
ABSTRACT
Oncogenic KRAS mutation-driven pancreatic ductal adenocarcinoma is currently the fourth-leading cause of cancer-related deaths in the United States. Macroautophagy (hereafter “autophagy”) is one of the lysosome-dependent degradation systems that can remove abnormal proteins, damaged organelles, or invading pathogens by activating dynamic membrane structures (e.g., phagophores, autophagosomes, and autolysosomes). Impaired autophagy (including excessive activation and defects) is a pathological feature of human diseases, including pancreatic cancer. However, dysfunctional autophagy has many types and plays a complex role in pancreatic tumor biology, depending on various factors, such as tumor stage, microenvironment, immunometabolic state, and death signals. As a modulator connecting various cellular events, pharmacological targeting of nonselective autophagy may lead to both good and bad therapeutic effects. In contrast, targeting selective autophagy could reduce potential side effects of the drugs used. In this review, we describe the advances and challenges of autophagy in the development and therapy of pancreatic cancer.
Abbreviations: AMPK: AMP-activated protein kinase; CQ: chloroquine; csc: cancer stem cells; DAMP: danger/damage-associated molecular pattern; EMT: epithelial-mesenchymal transition; lncRNA: long noncoding RNA; MIR: microRNA; PanIN: pancreatic intraepithelial neoplasia; PDAC: pancreatic ductal adenocarcinoma; PtdIns3K: phosphatidylinositol 3-kinase; SNARE: soluble NSF attachment protein receptor; UPS: ubiquitin-proteasome system.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the main type of pancreatic cancer, accounting for approximately 3% of all cancers, ranks fourth in cancer-related deaths in the United States, and has a 5-year overall survival rate of 9% [Citation1]. Epidemiologically, risk factors for developing PDAC include smoking, heavy drinking, obesity, low physical activity, low intake of vegetables and fruits, and high intake of fat [Citation2]. These risk factors may jointly cause chronic inflammation of the pancreas, an important extrinsic cause of tumorigenesis. The intrinsic cause of pancreatic tumorigenesis is related to gene mutations, especially mutations in KRAS (KRAS proto-oncogene, GTPase), TP53 (tumor protein p53), SMAD4 (SMAD family member 4), and BRCA2 (BRCA2 DNA repair associated) [Citation2]. As a main driver of pancreatic cancer, KRAS mutations are observed in 99% of patients. The forced expression of KRASG12D mutations in the pancreas is sufficient to induce the development and progression of pancreatic intraepithelial neoplasia (PanIN) to PDAC with significant stromal responses in mice [Citation3]. Despite unremitting efforts, the interaction between extrinsic and intrinsic factors during tumor transformation is still the basis for understanding the molecular pathogenesis of PDAC.
As one of the most aggressive solid malignant tumors, the diagnosis of PDAC usually occurs at an advanced stage, and patients may lack the opportunity for surgical resection. Although radiation therapy and immunotherapy are also potential options, chemotherapy is currently the main method of treating PDAC patients who are not eligible for resection [Citation4]. For PDAC patients with metastatic disease, FOLFIRINOX (a combination of cancer drugs including folinic acid, fluorouracil, irinotecan, and oxaliplatin) and nab-paclitaxel plus gemcitabine, are currently two standard combination therapies, showing a mild survival advantage compared to gemcitabine-mediated monotherapy [Citation5,Citation6]. However, primary or secondary drug resistance is a common phenomenon that eventually leads to chemotherapy failure. Therefore, understanding the signals and regulation of cell survival and cell death of PDAC is essential to overcoming drug resistance.
Macroautophagy (hereinafter referred to as autophagy) is a conserved cellular homeostasis mechanism that degrades various cargoes (e.g., proteins, organelles, and pathogens) [Citation7,Citation8]. It mainly plays a role in promoting survival under environmental stress, but also causes cell death in some cases [Citation9]. In addition to regulating noncancer diseases (e.g., neurodegenerative and infectious diseases), the pathological role of autophagy in cancer also depends on the context. Autophagy may play a role in limiting the earliest stages of tumor development while promoting survival in established tumors [Citation10]. In this review, we first introduce the process of autophagy, and then focus on the function of autophagy in the development and treatment of PDAC in both preclinical and clinical research. Research in autophagy is rapidly developing, and the understanding we gain not only helps us understand the basic molecular machinery of homeostasis, but also can guide the clinical decision-making for targeted therapy that is autophagy related.
The basic process of autophagy
Cells need integrated degradation mechanisms, such as autophagy and the ubiquitin-proteasome system (UPS), to remove intracellular waste generated during metabolism or aging [Citation11]. Unlike the UPS, the process of autophagy is mediated by the formation and maturation of multiple membrane vesicles, especially phagophores, autophagosomes, and autolysosomes . Phagophores are sac-like structures whose membranes are produced from a variety of resources, such as endoplasmic reticulum (ER), mitochondria contact sites, the Golgi apparatus, and plasma membrane. The nucleation and expansion of phagophores result in the production of autophagosomes (a spherical structure with double-layer membranes), which engulf various types of cargoes, such as endogenous proteins or organelles and exogenous microorganisms. Later, autophagosomes deliver cytoplasmic components to lysosomes by forming autolysosomes, which ultimately lead to lysosomal enzyme-mediated degradation. Some components of degraded cargo can be recycled for protein synthesis and energy production, thereby promoting cell survival.
Figure 1. The process and modulation of autophagy. Autophagy is an intracellular lysosomal-dependent degradation pathway that occurs with the dynamic generation of specific membrane structures (e.g., phagophores, autophagosomes, and autolysosomes). This process is regulated by the ATG family and other proteins (particularly kinases) that form different protein complexes. Abbreviations: AMPK, AMP-activated protein kinase; ATG, autophagy related; BECN1, beclin 1; MTOR, mechanistic target of rapamycin kinase; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4, phosphoinositide 3-kinase regulatory subunit 4; RB1CC1, RB1 inducible coiled-coil 1; Ub-like, ubiquitin-like; ULK1, unc-51 like autophagy activating kinase 1
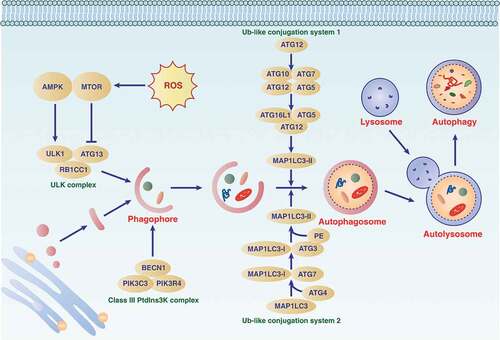
The autophagy process is fine-tuned by multiple signals or regulators at different levels. A series of ATG (autophagy-related) genes are responsible for the induction and execution of autophagy. To date, the ATG family includes more than 40 members; the first Atg protein was discovered in S. cerevisiae in the 1990s [Citation12–14], and half of the known Atg proteins have homologs in mammalian cells [Citation15]. The proteins encoded by ATG genes form the distinct complexes required for the dynamic regulation of autophagy membrane structures, and this process is further regulated by kinase-mediated phosphorylation modifications [Citation16].
AMP-activated protein kinase (AMPK) and MTOR (mechanistic target of rapamycin kinase) play opposite roles in autophagy by controlling the activation of ULK1 (unc-51 like autophagy activating kinase 1; a homolog of yeast Atg1) through phosphorylation. Unlike MTOR, which is a repressor of autophagy, the energy sensor AMPK is an autophagy promoter that controls the balance or metabolism between the production and consumption of adenosine triphosphate (ATP). In mammalian cells, MTOR is a component of MTOR complex 1 (MTORC1) and MTORC2, complexes with different functions. Active AMPK binds and inhibits MTORC1, and then stimulates the activating phosphorylation of ULK1, leading to the subsequent formation of the ULK1 complex (containing ULK1, ATG13 [autophagy related 13], ATG101 and RB1CC1 [RB1 inducible coiled-coil 1]). MTORC2 may indirectly suppress autophagy through the activation of MTORC1 [Citation17]. In addition to the ULK1 complex, the activation of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (containing PIK3C3 [phosphatidylinositol 3-kinase catalytic subunit type 3], PIK3R4 [phosphoinositide 3-kinase regulatory subunit 4], BECN1 [beclin 1], and ATG14), as well as ATG9 (a multispanning membrane protein), work together to control phagophore formation and maturation during autophagy induction.
After phagophore formation, cells further rely on two ubiquitin-like conjugation systems, namely ATG12 and MAP1LC3 (microtubule associated protein 1 light chain 3; also known as Atg8 in yeast), to drive the biogenesis of the autophagosomal membrane. The conjugation reactions and expansion of the phagophore are catalyzed and achieved by a series of ATGs (e.g., the E1 enzyme ATG7 and the E2 enzyme ATG3 or ATG10). Because MAP1LC3 is converted from MAP1LC3-I to MAP1LC3-II, recruited to phagophore membranes, and remains associated with completed autophagosomes, MAP1LC3-II is a widely-used marker for monitoring the numbers of autophagosomes [Citation18]. In addition to the MAP1LC3 subfamily (including MAP1LC3A, MAP1LC3B, MAP1LC3B2 and MAP1LC3C), the GABARAP (GABA type A receptor-associated protein) subfamily (including GABARAP, GABARAPL1, and GABARAPL2) also play a similar role in mediating autophagosome formation and/or maturation [Citation19]. The MAP1LC3 and GABARAP families are collectively referred to as Atg8-family proteins. Although they have different tissue or cell expression specificities, it is still unclear how cells selectively use different members of the MAP1LC3 or GABARAP subfamilies to induce autophagy.
The process of forming autolysosomes is mainly regulated by some non-ATG proteins, such as LAMP2 (lysosomal associated membrane protein 2), soluble NSF attachment protein receptors (SNAREs), and the homotypic fusion and protein sorting/vacuolar protein sorting complex. Finally, the degradation of autophagic substrates, including MAP1LC3-II itself, inside the lysosome requires proteases, acid phosphatases, lipases, and nucleases. Because lysosomes are the intersection of many transport pathways, there may be the possibility of other intracellular transport proteins regulating the formation of the autolysosome.
Overall, the term “autophagic flux” is used to describe the entire dynamic process of autophagy. One of the most common methods for monitoring autophagic flux is to measure the protein level of MAP1LC3-II in the absence or presence of early (e.g., 3-methyladenine and spautin-1) or late (e.g., bafilomycin A1 and chloroquine [CQ]) autophagy inhibitors [Citation20]. Several fluorescent biosensors, such as mCherry-GFP-LC3, mRFP-GFP-LC3, and GFP-LC3-RFP-LC3∆G, are also widely used in vitro to measure autophagic flux [Citation21]. However, testing autophagy flux in real-time in vivo remains a challenge.
Depending on whether the degradation of autophagy substrates requires a specific autophagy receptor and a ligand to mark the cargo, autophagy is divided into nonselective and selective types [Citation22,Citation23]. The autophagy receptors (e.g., SQSTM1/p62 [sequestosome 1], CALCOCO2/NDP52 [calcium binding and coiled-coil domain 2], and OPTN [optineurin]) act by interacting simultaneously with cargoes and Atg8-family proteins. Generally, the autophagy receptors interact with lipidated MAP1LC3 or GABARAP subfamily proteins by LC3-interacting region (LIR) motifs that bind to the LIR docking site of Atg8-family proteins in a cargo-dependent manner [Citation24]. The increasing identification of different types of selective autophagy indicates that cells may use special pathways to degrade different substrates without affecting other functions. This selectivity may be important for targeting autophagy to treat diseases.
The mechanism of autophagy upregulation in PDAC
PDAC tissues or cells have a higher level of ATG expression or autophagy activity [Citation25] through transcription or post-translational modification. On the one hand, PDAC cells maintain high levels of autophagy and lysosomal activity by decoupling the regulation of the microphthalmia/transcription factor E (MiT/TFE) family of transcription factors from MTOR signaling [Citation26]. The MiT/TFE family can control the expression of several genes involved in autophagy, including genes related to autophagy initiation (e.g., BECN1, UVRAG, and ATG9B), extension (e.g., GABARAP, MAP1LC3B, and ATG5), substrate capture (e.g., SQSTM1), and autophagosome transport and fusion with lysosomes (e.g., RAB7) [Citation27]. On the other hand, PDAC cells effectively dephosphorylate ULK1 through PTPA/PP2A (protein phosphatase 2 phosphatase activator), thereby maintaining a high degree of basal autophagy [Citation28].
Autophagy in pancreatic cancer initiation
The etiology of PDAC is multifaceted, involving chemical exposure, inflammation induction, and gene mutation. A dysfunction in autophagy plays a dual role in inhibiting or promoting pancreatic tumorigenesis mediated by these factors, as described below.
Pancreatic acinar cells are functional units of the exocrine pancreas. Although the origin of PDAC is controversial, acinar cells are the most likely candidate cells for pancreatic tumorigenesis. Since the 1980s, electron microscopy and autoradiography studies have shown that after exposure to carcinogenic nitrosamines, such as methylnitrosourea/MNU, N-methyl-N’-nitro-N-nitrosoguanidine/MNNG, and N-nitrosobis(2-hydroxypropyl)amine, increased autophagy is related to cell damage, dysplastic morphology, and metaplasia in pancreatic acinar cells [Citation29,Citation30]. In a chemical carcinogenic model in which Wistar rats were fed with azamatrine, the autophagy capacity is also increased in pancreatic premalignant atypical acinar nodule cells [Citation31]. Later, short-term exposure to cigarette smoke extract or smoking compounds, such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone/NNK, were shown to induce autophagy by the activation of AMPK to protect pancreatic ductal cells from damaged organelles or gene alterations during tumorigenesis [Citation32]. However, long-term exposure to smoking compounds limits apoptosis and autophagy, therefore promoting tumor formation in mice [Citation32]. Similarly, MAGEA6 (MAGE family member A6; a bioenergy sensor) suppresses autophagy and then accelerates the initiation of PDAC in mice [Citation33]. In contrast, when nutrients are depleted, the UPS-mediated degradation of MEGA6 may restore autophagy, leading to the subsequent development of PDAC in vitro and in vivo [Citation33]. The restriction of autophagy can be eliminated upon nutrient depletion by degrading MAGEA6 through the ubiquitin-proteasome system, thereby further supporting tumor progression [Citation33]. These studies highlight the effect of autophagy on the development of pancreatic tumors caused by different chemical risk factors or mutant protein expression that can promote cancer depending on the exposure time or tumor stage.
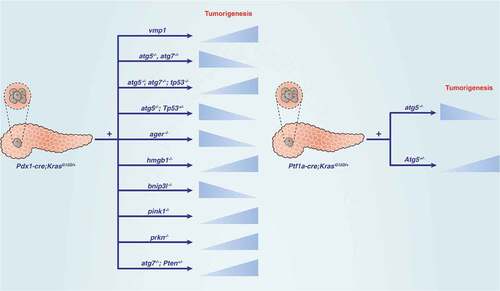
Inflammation is considered an extrinsic factor and plays an indispensable role in the development of pancreatic tumors. Autophagy has a complex role in regulating inflammation during pancreatic cancer initiation. During the activation of oncogenic signaling (e.g., gene mutation and inflammation), a pathological change called acinar-ductal metaplasia (ADM) is the precursor of PanIN. The conditional knockout of Atg7 in pancreatic epithelial cells (Pdx1-Cre;atg7fl/fl) leads to severe degeneration of acinar cells [Citation34], accompanied by the formation of ADM through ER stress, mitochondrial dysfunction, and impaired protein synthesis [Citation34]. Atg7 depletion in the pancreas (Pdx1-Cre;atg7fl/fl) also leads to inflammation and fibrosis in mice by regulating cell death signals [Citation35], indicating the potential role of ATG7-mediated inflammation regulation in the initiation of pancreatic cancer.
Autophagy plays a carcinogenic role in KRAS-driven pancreatic tumorigenesis by increasing the TBK1 (TANK binding kinase 1)-mediated dysplasia [Citation36,Citation37]. However, autophagy inhibitors or Atg5 deletion (Pdx1-Cre;atg5fl/fl) exacerbate pancreatitis and dysplasia because increased autophagy reduces TBK1-mediated inflammation in mice [Citation37]. Therefore, autophagy plays a dual role in the process of inflammation-related pancreatic tumorigenesis. HMGB1 (high mobility group box 1) is a DNA-binding protein and damage-associated molecular pattern (DAMP) molecule that promotes autophagy through a variety of mechanisms [Citation38]. Similar to the conditional knockout of Atg7 in the pancreas, the conditional knockout of Hmgb1 in the pancreas (Pdx1-Cre;hmgb1fl/fl) also accelerates inflammatory injury and inflammation-mediated KRAS activation and pancreatic tumors in mice [Citation39].
Some proteins control autophagic flux through a protein-protein interaction to regulate the occurrence of pancreatic cancer. For example, the expression of VMP1 (vacuolar membrane protein 1), a KRAS-mediated ER-related membrane protein, promotes the initiation of PDAC, which is related to enhanced autophagy through the formation of the VMP1-BECN1 complex in cells [Citation40]. In contrast, the administration of CQ not only inhibits autophagy flux, but also reverses PanIN formation induced by VMP1 overexpression in Pdx1-Cre;KrasG12D;vmp1 mice [Citation41], although CQ may have other functions independent of autophagy inhibition [Citation42]. Similarly, accumulation of the autophagy receptor SQSTM1 accelerates PanIN to PDAC progression and results in shorter survival in mice [Citation43]. The activation of NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) is regulated by SQSTM1-mediated KEAP1 (kelch like ECH associated protein 1) degradation in an autophagy-dependent manner [Citation44,Citation45]. SQSTM1-mediated NFE2L2 activation further promotes MDM2 (MDM2 proto-oncogene) expression, which leads to cell proliferation by stimulating NOTCH (notch receptor) and suppressing TP53 signaling in vitro and in vivo [Citation43]. In addition to NFE2L2, it remains unknown whether other types of SQSTM1-mediated protein degradation contribute to this KRAS-driven PDAC.
Although TP53 mutations accelerate KRAS-mediated pancreatic tumorigenesis, the function of TP53 in autophagy depends on the tumor stage [Citation46]. Genetic ablation of pancreatic autophagy leads to increased tumor cell initiation, whereas inhibition of autophagy by CQ or RNAi limits the pancreatic tumor growth in a TP53-independent manner [Citation47]. The conditional knockout of Atg7 in mice accelerates the formation of KRAS-driven PanINs in the early stages of progression but limits the development of PDAC by inducing apoptosis in the later stages [Citation48], indicating a different role for autophagy in tumor formation and development. In addition, combined knockout of Tp53 (we refer to both the human TP53 and mouse Trp53 gene as TP53 for simplicity) and the inhibition of autophagy (by knockout of Atg7 or Atg5, or using CQ) lead to the rapid death of mice with KRAS mutations [Citation48]. The onset of tumors in these mice accelerate, and the evolving tumors are metabolically rewired [Citation48]. When autophagy is insufficient, PDAC genetically engineered mouse models with homozygous Tp53 deletion or loss of heterozygosity/LOH may exhibit a tumor-promoting phenotype [Citation49]. Similar to TP53, PTEN (phosphatase and tensin homolog) also functions as a tumor suppressor, and its loss has been observed in PDAC. The conditional knockout of Atg7 with Pten hemizygosity in the pancreas promotes tumor formation and earlier death in mice [Citation50]. Collectively, these findings indicate a complex relationship between KRAS, TP53, PTEN, autophagy, and apoptosis, which affects tumor development [Citation48,Citation50] .
Figure 2. Dual role of autophagy regulators in KRAS-driven pancreatic tumorigenesis in mice. Abbreviations: ATG, autophagy related; AGER, advanced glycosylation end-product specific receptor; BNIP3L/NIX, BCL2 interacting protein 3 like; HMGB1, high mobility group box 1; PINK, PTEN induced kinase 1; PRKN, parkin RBR E3 ubiquitin protein ligase; PTEN, phosphatase and tensin homolog; TP53, tumor protein p53
Mitophagy is the selective degradation of mitochondria by autophagy, and this process is regulated by different mitochondrial or cytosolic proteins, such as PINK1 (PTEN induced kinase 1), PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase), and BNIP3L/NIX (BCL2 interacting protein 3 like) [Citation51]. Interestingly, the knockout of Pink1 or Prkn accelerates, and the depletion of Bnip3l prevents, KRAS-driven PDAC in mice by regulating iron or redox homeostasis, respectively [Citation52,Citation53]. These proteins also participate in mitochondrial quality control in a mitophagy-independent manner. The differences in the phenotypes of PINK1, PRKN, and BNIP3L in KRAS-driven PDAC animal models indicate that their regulation of pancreatic tumorigenesis may depend on their non-mitophagic function.
Cancer stem cells (CSC) are a small part of cells that have the ability to self-renew and differentiate in tumors. KRASG12D ablation-resistant pancreatic cancer cells exhibit CSC-like phenotype by increasing mitochondrial function, autophagy, and lysosome activity [Citation54]. Mitophagy appears to affect CSC function in the PDAC tumor microenvironment. This process is partially mediated by ISG15 (ISG15 ubiquitin like modifier), a regulator for mitochondrial dynamics and metabolism in CSCs [Citation55]. The loss of ISG15 leads to impaired mitophagy, resulting in mitochondrial dysfunction and reduced energy production, which ultimately reduces the stemness and metabolic plasticity of CSCs in vitro and xenograft models [Citation55]. However, whether ISG15 plays a role in the regulation of mitophagy in non-CSCs is unclear.
Autophagy in pancreatic cancer progression
Autophagy also plays dual roles in the progression of PDAC by modulating proliferation, invasion and metastasis, metabolism, cell death, or immunity. Established pancreatic cancer has a high level of autophagy in the basal state in pancreatic cancer cell lines and patient tumor tissues [Citation56]. Early studies show that the genetic (e.g., atg7−/- or hmgb1−/-) and pharmacological inhibition (e.g., using CQ) of autophagy lead to the generation of reactive oxygen species (ROS), increased DNA damage and metabolic dysfunction, which ultimately inhibits the development of pancreatic cancer in vitro and in vivo, indicating that increased autophagy contributes to the PDAC progression [Citation39,Citation56]. Recent studies suggest that the enhancement of autophagy may also inhibit the progress of PDAC. Below, we discuss the complex autophagy signal network that regulates the features associated with PDAC progression.
Pancreatic stroma
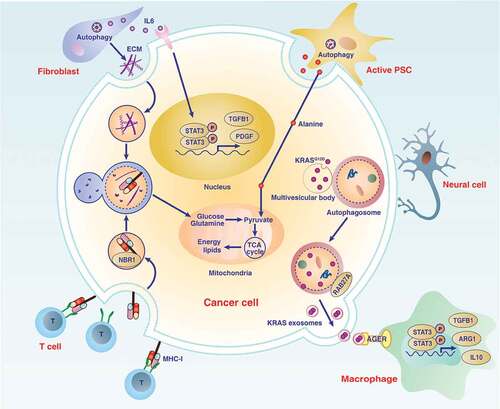
The progression of pancreatic cancer is accompanied by a fibrotic stromal (desmoplastic) response, and pancreatic stellate cells (PSCs) are the main source of collagen in pancreatic stroma. In addition to acinar cells, an increase in autophagy is related to the function of PSCs for PDAC progression in preclinical models or clinical samples [Citation57]. The autophagy level of PSCs in tumor tissue is higher than that of pancreatitis, which may lead to higher expression of extracellular matrix and pro-inflammatory IL6 (interleukin 6) in PSCs [Citation58] . Autophagy inhibitors put PSCs in a quiescent state and reduce the expression of extracellular matrix and IL6, thereby inhibiting inflammation and pancreatic tumor growth in cell culture and mice [Citation58]. IL6 is elevated in the serum of patients with pancreatic cancer and is an important proinflammatory and growth signal that activates the STAT3 (signal transducer and activator of transcription 3) pathway during the initiation and development of pancreatic tumors [Citation59,Citation60]. In addition, autophagy-mediated secretion of nutrients from PSCs promotes the metabolism of PDAC in xenograft models [Citation61], highlighting that autophagy mediates metabolic crosstalk between cancer and non-cancer cells in the pancreatic tumor microenvironment.
Figure 3. Role of autophagy in the pancreatic cancer microenvironment. The microenvironment of pancreatic cancer has unique characteristics and increased desmoplastic reactions, and is infiltrated by various immune or neural cells. Communication between cells in the microenvironment of pancreatic tumors depends on increased autophagy-dependent release or degradation of mediators (e.g., IL6, ECM, alanine, MHC-I, and exosomes), which ultimately promote cancer cell proliferation, invasion, or immune escape. Abbreviations: ARG1, arginase 1; ECM, extracellular matrix; IL6, interleukin 6; IL10, interleukin 10; KRAS, KRAS proto-oncogene, GTPase; MHC-I, major histocompatibility complex-I; NBR1, NBR1 autophagy cargo receptor; PDAC, pancreatic ductal adenocarcinoma; PDGF, platelet derived growth factor; PSC, pancreatic stellate cell; RAB27A, RAB27A, member RAS oncogene family; STAT3, signal transducer and activator of transcription 3; T cell, T lymphocyte cell; TCA cycle, tricarboxylic acid cycle; TGFB1, transforming growth factor beta 1
Cell proliferation
Two hallmarks of PDAC are that its proliferation signals are maintained and the growth of cancer cells is promoted. MicroRNAs (MIRs), such as MIR506 and MIR372, inhibit pancreatic cancer growth through targeting BECN1 or ULK1 expression in vitro, respectively [Citation62,Citation63], indicating the importance of MIRs in the modulation of autophagy-related PDAC. This process is further regulated by long noncoding RNAs (lncRNAs), a type of RNA that exceeds 200 nucleotides in length. For example, lncRNA PVT1 competitively replaces the MIR20A-5p sponge, thereby upregulating ULK1, and subsequently induces proliferation and development of PDAC by stimulating autophagy in cell culture and xenograft animal models [Citation64]. MDH1 (malate dehydrogenase 1), an enzyme that catalyzes the oxidation of malate to oxaloacetate in many metabolic pathways, promotes the proliferation of PDAC cells by maintaining ULK1 expression and subsequent autophagy in a hypoxic environment [Citation65]. Unlike MDH1, MMP7 (matrix metallopeptidase 7), a member of a family of zinc-containing enzymes capable of degrading various components of the extracellular matrix, increases autophagic flux and cell proliferation in hypoxic PDAC cells by activating YAP1 (Yes-associated protein 1) [Citation65], indicating that the Hippo signaling pathway may sustain proliferation signals through autophagy under hypoxic conditions.
The regulation of ULK1 protein stability by the UPS affects proliferation signals during PDAC development. The NEDD4L (NEDD4 like E3 ubiquitin protein ligase), an E3 ubiquitin protein ligase, is responsible for ULK1 degradation in PDAC cells [Citation66]. In contrast, the knockout of NEDD4L maintains the level of ULK1, leading to autophagy activation, followed by enhanced mitochondrial respiratory and PDAC cell proliferation [Citation66]. These findings further indicate that ULK1 is an important regulator of autophagy-related cell proliferation in PDAC cells. Collectively, the expression of ULK1 is regulated not only by gene transcription but also by protein degradation pathways, thereby affecting the formation of autophagy-inducing signals.
Immune receptors are also involved in the regulation of autophagy-related proliferation. AGER/RAGE (advanced glycosylation end-product specific receptor) is a member of the immunoglobulin superfamily of cell surface molecules [Citation67]. AGER promotes autophagy by activating the class III PtdIns3K complex, thereby promoting the survival and anti-apoptotic ability of PDAC cells [Citation68]. Moreover, AGER expression promotes ATG5-dependent autophagy, leading to proinflammatory IL6 production and subsequent activation of mitochondrial STAT3 and the production of ATP in vitro [Citation69]. Consequently, the absence of AGER restricts autophagy and STAT3 signaling, thereby inhibiting the development of KrasG12D/+-driven PDAC in mice [Citation69] , indicating that AGER-dependent autophagy promotes PDAC growth by maintaining energy production. AGER mediates the MAPK (mitogen-activated protein kinase) cascade, including MAP2K/MEK (mitogen-activated protein kinase kinase) and MAPK1/ERK1 kinase (mitogen-activated protein kinase 1), which may directly sustain KRAS-mediated proliferative capacity in PDAC cells and mouse models by activating autophagy [Citation70,Citation71]. Additionally, AGER-dependent autophagy increases the formation of neutrophil extracellular traps in animal models of PDAC and clinical patients, which may sustain a hypoxic and inflammatory tumor microenvironment [Citation72]. These findings support a pathological role of AGER in PDAC by regulating autophagy, metabolism, and inflammation.
SHOC2 (SHOC2 leucine rich repeat scaffold protein) is a positive regulator of RAS-MAPK signaling. SHOC2 inhibits MTOR1 activation through RPTOR/raptor (regulatory associated protein of MTOR complex 1), which ultimately causes autophagy and subsequent proliferation of PDAC cells. Excessive RPTOR that competitively binds SHOC2 also blocks MAPK activation, thereby limiting tumor growth [Citation73], indicating that the balance between SHOC2 and RPTOR affects RAS signaling and autophagy during the proliferation of PDAC cells. The tumor suppressor EI24 (EI24 autophagy associated transmembrane protein) is a component of the autophagy pathway that promotes proliferation of PDAC cells in vitro and in vivo [Citation74]. These findings indicate that both oncogenes and tumor suppressors can regulate the autophagy-related proliferation process.
MALAT1 (metastasis associated lung adenocarcinoma transcript 1) is an lncRNA that promotes the proliferation and metastasis of PDAC cells by regulating TIA1 (TIA1 cytotoxic granule associated RNA binding protein) or ELAVL1/HuR (ELAV like RNA binding protein 1) function to induce autophagic flux [Citation75]. Unlike AGER- or MALAT1-mediated autophagy activation, TP53BP2/ASPP2 (tumor protein p53 binding protein 2)-mediated autophagy inhibition limits cell proliferation in various PDAC cell lines by modulating the AMPK-MTOR pathway [Citation76], although the role of TP53BP2 in autophagy in vivo remains unclear. Autophagy inhibition mediated by MAGEA3 (MAGE family member A3; a tumor-specific antigen extensively expressed in solid and hematological malignancies) may promote PDAC development through an unknown mechanism in vitro and in vivo [Citation77]. These findings further support a dual role of autophagy in tumor biology.
Other cells in the tumor microenvironment, such as CSCs, also promote tumor proliferation and development by activating autophagy [Citation58,Citation78]. A high level of autophagy seems to be widely associated with the presence of a stem cell-like phenotype in PDAC, namely stemness [Citation79]. The exact mechanism by which autophagy regulates the stemness, such as through the presence of specific degradation substrates, requires further study.
Cell invasion and metastasis
Cancer cells have a strong ability to invade and metastasize, which is not only an indispensable biological process for tumor progression, but also a major cause of cancer-related death in patients. A large amount of evidence indicates that abnormal autophagy is related to the invasion and metastasis of PDAC in various models. For example, the protein level of ATG5 is positively related to the invasiveness of human pancreatic cancer, whereas the deletion of Atg5 inhibits tumor proliferation and metastasis in PDAC animal models [Citation80]. Unlike the core autophagy component ATG5, TGM2/TG2 (transglutaminase 2; a calcium-dependent enzyme of the transglutaminase family) is a negative regulator of autophagy involved in promoting metastasis in PDAC cells [Citation81]. The increase of TGM2 expression in PDAC cells is mediated by the activation of PRKCD/PKCδ (protein kinase C delta), and the inhibition of TGM2 by PRKCD inhibitors leads to the excessive activation of BECN1-dependent autophagy, thus causing cell death and metastasis inhibition in mice [Citation81,Citation82].
TGFB (transforming growth factor beta) plays an important role in advanced tumors, including promoting tumor metastasis and drug resistance. The autophagy induction effects of TGFB1 (transforming growth factor beta 1) are mediated by TFEB (transcription factor EB), which promotes the migration and metastasis in SMAD4-positive PDAC cells, but not SMAD4-negative cells [Citation83]. The role of TGFB1 in autophagy also depends on the SMAD4 expression level [Citation84]. TGFB1-induced autophagy promotes the proliferation of SMAD4-positive PDAC cells and inhibits their migration [Citation84]. In contrast, TGFB1-induced autophagy inhibits the proliferation of SMAD4-negative PDAC cells and promotes their migration by activating MAPK1 [Citation84]. These findings suggest that SMAD4 may be a checkpoint that distinguishes the different roles of autophagy in TGF family-induced proliferation and migration.
RALGAP (Ral GTPase activating protein) and UBL4A (ubiquitin like 4A) also regulate autophagy-related invasion and metastasis in PDAC through different mechanisms [Citation85,Citation86]. RALGAP is a negative regulator of autophagy by activating MTOR, leading to a decrease in the ability of PDAC cells to migrate and invade [Citation85]. UBL4A inhibits the proliferation and metastasis of PDAC cells by binding to LAMP1 (lysosomal associated membrane protein 1), resulting in lysosomal dysfunction and autophagy inhibition [Citation86]. In addition, QKI (QKI, KH domain containing RNA binding) and MAPK1 play a cell-dependent role in the activation of autophagy in fibroblasts and PSCs, respectively, mediating the mutual interference between stromal cells and cancer cells, which ultimately leads to proliferation, metastasis, and invasion of PDAC cells in vitro, organoid cultures, and xenograft mouse models [Citation87,Citation88]. These findings further suggest that increased autophagy in cells of the tumor microenvironment regulates PDAC invasion.
The epithelial-mesenchymal transition (EMT) is a process in which epithelial cells acquire interstitial characteristics that participate in promoting metastatic events. It is worth noting that, in some cases, the inhibition of autophagy may specifically activate the EMT program in KRAS-mutated PDAC cells [Citation89]. Mechanistically, autophagy inhibition promotes the upregulation of SQSTM1-mediated NFKB/NF-κB (nuclear factor kappa B) activation, thereby increasing the expression of EMT promoters, such as SNAI2 (snail family transcriptional repressor 2) and ZEB1 (zinc finger E-box binding homeobox 1) [Citation89]. These studies also indicate a dual role of autophagy in invasion and metastasis of PDAC.
Cell metabolism
PDAC cells are exposed to an environment with insufficient blood vessels. Therefore, when oxygen and nutrients are exhausted, it is critical for cells to obtain sufficient energy through certain metabolic pathways. In fact, PDAC relies on autophagy to provide enough bioenergy intermediates to maintain metabolism and energy homeostasis through fine-tuned mechanisms. On the one hand, MTORC1 promotes biosynthetic processes and inhibits catabolic pathways, such as autophagy, which is considered a key process for cells to adapt to an environment [Citation90]. TFEB promotes the recruitment of MTORC1 to lysosomes both in vitro and in vivo through regulating the expression of RRAGD (Ras related GTP binding D) [Citation91]. TFEB also promotes gene expression to maintain the metabolic function of lysosomes, which is critical for metabolic adaptation to the availability of nutrients and to support the metabolic requirements during PDAC proliferation [Citation26,Citation91]. On the other hand, excessive autophagy may trigger stress-mediated cell death by activating the NUPR1 (nuclear protein 1, transcriptional regulator)-AURKA (aurora kinase A) pathway during hypoxia and glucose starvation in MiaPACA2, PANC1, and BxPC3 cells [Citation92]. These studies describe the complex relationship between autophagy and PDAC metabolism, and highlight new areas where drug combinations targeting metabolism and autophagy may yield greater benefits. Below, we focus on how autophagy regulates oxidative stress, glucose metabolism, and amino acid metabolism in PDAC cells .
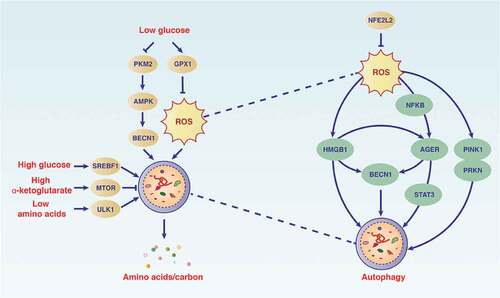
Figure 4. Autophagy maintains the metabolism and function of PDAC cells. The autophagy pathway is modulated by different metabolic conditions (e.g., oxidative stress, low glucose, high glucose, and low amino acids) in which cellular components are degraded. During this process, bioenergy intermediates are reused and oxidants are elimated, thereby promoting cell survival. Abbreviations: AGER, advanced glycosylation end-product specific receptor; AMPK, AMP-activated protein kinase; BECN1, beclin 1; GPX1, glutathione peroxidase 1; HMGB1, high mobility group box 1; MTOR, mechanistic target of rapamycin kinase; NFE2L2, nuclear factor, erythroid 2 like 2; NFKB, nuclear factor kappa B; PKM2, M2 splice isoform of PKM (pyruvate kinase M1/2); PINK, PTEN induced kinase 1; PRKN, parkin RBR E3 ubiquitin protein ligase; ROS, reactive oxygen species; SREBF1, sterol regulatory element binding transcription factor 1; STAT3, signal transducer and activator of transcription 3; ULK1, unc-51 like autophagy activating kinase 1
Oxidative stress
Oxidative stress refers to increased levels of ROS in cells, resulting in damage to lipids, proteins, and DNA. It has been associated with various pathologies, including PDAC progression. NFE2L2 is the main antioxidant transcription factor of cells in response to oxidative stress. Because ROS is the initiation signal of autophagy by suppressing MTORC1 activation [Citation93], NFE2L2 plays a role in the modulation of autophagic flux in PDAC cells [Citation94]. Blocking autophagy leads to increased stability of NFE2L2, whereas silencing NFE2L2 enhances the autophagic flux by promoting the formation of autolysosomes under conditions of ROS stimulation [Citation94]. As a danger signal, the release of HMGB1 during oxidative stress can trigger autophagy by binding to AGER in PDAC cells, which contributes to energy metabolism, inflammation, and tumor growth [Citation38,Citation95].
The mucin family consists of glycoproteins that form a physical barrier and protects cancer cells from various types of damage. The hypoxia-mediated abnormal expression of MUC1 (mucin 1, cell surface associated) and MUC4 (mucin 4, cell surface associated) is implicated in PDAC cells growth in vitro [Citation96]. However, ROS-induced autophagy mediates MUC4 degradation and may provide metabolites for tumor cell growth [Citation97]. Although the metal ion-induced Fenton response may increase ROS production, preventing copper absorption also promotes mitochondrial ROS production, which may activate autophagy to protect PDAC cells from death [Citation98]. The PINK1-PRKN pathway protects against mitochondrial oxidative injury via the autophagic degradation of mitochondrial iron importers, such as SLC25A37/mitoferrin-1 (solute carrier family 25 member 37) and SLC25A28/mitoferrin-2 (solute carrier family 25 member 28), thereby inhibiting the occurrence of pancreatic tumors [Citation52]. In addition, the depletion of MYOF (myoferlin; a ferlin family member protein overexpressed in many types of cancer) causes mitochondrial fission, which reduces energy production and triggers autophagy in PDAC cells [Citation99]. Overall, there is an integrated network between mitochondria, ROS, and autophagy in PDAC.
Glucose metabolism
The pancreatic cancer microenvironment has limited nutrients because of its dense stroma tissues and poor blood vessel formation. This characteristic of the microenvironment makes glucose an essential nutrient for PDAC under hypoxic conditions because it can be used as a basic fuel for anaerobic metabolism. Therefore, PDAC cells use autophagy-mediated glycolysis to resist death caused by glucose deprivation [Citation100]. For example, MIR7 may decrease autophagy-derived pools of glucose in PDAC cells by modulation of the activation of the STK11/LKB1 (serine/threonine kinase 11)-AMPK-MTOR pathway [Citation101]. Glucose deprivation also induces autophagy in PDAC cells through ROS, thereby maintaining cancer cell survival [Citation102]. In contrast, GPX1 (glutathione peroxidase 1) inhibits glucose deprivation-induced autophagy by limiting ROS production [Citation102]. Although these findings indicate that GPX1 acts as an anti-autophagic protein in PDAC, the role of other members of the GPX family in PDAC remains unknown.
Several glycolytic genes play a role in the regulation of metabolism-related autophagy. PKM2 (an M2 splice isoform of PKM [pyruvate kinase M1/2]) catalyzes the last step of glycolysis under aerobic conditions, and its expression is downregulated by low glucose in PDAC cells or the subvascular area of PDAC tissues [Citation103]. As a survival mechanism, reduced PKM2 may promote metabolic flux by increasing AMPK expression, thereby promoting low glucose-induced autophagy in PDAC cells [Citation103]. Autophagy positively regulates the expression of mitochondrial GFM1 (G elongation factor mitochondrial 1), providing essential nutrients for cells lacking glycolysis [Citation104,Citation105]. In addition to hypoglycemia, hyperglycemia also triggers autophagy for PDAC tumor growth by upregulating the expression of SREBF1/SREBP1 (sterol regulatory element binding transcription factor 1; an intracellular cholesterol sensor) in vitro and in vivo [Citation106]. These findings indicate that glucose or lipid abnormalities are important signals for autophagy induction.
In addition, KRAS-mediated expression of the lysosomal protein PLAC8 (placenta associated 8) is involved in the formation of autolysosomes, which may maintain the metabolic requirement of PDAC cells during tumor progression [Citation107]. Of note, the acute inhibition of KRAS or its downstream effector MAPK1 also increases the autophagy flux in KRAS-mutant human pancreatic cancer lines and KrasG12D-driven mouse models [Citation108], indicating that any changes in KRAS levels may lead to autophagy in PDAC. Therefore, even under low-glucose or hypoxic conditions, the combination of inhibiting KRAS signaling (e.g., using MAPK1 inhibitors) and autophagy signaling (e.g., using CQ) may become a powerful anticancer strategy for PDAC [Citation71,Citation108]. The functional role of HIF1A (hypoxia inducible factor 1 subunit alpha; the main regulator of hypoxia) is also expected to regulate autophagy by modulating KRAS and glucose signaling as a feedback mechanism.
Amino acid metabolism
During the development of PDAC, certain amino acids (e.g., branched-chain amino acids and glutamine) undergo significant changes, providing nutrition for cancer cell proliferation and invasion. Compared with the pharmacological inhibition of MTORC1, amino acid starvation is a powerful condition to induce autophagy in PDAC cells by activating ULK1 [Citation28]. The increase in autophagy plays a protective role in preserving the energy deficiency caused by amino acid starvation.
Glutamine is an important source of nitrogen and carbon in cancer cells and produces energy through mitochondria [Citation109,Citation110]. Glutamine is used to produce the tricarboxylic acid cycle intermediate alpha-ketoglutarate (αKG) through a series of mitochondrial enzymes (e.g., GLUD [glutamate dehydrogenase], GOT1 [glutamic-oxaloacetic transaminase 1], and GOT2 [glutamic-oxaloacetic transaminase 2]) to maintain the mitochondrial carbon pool. This biochemical process is called glutaminolysis and is one of the anti-aging strategies of metabolic stress [Citation111]. Human PDAC cells rely on GOT1-mediated glutaminolysis to obtain glutamine to promote tumor growth and development [Citation112]. Reprogramming of glutamine metabolism in PDAC is further regulated by oncogenic KRAS-mediated transcriptional regulation [Citation112]. Increased glutaminolysis-mediated αKG production leads to MTORC1 lysosomal translocation and activation by stimulating GTP loading of RRAGB (Ras related GTP binding B) and lysosomal translocation, thereby inhibiting autophagy [Citation113]. As a defense mechanism, the autophagy-mediated degradation of cellular materials provides amino acids (including glutamine) for reuse in PDAC cells during glutamine deprivation [Citation114]. Under the latter conditions, autophagy activation is further regulated by TFEB-induced macropinocytosis (a type of endocytosis involving a nonspecific uptake of extracellular materials, such as nutrients) [Citation114,Citation115]. Abnormal glutamine enhanced by glutaminolysis can also lead to proliferation inhibition and apoptosis of PDAC cell lines by activating MTORC1 and consequently inhibiting autophagy [Citation116]. These findings provide a framework for explaining the dynamic balance between glutamine metabolism, autophagy, and apoptosis in PDAC cells.
Branched-chain amino acids contain three essential amino acids (leucine, isoleucine, and valine), which cannot be synthesized in cells, so they must be obtained from food. Leucine is a branched-chain amino acid and is described as an MTOR agonist that promotes PDAC growth. Leucine reuse depends on the cooperation between autophagy and lysosomal proteolysis in human PDAC cell lines and murine pancreatic tumor cells isolated from KrasG12D/+;tp53−/− mice [Citation117]. SLC38A9 (solute carrier family 38 member 9) is not only a lysosomal arginine sensor, but also a high-affinity transporter for lysosomal proteolysis-produced leucine to exit lysosomes and activate MTORC1 in PDAC cells [Citation117]. Because SLC38A9 is a part of the lysosomal amino acid-sensing machinery, it is unclear whether other components play a similar role in the regulation of PDAC leucine metabolism.
PSCs in the tumor microenvironment also participate in the interaction between autophagy and amino acid metabolism to promote the growth of PDAC cells [Citation61]. Autophagy-mediated alanine release in PSCs can be used by PDAC cells, which promotes the tricarboxylic acid cycle or the biosynthesis of nonessential amino acids and lipids, possibly through the transamination of alanine into pyruvate [Citation61]. This autophagy-mediated feedback mechanism between PSCs and cancer cells supports the nutritional requirements of pancreatic cancer and promotes tumor growth [Citation61]. Autophagy can also mediate protein release in a nonclassical manner under certain circumstances. Whether autophagy releases endogenous proteins to mediate crosstalk between cells within the tumor microenvironment remains to be understood.
Cell death
Historically, autophagy has been defined as a type of cell death based on morphological classification [Citation118]. Currently, autophagy plays a dual role in cell death and survival, depending on whether the degradation substrate is an anti-injury or pro-injury protein [Citation119]. Induced autophagy is common and is triggered by various cell death stimuli, such as metabolic stress, oxidative damage, and anticancer drugs. It is worth noting that the term “autophagic cell death” is used to describe the phenomenon of increased autophagy accompanied by cell death, and the term “autophagy-dependent cell death” is a type of cell death that depends on the autophagy machinery [Citation120]. The ability to resist cell death is a hallmark of cancer and can be regulated by autophagy that depends on stimulation [Citation10].
Apoptosis
Apoptosis represents the main type of regulated cell death, including death due to the classical caspase family-mediated cell death receptor or mitochondrial pathway. Although targeted apoptosis (e.g., using gemcitabine) is the most commonly used nonsurgical treatment for PDAC, the PDAC cells can develop different mechanisms, including autophagy, to resist apoptosis [Citation68]. For example, 3-methyladenine (3 MA), an inhibitor of both the class I phosphoinositide 3-kinase and the class III PtdIns3K, inhibits autophagy and induces apoptosis following H2O2 exposure in PDAC cells [Citation121], indicating an anti-apoptosis role of autophagy. As mentioned above, AGER is an autophagy promoter in PDAC cells. NFKB pathway-mediated AGER upregulation increases autophagy flux and limits apoptosis in PDAC cells in response to oxidative stress or chemotherapy (e.g., gemcitabine and oxaliplatin) [Citation68]. In contrast, the knockdown of AGER inhibits autophagy in PDAC cells by activating MTORC1 or inhibiting BECN1, while increasing mitochondrial apoptosis by inducing TP53 phosphorylation [Citation68]. AGER also binds to mutant KRAS to maintain HIF1A activation in PDAC cells under hypoxic conditions [Citation122]. The inhibition of AGER expression by the compound scalarin inhibits autophagy and limits pancreatic tumor growth in PANC1 and MiaPACA2 cell lines [Citation123]. Although these studies indicate that AGER is a potential molecular switch between autophagy and apoptosis in a hypoxic pancreatic tumor microenvironment, it is unclear whether AGER-dependent autophagy inhibits apoptosis by directly degrading caspase effectors.
UCPs (uncoupling proteins) are a superfamily of mitochondrial carrier proteins anchored on the inner mitochondrial membrane. These proteins are highly expressed in various tumors (including PDAC) and mediate tumorigenesis and development [Citation124]. UCP2 promotes autophagy by inhibiting the AKT-MTOR pathway and activating BECN1, thereby inhibiting apoptosis or other types of cell death caused by ROS accumulation [Citation125]. The proteasome inhibitor bortezomib used clinically shows anticancer activity by inducing ROS-dependent apoptosis [Citation126]. Similar to blocking NFE2L2, autophagy inhibitors also enhance bortezomib-induced apoptosis in PDAC cells [Citation127], indicating that autophagy plays a pro-survival role in the regulation of bortezomib activity.
Epigenetic regulators, such as USP22 (ubiquitin specific peptidase 22) and PTBP3 (polypyrimidine tract binding protein 3), are implicated in autophagy-mediated gemcitabine resistance [Citation128,Citation129]. USP22 is a component of Spt-Ada-Gcn5-acetyltransferase/SAGA, a transcription regulation histone acetylation complex that regulates gene transcription at the epigenetic level, thereby promoting cell survival and apoptosis resistance [Citation130]. In PDAC patients, USP22 expression in tumors is associated with increased MAP1LC3 expression and poor prognosis. In PDAC cell lines, USP22 contributes to cell proliferation and gemcitabine resistance by activating MAPK1-mediated autophagy [Citation128]. Similarly, the RNA-splicing protein PTBP3 is also upregulated in tumors of PDAC patients as well as in animal models [Citation129]. PTBP3-mediated autophagy under hypoxic conditions leads to subsequent gemcitabine resistance of PDAC cells [Citation129]. However, the direct target genes of these epigenetic regulators mediate autophagy, and subsequent resistance to chemotherapy remains unknown.
Compared with cancer cells, pancreatic CSCs are more resistant to gemcitabine-induced apoptosis [Citation131]. This process may be mediated by increased autophagic flux under hypoxic conditions [Citation132]. In contrast, the administration of autophagy inhibitors may trigger apoptosis in hypoxic CSCs, and ultimately suppresses the development of PDAC in mouse models [Citation132]. In addition to CSCs, blocking autophagy also enhances apoptosis in PSCs during chemotherapy in vitro and in vivo [Citation133]. Therefore, targeting autophagy is an attractive strategy to inhibit self-renewed CSCs and activated fibroblasts in the pancreatic tumor microenvironment.
Necroptosis
Necroptosis is an alternative type of regulated cell death, mimicking the morphological characteristics of apoptosis and necrosis [Citation134]. During classic necroptosis, activated RIPK1 (receptor interacting serine/threonine kinase 1) and RIPK3 (receptor interacting serine/threonine kinase 3) interact with other proteins to produce the necrosome, which then induces MLKL (mixed lineage kinase domain like pseudokinase) to form large octamers. The oligomeric MLKL translocates to the plasma membrane and forms pores that damage the membrane, and then releases intracellular contents (namely DAMPs), affecting the surrounding cells and environment [Citation135]. These necroptosis regulators are not only highly expressed in human and murine PDAC, but can also be induced by the chemotherapeutic agent gemcitabine.
Dysfunctional necroptosis plays a dual role in PDAC, which may be affected by the status of autophagy [Citation136]. On the one hand, RIPK1-RIPK3-driven necroptosis induces adaptive immunosuppression by the release of DAMPs (e.g., CXCL1 [C-X-C motif chemokine ligand 1], CXCL5 [C-X-C motif chemokine ligand 5], and SAP130 [Sin3A associated protein 130]) that mediate pancreatic tumorigenesis [Citation137]. In addition, the depletion of RIPK3 results in mice with conditional deletion of the essential autophagy gene Atg7 being more sensitive to experimental pancreatitis [Citation35]. Although pancreatitis is a recognized risk factor for pancreatic cancer, it is unclear whether knocking out RIPK3 or other necroptotic regulators will affect pancreatic ATG7 depletion-mediated tumorigenesis. On the other hand, necroptosis-based cancer treatment has been proposed as a strategy to eliminate PDAC when caspase is inhibited or defective. For example, shikonin (a naphthoquinone extracted from Lithospermum) is an inhibitor of PKM2 and can trigger different types of cell death, including RIPK3-dependent necroptosis in PDAC cells [Citation138]. In the absence of glucose, decreased PKM2 promotes survival by inducing autophagy in PDAC [Citation103]. These findings support the notion that autophagy defects can enhance necroptosis, and this process is regulated by PKM2-mediated glycolysis.
In addition to regulating mitotic spindle formation and chromosome segregation, AURKA is also a positive regulator of autophagy in several diseases, including cancer. Pharmacological inhibition of AURKA is one of the strategies for treating tumors and shows promising clinical efficacy. CCT137690 is an AURKA inhibitor and exhibits strong anticancer activity in various PDAC tumor models by inducing necroptosis, but not other types of cell death [Citation139]. AURKA acts as a negative regulator of necroptosis in PDAC cells through the phosphorylation of GSK3B (glycogen synthase kinase 3 beta), thereby inhibiting the phosphorylation of RIPK3 and MLKL [Citation139]. The release of DAMPs (e.g., HMGB1 and ATP) by CCT137690 also improves the antitumor T-cell immunity in the established KRAS-driven PDAC [Citation139]. Thus, DAMPs may play a dual role in the microenvironment of pancreatic tumors, not only inhibiting but also enhancing antitumor immunity.
Ferroptosis
Activated autophagy generally promotes survival in response to stimulation of apoptosis or necrosis. However, in cancer and noncancer cells under the treatment of ferroptosis inducers, the enhancement of autophagy may switch from a pro-survival function into a pro-death function [Citation140–142]. Ferroptosis was initially identified as a mutant RAS-dependent cancer cell death [Citation143], relying on iron accumulation and lipid peroxidation [Citation144]. Although the exact effector of ferroptosis is unknown, it can be specifically induced by small-molecule compounds (e.g., erastin or RSL3) targeting antioxidant SLC7A11 (solute carrier family 7 member 11) or GPX4 (glutathione peroxidase 4), respectively [Citation145]. Consequently, increased ROS production triggers oxidative damage, including lipid peroxidation, which finally causes membrane damage and the release of DAMPs [Citation146]. The interplay between autophagy and lipid metabolism is complex [Citation147]. Several types of selective autophagy (e.g., ferritinophagy [Citation148], lipophagy [Citation149], and clockophagy [Citation150,Citation151]) and chaperone-mediated autophagy [Citation152] contribute to ferroptosis by the degradation of antiferroptosis regulators (e.g., ferritin, lipid droplets, ARNTL/BMAL1 [aryl hydrocarbon receptor nuclear translocator like], and GPX4), thereby promoting oxidative damage .
Figure 5. The mechanism of autophagy-dependent ferroptosis in PDAC. Several types of selective autophagy (e.g., ferritinophagy or lipophagy) contribute to ferroptosis in PDAC cells by mediating iron accumulation or lipid peroxidation. Autophagy-dependent ferroptosis plays a dual role in PDAC. On the one hand, small molecule compounds (e.g., erastin or RSL3) or drugs (e.g., rapamycin or zalcitabine) can induce autophagy-dependent ferroptosis for tumor therapy. On the other hand, oxidative stress induces autophagy-dependent ferroptosis in PDAC cells, which leads to the release of mutated KRAS and subsequent macrophage polarization, thereby promoting tumor growth. Abbreviations: AA, arachidonic acid; ACSL4, acyl-CoA synthetase long chain family member 4; ALOX5, arachidonate 5-lipoxygenase; ALOXs, ALOX: arachidonate lipoxygenases; ATF4, activating transcription factor 4; cGAMP, cyclic GMP-AMP; CGAS, cyclic GMP-AMP synthase; CoA, coenzyme A; CTSB, cathepsin B; DUSP1, dual specificity phosphatase 1; FA, fatty acid; Fe2+, ferrous iron; Fe3+, ferric iron; GPX4, glutathione peroxidase 4; GSH, glutathione; HSPA5, heat shock protein family A (hsp70) member 5; KRAS, KRAS proto-oncogene, GTPase; LPCAT3, lysophosphatidylcholine acyltransferase 3; mtDNA, mitochondrial DNA; MTOR, mechanistic target of rapamycin kinase; NCOA4, nuclear receptor coactivator 4; PE, phosphatidylethanolamine; POLG, DNA polymerase gamma, catalytic subunit; RAB7A, RAB7A, member RAS oncogene family; RAB27A, RAB27A, member RAS oncogene family; STING1/TMEM173, stimulator of interferon response cGAMP interactor 1; system xc−, the cystine/glutamate antiporter; TF, transferrin; TFAM, transcription factor A, mitochondrial; TFRC, transferrin receptor
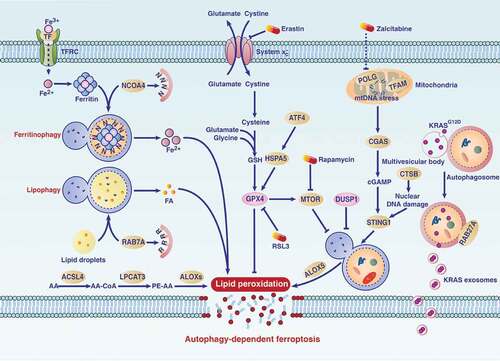
As with necroptosis, ferroptosis seems to play a complex role in the modulation of pancreatic tumor formation and therapy [Citation153–160]. SLC7A11 is a component of the cysteine-glutamic acid transporter, which plays a vital role in the uptake of cysteine and subsequent production of the antioxidant glutathione (GSH). The conditional knockout of Slc7a11 in pancreas, or treatment with cysteine-depletion drugs, inhibits mutant Kras- and Tp53-driven PDAC in mice [Citation156]. However, oxidative stress-related ferroptotic PDAC cells can release mutant KRAS into the extracellular space through autophagy-dependent exosome biogenesis and then be taken up by AGER expressed in macrophages [Citation153]. This ferroptotic cancer cell death-induced cell-to-cell communication eventually leads to macrophage polarization into a pro-tumor type [Citation153]. Therefore, ferroptosis not only inhibits the occurrence of pancreatic tumors by the consumption of cysteine in cancer cells, but also promotes the growth of tumors by DAMP-mediated macrophage polarization in the tumor microenvironment. Given that high iron is a risk factor for many tumors, it is unclear whether chronic iron accumulation-mediated oxidative damage plays a role similar to cysteine starvation in regulating pancreatic tumorigenesis [Citation161].
For established PDAC, the induction of ferroptosis by compounds or drugs significantly inhibits tumor growth. For example, zalcitabine is used to treat human immunodeficiency virus infection. It can also cause autophagy-dependent ferroptosis in PDAC cells by activating mitochondrial DNA stress-mediated STING1/STNG/TMEM173 (stimulator of interferon response cGAMP interactor 1) activation [Citation162]. Moreover, the translocation of lysosomal cysteine protease CTSB (cathepsin B) into the nucleus triggers DNA damage-related STING1 activation and subsequent autophagy-dependent ferroptosis in human PDAC cells [Citation163]. HSPA5/BIP (heat shock protein family A [hsp70] member 5)-mediated GPX4 stability is related to gemcitabine resistance in PDAC cells. Thus, inducing ferroptosis by targeting HSPA5 or GPX4 function may overcome gemcitabine resistance [Citation164]. The activation of DUSP1 (dual specificity phosphatase 1), a phosphatase that plays a role in inhibiting RAS signaling, limits autophagy-dependent ferroptosis in human PDAC cells [Citation160]. In addition, the interaction between MTOR and GPX4 signaling regulates autophagy-dependent ferroptosis in PDAC cells [Citation165]. High-dose rapamycin suppresses the growth of PDAC in vitro or in xenograft models by activating autophagy-dependent ferroptosis [Citation165]. Given that autophagy is highly upregulated in PDAC, pharmacological induction of autophagy-dependent ferroptosis may be a safe target therapy for PDAC.
Immune escape
The host’s immune system can recognize tumor antigens by presenting small antigen peptides to the receptors of cytotoxic T lymphocytes (CTLs) and reject new tumors [Citation166]. However, cancer cells may develop different systems (e.g., decreased antigenicity and impaired immunogenicity) to avoid destruction from the immune system [Citation167]. In addition to the DAMP release discussed above that is regulated by the interaction between autophagy and cell death, dysfunctional autophagy also participates in the formation of tumor immune escape through the degradation of immunomodulators, such as major histocompatibility complex class I (MHC-I), in PDAC [Citation168]. MHC-I on the surface of cancer cells plays a key role in antigen presentation, and immune checkpoints expressed in cancer cells and immune cells may limit CTL. Elevated autophagic degradation of MHC-I through the specific autophagy receptor NBR1 significantly reduces antigenicity [Citation168]. In contrast, pharmacological or genetic inhibition of autophagy can restore the expression of MHC-I in PDAC, thereby increasing antigenicity and subsequent immunogenicity during combined immune checkpoint blockade therapy in vitro and in orthotopic models [Citation168]. These findings suggest that inhibiting selective autophagy-mediated degradation of MHC-I may enhance the anticancer activity of immune checkpoint inhibitors in PDAC by inducing CTL. It is unclear whether other autophagic degradation substrates, especially expressed in antigen-presenting cells (e.g., dendritic cells [DCs] and macrophages) or effector cells (e.g., T and NK cells), are involved in tumor immune regulation. In addition, tumor nerve infiltration is associated with increased autophagy in PDAC patients [Citation169]. An interesting possibility is that this crosstalk between autophagy and the nervous system may allow amplification of immune escape in the microenvironment of PDAC.
Autophagy in pancreatic cancer therapy
Preclinical studies
Chemotherapy
As discussed above, the increase in autophagy in PDAC cells has led to apoptosis resistance to some first-line treatment drugs for PDAC patients, especially gemcitabine and oxaliplatin. The inhibition of autolysosome formation by CQ (a lysosomotropic agent) [Citation56], bafilomycin A1 (a V-ATPase inhibitor) [Citation56], UPSM (ultra pH-sensitive micelles) [Citation170], or withaferin-A (a SNARE inhibitor) [Citation171] suppress PDAC growth alone or together with other drugs. Natural products (e.g., ancistrolikokine E3) [Citation172], heavy metal chelators (e.g., N,N,N’,N’-tetrakis[2-pyridylmethyl]-ethylenediamine/TPEN) [Citation173], or organic compounds (e.g., EAD1) [Citation174] also show anticancer activity in PDAC cells by inhibiting autophagy. Together, these findings support the conclusion that the inhibition of autophagy is beneficial for PDAC treatment.
In contrast, the pharmacological induction of autophagy-dependent ferroptosis by targeting SLC7A11 (e.g., erastin and sorafenib), GPX4 (e.g., RSL3), or STING1 (e.g., zalcitabine) pathways also exhibits anticancer effects on PDAC cells in vitro or in xenograft models [Citation155,Citation162,Citation175]. In addition to ferroptosis, the induction of other types of autophagy-dependent cell death in PDAC cells or PSCs by drugs (e.g., tocotrienol, oleandrin, ascorbate, triptolide, and rottlerin) also suppress PDAC growth in vitro or in vivo [Citation176–181] . The mechanism and effect of these drugs are related to the activation of AMPK, the inhibition of MTOR or the induction of BECN1 expression.
Table 1. Monotherapies that affect autophagy in PDAC
In most cases (especially as adjuvant or neoadjuvant therapy), combination therapy is more effective than monotherapy for PDAC treatment. Combination therapy inhibits PDAC by decreasing autophagy activation or increasing autophagy activation . In addition to agents that target lysosomes (e.g., CQ or bafilomycin A1), the pharmacological blocking of autophagy by inhibiting AMPK (using lumican) [Citation207] or increasing the accumulation of lysosomal-associated palmitoylated proteins (using DQ661) [Citation208] enhance the anticancer activity of gemcitabine or other chemotherapy drugs. Alternatively, excessive autophagy activation may lead to tumor suppression through autophagy-dependent metabolic stress or injury. For example, combined treatment with adrenergic A and phenethyl isothiocyanate activates autophagy-dependent ferroptosis in human PDAC cells [Citation209]. Combined treatment with troglitazone and IFNB/interferon-β induces autophagy-dependent tumor suppression in PDAC cells by inhibiting the phosphorylation of EIF4E (eukaryotic translation initiation factor 4E) [Citation210]. The use of CP-31,398-RITA, gossypol-BRD4770 or dugrin-doxorubicin combination therapy also inhibits PDAC growth through activating autophagy-dependent cell death [Citation211–213]. These findings suggest the complexity of applying autophagy-related drugs in pancreatic tumor chemotherapy.
Table 2. Combination therapies that affect autophagy in PDAC
Radiotherapy
Radiotherapy uses radiation to destroy cancer cells, and this process is regulated by SMAD4-related autophagy in PDAC cells. In particular, the depletion of SMAD4 increases autophagy, which helps PDAC cells to resist ionizing radiation in vitro or in vivo [Citation235]. The downregulation of MIR23B also contributes to radiotherapy resistance in PDAC cells by activating ATG12-dependent autophagy [Citation236]. In contrast, the overexpression of MIR23B suppresses the expression of ATG12, thus restoring ionizing radiation sensitivity by inhibiting autophagy [Citation236]. These findings support a pro-survival role of autophagy during radiotherapy.
However, the combination of radiotherapy and chemotherapy (e.g., using gemcitabine or rapamycin) can suppress PDAC by activating excessive autophagy-dependent cell death in xenografts and orthotopic tumor models [Citation237,Citation238]. Similarly, the combined use of MG132 (a proteasome inhibitor) and radiation also triggers autophagy-dependent cell death in PDAC cells, and this process is mediated by the inhibition of pro-survival TRAF6 (TNF receptor associated factor 6; an adaptor protein that mediates a wide array of protein-protein interactions) [Citation239]. These findings indicate that feedback between protein degradation and interaction pathways modulate the sensitivity of radiotherapy in PDAC. Interestingly, the combination of seaweed polyphenols and radiation fractionation may inhibit autophagy through unknown molecular mechanisms to protect radiation-resistant PDAC cells [Citation240]. All these studies provide a theoretical basis for targeting autophagy to enhance the response to radiotherapy.
Immunotherapy
As mentioned above, the direct evidence of autophagy-modulated immunotherapy is that autophagy mediates the degradation of MHC-I in PDAC cells [Citation168]. Inhibiting this type of selective autophagy ultimately enhances the anticancer activity of immune checkpoint inhibitors by activating classic CD8+ T-cell-mediated antitumor immunity [Citation168]. It is still necessary to know how the different autophagy levels of immune cells (e.g., NK, B, and T cells as well as DCs and macrophages) in the tumor microenvironment regulate antitumor immunity under different conditions. How autophagy regulates immunometabolism, neuroimmunity, and microbial homeostasis in the tumor microenvironment remains an open question. In addition, autophagy-mediated protein release may play a role in mediating the communication between immune and cancer cells.
Clinical studies
Hydroxychloroquine (HCQ) is a derivative of CQ and is used to treat patients with malaria or autoimmune diseases. Both HCQ and CQ enhance the efficacy of many anticancer regimens by disrupting lysosomal function and impairing autophagy in preclinical animal models [Citation241]. Although they are all used as potential autophagy inhibitors, HCQ is only half as toxic as CQ in humans. Thus, in the past few years, clinical research has greatly expanded into cancer-related autophagy using HCQ combined with other chemotherapy for PDAC . Two randomized trials showed that, compared with nab-paclitaxel and gemcitabine treatment group, additional HCQ combination therapy can improve the response rate [Citation242–244]. Although survival rates are not affected, these studies show that this autophagy inhibition approach can hold promise [Citation242–244]. Some case reports also show that the combined use of HCQ and other drugs (e.g., the MAP2K/MEK-MAPK/ERK inhibitor) has potential benefits for some PDAC patients [Citation71,Citation108,Citation245], but it is still uncertain whether these combination therapies are useful for most patients.
Table 3. Published clinical studies on using HCQ to inhibit autophagy in PDAC
Conclusion and perspectives
In the past decade, we have made significant progress in our understanding of autophagy as a fundamental cellular homeostasis program, not only in comprehending its regulation networks and signaling pathways, but also in the wide range of pathophysiological effects of autophagy in health and disease. Similarly, our understanding of autophagy in PDAC is increasing dramatically. Impaired autophagy involves various biological processes involved in the occurrence and development of pancreatic cancer, which play complex and important roles not only in cancer cells but also in noncancer cells within the tumor microenvironment. The complex functions of autophagy in PDAC are further regulated by oncogenes (e.g., KRAS) and tumor suppressors (e.g., TP53 and PTEN).
While preclinical studies have achieved promising results in targeting autophagy to treat PDAC, clinical research involving this strategy is uncertain and even frustrating. Although the exact cause of this difference in preclinical and clinical studies is not clear, at least the following four aspects could explain it: First, animals and humans have different genetic backgrounds, which indicate that the model of mouse PDAC may not be able to mimic the pathological process of human PDAC. Second, most of the experimental mice are fed in a sterile environment, which may result in a significant difference in the intestinal microbial composition and immune response between mice and humans. This difference not only affects the activation of the autophagy pathway, but also leads to different microenvironments in pancreatic tumors. Third, the inhibition of autophagy is not the only effect of HCQ. Its potential off-target and even toxic side effects (e.g., causing liver and heart problems) may affect the interpretation of clinical trial results. Fourth, autophagy is also beneficial to normal tissue function in terms of overall maintenance of cellular homeostasis; the inhibition of nonselective autophagy by HCQ may cause normal tissue dysfunction or damage.
Autophagy plays a dual role in the regulation of pancreatic tumorigenesis and treatment, depending on the type of autophagy, the stage of cancer, and the activation by drugs [Citation10]. The duality of autophagy is ultimately reflected in its pro-survival and pro-death functions, which indicates that both autophagy inducers and autophagy inhibitors can be used to treat cancer, including PDAC. Therefore, the development of drugs targeting signaling factors or modulators of selective autophagy, such as specific autophagy receptors or cargos, may be a reasonable and safe clinical strategy to inhibit tumor growth. The accomplishment of this arduous goal requires multidisciplinary cooperation as well as the development of reliable and convenient autophagy detection methods in patients.
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript. This work was supported by grants from the US National Institutes of Health (R01CA160417 [D.T.], R01CA211070 [R.K.] and GM131919 [D.J.K.]).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
- Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022.
- Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450.
- Neoptolemos JP, Kleeff J, Michl P, et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–348.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703.
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542.
- Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822.
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174.
- Thumm M, Egner R, Koch B, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280.
- Harding TM, Morano KA, Scott SV, et al. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45.
- Paquette M, El-Houjeiri L, Pause A. mTOR pathways in cancer and autophagy. Cancers (Basel). 2018;10:18 1 doi:10.3390/cancers10010018
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728.
- Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812.
- Pasquier B. Autophagy inhibitors. Cell Mol Life Sci. 2016;73:985–1001.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326.
- Reggiori F, Komatsu M, Finley K, et al. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012;2012:219625.
- Kirkin V, McEwan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269.
- Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432:80–103.
- Ko YH, Cho YS, Won HS, et al. Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas. 2013;42:829–835.
- Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433.
- Wong PM, Feng Y, Wang J, et al. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048.
- Resau JH, Cottrell JR, Hudson EA, et al. Studies on the mechanisms of altered exocrine acinar cell differentiation and ductal metaplasia following nitrosamine exposure using hamster pancreatic explant organ culture. Carcinogenesis. 1985;6:29–35.
- Flaks B, Moore MA, Flaks A. Ultrastructural analysis of pancreatic carcinogenesis. IV. Pseudoductular transformation of acini in the hamster pancreas during N-nitroso-bis(2-hydroxypropyl)amine carcinogenesis. Carcinogenesis. 1981;2:1241–1253.
- Rez G, Toth S, Palfia Z. Cellular autophagic capacity is highly increased in azaserine-induced premalignant atypical acinar nodule cells. Carcinogenesis. 1999;20:1893–1898.
- Park CH, Lee IS, Grippo P, et al. Akt kinase mediates the prosurvival effect of smoking compounds in pancreatic ductal cells. Pancreas. 2013;42:655–662.
- Tsang YH, Wang Y, Kong K, et al. Differential expression of MAGEA6 toggles autophagy to promote pancreatic cancer progression. Elife. 2020;9:e48963
- Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A. 2015;112:E6166–74.
- Zhou X, Xie L, Xia L, et al. RIP3 attenuates the pancreatic damage induced by deletion of ATG7. Cell Death Dis. 2017;8:e2918.
- Mathew R, Khor S, Hackett SR, et al. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell. 2014;55:916–930.
- Yang S, Imamura Y, Jenkins RW, et al. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol Res. 2016;4:520–530.
- Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892.
- Kang R, Xie Y, Zhang Q, et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 2017;27:916–932.
- Molejon MI, Ropolo A, Re AL, et al. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci Rep. 2013;3:1055.
- Loncle C, Molejon MI, Lac S, et al. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes Kras(G12D)-mediated pancreatic cancer initiation. Cell Death Dis. 2016;7:e2295.
- Maes H, Kuchnio A, Peric A, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190–206.
- Todoric J, Antonucci L, Di Caro G, et al. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell. 2017;32:824–39 e8.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223.
- Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631.
- Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–708.
- Yang A, Rajeshkumar NV, Wang X, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–913.
- Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300.
- Amaravadi R, Debnath J. Mouse models address key concerns regarding autophagy inhibition in cancer therapy. Cancer Discov. 2014;4:873–875.
- Rosenfeldt MT, O’Prey J, Flossbach L, et al. PTEN deficiency permits the formation of pancreatic cancer in the absence of autophagy. Cell Death Differ. 2017;24:1303–1304.
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
- Li C, Zhang Y, Cheng X, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46:441–55 e8.
- Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019;9:1268–1287.
- Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632.
- Alcala S, Sancho P, Martinelli P, et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun. 2020;11:2682.
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729.
- Hwang RF, Moore T, Arumugam T, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926.
- Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152:1492–506 e24.
- Okada S, Okusaka T, Ishii H, et al. Elevated serum interleukin-6 levels in patients with pancreatic cancer. Jpn J Clin Oncol. 1998;28:12–15.
- Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469.
- Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–483.
- Sun L, Hu L, Cogdell D, et al. MIR506 induces autophagy-related cell death in pancreatic cancer cells by targeting the STAT3 pathway. Autophagy. 2017;13:703–714.
- Chen H, Zhang Z, Lu Y, et al. Downregulation of ULK1 by microRNA-372 inhibits the survival of human pancreatic adenocarcinoma cells. Cancer Sci. 2017;108:1811–1819.
- Huang F, Chen W, Peng J, et al. LncRNA PVT1 triggers Cyto-protective autophagy and promotes pancreatic ductal adenocarcinoma development via the miR-20a-5p/ULK1 Axis. Mol Cancer. 2018;17:98.
- New M, Van Acker T, Sakamaki JI, et al. MDH1 and MPP7 regulate autophagy in pancreatic ductal adenocarcinoma. Cancer Res. 2019;79:1884–1898.
- Lee DE, Yoo JE, Kim J, et al. NEDD4L downregulates autophagy and cell growth by modulating ULK1 and a glutamine transporter. Cell Death Dis. 2020;11:38.
- Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17.
- Kang R, Tang D, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676.
- Kang R, Loux T, Tang D, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012;109:7031–7036.
- Collisson EA, Trejo CL, Silva JM, et al. A central role for RAF–>MEK–>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–693.
- Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–627.
- Boone BA, Orlichenko L, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22:326–334.
- Xie CM, Tan M, Lin XT, et al. The FBXW7-SHOC2-raptor axis controls the cross-talks between the RAS-ERK and mTORC1 signaling pathways. Cell Rep. 2019;26:3037–50 e4.
- Hwang M, Jun DW, Kang EH, et al. EI24, as a component of autophagy, is involved in pancreatic cell proliferation. Front Oncol. 2019;9:652.
- Li L, Chen H, Gao Y, et al. Long noncoding RNA MALAT1 promotes aggressive pancreatic cancer proliferation and metastasis via the stimulation of autophagy. Mol Cancer Ther. 2016;15:2232–2243.
- Song B, Bian Q, Zhang YJ, et al. Downregulation of ASPP2 in pancreatic cancer cells contributes to increased resistance to gemcitabine through autophagy activation. Mol Cancer. 2015;14:177.
- Das B, Senapati S. Functional and mechanistic studies reveal MAGEA3 as a pro-survival factor in pancreatic cancer cells. J Exp Clin Cancer Res. 2019;38:294.
- Gong C, Bauvy C, Tonelli G, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–72,72e 1–11.
- Yang MC, Wang HC, Hou YC, et al. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer. 2015;14:179.
- Gorgulu K, Diakopoulos KN, Ai J, et al. Levels of the autophagy-related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology. 2019;156:203–17 e20.
- Akar U, Ozpolat B, Mehta K, et al. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol Cancer Res. 2007;5:241–249.
- Ozpolat B, Akar U, Mehta K, et al. PKC delta and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy. 2007;3:480–483.
- He R, Wang M, Zhao C, et al. TFEB-driven autophagy potentiates TGF-beta induced migration in pancreatic cancer cells. J Exp Clin Cancer Res. 2019;38:340.
- Liang C, Xu J, Meng Q, et al. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy. 2020;16:486–500.
- Martin TD, Chen XW, Kaplan RE, et al. Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol Cell. 2014;53:209–220.
- Chen H, Li L, Hu J, et al. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J Exp Clin Cancer Res. 2019;38:297.
- Chu L, Hu Y, Jiang YH, et al. Effects of RNA binding protein QKI on pancreatic cancer ductal epithelial cells and surrounding activation fibroblasts. J Cell Biochem. 2019;120:11551–11561.
- Yan Z, Ohuchida K, Fei S, et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J Exp Clin Cancer Res. 2019;38:221.
- Wang Y, Xiong H, Liu D, et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy. 2019;15:886–899.
- Alexander J, Valvezan BDM. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab. 2019;1:321–333.
- Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188–1192.
- Hamidi T, Cano CE, Grasso D, et al. Nupr1-aurora kinase A pathway provides protection against metabolic stress-mediated autophagic-associated cell death. Clin Cancer Res. 2012;18:5234–5246.
- Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158.
- Zhang L, Li J, Ma J, et al. The relevance of Nrf2 pathway and autophagy in pancreatic cancer cells upon stimulation of reactive oxygen species. Oxid Med Cell Longev. 2016;2016:3897250.
- Kang R, Tang D, Schapiro NE, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33:567–577.
- Singh AP, Moniaux N, Chauhan SC, et al. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004;64:622–630.
- Joshi S, Kumar S, Ponnusamy MP, et al. Hypoxia-induced oxidative stress promotes MUC4 degradation via autophagy to enhance pancreatic cancer cells survival. Oncogene. 2016;35:5882–5892.
- Yu Z, Zhou R, Zhao Y, et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019;52:e12568.
- Rademaker G, Hennequiere V, Brohee L, et al. Myoferlin controls mitochondrial structure and activity in pancreatic ductal adenocarcinoma, and affects tumor aggressiveness. Oncogene. 2018;37:4398–4412.
- Bryant KL, Mancias JD, Kimmelman AC, et al. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100.
- Gu DN, Jiang MJ, Mei Z, et al. microRNA-7 impairs autophagy-derived pools of glucose to suppress pancreatic cancer progression. Cancer Lett. 2017;400:69–78.
- Meng Q, Xu J, Liang C, et al. GPx1 is involved in the induction of protective autophagy in pancreatic cancer cells in response to glucose deprivation. Cell Death Dis. 2018;9:1187.
- Li X, Deng S, Liu M, et al. The responsively decreased PKM2 facilitates the survival of pancreatic cancer cells in hypoglucose. Cell Death Dis. 2018;9:133.
- Shiratori R, Furuichi K, Yamaguchi M, et al. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci Rep. 2019;9:18699.
- Maertin S, Elperin JM, Lotshaw E, et al. Roles of autophagy and metabolism in pancreatic cancer cell adaptation to environmental challenges. Am J Physiol Gastrointest Liver Physiol. 2017;313:G524–G36.
- Zhou C, Qian W, Li J, et al. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J Exp Clin Cancer Res. 2019;38:302.
- Kinsey C, Balakrishnan V, O’Dell MR, et al. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep. 2014;7:1143–1155.
- Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–640.
- Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–433.
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548.
- Eng CH, Abraham RT. Glutaminolysis yields a metabolic by-product that stimulates autophagy. Autophagy. 2010;6:968–970.
- Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105.
- Duran RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358.
- Seo JW, Choi J, Lee SY, et al. Autophagy is required for PDAC glutamine metabolism. Sci Rep. 2016;6:37594.
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637.
- Jeong SM, Hwang S, Park K, et al. Enhanced mitochondrial glutamine anaplerosis suppresses pancreatic cancer growth through autophagy inhibition. Sci Rep. 2016;6:30767.
- Wyant GA, Abu-Remaileh M, Wolfson RL, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171:642–54 e12.
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364.
- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–1518.
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
- Kang R, Tang D, Livesey KM, et al. The receptor for advanced glycation end-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid Redox Signal. 2011;15:2175–2184.
- Kang R, Hou W, Zhang Q, et al. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014;5:e1480.
- Guzman EA, Pitts TP, Diaz MC, et al. The marine natural product Scalarin inhibits the receptor for advanced glycation end products (RAGE) and autophagy in the PANC-1 and MIA PaCa-2 pancreatic cancer cell lines. Invest New Drugs. 2019;37:262–270.
- Donadelli M, Dando I, Fiorini C, et al. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci. 2014;71:1171–1190.
- Dando I, Pacchiana R, Pozza ED, et al. UCP2 inhibition induces ROS/Akt/mTOR axis: role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic Biol Med. 2017;113:176–189.
- Perez-Galan P, Roue G, Villamor N, et al. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107:257–264.
- Li X, Liang M, Jiang J, et al. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int J Biol Sci. 2018;14:1291–1305.
- Liang JX, Ning Z, Gao W, et al. Ubiquitinspecific protease 22induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol Rep. 2014;32:2726–2734.
- Ma J, Weng L, Jia Y, et al. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in pancreatic cancer cells by ATG12 up-regulation. J Cell Mol Med. 2020;24:2917–2930.
- Zhang XY, Varthi M, Sykes SM, et al. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29:102–111.
- Wang D, Zhu H, Zhu Y, et al. CD133(+)/CD44(+)/Oct4(+)/Nestin(+) stem-like cells isolated from Panc-1 cell line may contribute to multi-resistance and metastasis of pancreatic cancer. Acta Histochem. 2013;115:349–356.
- Rausch V, Liu L, Apel A, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227:325–335.
- Zhang X, Schonrogge M, Eichberg J, et al. Blocking autophagy in cancer-associated fibroblasts supports chemotherapy of pancreatic cancer cells. Front Oncol. 2018;8:590.
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465.
- Rickard JA, O’Donnell JA, Evans JM, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188.
- Goodall ML, Fitzwalter BE, Zahedi S, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–349.
- Seifert L, Werba G, Tiwari S, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–249.
- Chen C, Xiao W, Huang L, et al. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am J Transl Res. 2017;9:5507–5517.
- Xie Y, Zhu S, Zhong M, et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153:1429–43 e5.
- Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–435.
- Kang R, Tang D. Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep. 2017;5:153–159.
- Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072.
- Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2020;1–28. doi:10.1080/15548627.2020.1810918
- Kuang F, Liu J, Kang R, et al. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8. DOI:10.3389/fcell.2020.586578.
- Wen Q, Liu J, Kang R, et al. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–283.
- Xie Y, Li J, Kang R, et al. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431.
- Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428.
- Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
- Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
- Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15:2033–2035.
- Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116:2996–3005.
- Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020; 16:2069–2083
- Wang H, Liu C, Zhao Y, et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun. 2020;522:1063–1068.
- Daher B, Parks SK, Durivault J, et al. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019;79:3877–3890.
- Badgley MA, Kremer DM, Maurer HC, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89.
- Wang Y, Liua Y, Liua J, et al. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020; 531:581–587
- Wang K, Zhang Z, Wang M, et al. Role of GRP78 inhibiting artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells. Drug Des Devel Ther. 2019;13:2135–2144.
- Eling N, Reuter L, Hazin J, et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–532.
- Xie Y, Kuang F, Liu J, et al. DUSP1 blocks autophagy-dependent ferroptosis in pancreatic cancer. J pancreatol. 2020;3:154–160. DOI:10.1097/JP9.0000000000000054.
- Chen X, Xu C, Kang R, et al. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:1–14.
- Li C, Zhang Y, Liu J, et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2020; Mar 18; 1–13. doi:10.1080/15548627.2020.1739447
- Kuang F, Liu J, Li C, et al. Cathepsin B is a mediator of organelle-specific initiation of ferroptosis. Biochem Biophys Res Commun. 2020. DOI:10.1016/j.bbrc.2020.10.035
- Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–2077.
- Liu Y, Wang Y, Liu J, et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2020. DOI:10.1038/s41417-020-0182-y
- Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195:346–355.
- Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48:417–433.
- Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581:100–105.
- Yang YH, Liu JB, Gui Y, et al. Relationship between autophagy and perineural invasion, clinicopathological features, and prognosis in pancreatic cancer. World J Gastroenterol. 2017;23:7232–7241.
- Kong C, Li Y, Liu Z, et al. Targeting the oncogene KRAS mutant pancreatic cancer by synergistic blocking of lysosomal acidification and rapid drug release. ACS Nano. 2019;13:4049–4063.
- Li X, Zhu F, Jiang J, et al. Simultaneous inhibition of the ubiquitin-proteasome system and autophagy enhances apoptosis induced by ER stress aggravators in human pancreatic cancer cells. Autophagy. 2016;12:1521–1537.
- Awale S, Dibwe DF, Balachandran C, et al. Ancistrolikokine E3, a 5,8ʹ-coupled naphthylisoquinoline alkaloid, eliminates the tolerance of cancer cells to nutrition starvation by inhibition of the akt/mtor/autophagy signaling pathway. J Nat Prod. 2018;81:2282–2291.
- Yu Z, Yu Z, Chen Z, et al. Zinc chelator TPEN induces pancreatic cancer cell death through causing oxidative stress and inhibiting cell autophagy. J Cell Physiol. 2019;234:20648–20661.
- Nordstrom LU, Sironi J, Aranda E, et al. Discovery of autophagy inhibitors with antiproliferative activity in lung and pancreatic cancer cells. ACS Med Chem Lett. 2015;6:134–139.
- Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245.
- Rickmann M, Vaquero EC, Malagelada JR, et al. Tocotrienols induce apoptosis and autophagy in rat pancreatic stellate cells through the mitochondrial death pathway. Gastroenterology. 2007;132:2518–2532.
- Vaquero EC, Rickmann M, Molero X. Tocotrienols: balancing the mitochondrial crosstalk between apoptosis and autophagy. Autophagy. 2007;3:652–654.
- Newman RA, Kondo Y, Yokoyama T, et al. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr Cancer Ther. 2007;6:354–364.
- Du J, Martin SM, Levine M, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16:509–520.
- Mujumdar N, Mackenzie TN, Dudeja V, et al. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology. 2010;139:598–608.
- Singh BN, Kumar D, Shankar S, et al. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharmacol. 2012;84:1154–1163.
- Shen Q, Wang J, Liu CX, et al. Synthesis and evaluation of tetrahydroquinolin-2(1H)-one derivatives as novel anti-pancreatic cancer agents via targeting autophagy. Eur J Med Chem. 2019;170:28–44.
- Li C, Xu H, Chen X, et al. Aqueous extract of clove inhibits tumor growth by inducing autophagy through AMPK/ULK pathway. Phytother Res. 2019;33:1794–1804.
- Fukui M, Yamabe N, Choi HJ, et al. Mechanism of ascorbate-induced cell death in human pancreatic cancer cells: role of bcl-2, beclin 1 and autophagy. Planta Med. 2015;81:838–846.
- Pathania AS, Guru SK, Kumar S, et al. Interplay between cell cycle and autophagy induced by boswellic acid analog. Sci Rep. 2016;6:33146.
- Dando I, Donadelli M, Costanzo C, et al. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013;4:e664.
- Li H, Chen H, Li R, et al. Cucurbitacin I induces cancer cell death through the endoplasmic reticulum stress pathway. J Cell Biochem. 2018. doi:10.1002/jcb.27570
- Ovadje P, Chochkeh M, Akbari-Asl P, et al. Selective induction of apoptosis and autophagy through treatment with dandelion root extract in human pancreatic cancer cells. Pancreas. 2012;41:1039–1047.
- Zhang X, Hu P, Ding SY, et al. Induction of autophagy-dependent apoptosis in cancer cells through activation of ER stress: an uncovered anti-cancer mechanism by anti-alcoholism drug disulfiram. Am J Cancer Res. 2019;9:1266–1281.
- Krishan S, Sahni S, Leck LYW, et al. Regulation of autophagy and apoptosis by Dp44mT-mediated activation of AMPK in pancreatic cancer cells. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165657.
- Jia S, Xu X, Zhou S, et al. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019;10:142.
- Pardo R, Lo Re A, Archange C, et al. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology. 2010;10:19–26.
- Akimoto M, Iizuka M, Kanematsu R, et al. Anticancer effect of ginger extract against pancreatic cancer cells mainly through reactive oxygen species-mediated autotic cell death. PLoS One. 2015;10:e0126605.
- Liu L, Zhang N, Dou Y, et al. Lysosomal dysfunction and autophagy blockade contribute to IMB-6G-induced apoptosis in pancreatic cancer cells. Sci Rep. 2017;7:41862.
- Yu M, Nguyen ND, Huang Y, et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight. 2019;5:e126915
- Xu C, Wang Y, Tu Q, et al. Targeting surface nucleolin induces autophagy-dependent cell death in pancreatic cancer via AMPK activation. Oncogene. 2019;38:1832–1844.
- Gomez de Cedron M, Mouhid L, Garcia-Carrascosa E, et al. Marigold supercritical extract as potential co-adjuvant in pancreatic cancer: the energetic catastrophe induced via bmp8b ends up with autophagy-induced cell death. Front Bioeng Biotechnol. 2019;7:455.
- Cho YR, Lee JH, Kim JH, et al. Matrine suppresses KRAS-driven pancreatic cancer growth by inhibiting autophagy-mediated energy metabolism. Mol Oncol. 2018;12:1203–1215.
- Millet A, Plaisant M, Ronco C, et al. Discovery and Optimization of N-(4-(3-Aminophenyl)thiazol-2-yl)acetamide as a Novel Scaffold Active against Sensitive and Resistant Cancer Cells. J Med Chem. 2016;59:8276–8292.
- Liu W, Wang X, Sun J, et al. Parthenolide suppresses pancreatic cell growth by autophagy-mediated apoptosis. Onco Targets Ther. 2017;10:453–461.
- Ranjan A, Srivastava SK. Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci Rep. 2016;6:26165.
- Liao G, Gao B, Gao Y, et al. Phycocyanin inhibits tumorigenic potential of pancreatic cancer cells: role of apoptosis and autophagy. Sci Rep. 2016;6:34564.
- Ogata A, Yanagie H, Ishikawa E, et al. Antitumour effect of polyoxomolybdates: induction of apoptotic cell death and autophagy in in vitro and in vivo models. Br J Cancer. 2008;98:399–409.
- Bello C, Bai J, Zambron BK, et al. Induction of cell killing and autophagy by amphiphilic pyrrolidine derivatives on human pancreatic cancer cells. Eur J Med Chem. 2018;150:457–478.
- Dai ZJ, Gao J, Ma XB, et al. Antitumor effects of rapamycin in pancreatic cancer cells by inducing apoptosis and autophagy. Int J Mol Sci. 2012;14:273–285.
- Li X, Xu H, Li C, et al. Zinc-doped copper oxide nanocomposites inhibit the growth of pancreatic cancer by inducing autophagy through AMPK/mTOR pathway. Front Pharmacol. 2019;10:319.
- Li X, Roife D, Kang Y, et al. Extracellular lumican augments cytotoxicity of chemotherapy in pancreatic ductal adenocarcinoma cells via autophagy inhibition. Oncogene. 2016;35:4881–4890.
- Rebecca VW, Nicastri MC, McLaughlin N, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017;7:1266–1283.
- Kasukabe T, Honma Y, Okabe-Kado J, et al. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. 2016;36:968–976.
- Vitale G, Zappavigna S, Marra M, et al. The PPAR-gamma agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-beta treated pancreatic cancer cells. Biotechnol Adv. 2012;30:169–184.
- Xu XD, Zhao Y, Zhang M, et al. Inhibition of autophagy by deguelin sensitizes pancreatic cancer cells to doxorubicin. Int J Mol Sci. 2017;18:370
- Fiorini C, Menegazzi M, Padroni C, et al. Autophagy induced by p53-reactivating molecules protects pancreatic cancer cells from apoptosis. Apoptosis. 2013;18:337–346.
- Yuan Y, Tang AJ, Castoreno AB, et al. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 2013;4:e690.
- He R, Shi X, Zhou M, et al. Alantolactone induces apoptosis and improves chemosensitivity of pancreatic cancer cells by impairment of autophagy-lysosome pathway via targeting TFEB. Toxicol Appl Pharmacol. 2018;356:159–171.
- Xu C, Wang X, Zhou Y, et al. Synergy between arsenic trioxide and JQ1 on autophagy in pancreatic cancer. Oncogene. 2019;38:7249–7265.
- Donadelli M, Dando I, Zaniboni T, et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011;2:e152.
- Sleightholm R, Yang B, Yu F, et al. Chloroquine-modified hydroxyethyl starch as a polymeric drug for cancer therapy. Biomacromolecules. 2017;18:2247–2257.
- Zhang X, Kumstel S, Jiang K, et al. LW6 enhances chemosensitivity to gemcitabine and inhibits autophagic flux in pancreatic cancer. J Adv Res. 2019;20:9–21.
- Mukai S, Moriya S, Hiramoto M, et al. Macrolides sensitize EGFR-TKI-induced non-apoptotic cell death via blocking autophagy flux in pancreatic cancer cell lines. Int J Oncol. 2016;48:45–54.
- Chen CH, Hsieh TH, Lin YC, et al. Targeting autophagy by MPT0L145, a highly potent PIK3C3 Inhibitor, provides synergistic interaction to targeted or chemotherapeutic agents in cancer cells. Cancers (Basel). 2019;11:1345
- Wu Y, Tang Y, Xie S, et al. Chimeric peptide supramolecular nanoparticles for plectin-1 targeted miRNA-9 delivery in pancreatic cancer. Theranostics. 2020;10:1151–1165.
- Chen W, Zhou Y, Zhi X, et al. Delivery of miR-212 by chimeric peptide-condensed supramolecular nanoparticles enhances the sensitivity of pancreatic ductal adenocarcinoma to doxorubicin. Biomaterials. 2019;192:590–600.
- Lan CY, Chen SY, Kuo CW, et al. Quercetin facilitates cell death and chemosensitivity through RAGE/PI3K/AKT/mTOR axis in human pancreatic cancer cells. J Food Drug Anal. 2019;27:887–896.
- Zhao C, He R, Shen M, et al. PINK1/parkin-mediated mitophagy regulation by reactive oxygen species alleviates rocaglamide a-induced apoptosis in pancreatic cancer cells. Front Pharmacol. 2019;10:968.
- Thakur PC, Miller-Ocuin JL, Nguyen K, et al. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J Transl Med. 2018;16:190.
- Lund K, Olsen CE, Wong JJW, et al. 5-FU resistant EMT-like pancreatic cancer cells are hypersensitive to photochemical internalization of the novel endoglin-targeting immunotoxin CD105-saporin. J Exp Clin Cancer Res. 2017;36:187.
- Chen X, Yu Q, Liu Y, et al. Synergistic cytotoxicity and co-autophagy inhibition in pancreatic tumor cells and cancer-associated fibroblasts by dual functional peptide-modified liposomes. Acta Biomater. 2019;99:339–349.
- Lin JH, Chen SY, Lu CC, et al. Ursolic acid promotes apoptosis, autophagy, and chemosensitivity in gemcitabine-resistant human pancreatic cancer cells. Phytother Res. 2020;34:2053–2066.
- Elliott IA, Dann AM, Xu S, et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci U S A. 2019;116:6842–6847.
- Donohue E, Thomas A, Maurer N, et al. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J Cancer. 2013;4:585–596.
- Ma T, Chen W, Zhi X, et al. USP9X inhibition improves gemcitabine sensitivity in pancreatic cancer by inhibiting autophagy. Cancer Lett. 2018;436:129–138.
- Mirzoeva OK, Hann B, Hom YK, et al. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl). 2011;89:877–889.
- Wang F, Tian X, Zhang Z, et al. Demethylzeylasteral (ZST93) inhibits cell growth and enhances cell chemosensitivity to gemcitabine in human pancreatic cancer cells via apoptotic and autophagic pathways. Int J Cancer. 2018;142:1938–1951.
- Dai ZJ, Gao J, Kang HF, et al. Targeted inhibition of mammalian target of rapamycin (mTOR) enhances radiosensitivity in pancreatic carcinoma cells. Drug Des Devel Ther. 2013;7:149–159.
- Wang F, Xia X, Yang C, et al. SMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin Cancer Res. 2018;24:3176–3185.
- Wang P, Zhang J, Zhang L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145:1133–43 e12.
- Koshkina NV, Briggs K, Palalon F, et al. Autophagy and enhanced chemosensitivity in experimental pancreatic cancers induced by noninvasive radiofrequency field treatment. Cancer. 2014;120:480–491.
- Mukubou H, Tsujimura T, Sasaki R, et al. The role of autophagy in the treatment of pancreatic cancer with gemcitabine and ionizing radiation. Int J Oncol. 2010;37:821–828.
- Chiu HW, Lin SW, Lin LC, et al. Synergistic antitumor effects of radiation and proteasome inhibitor treatment in pancreatic cancer through the induction of autophagy and the downregulation of TRAF6. Cancer Lett. 2015;365:229–239.
- Aravindan S, Ramraj SK, Somasundaram ST, et al. Novel adjuvants from seaweed impede autophagy signaling in therapy-resistant residual pancreatic cancer. J Biomed Sci. 2015;22:28.
- Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017;18:1279
- Boone BA, Bahary N, Zureikat AH, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015;22:4402–4410.
- Zeh H, Bahary N, Boone BA, et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin Cancer Res. 2020;26:3126–3134.
- Karasic TB, O’Hara MH, Loaiza-Bonilla A, et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:993–998.
- Kinzya Grant HZ, Miller J, Laymon C, et al. FDG PET analysis of treatment response in patients with pancreatic adenocarcinoma randomized to preoperative therapy with or without hydroxychloroquine. J Nucl Med. 2015;56:1408
- Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19:637–638.