
ABSTRACT
Dominant de novo mutations in the co-chaperone BAG3 cause a severe form of myofibrillar myopathy, exhibiting progressive muscle weakness, muscle structural failure, and protein aggregation. To elucidate the mechanism of disease in, and identify therapies for, BAG3 myofibrillar myopathy, we generated two zebrafish models, one conditionally expressing BAG3P209L and one with a nonsense mutation in bag3. While transgenic BAG3P209L-expressing fish display protein aggregation, modeling the early phase of the disease, bag3-/- fish exhibit exercise dependent fiber disintegration, and reduced swimming activity, consistent with later stages of the disease. Detailed characterization of the bag3-/- fish, revealed an impairment in macroautophagic/autophagic activity, a defect we confirmed in BAG3 patient samples. Taken together, our data highlights that while BAG3P209L expression is sufficient to promote protein aggregation, it is the loss of BAG3 due to its sequestration within aggregates, which results in impaired autophagic activity, and subsequent muscle weakness. We therefore screened autophagy-promoting compounds for their effectiveness at removing protein aggregates, identifying nine including metformin. Further evaluation demonstrated metformin is not only able to bring about the removal of protein aggregates in zebrafish and human myoblasts but is also able to rescue the fiber disintegration and swimming deficit observed in the bag3−/- fish. Therefore, repurposing metformin provides a promising therapy for BAG3 myopathy.
Abbreviations:ACTN: actinin, alpha; BAG3: BAG cochaperone 3; CRYAB: crystallin alpha B; DES: desmin; DMSO: dimethyl sulfoxide; DNAJB6: DnaJ heat shock protein family (Hsp40) member B6; dpf: days post fertilization; eGFP: enhanced green fluorescent protein; FDA: Food and Drug Administration; FHL1: four and a half LIM domains 1; FLNC: filamin C; hpf: hours post-fertilization; HSPB8: heat shock protein family B [small] member 8; LDB3/ZASP: LIM domain binding 3; MYOT: myotilin; TTN: titin; WT: wild-type.
Introduction
Myofibrillar myopathies are a group of chronic muscle diseases characterized at the cellular level by accumulation of protein aggregates and structural failure of the muscle fiber. There is significant variability in the presentation of these diseases, with onset ranging from infantile to late seventies and muscle weakness ranging from mild reductions to severe impairment of skeletal, cardiac, and respiratory muscles resulting in early death. Causative mutations for myofibrillar myopathies have been identified in 10 genes: DES (desmin) [Citation1], CRYAB/αB-crystallin [Citation2], MYOT (myotilin) [Citation3], LDB3 (LIM domain binding 3)/ZASP (Z-band alternatively spliced PDZ motif-containing protein) [Citation4], FLNC (filamin C) [Citation5], BAG3 (BAG cochaperone 3) [Citation6], FHL1 (four and a half LIM domains 1) [Citation7], TTN (titin) [Citation8], DNAJB6 (DnaJ heat shock protein family [Hsp40] member B6) [Citation9], and HSPB8 (heat shock protein family B [small] member 8) [Citation10]. All of these genes encode proteins found at the Z-disk, a key structure involved in the transmission of tension and contractile forces along the muscle fiber.
While structural failure of the muscle fiber is a feature of myofibrillar myopathies, not all of the proteins associated with the disease have a direct structural role. One such protein is BAG3, a multi domain co-chaperone that is predominantly expressed in skeletal and cardiac muscle, where it co-localizes with FLNC and ACTN (actinin, alpha) at the Z-disk [Citation11]. In muscle, BAG3 regulates several cellular processes including inhibiting apoptosis and promoting cell survival [Citation12,Citation13], regulating protein turnover by stimulating macroautophagy/autophagy and inhibiting proteasomal degradation [Citation14–19], and regulating cellular mechanotransduction [Citation18]. Given such important functions of BAG3, it is unsurprising that mutations in it result in disease with two mutations identified to cause myofibrillar myopathy [Citation6,Citation20] and nine in dilated cardiomyopathy [Citation21–24].
The dominant de novo myofibrillar myopathy causing BAG3P209L mutation is not only the first identified but also the most well-characterized BAG3 mutation [Citation6,Citation20]. This is the most severe form of myofibrillar myopathy described, patients presenting with early onset (6–8 years of age) and rapidly progressing limb and axial muscle weakness that is often followed by cardiomyopathy, respiratory failure, and/or neuropathy [Citation6,Citation20]. At a cellular level, abnormal accumulation of sarcomeric and extracellular matrix proteins is evident, along with presence of granular-filamentous aggregates, Z-disk thickening, and myofibrillar disintegration [Citation6,Citation20,Citation25].
To examine the mechanistic basis of BAG3 myofibrillar myopathy, we previously generated zebrafish models that transiently express BAG3P209L in a subset of muscle cells, or have reduced bag3 expression [Citation26]. Using these models, we demonstrated that while the protein aggregation is due to the presence of BAG3P209L, the structural failure of the muscle fiber is due to protein insufficiency following sequestration of both wildtype and mutant protein in the aggregates. Our data suggested that strategies that promote the clearance of aggregates, and subsequently prevent the depletion of functional BAG3 may be highly beneficial for the treatment of myofibrillar myopathy [Citation26]. The identification of treatments for rare disease, such a BAG3 myopathy, is a challenge as the small patient population limits the studies that may be conducted on novel compounds. As such, in the current study, we explored the potential of known autophagy inducing compounds, many of which are already approved by the US Food and Drug Administration (FDA) for clinical use, as potential therapies for BAG3 myofibrillar myopathy.
We established zebrafish models for BAG3 myofibrillar myopathy and completed a screen of known autophagy promoting drugs identifying several effective at removing protein aggregates. We further identified a defect in autophagy in these fish, which was subsequently confirmed in patients. Our study suggests treatment of BAG3 myofibrillar myopathy would be achieved through stimulation of autophagy, removing protein aggregates and overcoming defects in autophagy.
Results
Expression of BAG3P209L results in the formation of protein aggregates
To determine the consequence of BAG3P209L expression, we generated zebrafish conditionally expressing fluorescently tagged, full-length, human wild-type (WT) BAG3 (Tg [BAG3-eGFP]) or BAG3P209L (Tg [BAG3P209L-eGFP]) in skeletal muscle (). Both transgenes are expressed at comparable levels in the zebrafish myotome (). At 48 h post-fertilization (hpf) confocal and super-resolution microscopy revealed that while both WT BAG3-eGFP and BAG3P209L-eGFP localized to the Z-disk of the sarcomere, BAG3P209L-eGFP additionally localized to the myosepta and formed protein aggregates throughout the muscle cell ( and S1A–C). Quantification of the number of aggregates revealed a significant increase in BAG3P209L-eGFP compared to WT BAG3-eGFP embryos (Figure S1D). By 144 hpf the BAG3P209L-eGFP aggregates became more evident at the myoseptal boundaries ( and S1E,F). Tg [BAG3P209L-eGFP] fish therefore recapitulated one of the key hallmark features of the disease, which is the formation of protein aggregates.
Figure 1. Expression of BAG3P209L results in protein aggregation. (A,B) Live images of 48 hpf transgenic zebrafish expressing BAG3-eGFP or BAG3P209L-eGFP specifically in skeletal muscle, with lens GFP expression indicating presence of Cre protein. (C) Western blot showing differential levels of transgene expression in the multiple Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] strains. Strains with comparable transgene expression (boxed) are used in all subsequent experiments. (D,G) Live confocal images of WT Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] embryos at 48 hpf and 144 hpf. Both, WT BAG3-eGFP and BAG3P209L-eGFP localize to the sarcomere but BAG3P209L-eGFP additionally localizes to the myoseptal boundary, and forms protein aggregates (arrowheads) along the length of the muscle fiber, and around the myoseptal boundaries. (Di and Gi) Enlarged images of regions denoted in (D–G)
![Figure 1. Expression of BAG3P209L results in protein aggregation. (A,B) Live images of 48 hpf transgenic zebrafish expressing BAG3-eGFP or BAG3P209L-eGFP specifically in skeletal muscle, with lens GFP expression indicating presence of Cre protein. (C) Western blot showing differential levels of transgene expression in the multiple Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] strains. Strains with comparable transgene expression (boxed) are used in all subsequent experiments. (D,G) Live confocal images of WT Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] embryos at 48 hpf and 144 hpf. Both, WT BAG3-eGFP and BAG3P209L-eGFP localize to the sarcomere but BAG3P209L-eGFP additionally localizes to the myoseptal boundary, and forms protein aggregates (arrowheads) along the length of the muscle fiber, and around the myoseptal boundaries. (Di and Gi) Enlarged images of regions denoted in (D–G)](/cms/asset/a8b632ea-25de-4900-a453-6850827261a7/kaup_a_1833500_f0001_oc.jpg)
Reduced Bag3 results in contraction dependent fiber disintegration
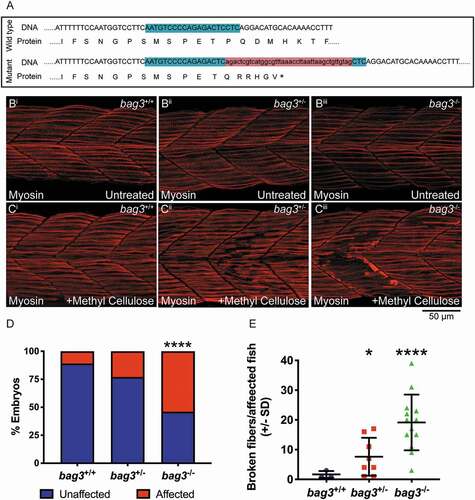
Given the hypothesis that protein aggregates ultimately lead to BAG3 loss of function, using CRISPR-Cas9 genome editing, we generated a bag3 mutant with a 41 base pair insertion in exon 2, which resulted in a frameshift and incorporation of a premature stop codon, and subsequent nonsense-mediated decay of the mRNA (). While under normal conditions the muscle structure in bag3 heterozygous (bag3+/-) and homozygous mutants (bag3-/-) was indistinguishable from that of WT embryos (bag3+/+), bag3+/- and bag3-/- embryos displayed a structural failure of the myofiber following incubation in methyl cellulose, a viscous solution that increases the load on the muscle (). A reduction in, or loss of Bag3, therefore resulted in contraction dependent fiber disintegration, which is the second defining feature of myofibrillar myopathy.
Figure 2. Bag3-deficient zebrafish embryos display fiber disintegration. (A) Schematic of bag3 WT and mutant exon 2 genomic DNA sequence, and resulting protein, with the mutant predicted to incorporate a premature stop (*). The mutant was generated using CRIPSR/Cas9 genome editing. The CRISPR target site is highlighted in green and the 41 bp insert in red. (B) Muscle fibers span the entire length of the somite in the 26 hpf wildtype (bag3+/+), bag3 heterozygous (bag3+/-) and bag3 mutant (bag3-/-) embryos as seen by Myosin antibody labeling. (C) Incubation of 26 hpf bag3+/- and bag3-/- in methyl cellulose results in fiber disintegration, which is not evident in bag3+/+ embryos. (D) Graph showing the percentage of affected bag3+/+, bag3+/- and bag3-/- embryos the latter two genotypes having a significant increase in the proportion of fish displaying the fiber disintegration phenotype (p < 0.05 based on a binomial test). (E) Quantification of the number of broken fibers in affected embryos following incubation in methyl cellulose with bag3-/- showing a significant increase. Error bars represent ± SD from three biological replicates. The total number of fish examined in each replicate is documented in Table S2. *p < 0.05, ****p < 0.0001 based on a generalized linear mixed model
Reduced Bag3, but not the presence of BAG3P209L, results in muscle weakness
To assess the pathogenicity of BAG3P209L and the loss of bag3 on skeletal muscle, we examined muscle function in both the transgenic overexpression models and the loss of function bag3-/- mutant. At 2 dpf we performed touch evoked response assays to measure the maximum acceleration during a burst swimming motion, with acceleration being directly proportional to the force produced by the muscle. We revealed no significant differences in the maximum acceleration in Tg [BAG3-eGFP] () and Tg [BAG3P209L-eGFP] () when compared to their respective sibling controls, and in bag3-/- mutants when compared to bag3+/+ and bag3+/- embryos ().
Figure 3. Quantification of muscle function in BAG3 myofibrillar myopathy models. (A,B) Quantification of the maximum acceleration, in m/s2, recorded from touch-evoked response assays in 48 hpf Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP]. (C,D) Quantification of distance, in mm, traveled by 6 dpf Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP]. (E) Maximum acceleration, in m/sec2, of bag3+/+, bag3+/-, and bag3-/-. (F) Distance traveled, in mm, by bag3+/+, bag3+/-, and bag3-/-. (G,H) Distance traveled, in mm, by Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] on bag3+/+, bag3+/-, and bag3-/- background. Error bars represent ± SEM for 3–4 biological replicates. The number of fish in each replicate is documented in Table S3. *p < 0.05, ***p < 0.01 calculated using a mixed linear model
![Figure 3. Quantification of muscle function in BAG3 myofibrillar myopathy models. (A,B) Quantification of the maximum acceleration, in m/s2, recorded from touch-evoked response assays in 48 hpf Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP]. (C,D) Quantification of distance, in mm, traveled by 6 dpf Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP]. (E) Maximum acceleration, in m/sec2, of bag3+/+, bag3+/-, and bag3-/-. (F) Distance traveled, in mm, by bag3+/+, bag3+/-, and bag3-/-. (G,H) Distance traveled, in mm, by Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] on bag3+/+, bag3+/-, and bag3-/- background. Error bars represent ± SEM for 3–4 biological replicates. The number of fish in each replicate is documented in Table S3. *p < 0.05, ***p < 0.01 calculated using a mixed linear model](/cms/asset/16e219f7-1e58-4184-83a9-139201c743cf/kaup_a_1833500_f0003_oc.jpg)
We also measured, at 6 dpf, the distance they have swum, which is an indicator of muscle performance. While the distance swam by Tg [BAG3-eGFP] () and Tg [BAG3P209L-eGFP] () was comparable to their respective sibling controls, both bag3+/- and bag3-/- were found to have significant reductions in distance swam (). A 50% reduction or complete loss of bag3 is therefore sufficient to cause impaired muscle performance.
We next examined the capacity of WT BAG3 and BAG3P209L to rescue the muscle impairment observed in the loss of function model. Remarkably, expression of WT BAG3-eGFP or BAG3P209L-eGFP in bag3+/- and bag3-/- larvae was sufficient to rescue the phenotype resulting from reduction of Bag3 (). These results not only confirmed that the muscle weakness seen in bag3−/- larvae is due to loss of Bag3, but they also demonstrated that BAG3P209L-eGFP is functional and capable of preventing muscle weakness associated with the loss of Bag3.
bag3 mutants have impaired autophagy, potentially explaining their decreased muscle function
Given that fiber disintegration was seen in 24 hpf bag3−/- embryos, we examined if this structural failure was responsible for the muscle weakness observed at 6 dpf. To visualize the muscle structure, we crossed the bag3 mutant to a transgenic strain in which GFP tagged FLNC is specifically expressed in muscle [Citation27]. Live imaging of the muscle structure in larvae revealed no gross muscle defects in bag3-/- larvae (Figure S2A), indicating that fiber disintegration, as seen in the methyl cellulose-treated embryos at 24 hpf, was not responsible for muscle weakness in the loss of function model. The 6 dpf fish were, however, still susceptible to muscle damage following increased load as 6 dpf FLNC-eGFP expressing bag3-/- larvae incubated in methyl cellulose, displayed infrequent loss of structural integrity, which was not observed in bag3+/+ and bag3+/- larvae (Figure S2A).
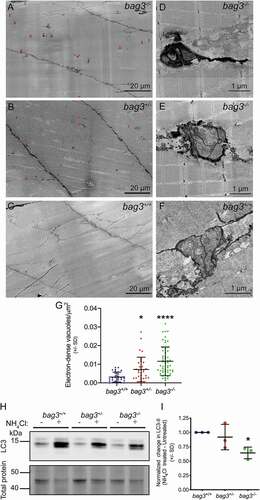
Since we identified no gross defects in the bag3-/- larvae not incubated in methyl cellulose, we further examined muscle structure using electron microscopy. While no sarcomeric defects were evident at an ultrastructural level, we observed large, electron-dense, double membranous, autophagic vacuoles at the sarcolemma of bag3-/- larvae compared to bag3+/+ and bag3+/- larvae (). The structure of the vacuoles seen in bag3-/- fish was similar to that of bag3+/+ fish however, in bag3-/- larvae, the compartments were more darkly stained and often had protrusions extending into the muscle fiber (). Quantification of the vacuoles revealed a small but significant increase in the density of autophagic vacuoles in bag3+/- larvae compared to bag3+/+, and dramatic increase in the density of vacuoles in bag3-/- larvae (). We also examined muscle ultrastructure in 6 dpf Tg [BAG3P209L-eGFP] embryos. We observed no defects in sarcomeric structure, and the density of autophagic vacuoles was comparable to that of Tg [BAG3-eGFP] embryos (Figure S3). Taken together, these results suggested that the loss of Bag3, but not the expression of BAG3P209L, results in an impairment in autophagy. We therefore examined autophagic flux in 6 dpf bag3+/+, bag3+/-, bag3-/- larvae by assessing the difference in LC3 accumulation between untreated and NH4Cl-treated larvae. We observed a significant reduction in LC3 accumulation in bag3-/- compared to bag3+/+ larvae (), demonstrating that loss of Bag3 resulted in reduced autophagic capacity, which may have contributed to the functional deficit observed following loss of Bag3.
Figure 4. Ultrastructural characterization of muscle in 6 dpf Bag3-deficient zebrafish embryos. While the sarcomeric structure of bag3−/- (A) is undistinguishable to that of bag3+/- (B) and bag3+/+ (C), bag3-/- additionally have an increased prevalence in sarcolemma associated, double membrane, electron-dense autophagic vacuoles (arrowhead). (D,E) Autophagic vacuoles in bag3-/- embryo are darkly stained, have multiple compartments and occasionally have protrusions extending into the muscle fiber. (F) Autophagic vacuoles in bag3+/+ embryo have multiple compartments but are not electron-dense. (G) Density of autophagic vacuoles in bag3+/+, bag3+/-, and bag3-/- embryos depicted as number of vacuoles/μm2. Error bars indicate SD. * p < 0.05, ****p < 0.0001 calculated using a Kruskal-Wallis test with Dunnett’s post-hoc multiple comparison correction test. (H) Western blot for LC3 and Direct blue stain for total protein (loading control) on protein lysates obtained from bag3+/+, bag3+/-, and bag3-/- with or without ammonium chloride (NH4Cl) treatment. (I) The autophagic flux determined by normalizing LC3-II levels to that of total protein. The change in LC3-II levels following NH4Cl treatment is presented relative to the levels in the respective untreated groups. Error bars indicate SD. * p < 0.05 calculated using a one-way ANOVA with Dunnett’s post-hoc multiple comparison correction test
Myofibrillar myopathy patients also have an impairment in autophagy
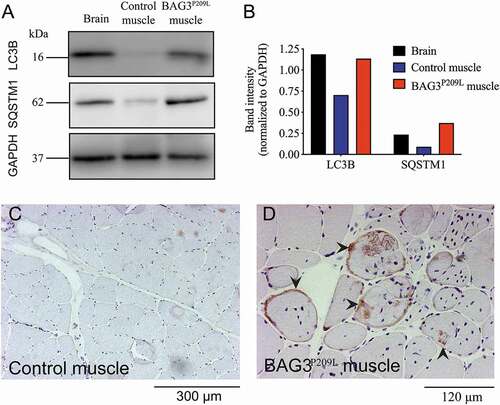
Given that we observed impaired autophagy in our zebrafish model of BAG3 myofibrillar myopathy, we sought to investigate if autophagy was also impaired in myofibrillar myopathy patients. Western blot experiments revealed accumulation of SQSTM1/p62 and LC3 in BAG3 myofibrillar myopathy patient muscle compared to a healthy control (). Additionally, using immunohistochemistry, we observed abnormal accumulation of SQSTM1 within muscle cells of BAG3P209L patients () and in myofibrillar myopathy patients carrying mutations in DES, MYOT, or LDB3 (Figure S4). Taken together, as identified in the zebrafish model, impaired autophagy is a feature of BAG3-related and other myofibrillar myopathies.
Figure 5. Defective autophagy is evident in BAG3P209L myofibrillar myopathy patients. (A) Western blot for LC3 and SQSTM1 in control and BAG3P209L patient. Quantification of LC3 and SQSTM1 (B) reveals accumulation in a BAG3P209L patient but not in control muscle. Abnormal SQSTM1 accumulation (arrowheads) is observed in patient (D) muscle cells but not in control samples (C)
Increasing autophagy significantly reduces BAG3P209L protein aggregates
Our data suggests that the expression of BAG3P209L and the formation of protein aggregates does not directly cause muscle weakness, with loss of BAG3 due to the sequestration of BAG3 within aggregates resulting in impaired autophagy and subsequent muscle weakness. As such, we hypothesized that upregulation of autophagy might be beneficial for the treatment of BAG3 myofibrillar myopathy by not only promoting the clearance of aggregates and preventing any further sequestration of functional protein, but also by compensating for the reduced levels of basal autophagy in affected muscle fibers. We therefore performed a screen of 71 known autophagy promoting drugs to identify the most effective at removing aggregates in our BAG3P209L-eGFP expressing fish. 32 hpf Tg [BAG3P209L-eGFP] embryos were incubated in one of 71, randomly assigned, drugs or in unsupplemented E3 embryo medium or DMSO-supplemented E3 embryo medium. Following 16 h of drug treatment the embryos were fixed, imaged, and the number of aggregates was quantified using an automated aggregate quantification tool. Eight of the 71 compounds tested were toxic at the dose used and were not further characterized (Figure S5). Six compounds, although predicted to increase autophagy, were found to significantly increase the number of aggregates when compared to DMSO control-treated embryos (Figure S4). We identified nine drugs that were highly effective at reducing aggregate numbers when compared to DMSO treatment ( and S5).
Figure 6. Pharmacological stimulation of autophagy promotes the clearance of aggregates. (A) Graph depicting normalized aggregate number of the nine drugs that significantly (p < 0.05) reduce protein aggregation in Tg [BAG3P209L-eGFP] embryos, compared to DMSO control. Amiodarone and metformin are the two drugs currently approved for clinical use by the Federal Drug Association. (B–E) Representative confocal images of untreated 48 hpf Tg [BAG3P209L-eGFP] embryos or treated with DMSO, amiodarone or metformin, the latter two resulting in reduced protein aggregation. Error bars represent ±SEM with three independent biological replicates. The number of fish in each replicate is documented in Table S4
![Figure 6. Pharmacological stimulation of autophagy promotes the clearance of aggregates. (A) Graph depicting normalized aggregate number of the nine drugs that significantly (p < 0.05) reduce protein aggregation in Tg [BAG3P209L-eGFP] embryos, compared to DMSO control. Amiodarone and metformin are the two drugs currently approved for clinical use by the Federal Drug Association. (B–E) Representative confocal images of untreated 48 hpf Tg [BAG3P209L-eGFP] embryos or treated with DMSO, amiodarone or metformin, the latter two resulting in reduced protein aggregation. Error bars represent ±SEM with three independent biological replicates. The number of fish in each replicate is documented in Table S4](/cms/asset/dbd6fee6-ebc6-45b9-900e-095c7caf1c10/kaup_a_1833500_f0006_oc.jpg)
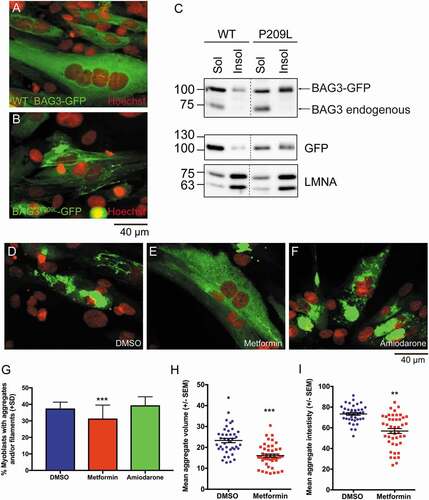
Two of these drugs, metformin and amiadarone are currently approved by the FDA, which would allow for rapid translation to clinical use, therefore we further investigated their efficacy in human myoblasts. Similar to the zebrafish experiments, overexpression of BAG3P209L-GFP, but not WT BAG3-GFP, resulted in protein aggregation (). While the majority of WT BAG3-GFP localized to the soluble protein fraction, the majority of BAG3P209L-GFP was in the insoluble fraction (). Having shown that BAG3P209L forms protein aggregates in human myoblasts, we examined the ability of metformin and amiodarone to reduce them. While amiodarone treatment had no effect on protein aggregation, metformin treatment resulted in a significant reduction in the number of aggregate-containing myoblasts, with the aggregate volume and intensity also significantly reduced ().
Figure 7. Metformin reduces protein aggregation in human myoblasts. (A,B) Confocal images of human myoblasts transfected with BAG3-eGFP or BAG3P209L-eGFP with protein aggregation evident in the later. (C) Western blot showing that while majority of BAG3-eGFP is found within the soluble lysate, a large proportion of BAG3P209L-eGFP is localized to the insoluble fraction. (D-F) Confocal images showing protein aggregation in human myoblasts following 48 h treatment with DMSO (n = 1719), metformin (n = 1664) or amiodarone (n = 970). (G) Metformin but not amiodarone results in a significant reduction in the percentage of myoblasts with aggregates and/or filaments. ***p < 0.001 as determined using a chi-square test (p = 0.0001, chi-square = 17.88, df = 2). Metformin (n = 44) additionally significantly reduces aggregate volume (H) and intensity (I) compared to DMSO (n = 41) treated myoblasts, as determined using a T-test. (****p < 0.001, t = 5.561, df = 83)
Metformin reduces fiber disintegration, and rescues the functional deficit observed in bag3 mutants
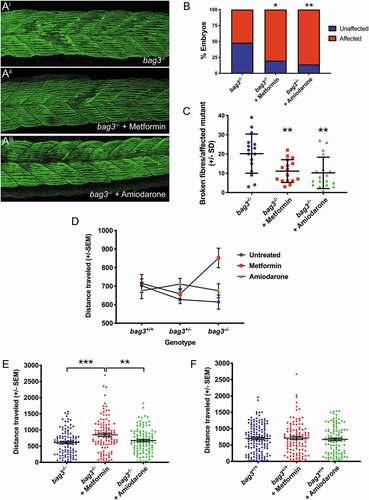
As amiodarone and metformin significantly ameliorated the protein aggregate aspect of myofibrillar myopathy in zebrafish or zebrafish and human myoblasts respectively, we wished to further examine their potential to prevent fiber disintegration and improve muscle function, which is ultimately the desired outcome. As such, 5-somite staged bag3-/- mutant embryos were dechorionated and incubated in unsupplemented E3 embryo medium or in E3 medium containing metformin or amiodarone. Following a 12 h treatment, the embryos were placed in methyl cellulose, with or without the drug, for 2 h to promote fiber disintegration, and subsequently fixed and labeled to examine fiber integrity (). While there was a significant increase in the proportion of embryos displaying fiber disintegration following metformin or amiodarone treatment (), both drugs resulted in a striking reduction in the frequency of fiber disintegration, when compared to untreated bag3-/- embryos ().
Figure 8. Metformin prevents fiber disintegration and improves muscle function in bag3 mutant embryos. (A) Representative maximum projection images of Myosin-labeled 26 hpf; untreated, metformin-treated, and amiodarone treated embryos following incubation in methyl cellulose. (B) Metformin or amiodarone treatment results in an increase in the proportion of bag3-/- embryos that display damaged muscle fibers, as determined using a binomial test. (C) Quantification of disintegrated fibers in vehicle-, metformin-, and amiodarone-treated bag3-/- embryos fish reveals that both drug treatments result in a significant reduction as calculated using a mixed linear model. (D) Analysis of distance traveled (in mm) using a linear mixed model identifies a significant interaction between genotype and drug treatment (p = 0.005). (E) Metformin, but not amiodarone, significantly increases the distance swam (in mm) by bag3-/- fish, compared to untreated bag3-/- controls. (F) The distance swam (in mm) by metformin- or amiodarone-treated bag3+/+ fish is not significantly different to that of untreated bag3+/+ controls. Error bars represent ±SEM with four-six independent biological replicates. The number of fish in each replicate is documented in Table S5. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 based on a linear mixed model
To confirm that this reduction was due to an increase in autophagy, we examined LC3 accumulation in 28 hpf untreated, NH4Cl-treated, metformin & NH4Cl-treated, and amiodarone & NH4Cl-treated WT embryos. We observed an increase in LC3 accumulation in both metformin & NH4Cl-treated and amiodarone & NH4Cl-treated WT embryos, compared to embryos treated with NH4Cl alone (Figure S6A,B), demonstrating that a 14 h treatment with metformin or amiodarone was sufficient to upregulate autophagy in vivo, and subsequently reduce fiber disintegration.
We further validated the therapeutic potential of metformin and amiodarone by examining their capacity to improve muscle function. Our initial studies revealed that the drug doses used at the 36 hpf and 5-somite stages were toxic at 6 dpf. We therefore conducted a drug toxicity assay to determine a well-tolerated, nontoxic dose, to use on 6 dpf larvae for our functional assays. We tested the effect of 5 concentrations of metformin and amiodarone on fish survival and swimming performance. While larvae treated with the highest concentration of metformin (10 μM) showed no signs of toxicity, an equivalent 10 μM dose of amiodarone resulted in larval death. Treatment with 1 μM of amiodarone had no toxic effects and as such, this concentration was used for subsequent experiments (Figure S7).
Having identified suitable nontoxic levels, we treated 5 dpf old bag3-/- with 1 μM amiodarone, 10 μM metfromin, or unsupplemented E3 embryo medium, for 16 h after which the locomotion assays were performed. A mixed linear model revealed a significant interaction between genotype and drug treatment (p = 0.005; , Table S5). While amiodarone treatment had no significant effect on muscle function of bag3−/- larvae, metformin treatment dramatically improved the distance swam by bag3-/- fish compared to untreated and amiodarone-treated bag3−/- (). Remarkably, the mean distance swam by metformin-treated bag3-/- larvae (852 ± 52.9 mm SEM) was no longer different from untreated bag3+/+ larvae (702 ± 36.7 mm SEM) indicating that metformin completely restored muscle function in the mutant fish. To examine if this increased swimming performance in metformin-treated bag3−/- larvae was due to a general increase in motility of the fish, irrespective of the bag3 mutation, we compared the locomotion of untreated and metformin-treated bag3+/+ larvae. Treatment of bag3+/+ with metformin had no effect on their swimming distance, demonstrating that the improvement in muscle function seen in bag3−/- was specific, and not a generalized response (). Together, these results demonstrated that metformin specifically improves swimming performance in bag3-/- larvae. To determine if this improvement was due to increased autophagy, we assessed the autophagic flux in 6 dpf bag3+/+, bag3+/-, and bag3−/- embryos with metformin treatment. While the accumulation of LC3 in bag3+/+ and bag3+/- embryos treated with NH4Cl alone were comparable to those treated with metformin and NH4Cl, LC3 levels in bag3-/- embryos treated with metformin and NH4Cl was higher than bag3−/- embryos treated with NH4Cl alone (Figure S6C,D). These results highlighted that metformin stimulates autophagy resulting in increased muscle function in bag3−/- larvae.
Discussion
In the current study, we have generated novel zebrafish models of BAG3 myofibrillar myopathy, which recapitulate the hallmark features of the disease. Our data collectively demonstrated that the FDA-approved, autophagy stimulating drug, metformin reduced protein aggregation, prevented fiber disintegration, and rescued muscle weakness in our model, making it a very promising therapy for the treatment of BAG3 myofibrillar myopathy, BAG3 cardiomyopathy, and the wider group of myofibrillar myopathies.
Using the models we have generated, we revealed that the overexpression of BAG3P209L results in protein aggregation, with the majority of BAG3P209L being in the insoluble fraction. Despite this, BAG3P209L was functional and could rescue the loss of function. This is consistent with the proposal that the formation of protein aggregates results in sequestration of functional BAG3, both BAG3P209L and WT BAG3, ultimately causing insufficiency [Citation26] and analyses showing BAG3P209L is correctly folded and functional [Citation28]. While the aggregation of BAG3P209L is the initial trigger for the disease, its overexpression did not impair autophagy or result in muscle weakness in the early embryo. The loss of Bag3, on the other hand, resulted in fiber disintegration and impaired muscle function, due to reduced autophagic activity. We further confirmed that this reduced autophagic activity is evident in skeletal muscle from BAG3 MFM patients, consistent with previous reports of defective autophagy in BAG3 cardiomyopathy [Citation29,Citation30]. The BAG3P209L-expressing fish therefore provides a model for the early stages of BAG3 myofibrillar myopathy, where aggregates are present, but muscle performance is unaffected, and the loss of function bag3 mutant provides a model for the later stages of the disease in which fiber disintegration and muscle weakness are evident.
Given that the presence of aggregates triggers the loss of Bag3 and associated muscle weakness, we examined autophagy upregulation as a strategy to promote aggregate clearance. We tested 71 compounds all of which have been previously shown to stimulate autophagy. Despite this, only 13% of compounds significantly reduced protein aggregates. While this relatively low number of successful hits may be attributed to the use of all compounds at a single concentration of 10 µm, it may also be explained by the selective nature of autophagy stimulating drugs and their mechanism of action. For example, lithium [Citation31], rilmenedine [Citation32], and trehalose [Citation33,Citation34] have previously been shown to reduce protein aggregation in models of neurodegenerative diseases, although they have failed to show beneficial effects in patients, potentially due to difficulties in achieving therapeutic levels. However, in the current study, none of these drugs had an effect on the number of BAG3P209L aggregates, suggesting that autophagy stimulating drugs may be selective in the type of aggregates they target and/or the tissue in which they act. There is, therefore, a need for specific models to evaluate therapies for each of the protein aggregate disorders.
Of the 9 drugs that successfully reduced protein aggregates in the zebrafish BAG3P209L-expressing fish, two compounds, amiodarone and metformin, are currently approved for clinical use by the US FDA. Amiodarone is known to induce neuropathy in some cases and, even more rarely, myopathy [Citation35,Citation36]. As a result, amiodarone would not be indicated for treating BAG3 myopathy. Metformin, on the other hand, is the most effective compound we identified and is the most widely prescribed drug for the treatment of diabetes, with extensive documentation on its tolerability and safety and an outstanding risk to benefit profile. Metformin has been shown to exert its therapeutic benefits through several different pathways including AMP protein kinase-dependent and independent mechanisms, modulating mitochondrial oxidative phosphorylation (reviewed in [Citation37]), and most recently by altering the gut microbiome [Citation38]. While we believe the increase in autophagy flux following metformin treatment is responsible for the improvement in muscle pathology and function that was seen in our BAG3 myofibrillar myopathy models, the modulation of these other pathways may also be contributing.
Remarkably, in addition to clearing protein aggregates, metformin prevented the fiber disintegration and muscle weakness seen in the loss of function bag3 mutant. Surprisingly, metformin did not rescue muscle function in bag3 heterozygous fish. This is consistent with the analysis of autophagic flux following metformin treatment, which identified that only mutant fish showed a dramatic increase in autophagy. We have previously shown that the presence of Bag3 can inhibit other forms of autophagy [Citation39]. It is therefore possible that the reduction in Bag3 in heterozygous fish was sufficient to impact muscle function and cause a mild reduction in fiber integrity, but the remaining Bag3 could block other forms of autophagy, thus preventing the response to metformin. In bag3 mutants, however, the block on autophagy was relieved, and as a result, metformin could efficiently increase autophagy and significantly improve muscle function and integrity. Metformin treatment in BAG3 myofibrillar myopathy patients could therefore have dual benefits: increasing autophagy, promoting aggregate clearance and preventing loss of functional BAG3 by sequestration; additionally, the increase in autophagy levels would compensate for the reduced autophagic levels due to reduction in BAG3. Metformin therefore has potential to address both of the muscle phenotypes associated with the disease and is readily applicable to the treatment of BAG3 myofibrillar myopathy. In addition, given the conserved impairment in autophagy, metformin may also be suitable for the treatment of cardiomyopathy resulting from mutations in BAG3, and other myofibrillar and aggregate myopathies.
Materials and methods
Fish maintenance
All experiments were carried out on embryos of TU/TL background. Fish were anesthetized using Tricaine methanesulfonate (3-amino benzoic acidethylester; Sigma Aldrich, E10521) at a final concentration of 0.16% in E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl, 0.33 mM MgSO4, 0.00004% [v/v] methylene blue in water, pH 7.2).
Generation of transgenic and mutant strains
Transgenic constructs were assembled using the multisite gateway cloning kit [Citation40] with the actb1 (actin, beta 1) promoter in the 5ʹ entry position, loxP-mCherry-pA-loxP (Genbank accession: KF753698) in the middle entry position, and C-terminal, eGFP tagged, human full-length WT BAG3 or BAGP209L [Citation26] in the 3ʹ entry position. The constructs were injected at 25 ng/μl into one-cell-stage embryos along with transposase RNA (25 ng/μl) that was synthesized from the pcs2FA-transposase vector [Citation40] using the mMessage machine Sp6 kit (Ambion, AM1340). Transgenic strains generated were Tg(actb1:loxP-mCherry-pA-loxP:Hs.BAG3-eGFP), Tg(actb1:loxP-mCherry-pA-loxP:Hs.BAG3P209L-eGFP). Crossing of either of these two strains to the Tg(actc1b:iCre) [Citation41] strain results in the excision of the loxP-mCherry-pA-loxP cassette and subsequent generation of Tg(actb1:Hs.BAG3-eGFP) and Tg(actb1:Hs.BAG3P209L-eGFP) hereafter referred to as Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] respectively. The Tg [FLNC-eGFP] line is described in [Citation27]. The bag3 mutant strain was generated using CRISPR-Cas9 technology as per [Citation42] with the following amendments: the guide RNA was synthesized according to [Citation43]. The guide RNA was injected at 120 ng/μl into the one-cell-stage embryos, along with Cas9 protein and Cascade Blue dye (Molecular Probes, D1976). All primers used for the generation and genotyping of the bag3 mutant strain are listed in Table S1.
Immunofluorescence and western blot experiments
Zebrafish immunofluorescence experiments were performed accordingly to previously published protocols [Citation44]. Primary antibodies used in this study were anti-eGFP (Invitrogen, A-11122; 1:150), anti-MYH1 (Developmental Studies Hybridoma Bank, A4.1025, 1:10), and anti-ACTN2/Actinin2 (Sigma Aldrich, A7811; 1:100). Stained embryos were dehydrated in 100% ethanol, following which they were cleared in benzyl alcohol/benzyl benzoate (2:1) and mounted in DPX (Sigma Aldrich, 44581). Super-resolution images were subsequently acquired on a Zeiss LSM 980 Airyscan 2 microscope. For in vivo confocal microscopy, live embryos were mounted in 1% low melting point agarose and imaged using the Zeiss LSM 710 confocal microscope. The maximum intensity projections were obtained using Fiji (http://fiji.sc).
For zebrafish western blot assays, protein lysates were obtained as per [Citation45] from independent biological replicates, each containing multiple embryos, and quantified using the Qubit fluorometric quantification (Thermo Fisher Scientific). A 30 μg aliquot of each sample, along with reducing agent (Life Technologies, B0009) and protein loading dye (Life Technologies, NP0007), was heated at 70°C for 10 min, separated by SDS-PAGE on NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific, NP0335BOX), and transferred onto PVDF membrane (Millipore, IPVH00010). Following transfer, the membrane was blocked with 5% skimmed milk in PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 and 0.1% [w:v] Tween 20 [Sigma Aldrich, P9416]) and subsequently probed with anti-LC3 (Cell Signaling Technology, 12741; 1:2000), anti-TUBB/beta-tubulin (Abcam, ab6046, 1:5000), or anti-GFP (Sigma Aldrich, 11814460001, 1:1000), washed and incubated with HRP-conjugated secondary antibody (1:10 000; Southern Biotech, 4010-05). Immunoblots were developed using ECL prime (GE Healthcare, GERPN2232) and imaged using a chemiluminescence detector (Vilber Lourmat). The membrane was subsequently stripped by incubating in 1X stripping buffer (200 mM glycine, 0.1% SDS, 1% Tween 20 [Sigma Aldrich, P9416], pH 2.2) twice for 10 min, washed in PBST, and stained with Direct blue (Sigma Aldrich, 212407) as described previously [Citation46] to detect total protein. The blot images were quantified using Image Lab software (Bio-Rad), and values for each genotype were normalized to WT controls to allow comparison between independent biological replicates. A one-way ANOVA was used to test for significant changes in LC3 accumulation.
Details on human muscle biopsies are provided in Table S7. For immunohistochemistry, sections were re-cut from stored frozen muscle (−80°C). Cryo-sections (8-μm thick) were air-dried and fixed in cold acetone at −20°C for 10 min. Staining was carried out using an automated immunostainer (BenchMark XT, Ventana medical systems) as per manufacturer’s instructions with anti-SQSTM1/p62 antibody (1:500; BD Biosciences, 610833). For western blot the biopsy samples were isolated from the patient and homogenized in a lysis buffer containing 20 mM Tris-HCl, pH 7.8, 140 mM NaCl, 1 mM EDTA, 0.5% NP40 (MP Biomedicals INC, 198596), 1 mM phenylmethylsulfonyl fluoride (Sigma Aldrich, 11359061001), and complete protease inhibitor mixture (Roche Diagnostics, 11836153001), with a POTTER S Homogenizer (B.Braun Biotech International-Sartorius group). Samples were pulsed 5 times for 5 s each at 110 g. Samples were then passed 5 times through a 30.5-gauge needle to disrupt the nuclei, then incubated at 4°C for 15 min and finally centrifuged at 17,900 g for 15 min at 4°C. Total protein concentration was determined according to Bradford’s method. Samples were resolved on 12% polyacrylamide gel for LC3B (1:1000, Cell Signaling Technology) and SQSTM1/p62 (1:1000, Millipore, Temecula, CA, USA) antibodies and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). We also determined the expression for each sample of anti-GAPDH (1:600; Sigma Aldrich, G9545) housekeeping protein. Detection was performed with anti-rabbit and anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies, respectively (DakoCytomation, P0217 and P0260), followed by enhanced chemiluminescence (ECL) development (Amersham Biosciences, Piscataway, NJ, USA). Bands were visualized by Odyssey system. Each sample was evaluated in three independent experiments.
Movement assays
Touch-evoked response assay to examine maximum acceleration and assays to determine distance swam were performed on 2 dpf embryos and 6 dpf larvae respectively, as per [Citation41,Citation47].
Drug screen and automated aggregate quantification
The SCREEN-WELL Autophagy library (ENZO Life Sciences, BML-2837-0100) containing 71-autophagy inducing compounds was used in this study. 32 hpf Tg [BAG3P209L-eGFP] embryos were placed in wells of a 24-well plate (5 embryos per well) following which drug was added to a final concentration of 10 μM in 1 ml volume. E3 embryo media without any drug supplementation, and E3 embryo media containing 0.1% DMSO were used as controls. Embryos were incubated in their respective drugs (or control solutions) for 16 h following which, fixed in 4% paraformaldehyde for 2 h, washed into PBST and mounted in 1% low melting point agarose for imaging using the Zeiss LSM 710 confocal microscope.
A semi-automated workflow for detecting aggregates in three-dimensional confocal image stacks was developed using ImageJ and the ImgLib2 library [Citation48,Citation49]. Briefly, a median filter with a radius of one pixel was applied to the image volume to reduce overall shot noise. To calculate overall fish muscle tissue volume, image stacks were smoothed further by applying a Gaussian filter with a radius of five pixels and then segmented using an Otsu automatic threshold [Citation50]. The fish muscle tissue was delineated as the largest connected component in the field of view resulting in exclusion of small spurious segmented particles. Aggregates were enhanced by applying a Difference of Gaussian (DoG) blob enhancing filter to the median filtered image volume, where the estimated radius of the blob (i.e., aggregate) was set at 1 μm. Aggregates were then detected in the DoG filtered image volume using a local neighborhood maxima detection algorithm with a minimum peak intensity cutoff of 2,500 (in a 12-bit grayscale intensity range). Detected aggregates were masked using the segmented muscle tissue region to select only the aggregates within the tissue. Aggregate density was calculated as the number of aggregates per unit volume of tissue. Source code and a current build for the ImageJ/Fiji Aggregate plugin can be found at: https://gitlab.erc.monash.edu.au/mmi/aggregate. All drugs were tested in triplicate with the treatment and imaging order randomized within each replicate, and the identity of treatment blinded. Fish for each replicate were derived from different parents. This semi-automated workflow was also used to quantify the number of aggregates in 2 dpf, untreated Tg [BAG3-eGFP] and Tg [BAG3P209L-eGFP] embryos (Figure S1D).
Drug concentrations
For the drug screen and fiber disintegration rescue experiments, all compounds were used at 10 µM. For swimming analysis at 6 dpf a dose-toxicity assay identified optimal concentrations of 10 µM for metformin (Abcam, ab120847) and 1 µM for amiodarone (Abcam, ab141444). To examine autophagic flux in drug-treated embryos at 24 hpf (fiber disintegration rescue experiments) ammonium chloride was used at 100 µM for 4 h. To examine autophagic flux in metformin-treated 6 dpf bag3 fish, 100 µM ammonium chloride and 10 µM metformin was added for 12–14 h.
Muscle integrity and swimming performance
For experiments on the effect of drug treatment on distance swam, 5 dpf larvae were placed in individual wells of a 24-well plate following which an epMotion liquid handling robot (Eppendorf) was used to automatically dispense drugs into individual wells to a total volume of 1 ml, in a randomized order. The fish were incubated in their respective drugs for 14–16 h after which the locomotor activity assays described above were performed. To examine the effect of drug treatment on muscle fiber integrity, 5 somite embryos were dechorionated and placed in 25 ml of E3 embryo medium, following which drugs were dispensed. After 12–14 h of incubation, the embryos were incubated in 1% methyl cellulose for 2 h and subsequently fixed and labeled for Myhc.
Electron microscopy
Zebrafish were fixed according to standard procedures in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer. Post-fixed with 1% OsO4, 1.5% K3Fe(III)(CN)6. Dehydration was done with ethanol and the zebrafish were flat embedded in Epon 812. Ultrathin sections of 70 nm were cut on a Leica Ultracut UCT7 and stained with uranyl acetate and lead citrate. Large area EM tile sets were taken on a FEI NovaNanoSEM 450 equipped with an ETD secondary electrons in-lens detector set at 10 kV and a STEM II (HAADF) detector set at 30 kV. MAPS 2.1 software was used to create the tile sets. High resolution EM imaging was done on a Hitachi 7500 TEM and a FEI Tecnai 12 TEM. Fiji was used to trace individual muscle cells and count the number of electron-dense autophagic vacuoles, following which the density of vacuoles was calculated.
Cell culture
Anonymous immortalized healthy control myoblast lines were provided by Eric A. Shoubridge (Department of Human Genetics, McGill University, Quebec, Canada). Briefly, these cell lines were established by cultivating human myoblasts from biopsy material and transducing them with retroviral vector expressing HPV16-E6/E7 and with another one expressing the catalytic component of human TERT (telomerase reverse transcriptase; hTERT), to increase their lifespan in vitro [Citation51,Citation52]. The resulting immortalized human myoblast cell line was cultivated in growth medium (GM, SkMAX skeletal muscle medium with supplements [Wisent, Kit #301-061-CL] containing 20% heat-inactivated fetal bovine serum [GIBCO, 12,483–020]) in a humidified incubator at 37°C, 5% CO2. Cells were passaged using 0.05% trypsin when they have reached 70–80% confluency (about every 3–4 d) and kept in culture not longer than passage 15.
Differentiation and transduction of immortalized human myoblasts
The immortalized human myoblasts were plated at a density of 1 × 106 cells per 35 mm dish on plastic in 2 ml growth medium. To induce differentiation into myotubes, cells were washed once with 1xPBS, and differentiation medium (DMEM containing 2% horse serum [GIBCO, 16050-122]) was added for the indicated time of differentiation. At day 2 (D2), cells were transduced with Ad-BAG3-GFP WT or Ad-BAG3P209L-GFP, or with 1 PFU of Ad-GFP only as control, as described [Citation53]. Briefly, cells were incubated with adenoviruses in 400 µl of medium for 1 h before adding 1.6 ml medium. The next day, cells were washed once with 1xPBS and 2 ml of differentiation medium was added. Different batches of horse serum were tested for transduction efficiency. The recombinant adenoviruses (Ad-BAG3-GFP, Ad-BAG3P209L-GFP and Ad-GFP) were custom made by Welgen Inc, Worcester, MA, USA and were amplified in the HEK293VR (a gift from Philip E. Branton, McGill University), as described before [Citation54]. All recombinant adenoviruses were confirmed by sequencing of the inserted sequences. Virus titers were determined using the AdenoX Rapid titer kit (Clontech Laboratories, 631028) following the manufacturer’s instructions.
Myoblast antibodies and chemicals
The following antibodies and drugs were used: rabbit anti-BAG3 LP10 was raised against a C-terminal peptide (SSMTDTPGNPAAP) of human BAG3 fused with glutathione S [Citation16]; metformin hydrochloride (Abcam, ab120847) and amiodarone (Abcam, ab141444) were used at 100 µM and 15 µM respectively for 48 h; Hoechst Bisbenzimide H33342 (Sigma, B2261).
Myoblast immunofluorescence and immunoblotting
Immortalized human myoblasts were washed twice with 1 ml Luftig buffer (0.2 M sucrose [Bio Basic Canada Inc, 57-50-1], 35 mM PIPES [Sigma Aldrich, P7657-1KG], pH 7.4, 5 mM EGTA, 5 mM MgSO4) and fixed in 4% paraformaldehyde in PBS for 25 min at 37°C. DNA was stained with a cell-permeable Hoechst in PBS for 15 min at room temperature. Specimen were washed 2x with PBS-M (1xPBS containing 1 mM MgCl2) and post-fixed in PBS+3.7% formaldehyde for 20 min at 37°C [Citation53]. Cells were subsequently washed twice with PBS-M, once with H2O and mounted. Epifluorescence images were acquired with an AxioObserver Z1 system using a 40x Plan-Neofluoar 0.6 NA objective and a charge-coupled device (CCD) camera Axiocam MRm controlled by the Zen software (Carl Zeiss). Confocal microscopy of fixed cells was performed with a Perkin Elmer UltraVIEW Spinning Disc Confocal (40x 0.75 NA), equipped with an EMCCD cooled charge-coupled camera at −50°C (Hamamatsu Photonics K.K) and driven by Volocity software version 6.01.
The Volocity software version 6.0 (Quorum Technologies) and Image J 1.48v (National Institute of Health) software were used for processing on entire images before cropping to emphasize the main point of the image. Processing was limited to background subtraction, brightness and contrast adjustment, and deconvolution.
For immunoblotting, equal amounts of proteins were loaded on SDS-PAGE and analyzed by western blot as described before [Citation55]. Protein concentrations were determined using the DCM Protein Assay Kit (Bio-Rad, #500-0116).
Triton-soluble and -insoluble cell fractionation
Immortalized human myoblasts were washed once with 2 ml pre-warmed 1xPBS and detached with 1 ml pre-warmed PBS-Citrate-EDTA (13.6 mM sodium citrate, 0.6 mM EDTA pH8 in 1xPBS) for 3–5 min in a humidified incubator at 37°C, 5% CO2. Cells were carefully transferred into a 1.5 ml plastic tube and centrifuged for 5 min at 100 g at 4°C. The supernatant was removed and the pelleted cells lysed by 3-cycles of freezing and thawing in 150 µl lysis buffer (20 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 mM EDTA, 0.1% IGEPAL [Sigma Aldrich, I3021], 1 mM DTT, 1x Complete Protease Inhibitor [Roche, 11873580001]). Cell extracts were centrifuged for 5 min at 13,400 g, 4°C and the triton-soluble fraction was collected in SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 2.3% SDS, 10% glycerol [FisherBiotech, BP229-4], 5% beta-mercaptoethanol [Bio Basic Canada Inc, MB0338], 0.005% bromophenol blue [Sigma Aldrich, B-6131], 1 mM phenylmethylsulfonyl fluoride [Sigma Aldrich, P7626]). The triton insoluble pellet was washed 1x with 150 µl lysis buffer, centrifuged for 5 min at 13,400 g at 4°C and resuspended in SDS sample buffer. After aspirating the supernatant, the pellet was resuspended in 150 µl lysis buffer and 75 µl 3xTEX was added. Protein quantification was performed on the soluble fraction and equal amounts of soluble and insoluble fractions were loaded on SDS-PAGE and analyzed by western blot.
Analysis of aggregate intensity and volume
Z-stacks of fixed cells were acquired using a Perkin Elmer UltraVIEW Spinning Disc Confocal (40x 0.75 NA). The background was removed for each acquisition before cropping representative cells for each treatment by drawing semi-automatically a line around the selected cells. The P209L aggregates volume and intensity were measured on the complete Z-stack using the Volocity software. The ratio between volume and intensity of aggregates versus the complete cell was quantified.
Analysis of aggregates versus diffuse staining
Epifluorescence images of 40 random fields per condition were acquired with an AxioObserver Z1 system using a 40x Plan-Neofluoar 0.6 NA objective. For each field, the percentage of cells or myotubes containing aggregates and/or filaments was counted semi-automatically using the Image J 1.48v software. Statistical calculations were performed using Prism 6.0 (GraphPad Software) statistical software. The statistical tests used are indicated in the figure legends.
Statistical analysis
GraphPad Prism and SPSS statistics packages were used to analyze data in this study. Note that the number of independent biological replicates examined for each experiment and the number of fish used within each replicate combined with the significance tests used and the associated t/F value, degrees of freedom (df) and exact p values obtained are detailed in Tables S2–6.
Drug screen
To identify drugs that significantly reduced protein aggregation in Tg [BAG3P209L-eGFP] embryos a one way ANOVA statistical analysis was performed with treatment as a fixed effect and replicate as a random effect, and Dunnett’s post-hoc test was used to correct for multiple comparisons.
Movement assays
Outliers, identified as being more than 1.5 times the interquartile range below the 1st quartile or above the 3rd quartile in each treatment group, were omitted, and then linear mixed model fitted to identify significant differences in measure of movement among groups. For experiments examining the ability of the drugs to rescue the movement deficit observed in the mutant fish, models included genotype, and drug where appropriate, as fixed effects and replicate as a random effect. Pairwise comparisons of groups were carried as necessary using least significant difference to correct for multiple comparisons.
Fiber disintegration experiments
To compare the proportions of fish of each genotype displaying fiber disintegration, the proportion of heterozygous and mutant fish demonstrating fiber damage was compared to that observed in WT fish following methyl cellulose treatment using a binomial test (). Similarly, the proportion of mutant fish displaying fiber disintegration in the methyl cellulose assay following amiodarone or metformin treatment was compared to that observed in untreated mutants using a binomial test (). Pairwise comparisons of groups were carried out using Bonferroni correction for multiple comparisons. To compare counts of broken fibers per affected fish among groups, a generalized linear mixed model was fitted using a Poisson distribution and log link, with drug treatment () or genotype () as a fixed effect and fish identification number as an observation-level random effect to handle overdispersion. Pairwise comparisons of groups were carried as necessary using least significant difference to correct for multiple comparisons.
Autophagic flux assays
Changes in LC3 levels between the different genotypes were normalized to the values for WT fish without drug treatment and were compared using a one way ANOVA followed by pairwise Dunnett’s test with the control correcting for multiple comparison correction test.
Compliance with ethical standards
Fish maintenance and handling were carried out as per standard operating procedures approved by the Monash Animal Services Ethics Committee. The generation of transgenic and mutant strains was approved by the School of Biological Sciences Animal Ethics Committee (BSCI/2013/27 and BSCI/2015/06 respectively). This study was performed according to the guidelines of the Committee on the Use of Human Subjects in Research of the Policlinico Hospital of Milan (Milan, Italy) or the Institute of Myology, Pitié-Salpêtriere hospital (Paris, France). Informed consent was obtained from all family members. The authors have no conflicts of interest to declare.
Supplemental Material
Download Zip (55.9 MB)Acknowledgments
We would like to thank Prof Bernard Brais for the myoblast cell lines utilized, Dr Aidan Sudbury for statistical advice, Coco Bayly-Jones for assistance with the maintenance of mutant and transgenic strains, Oleksandr Chernyavskiy and Monash Micro Imaging for assistance with super resolution imaging, and the patients who contributed samples to this study. The hybridoma product A4.1025 was developed by Blau, H.M., obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Raw data and files for statistical analysis are available at Bridges and can be accessed using the following link: https://bridges.monash.edu/projects/Metformin_rescues_muscle_function_in_BAG3_myofibrillar_myopathy_models/60959.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Goldfarb LG, Park KY, Cervenáková L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998 Aug;19:402–403.
- Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998 Sep;20:92–95.
- Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004 Apr;62:1363–1371.
- Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005 Feb;57:269–276.
- Vorgerd M, van der Ven PFM, Bruchertseifer V, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005 Aug;77:297–304.
- Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009 Dec;65:83–89.
- Selcen D, Bromberg MB, Chin SS, et al. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. 2011 Nov;77:1951–1959.
- Pfeffer G, Barresi R, Wilson IJ, et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J Neurol Neurosurg Psychiatry. 2014;85:331–338.
- Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 2012 Apr;44:450–5–S1–2.
- Ghaoui R, Palmio J, Brewer J, et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology. 2016 Jan;86:391–398. Lippincott Williams & Wilkins.
- Homma S, Iwasaki M, Shelton GD, et al. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006 Sep;169:761–773.
- Lee JH, Takahashi T, Yasuhara N, et al. Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene. 1999 Nov;18:6183–6190.
- Takayama S, Xie Z, Reed JC. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem. 1999 Jan;274:781–786.
- Fuchs M, Poirier DJ, Seguin SJ, et al. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J. 2010 Jan;425:245–255.
- Lünemann JD, Schmidt J, Schmid D, et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. 2007 May;61:476–483. Wiley Subscription Services, Inc., A Wiley Company.
- Carra S, Seguin SJ, Lambert H, et al. HspB 8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008 Jan;283:1437–1444.
- Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010 Jan;20:143–148.
- Ulbricht A, Eppler FJ, Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013 Feb;23:430–435.
- Gamerdinger M, Kaya AM, Wolfrum U, et al. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011 Feb;12:149–156.
- Lee H, Cherk S, Chan S, et al. BAG3-related myofibrillar myopathy in a Chinese family. Clin Genet. 2012 Apr;81:394–398.
- Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011 Mar;88:273–282. The American Society of Human Genetics.
- Feldman AM, Begay RL, Knezevic T, et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J Cell Physiol. 2014 Nov;229:1697–1702.
- Franaszczyk M, Bilinska ZT, Sobieszcza S-ME-MG, et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: four novel mutations and a genotype-phenotype correlation. J Transl Med. 2014 July;12:192.
- Hunt L, Atherton J, McGaughran J. Dilated cardiomyopathy - three brothers and a BAG3 mutation. Heart Lung Circ. 2014;23(Suppl 2):e11.
- Odgerel Z, Sarkozy A, Lee H-S, et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul Disord. 2010 July;20:438–442. Elsevier B.V.
- Ruparelia AA, Oorschot V, Vaz R, et al. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014 Oct;128:821–833. Springer Berlin Heidelberg.
- Leber Y, Ruparelia AA, Kirfel G, et al. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum Mol Genet. 2016 July;25:2776–2788.
- Meister-Broekema M, Freilich R, Jagadeesan C, et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat Commun. 2018 Dec;9:5342. Nature Publishing Group.
- Fang X, Bogomolovas J, Wu T, et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017 Aug;127:3189–3200.
- Schänzer A, Rupp S, Gräf S, et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol Genet Metab. 2018 Mar;123:388–399.
- Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2008 Feb;105:2052–2057.
- Rose C, Menzies FM, Renna M, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010 May;19:2144–2153.
- Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013 Sep;9:1308–1320. Taylor & Francis.
- Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010 Sep;39:423–438.
- Fernando Roth R, Itabashi H, Louie J, et al. Amiodarone toxicity: myopathy and neuropathy. Am Heart J. 1990 May;119:1223–1225.
- Heger JJ, Prystowsky EN, Jackman WM, et al. Clinical efficacy and electrophysiology during long-term therapy for recurrent ventricular tachycardia or ventricular fibrillation. N Engl J Med. 1981 Sep;305:539–545.
- Foretz M, Guigas B, Bertrand L, et al. Metformin: from mechanisms of action to therapies. Cell Metab. 2014 Dec;20:953–966. Elsevier.
- Wu H, Esteve E, Tremaroli V, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017 May;33:S62.
- Ruparelia AA, Oorschot V, Ramm G, et al. FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum Mol Genet. 2016 25:2131–2142. Oxford University Press.
- Kwan KM, Fujimoto E, Grabher C, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007 Nov;236:3088–3099.
- Sztal TE, Zhao M, Williams C, et al. Zebrafish models for nemaline myopathy reveal a spectrum of nemaline bodies contributing to reduced muscle function. Acta Neuropathol. 2015 Apr:130:389–406;1–18. Springer Berlin Heidelberg.
- Ablain J, Durand EM, Yang S, et al. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell. 2015 Mar;32:756–764.
- Hwang WY, Fu Y, Reyon D, et al. Brief communications. Nat Biotechnol. 2013 Jan;31:227–229. Nature Publishing Group.
- Ruparelia AA, Zhao M, Currie PD, et al. Characterization and investigation of zebrafish models of filamin-related myofibrillar myopathy. Hum Mol Genet. 2012 July;21:4073–4083.
- Boglev Y, Badrock AP, Trotter AJ, et al. Autophagy induction is a Tor- and Tp53-independent cell survival response in a zebrafish model of disrupted ribosome biogenesis. . PLoS Genetics. 2013 . 9:e1003279.
- Zeng L, Guo J, Xu H-B, et al. Direct Blue 71 staining as a destaining-free alternative loading control method for western blotting. Electrophoresis. 2013 Aug;34:2234–2239. John Wiley & Sons, Ltd.
- Sztal TE, Ruparelia AA, Williams C, et al. Using touch-evoked response and locomotion assays to assess muscle performance and function in zebrafish. J Vis Exp. 2016 Oct:116:e54431.
- Abràmoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004 Aug;11:36–42.
- Pietzsch T, Preibisch S, Tomancak P, et al. ImgLib 2–generic image processing in Java. Bioinformatics. 2012 Nov;28:3009–3011. Oxford University Press.
- Otsu N. A threshold selection method from gray-level histograms. 1979 IEEE Transactions on Systems, Man, and Cybernetics, 9:62–66.
- Sasarman F, Karpati G, Shoubridge EA. Nuclear genetic control of mitochondrial translation in skeletal muscle revealed in patients with mitochondrial myopathy. Hum Mol Genet. 2002 July;11:1669–1681.
- Lochmuller H, Johns T, Shoubridge EA. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp Cell Res. 1999 Apr;248:186–193.
- Fuchs M, Boulanger M-C, Lambert H, et al. Adenofection: a method for studying the role of molecular chaperones in cellular morphodynamics by depletion-rescue experiments. J Vis Exp. 2016 Sep;115:e54557.
- Landry M-C, Champagne C, Boulanger M-C, et al. A functional interplay between the small GTPase Rab11a and mitochondria-shaping proteins regulates mitochondrial positioning and polarization of the actin cytoskeleton downstream of Src family kinases. J Biol Chem. 2014 Jan;289:2230–2249. American Society for Biochemistry and Molecular Biology.
- Champagne C, Landry M-C, Gingras M-C, et al. Activation of adenovirus type 2 early region 4 ORF4 cytoplasmic death function by direct binding to Src kinase domain. J Biol Chem. 2004 June;279:25905–25915. American Society for Biochemistry and Molecular Biology.