
ABSTRACT
In less than eleven months, the world was brought to a halt by the COVID-19 outbreak. With hospitals becoming overwhelmed, one of the highest priorities concerned critical care triage to ration the scarce resources of intensive care units. Which patient should be treated first? Based on what clinical and biological criteria? A global joint effort rapidly led to sequencing the genomes of tens of thousands of COVID-19 patients to determine the patients’ genetic signature that causes them to be at risk of suddenly developing severe disease. In this commentary, we would like to consider some points concerning the use of a multifactorial risk score for COVID-19 severity. This score includes macroautophagy (hereafter referred to as autophagy), a critical host process that controls all steps harnessed by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus.
Abbreviation list: ATG5: autophagy related 5; BECN1: beclin 1; COVID-19: coronavirus infectious disease-2019; EGR1: early growth response 1; ER: endoplasmic reticulum; DMVs: double-membrane vesicles; IBV: infectious bronchitis virus; MAP1LC3: microtubule associated protein 1 light chain 3; LC3-I: proteolytically processed, non-lipidated MAP1LC3; LC3-II: lipidated MAP1LC3; MEFs: mouse embryonic fibroblasts; MERS-CoV: Middle East respiratory syndrome-coronavirus; MHV: mouse hepatitis virus; NSP: non-structural protein; PEDV: porcine epidemic diarrhea virus; PLP2-TM: membrane-associated papain-like protease 2; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TGEV: transmissible gastroenteritis virus.
The world is in a race against time to fight the COVID-19 pandemic. Since the first case reported in December 2019 and up to 20 November2020, SARS-CoV-2 has infected 57million people worldwide – probably even more – and 1,36million have died from COVID-19. In the face of this exponential progression, we need, more than ever, to gain control over the SARS-CoV-2 virus and identify the population at risk to ensure the best therapeutic management. Currently, only a symptomatic ineffective therapy against COVID-19 is provided, as we have little, if any, information on the infectious pathway of the virus. As scientists and clinicians, we go back to the essence of science and share our hypotheses in confronting our arguments. Any cross-disciplinary debate that will emerge will help combat this virus as quickly as possible.
Based on the extensive information available concerning other coronaviruses (SARS-CoV, Middle East respiratory syndrome-coronavirus [MERS-CoV], mouse hepatitis virus [MHV], infectious bronchitis virus [IBV], porcine epidemic diarrhea virus [PEDV], etc.) we “advocate” for a role for autophagy in SARS-CoV-2 infection: i) As an obligate intracellular pathogen, without machinery for self-replication, SARS-CoV-2 actively hijacks the host cell to produce viral particles and escape host defenses. In turn, cells have optimized rapid clearance of the virus by developing autophagy (specifically virophagy) for selective engulfment of the particles and its viral components within a double-membrane vesicle, the autophagosome, and rapid trafficking of these vesicles to the lysosomes for degradation [Citation1]. In addition to this innate defense, virophagy ensures the presentation of viral antigens to the adaptive immune system and limits the inflammatory wave (signalphagy of inflammasome components). By orchestrating overall antiviral defense, autophagy may safeguard the host against infection and the “cytokine storm,” which are out of control in the most severe COVID-19 patients ().
Figure 1. Overview of the putative pro-viral roles of virophagy in the life cycle of the coronavirus. The infection of cells of the lung epithelium starts with the binding of the SARS-CoV-2 particle to the cell surface receptor ACE2 (1) and subsequent cell entry either at the plasma membrane or in endosomes upon endocytosis (2). Subsequent lysosomal damage, release of the viral genome (3), and excessive viral glycoprotein synthesis in the endoplasmic reticulum (ER) induce autophagy. Several factors of virulence (depicted in ) hijack virophagy, providing SARS-CoV-2 with a niche for replication, the membrane of its envelope, immune escape, an intracellular shuttle for their exocytosis, and the production of an inflammatory storm. The non-canonical functions of the autophagy machinery in LC3-associated phagocytosis, the formation of double-membrane EDEMosomes, and secretory autophagy are also depicted
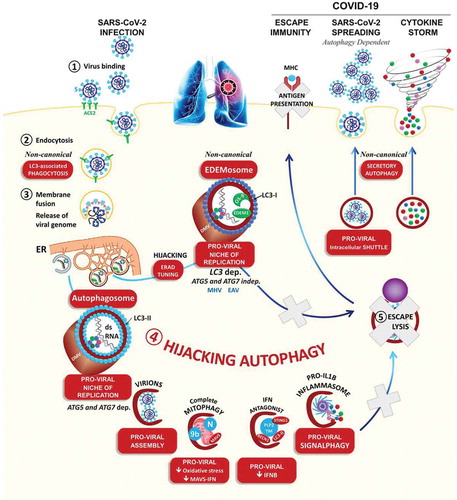
Figure 2. Molecular manipulation of the autophagic pathway by coronavirus-associated proteins. Upon infection of the host cells, lysosomal damage, release of the viral genome, and excessive ER and oxidative stress, all activate autophagy. Also shown are the viral proteins for which their overexpression in transfected cells is sufficient to activate and inhibit virophagy at late steps to avoid lysosome-dependent degradation
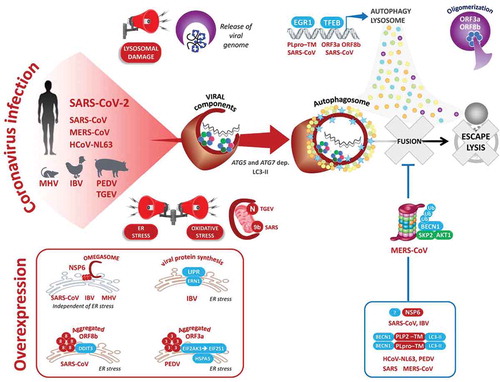
ii) Being a highly pathogenic virus, we presume that SARS-CoV-2 subverts the autophagy pathway: lysosomal damage [Citation2,Citation3], the release of the viral genome, and excessive viral protein synthesis in the endoplasmic reticulum [Citation3–6], all sound the alarm sirens, leading to the induction of autophagy and further transcription of autophagy components [Citation2,Citation3,Citation7,Citation8] (). Importantly, infection by all known members of the coronavirus family triggers the accumulation of double-membrane vesicles (DMVs), resembling autophagosomes (SARS-CoV, MHV [Citation9–11]). At the site of DMVs, the autophagy machinery, notably MAP1LC3, interacts with the viral replication and transcription complexes (RTCs) and the double-stranded RNA (dsRNA) replication intermediate [Citation9,Citation12–14]. There, the role of autophagy in DMV formation, and coronavirus replication has been clarified by the depletion of ATG5, ATG7, BECN1, or MAP1LC3 (MHV [Citation9], PEDV [Citation15–17]).
iii) From a molecular standpoint, a set of at least eight virulence factors was found to highjack the flux of host autophagy at different steps to escape elimination: NSP6, N, p4b, p5, PLP2 (papain-like protease 2)/PLpro-TM, ORF3a, ORF8b, and ORF9b (). The non-structural protein NSP6 generates autophagosomes from ER-derived omegasomes and inhibits their expansion (IBV, MHV, SARS-CoV, and MERS-CoV [Citation4,Citation18]). Similarly, a MERS-CoV infection or the overexpression of PLP2-TM induces autophagy, but decreases the level of BECN1, a common viral strategy that may result in greater membrane mobilization for viral replication without allowing clearance of viral particles (SARS-CoV [Citation19], and MERS-CoV [Citation20]). Likewise, the SARS-CoV ORF9b and transmissible gastroenteritis virus (TGEV) nucleocapsid N proteins localize to mitochondria where they induce mitophagy, to protect the virus from oxidative stress-induced cell death, and the MAVS-IFN antiviral response [Citation21,Citation22]. Finally, the overexpression of CoV ORF3a, ORF8b, or PLpro-TM is sufficient to activate the NLRP3 inflammasome, and both EGR1 (early growth response 1) and TFEB (transcription factor EB), two master transcription factors of autophagy, lysosomal biogenesis, and exocytosis [Citation2,Citation3,Citation7,Citation8].
In keeping with this scenario, we propose that hijacking virophagy may provide SARS-CoV-2 with a virus factory including a niche for replication, the membrane of its envelope, immune escape, an intracellular shuttle for exocytosis, and elements of an inflammatory storm, which are all associated with the most severe COVID-19 cases (). As for all scientific evidence, can we trust and generalize the role of virophagy as identified for the other CoV to the SARS-CoV-2 life cycle? The supporters of this comparison argue that the activation of autophagy in infected cells has been demonstrated for all members of the coronavirus family by a wide range of analyses: The formation of double-membrane vesicles by transmission electron microscopy, green fluorescent protein-MAP1LC3 (GFP-LC3)/LC3 puncta by fluorescence microscopy, production of lipidated LC3 (LC3-II) as well as global increases in the autophagy machinery [Citation3–6,Citation15,Citation16,Citation18–24]. Two preprint reports suggested that a SARS-CoV-2 infection may restrict autophagy in vitro [Citation25,Citation26]. However, for the detractors, the excitement of these connections was tamped down by the evidence that MHV, IBV, TGEV, or PEDV can replicate in mouse embryonic fibroblasts (MEFs) that lack Atg5 or Atg7 [Citation15,Citation27–31].
To referee between the pros and cons, it is challenging, if ever possible, to understand the CoV-autophagy battleground by using in vitro cellular models (such as HEK293, HeLa, Vero, MEFs, and MCF-7 cells) that are not relevant to the lung epithelial cells infected in vivo. Far from the real infection conditions, the ectopic overexpression of a single viral protein or GFP-LC3 can affect autophagy in unpredictable and variable ways in different studies [Citation3–6,Citation15,Citation16,Citation18–24]. It is also fair to consider that different coronaviruses may activate autophagy to favor (MHV [Citation9] and PEDV [Citation32]) or repress (TGEV [Citation15] and PEDV [Citation17]) virus replication.
Several arguments can be put forward to reconcile these controversies: i) The studies mentioned above that exploit the requirement of ATG5, ATG7, and BECN1 in MEFs, and HEK cells may leave open the possibility that the virus activates non-canonical autophagy independent of ATG5, ATG7 or BECN1 (IBV [Citation5]). ii) Remarkably, several ATG proteins, particularly LC3, carry out autophagy-independent functions in vesicular trafficking, which may favor CoV endocytosis/phagocytosis, replication, and release (). iii) Along this line, rather than autophagosomes, a few studies recognized that MHV [Citation27,Citation28] and the closely related arterivirus EAV [Citation33] can replicate within double-membrane EDEMosomes (LC3-I-positive endoplasmic reticulum-derived vesicles exporting short-lived ERAD regulators) coated with non-lipidated LC3-I for replication.
At the crossroads of these presumptions, and uncertainties, what can we propose? As a thoughtful attempt, harnessing autophagy pharmacologically is currently being tested in 58% of clinical trials on patients with COVID-19 [Citation26,Citation34–38]. While waiting for a treatment or a vaccine, the fact remains that the severity of COVID-19 varies widely from asymptomatic, to mild, to a deadly disease. How can we distinguish between them? The evidence to date suggests that the virus can infect people of any age, and most of the severe cases are elderly men (62%) with significant comorbidities (high blood pressure, obesity, diabetes, and chronic obstructive pulmonary disease [COPD]) that all involve a state of chronic inflammation () [Citation39]. Fighting the COVID-19 pandemic involves systematic PCR and serological screening to identify those infected and possibly immune, providing a “certificate of immunity” to exit quarantine [Citation40]. However, this yes-no answer to a SARS-CoV-2 infection is insufficient for go/no-go decision-making concerning patient care and admission into the intensive care unit [Citation41].
Figure 3. Multifactorial score risk for developing COVID-19. (A) Left, critical roles of autophagy in the host’s immunity and COVID-19 comorbidities. Autophagy is an essential lysosomal degradation pathway that protects the host against key COVID-19 risk factors, including infection, aging, diabetes, obesity, heart, and lung diseases. Right, multifactorial risk score incorporating the polymorphisms of ATG, together with that of SARS-CoV2 entry (its receptor ACE2 [angiotensin I converting enzyme 2] and two cell membrane ACE2 shedding proteases: ADAM17 [ADAM metalloproteinase domain 17] and TMPRSS2 [transmembrane serine protease 2]), and pathway-associated components (innate and adaptive immunities), to identify possible at-risk populations vulnerable to viral infection. (B) Right top, heatmap, and GSEA analysis of the autophagy reactome (p-values and normalized enrichment scores [NES] were calculated using the Phantasus and the GSEA websites) and the inflammatory response (GO_DEFENSE_ RESPONSE_TO_VIRUS) signature of lungs of COVID-19 patients and healthy controls (GSE147507 [Citation54]). Right bottom, the Smoking x Susceptibility EGR1 SNP relationship in COPD patients [Citation58] and the experimental evidence linking EGR1 knockdown to a downstream defect in ATG4B transcription, MAP1LC3B/LC3 conversion to LC3-II, and autophagosome formation [Citation59]. (See Figure S1)
![Figure 3. Multifactorial score risk for developing COVID-19. (A) Left, critical roles of autophagy in the host’s immunity and COVID-19 comorbidities. Autophagy is an essential lysosomal degradation pathway that protects the host against key COVID-19 risk factors, including infection, aging, diabetes, obesity, heart, and lung diseases. Right, multifactorial risk score incorporating the polymorphisms of ATG, together with that of SARS-CoV2 entry (its receptor ACE2 [angiotensin I converting enzyme 2] and two cell membrane ACE2 shedding proteases: ADAM17 [ADAM metalloproteinase domain 17] and TMPRSS2 [transmembrane serine protease 2]), and pathway-associated components (innate and adaptive immunities), to identify possible at-risk populations vulnerable to viral infection. (B) Right top, heatmap, and GSEA analysis of the autophagy reactome (p-values and normalized enrichment scores [NES] were calculated using the Phantasus and the GSEA websites) and the inflammatory response (GO_DEFENSE_ RESPONSE_TO_VIRUS) signature of lungs of COVID-19 patients and healthy controls (GSE147507 [Citation54]). Right bottom, the Smoking x Susceptibility EGR1 SNP relationship in COPD patients [Citation58] and the experimental evidence linking EGR1 knockdown to a downstream defect in ATG4B transcription, MAP1LC3B/LC3 conversion to LC3-II, and autophagosome formation [Citation59]. (See Figure S1)](/cms/asset/5abccdc7-3778-4529-9294-63f1f529041f/kaup_a_1844433_f0003_oc.jpg)
With the lack of the underlying susceptibility factors, it is difficult to predict who will develop the disease, its severity, and treatment response. Currently, several studies have been planned or are ongoing to study factors predisposing to COVID-19 [Citation42]. Some studies have proposed genetic testing for ACE2, the receptor that opens the door to SARS-CoV-2 [Citation43]. However, it is highly unlikely that the remarkable phenotypic variation of COVID-19 could be explained by a single ACE2 variant, which would underestimate the risk.
We therefore favor a polygenic model that is broad enough to encompass all steps harnessed by the virus to infect, replicate, and escape the host’s innate and adaptive immune defenses. Among the candidates, the autophagy machinery meets these requirements. In support of this: i) Several studies, as mentioned above, have shown the direct interaction of several components of the coronavirus with the autophagy machinery [Citation14,Citation19].
ii) During the pandemic, a new mutation was discovered in SARS-CoV-2 NSP6, an autophagy-inducing protein, likely supporting the concept of a subversion of autophagy in the aggravation of COVID-19 respiratory distress [Citation44,Citation45].
iii) Of interest, common and rare mutations in autophagy genes are associated with human conditions and diseases, particularly aging, obesity, diabetes, COPD, and infectious bacterial and viral diseases, many of which are COVID-19 comorbidities [Citation46–52] (, Figure S1).
iv) The cytokine storm is also a complication of various autoimmune diseases such as lupus, a condition associated with autophagy gene polymorphism [Citation53].
v) Using publicly available RNA-Seq data (downloaded from Gene Expression Omnibus, GSE147507 [Citation54]), we performed a Gene Set Enrichment Analysis (GSEA) and found a decrease in the expression of the central transcriptional regulator of autophagy, EGR1, and concomitantly a downstream downregulation of the autophagy machinery components in COVID-19 patient lungs. Interestingly, mounting lines of evidence have indeed shown that EGR1 is induced after infection by two coronaviruses (SARS-CoV-2 [Citation8] and MHV [Citation7]) and several other viruses (herpes simplex virus type-1/HSV-1, Kaposi sarcoma-associated herpes virus/KSHV, Epstein-Barr virus/EBV, human immunodeficiency virus/HIV, human foamy virus/HFV, human polyomavirus JC virus/JCV, human cytomegalovirus/HCMV, human T cell leukemia virus type 1/HTLV-1, and HTLV-2, among others). In turn, EGR1 emerges to be essential to control viral replication and disease progression in infected hosts. Mechanistically, EGR1 acts primarily as a transcription factor that binds to and activates viral promoters. Besides virus genes, EGR1 may change the infected cell phenotype, which could help to establish viral pathogenicity, as evidenced by SARS-CoV-induced pulmonary fibrosis [Citation8].
vi) Of interest, it should be emphasized that the deletion of the EGR1 gene was demonstrated to have no impact on vaccinia virus gene expression, but significantly decreases infectivity only in serum-starved cells [Citation55]. The observations that serum starvation promotes autophagy, and EGR1 controls the expression of the autophagy machinery are of interest [Citation56,Citation57]. Whether the decrease in virus infectivity in starved cells reflects autophagy degradation, turning off transcription, or both remains an issue to be pursued further.
vii) In line with the above, an EGR1 polymorphism has been associated with COPD in smokers [Citation58,Citation59]. By performing a comprehensive survey of the literature, a total of 90 SNPs in 31 autophagy genes were associated with inflammatory, cardiovascular, infectious, and lung diseases (Figure S1). It is noteworthy that more than 93% of the risk-associated alleles are localized to non-coding genomic regions (intron, intergenic, and 5ʹ and 3ʹ untranslated regions). These 84 ATG SNPs, including EGR1 SNPs [Citation58,Citation59], are regulatory SNPs, also called expression quantitative trait loci (eQTL) that control mRNA transcript abundance by influencing the binding of a transcription factor. In contrast, only six missense ATG variations change the amino acid sequence and are rare (from 0.1 to 5%). This is in line with the lethality of the atg7 knockout in mice, due to the survival role of autophagy. Likewise, the allelic losses of autophagy genes (Atg16l1, Irgm1, Atg4c, Atg5, Uvrag, Ambra1, etc.) render cells or mice prone to inflammatory disease, infection, and cancer, highly suggesting a dosage effect. With this in mind, we therefore propose that individuals with ATG eQTL with a low degree of penetrance, such as a decreased expression level, are extremely susceptible to SARS-CoV infections and comorbidities. Thus, a clear picture of host vulnerability can be found in autophagy-related polymorphisms that may predispose to key inflammation-related risk factors [Citation46] for death from COVID-19 ().
Therefore, as a complement to the detection of SARS-CoV-2 and evaluation of comorbidities, we propose that a multifactorial score including genetic variants of autophagy together with that of other key pathways involved in SARS-CoV-2 entry [Citation60], inflammation, adaptive and innate immunities should be developed and validated (). Computing such a patient’s polygenetic risk score would help expedite risk assessment within hours and accurately guide precision medical care, thereby providing a head-start against the next COVID-19 waves.
Supplemental Material
Download Zip (7.2 MB)Acknowledgments
The authors would like to thank Charles-Hugo Marquette, Gérard Milano, Sylvie Leroy, and Christiane Brahimi-Horn, and TRANSAUTOPHAGY: European Network for Multidisciplinary Research and Translation of Autophagy Knowledge, COST Action CA15138 for helpful comments and discussions.
Disclosure statement
All authors declare no competing interests: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16:341–354.
- Yue Y, Nabar NR, Shi CS, et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9:904.
- Shi CS, Nabar NR, Huang NN, et al. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101.
- Cottam EM, Maier HJ, Manifava M, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335–1347.
- Fung TS, Liu DX. The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology. 2019;533:34–44.
- Zou D, Xu J, Duan X, et al. Porcine epidemic diarrhea virus ORF3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet Microbiol. 2019;235:209–219.
- Cai Y, Liu Y, Zhang X. Induction of transcription factor Egr-1 gene expression in astrocytoma cells by murine coronavirus infection. Virology. 2006;355:152–163.
- Li SW, Wang CY, Jou YJ, et al. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep. 2016;6:25754.
- Prentice E, Jerome WG, Yoshimori T, et al. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–10141.
- Gosert R, Kanjanahaluethai A, Egger D, et al. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol. 2002;76:3697–3708.
- Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol. 2006;80:5927–5940.
- Prentice E, McAuliffe J, Lu X, et al. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J Virol. 2004;78:9977–9986.
- de Haan CA, Reggiori F. Are nidoviruses hijacking the autophagy machinery? Autophagy. 2008;4:276–279.
- V’Kovski P, Gerber M, Kelly J, et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. Elife. 2019;8: e42037.
- Guo L, Yu H, Gu W, et al. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci Rep. 2016;6:23864.
- Guo X, Zhang M, Zhang X, et al. Porcine epidemic diarrhea virus induces autophagy to benefit its replication. Viruses. 2017;9(3):E53.
- Ko S, Gu MJ, Kim CG, et al. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antiviral Res. 2017;146:86–95.
- Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10:1426–1441.
- Chen X, Wang K, Xing Y, et al. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014;5:912–927.
- Gassen NC, Niemeyer D, Muth D, et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-coronavirus infection. Nat Commun. 2019;10:5770.
- Shi CS, Qi HY, Boularan C, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. 2014;193:3080–3089.
- Zhu L, Mou C, Yang X, et al. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget. 2016;7:27122–27141.
- Sun D, Shi H, Guo D, et al. Analysis of protein expression changes of the Vero E6 cells infected with classic PEDV strain CV777 by using quantitative proteomic technique. J Virol Methods. 2015;218:27–39.
- Qin P, Du EZ, Luo WT, et al. Characteristics of the life cycle of porcine deltacoronavirus (PDCoV) in vitro: replication kinetics, cellular ultrastructure and virion morphology, and evidence of inducing autophagy. Viruses. 2019;11:455.
- Gassen NC, Papies J, Bajaj T, et al. Analysis of SARS-CoV-2-controlled autophagy reveals spermidine, MK-2206, and niclosamide as putative antiviral therapeutics. bioRxiv. 2020;04(15):997254.
- Gorshkov K, Chen CZ, Bostwick R, et al. The SARS-CoV-2 cytopathic effect is blocked with autophagy modulators. bioRxiv. 2020;05(16):091520.
- Zhao Z, Thackray LB, Miller BC, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585.
- Reggiori F, Monastyrska I, Verheije MH, et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508.
- Schneider M, Ackermann K, Stuart M, et al. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J Virol. 2012;86:10112–10122.
- Maier HJ, Britton P. Involvement of autophagy in coronavirus replication. Viruses. 2012;4:3440–3451.
- Maier HJ, Cottam EM, Stevenson-Leggett P, et al. Visualizing the autophagy pathway in avian cells and its application to studying infectious bronchitis virus. Autophagy. 2013;9:496–509.
- Ding N, Zhao K, Lan Y, et al. Induction of atypical autophagy by porcine hemagglutinating encephalomyelitis virus contributes to viral replication. Front Cell Infect Microbiol. 2017;7:56.
- Monastyrska I, Ulasli M, Rottier PJ, et al. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy. 2013;9:164–174.
- Brest P, Benzaquen J, Klionsky DJ, et al. Open questions for harnessing autophagy-modulating drugs in the SARS-CoV-2 war: hope or hype? Autophagy. 2020:1–4.
- Bonam SR, Muller S, Bayry J, et al. Autophagy as an emerging target for COVID-19: lessons from an old friend, chloroquine. Autophagy. 2020:1–7.
- Carmona-Gutierrez D, Bauer MA, Zimmermann A, et al. Digesting the crisis: autophagy and coronaviruses. Microb Cell. 2020;7:119–128.
- Yang N, Shen HM. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int J Biol Sci. 2020;16:1724–1731.
- Nabirotchkin S, Peluffo AE, Bouaziz J, et al. Focusing on the unfolded protein response and autophagy related pathways to reposition common approved drugs against COVID-19. MDPI Preprint. 2020.
- Halpin DMG, Faner R, Sibila O, et al. Do chronic respiratory diseases or their treatment affect the risk of SARS-CoV-2 infection? Lancet Respir Med. 2020;8:436–438.
- Wolfel R, Corman VM, Guggemos W, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581(7809):465–469.
- Wu JT, Leung K, Bushman M, et al. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat Med. 2020;26:506–10.
- Callaway E, Ledford H, Mallapaty S. Six months of coronavirus: the mysteries scientists are still racing to solve. Nature. 2020;583:178–179.
- Sawalha AH, Zhao M, Coit P, et al. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin Immunol. 2020;215:108410.
- Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J Infect. 2020;81:e24–e7.
- Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221–236.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
- Li N, Fan X, Wang X, et al. Autophagy-related 5 gene rs510432 polymorphism is associated with hepatocellular carcinoma in patients with chronic hepatitis B virus infection. Immunol Invest. 2019;48:378–391.
- Li N, Fan X, Wang X, et al. Genetic association of polymorphisms at the intergenic region between PRDM1 and ATG5 with hepatitis B virus infection in Han Chinese patients. J Med Virol. 2020;92:1198–1205.
- Mao JJ, Wu LX, Wang W, et al. Nucleotide variation in ATG4A and susceptibility to cervical cancer in Southwestern Chinese women. Oncol Lett. 2018;15:2992–3000.
- Reuken PA, Lutz P, Casper M, et al. The ATG16L1 gene variant rs2241880 (p.T300A) is associated with susceptibility to HCC in patients with cirrhosis. Liver Int. 2019;39:2360–2367.
- Wisetsathorn S, Tantithavorn V, Hirankarn N, et al. Gene polymorphisms of autophagy machinery and the risk of hepatitis B virus-related hepatocellular carcinoma in a Thai population. Sci Asia. 2017;43:362–368.
- Zhou Z, Sturgis EM, Liu Z, et al. Genetic variants of NOXA and MCL1 modify the risk of HPV16-associated squamous cell carcinoma of the head and neck. BMC Cancer. 2012;12:159.
- Qi YY, Zhou XJ, Zhang H. Autophagy and immunological aberrations in systemic lupus erythematosus. Eur J Immunol. 2019;49:523–533.
- Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–45e9.
- de Oliveira LC, Brasil B, Unger B, et al. The host factor early growth response gene (EGR-1) regulates vaccinia virus infectivity during infection of starved mouse cells. Viruses. 2018;10(4):140.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Peng WX, Xiong EM, Ge L, et al. Egr-1 promotes hypoxia-induced autophagy to enhance chemo-resistance of hepatocellular carcinoma cells. Exp Cell Res. 2016;340:62–70.
- Chen CZ, Ou CY, Wang RH, et al. Association of Egr-1 and autophagy-related gene polymorphism in men with chronic obstructive pulmonary disease. J Formos Med Assoc. 2015;114:750–755.
- Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3:e3316.
- Brest P, Refae S, Mograbi B, et al. Host polymorphisms may impact SARS-CoV-2 infectivity. Trends Genet. 2020;36(11):813–815.