induced autophagy selectively target peroxisomal proteins in bystander CD4+ T cells.
Env-induced autophagy targets peroxisomes in target cells
Based on the above results, we next wanted to analyze whether the entire peroxisomes were degraded by autophagy. We labeled peroxisomes in target cells with green fluorescent protein (GFP) by transfecting the GFP-SKL plasmid that contains a C-terminal tripeptide, serine-lysine-leucine (SKL), called a “peroxisomal targeting signal/PTS”, targeting the GFP into peroxisomes. We confirmed that this construct stains peroxisomes by labeling PEX14 in the GFP-SKL-transfected cells. The PEX14 puncta massively colocalized with the GFP signal, confirming that the GFP-SKL construct efficiently labels peroxisomes (). We thus co-cultured the GFP-SKL-transfected target cells with Env-expressing cells during 24 h and 48 h and quantified the GFP signal using flow cytometry. We observed a significant decrease in the GFP signal in target cells in contact with Env-expressing cells in a time-dependent manner (). Interestingly, the fluorescence signal was rescued by the addition of spautin 1, arguing that Env induces pexophagy. To further confirm this result, we analyzed the colocalization between GFP-SKL and MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta, here called LC3B), which is an ortholog of Atg8, by immunofluorescence. Target cells, transfected with the GFP-SKL construct, were co-cultured during 48 h with Env-expressing cells in presence of BafA1 to block the autophagic flux. By confocal microscopy, we observed several colocalizations between LC3B and GFP-SKL (). As the number of peroxisomes and autophagosomes was very high in the conditions used, we decided to quantify the colocalization events between GFP-SKL and LC3B in the total target cell volume. To this aim, several optical slices of target cells (Z = 1 µm) were acquired after their co-culture with effector cells expressing or not Env. We used the ImageJ Plugin “JaCOP” allowing the determination of the Mander’s colocalization score between LC3B puncta and peroxisomes. We observed a significant increase in the colocalization between GFP-SKL and LC3B in target cells after their contact with Env-expressing cells compared to the control (), confirming that peroxisomes were targeted to autophagy in cells exposed to Env. Furthermore, Env induced an increase in the colocalization events between GFP-SKL and the SQSTM1/p62 autophagic receptor ().
Figure 3. Env-induced autophagy targets mature peroxisomes. (A) The SKL motif drives GFP to peroxisomes. Target cells were transfected with the GFP-SKL construct. Twenty-four h later, peroxisomes were labeled using an anti-PEX14 antibody. Images were acquired with a confocal microscope. (B) The GFP-labeled peroxisomes decrease overtime in target cells after their co-culture with effector cells expressing Env. Target cells were transfected with the GFP-SKL construct. Twenty-four h later, the transfected cells were co-cultured with effector cells in presence or not spautin 1 (50 µM). The mean fluorescence intensity (MFI) of the GFP signal was analyzed in target cells using flow cytometry after 24 and 48 h of co-culture. Data are representative of at least 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. (C) The colocalization between the GFP labeled peroxisomes and LC3B increases in target cells upon Env exposure. Target cells were transfected with the GFP-SKL construct. Twenty-four h later, the transfected cells were co-cultured with effector cells in the presence of BafA1 (50 nM). Autophagosomes were labeled using an anti-LC3B by immunofluorescence. The graph represents the overlap of peroxisomes staining in the LC3B-positive structures. Data were acquired for the total volume of at least 15 cells and are representative of at least 3 independent experiments. ***p < 0.001. (D) The same experiment as C was done using an anti-SQSTM1/p62 antibody. The graph represents the overlap of peroxisomes staining in the SQSTM1/p62-positive structures. Data are representative of 3 independent experiments. *p < 0.5. (E) Target cells were stained by immunofluorescence with an anti-PEX14 antibody after 48 h of co-cultures with effector cells in presence or not of spautin 1 (50 µM). Total peroxisomes (in red) of each image were submitted to a computer-generated mask using the “Cell Profiler” software in order to identify only the peroxisomes for which the diameter is > 0.7 µm (in green). (F and G) In the same experiments as E, we quantified, respectively, (F) the mean number of total peroxisomes, and the number of large ones and (G) the ratio of the number of large peroxisomes over the number of total peroxisomes per cell, in each condition. The total volume of the target cells was imaged using a confocal microscope. Data obtained with the “Cell Profiler software” are representative of at least 3 independent experiments. ***p < 0.001
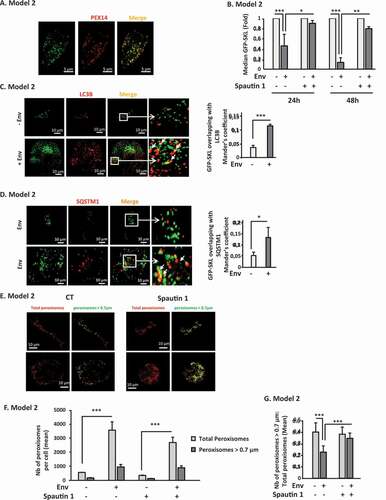
As peroxisomes respond to changes in the cellular environment by adapting their number and morphology [Citation36], we analyzed these last parameters in target cells after their contact with Env-expressing cells by immunofluorescence with a particular focus on their size. To this aim, we stained peroxisomes in target cells after 48 h of co-culture with Env-expressing cells using an anti-PEX14 antibody in presence or absence of spautin 1. Peroxisomes vary in size from 0.1 to 1 μm in diameter. Although they are capable of fission, the “mature” peroxisomes, which exert their antioxidant capacity, are larger than those undergoing biogenesis. The target cells were analyzed by confocal microscopy (). In order to distinguish large peroxisomes from small ones (in the process of biogenesis), we used the “Cell Profiler” software and designed a protocol allowing us to measure their area in target cells. Stacks of 1 μm between each optical slice were acquired to avoid capturing the same peroxisome twice. Each acquisition, with a confocal microscope, was performed on the total volume of the target cells. We set up a threshold of 0.7 μm in diameter to identify “mature” peroxisomes. We first quantified the mean number of total peroxisomes, and the number of large ones. We observed an increase in the total peroxisomes upon Env exposure (). This result is in accordance with the observed Env-induced production of ROS (). Moreover, the addition of spautin 1 slightly decreased the total number of peroxisomes, which is in accordance with the reduced production of ROS observed upon spautin 1 treatment in . Then, based on these measurements, we established, for each image analyzed, a ratio between the number of mature peroxisomes (diameter> 0.7 μm) and the number of total peroxisomes. The data were collected for each image constituting the volume of a target cell. Interestingly, this ratio was decreased in target cells after their contact with Env-expressing cells and the addition of spautin 1 restored the ratio close to the one of control cells (). As demonstrated in , C and D, Env-induced autophagy degraded PEX14. These results, together with the LC3B and SQSTM1/p62 colocalization experiments ( and D), strongly suggested that Env-induced autophagy degraded active peroxisomes.
Discussion
The interplay between autophagy and HIV-1 infection is complex and depends on the target cell type and on the infectious status of the cell (infected vs bystander cells) [Citation30,Citation37,Citation38]. We previously showed that Env, expressed on infected cells, induces autophagy in bystander CD4+ T cells, after its interaction with HIV-1 receptors present at their surface. Notably, this Env-induced autophagy leads to apoptosis of these target cells, mechanism likely contributing to CD4+ T cells depletion observed in HIV-1 associated physiopathology [Citation14]. As HIV-1 Env induces the production of ROS in bystander CD4+ T cells [Citation16], we first demonstrated that this oxidative stress is responsible for subsequent apoptosis. The cellular ROS levels are tightly regulated by the action of antioxidant enzymes in order to avoid their harmful effect. As we previously showed that Env-mediated autophagy is a prerequisite to apoptosis in bystander cells, we hypothesized that antioxidant pathways is targeted by autophagy and, consequently, deprive the cell of their protective action. Inhibition of autophagy by adding spautin 1 blocks apoptosis (), but also dramatically decreases the production of ROS in bystander cells cocultured with effector cells expressing Env (), strongly suggesting that Env-induced autophagy leads to an uncontrolled oxidative stress in bystander cells. ROS can be produced at different cellular sites and three major organelles are known to control it: mitochondria, endoplasmic reticulum and peroxisomes, forming the “redox triangle” [Citation39]. These three organelles are interconnected and cooperate to fight oxidative stress. Importantly, the inhibition of peroxisomal CAT, concomitantly with an increase of ROS, results in mitochondrial redox imbalance [Citation40]. Moreover, Env induces the mitochondrial pathway of apoptosis in bystander CD4+ T cells [Citation31]. In addition, a recent study showed that a subset of miRNAs, targeting peroxisomal proteins, are upregulated in the brain of HIV/AIDS patients with and without HIV-associated neurocognitive disorders/HAND. The authors found that these miRNAs significantly decreased peroxisome number and affect their morphology [Citation41]. In this context, we focused our work on peroxisomes and demonstrate that these organelles are targeted by Env-induced autophagy. Our results suggest that peroxisomes are insufficiently recognized targets of HIV-1 infection and open new routes in the characterization of the relationships between autophagy and this virus. Obviously, we cannot exclude a role of the mitochondria and the endoplasmic reticulum in our model and it would be important to test if these organelles, as well as other antioxidant pathways, are also targeted by autophagy in the same context.
By analyzing both peroxisomal proteins and entire peroxisomes, we were able to demonstrate, for the first time, that pexophagy plays a major role in a viral infection, probably by impairing the control of induced oxidative stress. Pexophagy, which occurs at the cellular basal level, is thus induced upon Env exposure. It would be important now to decipher the specific Env-induced signaling pathway triggered upstream of this phenomenon and particularly the role of the kinase ATM (ATM serine/threonine kinase). Indeed, this protein is activated by peroxisomal ROS, leading to pexophagy and involves the selective targeting of PEX5 by SQSTM1/p62 [Citation42].
Concerning HIV-1 infection, the degradation of cellular antioxidant system requires a special attention as a known side-effect of ART (Anti-Retroviral Treatment) is to induce an oxidative stress in patients [Citation43–46]. Moreover, oxidative stress is also clearly involved in the development of HIV-1-associated disorders such as neurotoxicity, dementia and immune imbalance [Citation47–52]. Several HIV-1-associated co-morbidities related to an increased risk of cardiovascular diseases development are favored by the oxidative stress induced by the presence of the virus, such as high blood pressure, atherosclerosis and myocarditis [Citation53–55]. Moreover, in this infectious context, the peroxisome depletion could be beneficial for pathogens and their escape from innate immunity. Indeed, peroxisomes have been identified as antiviral innate immunity signaling platforms since they harbor mitochondrial antiviral signaling protein at their surface and participate to type I interferon production in response to viral invasion [Citation56]. Beyond this, one of the major barriers against the eradication of HIV-1 in patients is the presence of cellular reservoirs that contain integrated, replication-competent provirus, resistant to ART. Even if these treatments maintain a very low level of viral replication and have considerably ameliorated the HIV-1 infected patient life, a rebound of viremia is observed upon treatment interruption. Interestingly, a new therapy, based on the oxidative stress response, has been proposed to target the HIV-1 reservoir. Auranofin (AF) is a gold compound with a pro-oxidant function [Citation57] capable of inducing apoptosis of memory T cells based on their oxidative status [Citation58]. AF combined with ART completely suppresses simian immunodeficiency virus/SIV viremia in macaques. More interestingly, the survival of treated macaque is maintained for 2 years after the treatment interruption without any sign of immunodeficiency [Citation59]. These results indicate that apoptosis-inducing drugs, acting on oxidative stress, could be considered to reduce the HIV-1 reservoir. It would be thus interesting to analyze if AF, combined to ART, could modulate memory T cells autophagy.
In conclusion, our results open new route for future investigations at multiple levels of HIV-1-host interactions and, to our knowledge, they demonstrate for the first time a role of pexophagy during a viral infection.
Materials and methods
Cell culture
The HEK293, HEK.Env [Citation31] and HEK/CD4.403/CXCR4 [Citation13] cell lines were cultured in DMEM (Life Technologies, 61,965–026) supplemented with 1% penicillin-streptomycin, 1% Glutamax, and 10% fetal calf serum (Sigma-Aldrich, F7524). The CEM T cell line was provided by the ATCC (ATCC® CCL119™). The CEM-derived A2.01/CD4.403 cell clone, which expresses a mutant form of CD4 truncated at position 403, was previously described [Citation13] and was provided by D.R. Littman (New York Medical College, New York, NY, USA). The 8.E5 cell line, a CEM-derived T cell line containing a single integrated copy of HIV-1 and unable to produce infectious virions, was provided by F. Barré-Sinoussi (Institut Pasteur, Paris, France). These T cell lines were cultured in RPMI 1640 medium (Life Technologies, 61,870–010) supplemented with 1% penicillin-streptomycin, 1% Glutamax, and 10% fetal calf serum. Primary CD4+ T cells were purified from the blood of healthy donors by negative selection using the Human CD4+ T Cell Enrichment cocktail (StemCell Technologies, 19,157). These primary cells were cultured in complete RPMI 1640 medium.
Reagents and antibodies
AMD3100, NAC, spautin 1, E64d, pepstatin A, bafilomycin A1 were purchased from Sigma-Aldrich (239,820, A9165, E8640, P5318, B1793, respectively). The anti-PEX14 and anti-BECN1 antibodies (Abs) were purchased from Proteintech (105,941-AP and 11,306-1-AP, respectively). The anti-SQSTM1/p62 Ab was purchased from Euromedex (GTX100685). The anti-CAT (catalase) Ab was purchased from Abcam (ab1877-10). The anti-LC3B and the anti-GAPDH Abs were purchased from Sigma-Aldrich (L7543 and G9295, respectively).
Transfections
RNA interference: ON-TARGET plus Human SMART Pool from Dharmacon were used for RNA interference targeting BECN1 (reference L-010552) and siGENOME Human siRNA – SMART Pool for SQSTM1/p62 (reference M-010230). The ON-TARGET plus Non-targeting Pool (reference D-001810-10) was used as a control. Briefly, 0.5 × 106 target cells were transfected using LipoRNAiMax (Invitrogen, P/N56531) with 20 nM siRNA according to the manufacturer’s instructions. Sixteen h after transfection, target cells were co-cultured with effector cells expressing, or not, Env (1.106/ml) for 48 h before analysis.
Plasmids: Target cells (0.5x106) were cultured in 6-well plates and transfected with pEGFP-SKL (Addgene, 53,450; from Jay Brenman Lab) plasmids using TurboFect Transfection Reagent (Thermo-Scientific, R0531) according to the manufacturer’s instructions. Transfected target cells were co-cultured with effector cells expressing or not Env for 48 h before analysis.
RT-PCR
Target cells were co-cultured with effector cells expressing, or not, Env for 48 h. RNA was purified from 106 target cells using the Nucleospin RNA plus (Macherey-Nagel, 740,984.50), and RT-PCR was performed using the QIAGEN OneStep RT-PCR kit according to the manufacturer’s instructions. The oligonucleotides used for PEX14 and GAPDH mRNA are: PEX14 FOR: 5ʹ-GGGCTGACAGATGAAGAGATTG-3ʹ and PEX14 BACK: 5ʹ-CCTGGATCTTCTGCTGCTGCTG −3ʹ GAPDH FOR: 5ʹ-CCCATCACCATCTTCCAG-3ʹ; GAPDH BACK: 5ʹ-CCTGCTTCACCACCTTCT-3ʹ.
Apoptosis and total cell death analysis
Target cells were co-cultured with effector cells expressing, or not, Env for 48 h. Apoptosis was analyzed in the target cells using “Cleaved PARP (Asp214) Cellular Kit” from CisBio (PerkinElmer, 64PARPEG) according to the manufacturer’s instructions. FRET analysis was measured using a SPARK 10 M TECAN. Propidium iodide staining was used to quantified the level of total cell death. Briefly, suspension cells were washed with phosphate-buffered saline (ThermoFisher Scientific, 14,190–094), 4 μl PI was added to the cells and they were then analyzed on a COULTER EPICS XL Flow Cytometer (Beckman Coulter).
ROS analysis
DHR123 assay: Target cells were co-cultured with effector cells during 48 h. Then, cells were incubated for 15 min at 37°C in HBSS containing 0.86 mM DHR 123 (ThermoFisher, D23806) and fluorescence intensity was rapidly measured at 543 nm by flow cytometry.
Oxyblot assay: After 48 h of coculture with effector cells, target cells were lysed and the carbonyl content present in cellular proteins was determined using the “OxyBlot Protein Oxidation Detection Kit” (Sigma-Aldrich, S7150) according to the manufacturer’s instructions. Equivalent amounts of each protein fraction were analyzed. Immunoblots were visualized by ECL as described in next section.
Western blot analysis
Target cells were co-cultured with effector cells during 48 h. Target cells were then harvested and lysed directly in Laemmli buffer 2x and heated for 10 min at 95°C. Cell lysates were loaded on 12% Prosieve 50 gels (Lonza, LON50618) and transferred to polyvinylidene (PVDF) membranes. After a blocking step in PBS containing 5% casein (Sigma-Aldrich, C7078) and 0.05% Tween 20 (Sigma-Aldrich, P9416) for 1 h at room temperature, blots were incubated overnight at 4°C with the primary Ab in the blocking buffer. After 3 washes with PBS+0.05% Tween 20, the blots were incubated for 1 h at room temperature with peroxidase-coupled antiserum (Sigma-Aldrich, goat anti-rabbit and goat anti-mouse, A0545 and A2304, respectively) diluted in blocking buffer. After further washes, the immune complexes were revealed by ECL (Clarity western ECL Substrate; Bio-Rad, 170–5061) and analyzed with a ChemiDoc camera (Bio-Rad). Quantification of protein expression was performed using the Image lab Software (Bio-Rad). Data were normalized with reference to the densitometry analysis of GAPDH.
Immunofluorescence studies
Target cells were cultured on glass slides and co-cultured with effector cells during 48 h. For the staining, target cells were first incubated with 1% paraformaldehyde for 10 min and then with methanol during 5 min for LC3B staining and incubated with 1% paraformaldehyde + 0.1% Triton X-100 (Sigma-Aldrich, T8787) for 15 min for SQSTM1/p62 staining. After a saturation step in a PBS, 0.1% saponin (Calbiochem, 558,255), 1% FBS buffer, the primary Ab was incubated during 2 h at room temperature. After several washes, the secondary Ab coupled with a fluorochrome was added during 1 h. Imaging was done using a Spinning Disk Dragonfly Andor microscope (Montpellier RIO Imaging platform). Acquired images were analyzed using the Fiji plugin JaCOP. The Mander’s coefficient, measuring the overlap of GFP-SKL in LC3B or SQSTM1/p62 staining, was determined on each z-stack of each acquisition before being averaged on all stacks of each cell [Citation60]. For the « Cell profiler » analysis, a procedure was set up to discriminate peroxisomes over their area size.
Statistical analysis
Differences were considered significant at *p < 0.05, **p < 0.01, and ***p < 0.001. Variances were measured using the Wilcoxon test on GraphPad Prism.
Supplemental Material
Download Zip (1.2 MB)Acknowledgments
This work was supported by the “Agence Nationale de Recherche sur le SIDA et les Hépatites Virales” (ANRS), by SIDACTION, by the Centre National de la Recherche Scientifique (CNRS) and by the University of Montpellier. MG was the recipient of a fellowship from the Labex EPIGENMED. We thank Dr. Matteo Bonazzi (IRIM CNRS UMR9004 Montpellier, France) for its help on imaging analysis.
Disclosure statement
No conflict of interest.
Supplemental material
Supplemental data for this article can be accessed here.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003;84:1649–1661.
- Fauci AS. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science. 1988;239:617–622.
- Finkel TH, Tudor-Williams G, Banda NK, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med. 1995;1:129–134.
- Herbeuval JP, Grivel JC, Boasso A, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106:3524–3531.
- Varbanov M, Espert L, Biard-Piechaczyk M. Mechanisms of T-cell depletion in HIV-1 infection triggered by viral proteins. AIDS Rev. 2006;8:221–236.
- Westendorp MO, Frank R, Ochsenbauer C, et al. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500.
- Ameisen JC, Capron A. Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunol Today. 1991;12:102–105.
- Jekle A, Keppler OT, De Clercq E, et al. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J Virol. 2003;77:5846–5854.
- Meyaard L, Otto SA, Jonker RR, et al. Programmed death of T cells in HIV-1 infection. Science. 1992;257:217–219.
- Terai C, Kornbluth RS, Pauza CD, et al. Apoptosis as a mechanism of cell death in cultured T lymphoblasts acutely infected with HIV-1. J Clin Invest. 1991;87:1710–1715.
- Garg H, Blumenthal R. Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell Mol Life Sci. 2008;65:3134–3144.
- Perfettini JL, Castedo M, Roumier T, et al. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005;12(Suppl 1):916–923.
- Denizot M, Varbanov M, Espert L, et al. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy. 2008;4:998–1008.
- Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172.
- Garg H, Blumenthal R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J Leukoc Biol. 2006;79:351–362.
- Molina L, Grimaldi M, Robert-Hebmann V, et al. Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics. 2007;7:3116–3130.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846.
- Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016;26:624–635.
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75.
- Johansen T, Lamark T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J Mol Biol. 2019.
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15.
- Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–388.
- Islinger M, Voelkl A, Fahimi HD, et al. The peroxisome: an update on mysteries 2.0. Histochem Cell Biol. 2018;150:443–471.
- Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766.
- Eberhart T, Kovacs WJ. Pexophagy in yeast and mammals: an update on mysteries. Histochem Cell Biol. 2018;150:473–488.
- Cho DH, Kim YS, Jo DS, et al. Pexophagy: molecular Mechanisms and Implications for Health and Diseases. Mol Cells. 2018;41:55–64.
- Honsho M, Yamashita S, Fujiki Y. Peroxisome homeostasis: mechanisms of division and selective degradation of peroxisomes in mammals. Biochim Biophys Acta. 2016;1863:984–991.
- Espert L, Denizot M, Grimaldi M, et al. [Autophagy and CD4 T lymphocyte destruction by HIV-1]. Med Sci (Paris). 2006;22:677–678.
- Espert L, Varbanov M, Robert-Hebmann V, et al. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One. 2009;4:e5787.
- Roggero R, Robert-Hebmann V, Harrington S, et al. Binding of human immunodeficiency virus type 1 gp120 to CXCR4 induces mitochondrial transmembrane depolarization and cytochrome c-mediated apoptosis independently of Fas signaling. J Virol. 2001;75:7637–7650.
- Blanco J, Barretina J, Ferri KF, et al. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology. 2003;305:318–329.
- Donzella GA, Schols D, Lin SW, et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4:72–77.
- Grant P, Ahlemeyer B, Karnati S, et al. The biogenesis protein PEX14 is an optimal marker for the identification and localization of peroxisomes in different cell types, tissues, and species in morphological studies. Histochem Cell Biol. 2013;140:423–442.
- Yamashita S, Abe K, Tatemichi Y, et al. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy. 2014;10:1549–1564.
- Ribeiro D, Castro I, Fahimi HD, et al. Peroxisome morphology in pathology. Histol Histopathol. 2012;27:661–676.
- Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268.
- Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. Aids. 2008;22:695–699.
- Yoboue ED, Sitia R, Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018;9:331.
- Ivashchenko O, Van Veldhoven PP, Brees C, et al. Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar cross-talk. Mol Biol Cell. 2011;22:1440–1451.
- Xu Z, Asahchop EL, Branton WG, et al. MicroRNAs upregulated during HIV infection target peroxisome biogenesis factors: implications for virus biology, disease mechanisms and neuropathology. PLoS Pathog. 2017;13:e1006360.
- Tripathi DN, Zhang J, Jing J, et al. A new role for ATM in selective autophagy of peroxisomes (pexophagy). Autophagy. 2016;12:711–712.
- Manda KR, Banerjee A, Banks WA, et al. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood-brain barrier endothelial cells. Free Radic Biol Med. 2011;50:801–810.
- Nagiah S, Phulukdaree A, Chuturgoon A. Inverse association between microRNA-124a and ABCC4 in HepG2 cells treated with antiretroviral drugs. Xenobiotica. 2016;46:825–830.
- Wang X, Chai H, Lin PH, et al. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti-human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am J Pathol. 2009;174:771–781.
- Weiss M, Kost B, Renner-Muller I, et al. Efavirenz causes oxidative stress, endoplasmic reticulum stress, and autophagy in endothelial cells. Cardiovasc Toxicol. 2016;16:90–99.
- Baier-Bitterlich G, Wachter H, Fuchs D. Oxidative stress and apoptosis in HIV infection. Science. 1996;271:582–583.
- Baruchel S, Wainberg MA. The role of oxidative stress in disease progression in individuals infected by the human immunodeficiency virus. J Leukoc Biol. 1992;52:111–114.
- Couret J, Chang TL. Reactive oxygen species in HIV infection. EC microbiol. 2016;3:597–604.
- Israel N, Gougerot-Pocidalo MA. Oxidative stress in human immunodeficiency virus infection. Cell Mol Life Sci. 1997;53:864–870.
- Ivanov AV, Valuev-Elliston VT, Ivanova ON, et al. Oxidative Stress during HIV Infection: mechanisms and Consequences. Oxid Med Cell Longev. 2016;2016:8910396.
- Legrand-Poels S, Vaira D, Pincemail J, et al. Activation of human immunodeficiency virus type 1 by oxidative stress. AIDS Res Hum Retroviruses. 1990;6:1389–1397.
- Ballocca F, D’Ascenzo F, Gili S, et al. Cardiovascular disease in patients with HIV. Trends Cardiovasc Med. 2017;27:558–563.
- Fan X, Staitieh BS, Jensen JS, et al. Activating the Nrf2-mediated antioxidant response element restores barrier function in the alveolar epithelium of HIV-1 transgenic rats. Am J Physiol Lung Cell Mol Physiol. 2013;305:L267–77.
- Spieker LE, Karadag B, Binggeli C, et al. Rapid progression of atherosclerotic coronary artery disease in patients with human immunodeficiency virus infection. Heart Vessels. 2005;20:171–174.
- Dixit E, Boulant S, Zhang Y, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681.
- Becker K, Gromer S, Schirmer RH, et al. Thioredoxin reductase as a pathophysiological factor and drug target. Eur J Biochem. 2000;267:6118–6125.
- Badley AD, Sainski A, Wightman F, et al. Altering cell death pathways as an approach to cure HIV infection. Cell Death Dis. 2013;4:e718.
- Shytaj IL, Nickel G, Arts E, et al. Two-Year Follow-Up of Macaques Developing Intermittent Control of the Human Immunodeficiency Virus Homolog Simian Immunodeficiency Virus SIVmac251 in the Chronic Phase of Infection. J Virol. 2015;89:7521–7535.
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232.