
ABSTRACT
Blood-brain barrier (BBB) disruption is a key event in triggering secondary damage to the central nervous system (CNS) under stroke, and is frequently associated with abnormal macroautophagy/autophagy in brain microvascular endothelial cells (BMECs). However, the underlying mechanism of autophagy in maintaining BBB integrity remains unclear. Here we report that in BMECs of patients suffering stroke, CLDN5 (claudin 5) abnormally aggregates in the cytosol accompanied by autophagy activation. In vivo zebrafish and in vitro cell studies reveal that BBB breakdown is partially caused by CAV1 (caveolin 1)-mediated redistribution of membranous CLDN5 into the cytosol under hypoxia. Meanwhile, autophagy is activated and contributes mainly to the degradation of CAV1 and aggregated CLDN5 in the cytosol of BMECs, therefore alleviating BBB breakdown. Blockage of autophagy by genetic methods or chemicals aggravates cytosolic aggregation of CLDN5, resulting in severer BBB impairment. These data demonstrate that autophagy functions in the protection of BBB integrity by regulating CLDN5 redistribution and provide a potential therapeutic strategy for BBB disorder-related cerebrovascular disease.
Abbreviations: BBB: blood-brain barrier; BECN1: beclin 1; BMEC: brain microvascular endothelial cell; CAV1: caveolin 1; CCA: common carotid artery; CLDN5: claudin 5; CNS: central nervous system; CQ: chloroquine; HIF1A: hypoxia inducible factor 1 subunit alpha; MCAO: middle cerebral artery occlusion-reperfusion; OCLN: occludin; ROS: reactive oxygen species; STED: stimulated emission depletion; TEER: trans-endothelial electrical resistance; TEM: transmission electron microscopy; TJ: tight junction; TJP1: tight junction protein 1; UPS: ubiquitin-proteasome system
Introduction
The blood-brain barrier (BBB) is a biological interface between the circulating blood and the central nervous system (CNS), and plays a protective role in facilitating the influx and efflux of molecules to maintain CNS homeostasis. Anatomically, BBB is formed by self-fusion of brain microvascular endothelial cells (BMECs) with tight junctions (TJs) and by interactions with other components, including astrocytes, pericytes, and perivascular microglia [Citation1]. BBB breakdown is not only a prodromal and common primary injury, but also the consequences of insults (e.g., in septic encephalopathy, neuroinflammation) in CNS diseases, because this decomposition could lead to leukocyte infiltration into the CNS and promote the lesion development [Citation2]. Therefore, pathological analysis of BBB disruption and assessment of the potential endogenous protection mechanism of BBB are indispensable. Regulating BBB permeability to protect the CNS from secondary damage may be a potential strategy for management of CNS disorders including stroke.
During the pathological process of stroke, a hypoxic microenvironment is inevitably formed in BMECs caused by interruption of blood supply, finally leads to a reduction in TJ protein levels and BBB dysfunction. Among the TJ proteins, CLDN5 (claudin 5) is highly expressed in the BMECs and is involved in constituting the backbone of TJ-strands to regulate BBB permeability. Our earlier reports demonstrate that in zebrafish Cldn5a is required for the development of a neuroepithelial barrier [Citation3], and that redistribution of CLDN5 from the membrane to the cytosol could result in increased BBB permeability [Citation4]. In vitro evidence reveals that following oxygen glucose deprivation and re-oxygenation insults, the translocated membranous CLDN5 accumulates in the cytosol and is finally eliminated by intracellular degradation pathways including autophagy [Citation5]. Multiple studies have demonstrated that self-propagating abnormal TJ proteins under the pathological processes are transported from the membrane into the cytosol via endocytosis [Citation6]. A failure of clearing these proteins may result in neurodegenerative diseases and aging, such as Alzheimer disease [Citation7]. Therefore, removal of such aggregated proteins in the cytosol timely and effectively is necessary to counteract the toxicity and ensure intracellular homeostasis [Citation8]. Even so, to date, the detailed mechanisms involved in the clearance of delocalized TJ proteins in the cytosol remain incompletely understood.
Autophagy is a conserved lysosomal degradation pathway that is stimulated by and involved in eliminating the breakdown of organelles and proteins in a variety of pathological processes, thereby avoiding excessive injury and cellular dysfunction in various organs and cells [Citation9]. This process is a primary protective response to several external stimuli for cell metabolism, as proposed in diverse models of diseases [Citation10,Citation11]. Our previous study has shown that a protective autophagy is stimulated in low serum-treated BMECs and functions in maintaining the integrity of the BBB via regulation of dynamic CLDN5 [Citation12]. Autophagy deficiency due to mutations in autophagy-related (ATG) genes or blockage by pharmacological inhibitors has been shown to accelerate apoptotic processes and lead to neurovascular disorders [Citation13] or embryonic lethality [Citation14]. In mice, an antioxidant role of autophagy was demonstrated to maintain the glomerular endothelial cell barrier under starvation by activating the cellular antioxidant system [Citation15], while autophagy-deficient mice display cerebrovascular barrier dysfunction along with a loss of TJ proteins [Citation16]. Even though, the direct in vivo evidences and the underlying mechanisms of autophagy on the maintaining of BBB permeability is still lacking.
Although mice are widely used to study autophagy on the tissue barrier, zebrafish shows unique advantages in the research of cerebrovascular pathological changes due to their optically translucent larval body and the masses of transgenic lines with tissue-specific reporters. Zebrafish BBB develops as early as 3 days post-fertilization (dpf) and can be used to evaluate the permeability of the BBB against small molecules [Citation17]. Zebrafish has also been proven as a powerful in vivo model to study autophagic activity in different biological and pathological processes, such as development [Citation18], tissue regeneration [Citation19], infection [Citation20], and neurodegeneration [Citation21]. However, the application of zebrafish model in the study of autophagy in vascular system is rarely reported.
In the current study, we combined in vivo zebrafish and multiple in vitro endothelial cell models to investigate the role of autophagy on the abnormal delocalization of endothelial membranous CLDN5 and BBB permeability in the process of hypoxia-induced cerebrovascular disorders. Our results revealed that the activation of autophagy in BMECs inhibits CLDN5 redistribution and clear the abnormally accumulated CLDN5 in the cytosol, which plays an important role on the maintaining of BBB integrity during the early hypoxia-induced cerebrovascular impairment.
Results
An increase of autophagic vacuoles is associated with the redistribution of CLDN5 (claudin 5) in brain endothelial cells after stroke
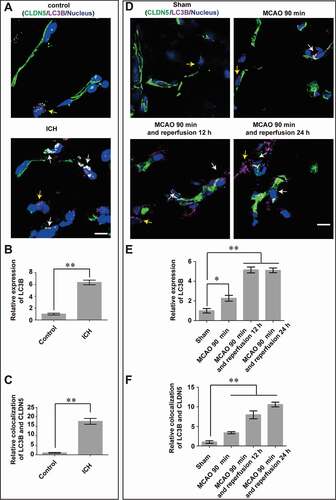
To investigate the involvement of autophagy in the pathogenesis of BBB disruption after stroke, the expression of LC3B, a marker of autophagy, was assessed in both brain tissues from patients with intracranial hemorrhage (ICH) stroke () and mice with middle cerebral artery occlusion-reperfusion (MCAO/R) injuries (; Fig. S1A,B). As a result, an upregulated expression of LC3B in brain microvascular endothelial cells (BMECs) from stroke groups (in both patients with ICH and mice with MCAO/R) was observed. Meanwhile, the tight junction (TJ) protein CLDN5 was detected to be abnormally aggregated and colocalized with LC3B in the cytosol, especially in the perinuclear space of BMECs in stroke group (white arrows, ; ; Fig. S1A,C). These suggest that autophagy may be involved in the pathological breakdown of BBB under stroke.
Figure 1. Autophagy correlates with the redistribution of endothelial CLDN5 in the stroke-induced BBB impairment. The expression of LC3B (purple) and the localization of CLDN5 (green) in cerebral endothelial cells was observed in brain tissues from patients with ICH (A-C) and male mice with MCAO/R (D-F). CLDN5 in brain microvascular endothelial cells (BMECs) from patients with ICH (n = 14) or mice after MCAO aggregated in the cytosol, especially in the perinuclear space (yellow arrows), which partly colocalized with the upregulated LC3B (white arrows). (B and C) Quantitative integrated optical density (IOD) of total LC3B, and colocalization of LC3B and CLDN5 in the cytosol of BMECs. P value indicates two-tailed unpaired t test. (E and F) Quantitative IOD of total LC3B, and colocalization of LC3B and CLDN5 in the cytosol of mice BMECs post MCAO/R treatment. Control: brain tissues from non-stroke patients (n = 12). LC3: microtubule-associated protein 1 light chain 3. ICH: intracranial hemorrhage. MCAO/R: middle cerebral artery occlusion/reperfusion. Sample sizes: sham, n = 12; MCAO, n = 9; MCAO and 12 h reperfusion, n = 8; MCAO and 24 h reperfusion, n = 8. Data were presented as mean ± SEM. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01. Scale bars: 10 μm
Disruption of BBB integrity induced by hypoxia treatment
In the lesion of stroke, hypoxic microenvironment is caused, which promotes further pathological development of stroke [Citation22]. Thus, to determine the role of autophagy on the BBB integrity and the pathological changes of BBB under stroke, a hypoxia-induced BBB damage model was generated in vitro and in vivo respectively to mimic cerebrovascular pathologic niches. The results showed that after 5 days of culture, a confluent monolayer of murine brain microvascular endothelial cells (bEnd.3) was formed, as evidenced by the stable trans-endothelial electrical resistance (TEER) value of 92 Ω·cm2 which indicates the formation of a tight endothelial barrier (Fig. S2A). CoCl2, a classic hypoxia inducer functions on inactivating hydroxylase enzymes and stabilizing HIF1A/HIF-1α (hypoxia inducible factor 1 subunit alpha) [Citation23], was applied to induce hypoxic conditions in the endothelial cells (ECs) and showed a dose- and time-dependent response (Fig. S2B,C). Upregulated expression of HIF1A further confirmed the hypoxic effects of CoCl2 on bEnd.3 cells (Fig. S2D). Next, the effect of CoCl2-induced hypoxia on the endothelial barrier was assayed by measurements of TEER and FITC-dextran infiltration respectively. The reduction of TEER and the increased FITC-dextran infiltration indicates a dose- and time-dependent pattern of impaired endothelial barrier function post CoCl2 treatment (). Reactive oxygen species (ROS) has been proved as a trigger for many downstream pathways that directly mediate BBB compromise such as oxidative damage, TJ modification and matrix metalloproteinases (MMP) activation [Citation24,Citation25]. Therefore, we next explored the generation of ROS in bEnd.3 cells after hypoxia treatment. As shown in Figure S2E, an increase of ROS was detected by flow cytometry analysis post CoCl2 treatment, which could be efficiently attenuated by ROS scavenger of CAT (catalase). These results reveal that the function of the endothelial barrier is impaired after hypoxia treatment.
Figure 2. Hypoxia treatment results in impairment of BBB. (A and B) After the formation of a monolayer of bEnd.3 on transwell membrane, the barrier function was reduced under CoCl2-induced hypoxia condition, as evaluated by the TEER and the infiltrated FITC-dextran (10 kDa) measurement. Hyp (100), Hyp (200) and Hyp (400) indicate hypoxia induction by CoCl2 with a concentration of 100, 200, 400 μmol/L, respectively. (C) Cerebral microangiograph on 5 dpf zebrafish to evaluate the BBB permeability after hypoxia induction. After injection of FSS (735 Da) or FITC-dextran (70 kDa) into the primary head sinus (schematic diagrams on the left), reconstructed 3-dimensional cerebrovascular images showed severe infiltration of the dye penetrant through BBB after N2-induced hypoxic treatment for 1 or 3 h (middle and right). Tg(kdrl:ras-cherry)s916 transgenic zebrafish in which vascular endothelial cells were labeled by cherry fluorescent protein was used. n = 12 fishes per group. Scale bar: 100 μm. (D) A reduced expression of TJ proteins including TJP1, OCLN and CLDN5 was detected in monolayer bEnd.3 cells after 200 μmol/L CoCl2 induction with a time-dependent manner (0, 6, 12, 24 and 48 h). n = 3 repeats. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. (E) TJ proteins, especially CLDN5, delocalized from membrane (yellow arrow) and aggregated in the cytosol (white arrows) of bEnd.3 after 200 μmol/L CoCl2-induced hypoxia treatment for 12 h. The integrated optical density (IOD) of the cytosolic proteins was quantified. n = 3. P value indicates two-tailed unpaired t test. Scale bar: 10 μm. TEER: trans-endothelial electrical resistance; Norm: normoxia; Hyp: hypoxia; FSS: fluorescein sodium salt; FITC-dextran: fluorescein-labeled dextran; Papp: apparent permeability coefficient. Data were presented as mean ± SEM. *, P < 0.05; **, P < 0.01
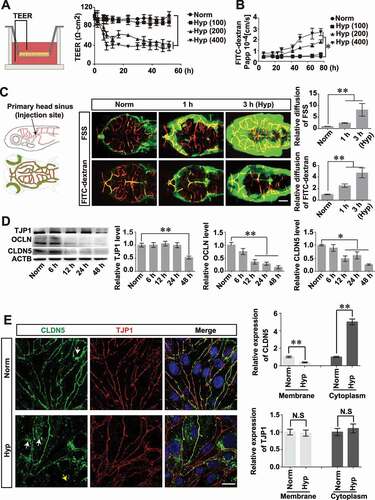
Hypoxia-induced BBB dysfunction was also demonstrated by in vivo analyses using Tg(kdrl:ras-cherry) zebrafish in which the vascular endothelial cells are specifically labeled by cherry fluorescent proteins. After hypoxia (N2, 99.99%) treatment for 1 h or 3 h, the dissolved oxygen concentration in the embryo water reduced to around 3.8 mg/L or 2.8 mg/L respectively (Fig. S2F). This global hypoxia treatment resulted in a mortality of zebrafish at 5 days post-fertilization (dpf) old in time-dependent manner (Fig. S2G). Meanwhile, the expression of Hif1a in the brain of zebrafish larvae post hypoxia treatment was detected to be increased within 1 h or 3 h accordingly (Fig. S2H). Subsequently, the tightness of zebrafish BBB after hypoxia treatment was evaluated by cerebrovascular dye diffusion assay (). As a result, the cerebrovasculature of hypoxia-induced zebrafish showed a significantly increased permeability to both Fluorescein sodium (375 Da) and FITC-dextran (70 kDa) when comparing with that of zebrafish under normoxia conditions ().
Above results provide both in vivo and in vitro evidences for an impairment of BBB function under hypoxia treatment. To explore whether this dysfunction is directly related to the breakdown of endothelial TJs, the expression and localization of TJ components were determined next. It was found that after hypoxia induction, the expression of CLDN5, OCLN (occludin), and TJP1/ZO-1 (tight junction protein 1), was reduced in a hypoxic time-dependent manner (). In addition, membrane-bound CLDN5 (yellow arrow, ), but not TJP1, delocalized from the cell membrane and aggregated in the cytosol (white arrows, ) due to hypoxia-induced injury (). These data indicate that hypoxia treatment promotes CLDN5 redistribution, therefore resulting in BBB impairment.
Autophagic vacuoles increase in BMECs under hypoxia treatment
As a general cellular mechanism of survival, autophagy is normally activated in hypoxic microenvironment [Citation26]. Blockage or activation of autophagy by inhibitors or enhancers leads to the direct change in the number of autophagic vesicles and the expression of autophagy-related proteins. Since a remarkable upregulation of LC3 was observed in the cerebrovascular ECs of both stroke patients and MCAO mice, we next asked for more evidences of the involvement of autophagy in BMECs post hypoxia induction. To this end, we first investigated the expression of autophagosome marker of LC3 in live bEnd.3 cells by transfecting GFP-LC3 plasmid. The results indicated an increased expression of LC3 in hypoxia-treated cells (Fig. S3A,B). When the autophagy inhibitor of chloroquine (CQ) was added to prevent lysosomal acidification and to interfere the degradation of autolysosomes, the number of GFP-LC3 punctate dots significantly increased and accumulated in the cytosol of cells post hypoxia treatment (Fig. S3A,B). Meanwhile, the autophagic flux was traced to investigate the fusion of autophagosomes in hypoxic cells (Fig. S3C). Real-time images of GFP-LC3 punctate dots revealed that the fusion of autophagosomes was significantly enhanced in the hypoxia treated group compared to that in the control group (Fig. S3C,D). Additionally, the expression level of autophagosome- or autolysosome-related proteins was analyzed by protein immunoblotting. The results revealed that hypoxia induction caused an upregulation of LAMP1 (lysosomal associated membrane protein 1), ATG5 (autophagy related 5) and LC3-II/-I, while additional treatment of 3-methyladenine (3-MA), a class III PtdIns3K inhibitor to prevent autophagosome formation, inhibited the expression of related proteins. In contrast, enhancement of autophagy by rapamycin (Rapa), an inhibitor of MTOR phosphorylation to assist the formation of autophagosomes, further enhanced the expression of the autolysosome-related proteins (Fig. S3E).
To find direct evidence of the involvement of autophagy in hypoxia-treated bEnd.3 cells, transmission electron microscopy (TEM) analyses were performed to visualize the autophagic vesicles. As shown in (), the number of autophagic vacuoles in ECs was obviously increased after hypoxia treatment, and was reduced when treated with 3-MA. Conversely, when enhancing autophagy by Rapa, the number of autophagic vesicles was significantly increased in comparison with that in the ECs induced by hypoxia ().
Figure 3. An increase of autophagosomes and autolysosomes in cerebrovascular endothelial cells after hypoxia induction. (A and B) Hypoxia induction caused an increase of autophagosomes and autolysosomes while blockage of autophagy by 3-MA inhibited the formation of autophagosomes in bEnd.3 cells (yellow arrowheads), as imaged by TEM. Conversely, activation of autophagy by Rapa treatment increased the number of autophagosomes (yellow arrowheads). The lower panel show high magnification of yellow square-labeled area in the up panel. Autophagic vacuoles were counted quantified from a 30 μm2-sized region per cell. n = 5. (C) BMECs of zebrafish embryos were scanned by TEM after cross section of the brain region (red line in the schematic diagram) and autophagosomes in BMECs of zebrafish post 1 h-hypoxia treatment were captured (D). The lower panel show high magnification scans of the area labeled by yellow squares in the upper panel. (E) Hypoxia treatment caused an increase of autophagosomes in BMECs of zebrafish while 3-MA inhibits the formation of autophagic vacuoles. In contrast, Rapa treatment increased the number of autophagosomes in hypoxia-induced zebrafish BMECs. Autophagic vacuoles were quantified in 10 μm2-sized region per endothelial cell. Yellow arrowheads indicate autophagosomes. n = 4 fishes per group. Data were presented as mean±SEM. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01. Norm: normoxia; Hyp: hypoxia; CoCl2: 200 μmol/L, treated for 12 h; CQ: Chloroquine, 30 μmol/L; 3-MA: 3-methyladenine, 10 mmol/L; Rapa: rapamycin, 50 nmol/L; L: cerebrovascular lumen; AP, autophagosome; AL, autolysosome; HEM, hematocyte. Scale bars: 1 μm
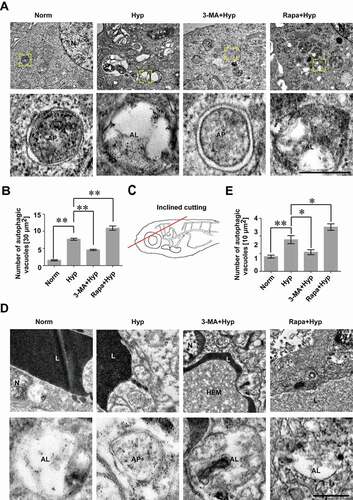
Besides the in vitro cellular evidence, we further sought occurrence of the autophagy in vivo by investigating the autophagic vesicles in the BMECs of zebrafish larvae after hypoxia treatment for 1 h, followed by a cross section trough the brain region (). The TEM analyses of zebrafish cerebrovascular ECs showed that number of autophagic vesicles significantly increased due to the hypoxia induction, while was decreased with 3-MA treatment and was further increased when treated with 3-MA, under hypoxic condition ().
Above in vitro and in vivo data demonstrate an increase of autophagosomes and upregulated expression of LC3 in BMECs post hypoxic treatment, which is consistent with the observation of the autophagy activation in the BMECs of stroke patient and MCAO mice. We next questioned the correlation of cerebrovascular autophagy and the maintenance of BBB function under hypoxia.
Autophagy alleviates the impairment of BBB under short-term hypoxia induction
As a conserved homeostatic process, autophagy is essential for cell survival in the pathologic process of stroke [Citation27], and defects of the autophagic process in brain after stroke promote improper homeostasis and neurodegeneration [Citation28]. To explore the role of endothelial autophagy in the hypoxia-induced BBB damage, the TEER and permeability of the endothelial barrier were measured first. The results showed that in comparison with the TEER value in the hypoxia group, abrogation of autophagy by 3-MA treatment resulted in a further decrease of tightness from 12 to 39 h (). However, enhancement of autophagy with Rapa could partially reverse the drop of TEER (). We also evaluated the effect of autophagy in the modulation of paracellular permeability by dye infiltration test in parallel. As shown in , inhibition of autophagy intensified the infiltration of FITC-dextran (10 kDa) through the endothelial monolayer with 3-MA treatment for 12 or 24 h when in comparison with that in the hypoxia-treated groups (). In contrast, enhancement of autophagy reduced the penetration after Rapa treatment for 12 or 24 h (). To explore the in vivo evidence confirming the role of autophagy in BBB permeability, the dye diffusion assay was performed using dye penetrant (fluorescein sodium: 375 Da and FITC-dextran: 70 kDa) to visualize the leakage of zebrafish cerebrovasculature. A severer dye leakage through BBB was observed after 3-MA treatment for 1 h when compared with that in zebrafish cerebrovasculature only under hypoxia treatment (). However, enhancement of autophagy by Rapa could partly rescue the BBB disruption caused by hypoxia treatment (). Above results indicate that autophagy activation could partly maintain BBB function after short-term hypoxia treatment.
Figure 4. The protective role of autophagy on maintaining BBB function. (A and B) The TEER value and the infiltration of FITC-dextran (10 kDa) across monolayer of bEnd.3 cells were measured for analyzing the role of autophagy on endothelial barrier function. Blockage of hypoxia-induced autophagy by 3-MA increased the permeability through the monolayer endothelial cells while Rapa treatment partly restored the tightness of the endothelial barrier caused by hypoxia for 12 h or 24 h induction respectively. (C and D) Blockage of autophagy by 3-MA aggravated hypoxia (evoked by N2 treatment for 1 h)-induced BBB breakdown in zebrafish larvae. A severer infiltration of dye penetrant (FSS, 735 Da or FITC-dextran, 70 kDa) through cerebrovascular wall of endothelial specific transgenic Tg(kdrl:ras-cherry)s916 zebrafish at 5 dpf in 3-MA-treated group was observed. In contrast, Rapa treatment partly restored the leakage of zebrafish BBB caused by hypoxia induction. n = 4 fishes for each group. (E and F) Hypoxia treatment caused early and late stage apoptosis in bEnd.3 cells. 3-MA treatment significantly enhanced this effect while Rapa induction had a counteraction effect of hypoxia-induced autophagy on apoptosis changes. Norm: normoxia; Hyp: hypoxia; 3-MA: 3-methyladenine, 10 mmol/L; Rapa: rapamycin, 50 nmol/L; FSS: fluorescein sodium salt; FITC-dextran: Fluorescein-labeled dextran; Papp: apparent permeability coefficient; PI: propidium iodide. Data were expressed as mean ± SEM, n = 4. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01. Scale bar: 100 μm
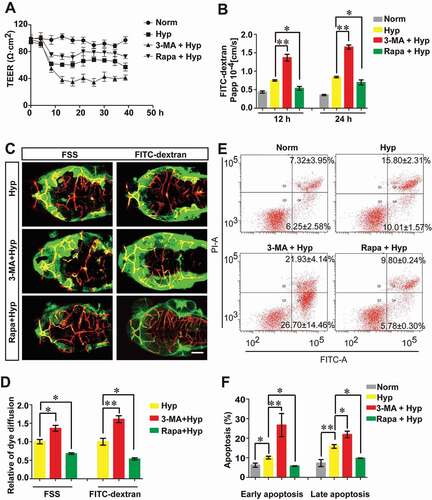
Additionally, we also checked the relevance between hypoxia-induced autophagy and endothelial cell apoptosis by analyzing early and late stage apoptosis in bEnd.3 cells. The results showed that inhibition of the autophagy with 3-MA promoted both early and late cell apoptosis while activation of autophagy by Rapa repressed cell apoptosis (). These data suggest that endothelial autophagy induced by hypoxia is conducive for cell viability, which may contribute to the maintenance of endothelial barrier function.
Autophagy participates in the degradation of aggregated CLDN5 in the cytosol
Aggregation of abnormal proteins in CNS has been demonstrated to result in neurodegenerative diseases [Citation29]. Hence, timely and effective removal of abnormal proteins is essential for the cell survival [Citation30]. Previous studies indicate that autophagy could mediate the degradation of TJ proteins [Citation31]. As a main component of TJ, membranous CLDN5 plays essential role in modulation and maintaining BBB permeability. Since we have shown an obvious colocalization of CLDN5 and autophagic marker of LC3 in cerebrovascular ECs of stroke patient and mice MCAO/R models, we speculate that autophagy might be involved in the redistribution and degradation of CLDN5 to maintain intracellular homeostasis. To confirm this, the effect of autophagy on the localization of endothelial CLDN5 after hypoxia treatment was analyzed first. Immunofluorescent staining revealed that CoCl2- or 1% O2-induced hypoxia treatment caused a cytosolic colocalization of CLDN5 and LC3 in both bEnd.3 (red arrows, Fig. S4A,B) and human brain capillary endothelial cells (hCMEC/D3) (red arrows, Fig. S4C). Moreover, inhibition of autophagy by 3-MA or CQ accelerated the delocalization of CLDN5 from endothelial membrane (yellow arrows, Fig. S4A,B) into cytosol and facilitated its accumulation in the cytosol (white and red arrows, Fig. S4A,B). However, this redistribution could be partly blocked by Rapa treatment, as shown by the amounts of CLDN5 clusters on the cell membrane (Fig. S4A,B). Quantificational immunoblotting analyses of CLDN5 revealed that CoCl2-induced hypoxia treatment led to loss of membranous and total CLDN5 in ECs, and CQ or 3-MA stimulation enhanced this membranous loss of CLDN5 into cytosol (; Fig. S4A). Notably, when hypoxia-induced autophagy was blocked by CQ, the degradation of total amount or cytosolic accumulated CLDN5 could be efficiently inhibited (). To better mimic ischemic stroke in vitro, oxygen-glucose deprivation (OGD)-induced model was also applied on bEnd.3 cells. Functionally, it was found that 6 h-induction of OGD on endothelial cells caused an increased permeability of bEnd.3 monolayer as measured by TEER and FITC-dextran diffusion assay (Fig. S5A,B). However, Rapa treatment under OGD conditions partially rescued the loss of endothelial tightness while 3-MA enhanced this leakage (Fig. S5A,B). Expression analyses of LC3 and CLDN5 in bEnd.3 cells revealed that OGD induction caused upregulated expression of autophagic marker of LC3 and promoted the internalization of membranous CLDN5 into cytosol for further degradation (Fig. S5C). Blockage of autophagic pathway by 3-MA or CQ in OGD-induced bEnd.3 cells led to severer delocalization of membranous CLDN5 and accumulation of cytosolic CLDN5 (Fig. S5C). In contrast, Rapa treatment partly restored this redistribution of CLDN5 caused by OGD induction (Fig. S5C). Together, these results suggest an involvement of autophagy in the redistribution of CLDN5 from membrane to the cytosol and its further degradation process under hypoxia conditions.
Figure 5. Autophagy mediates the degradation of endothelial CLDN5 in the cytosol. (A) After hypoxia treatment, the membranous, cytosolic and total CLDN5 in bEnd.3 cell was quantified by western blot analyses. Blockage of autophagy by CQ caused an accumulation of CLDN5 in the cytosol while Rapa-induction partly retarded the redistribution of membranous CLDN5. (B) Western blotting analyses showed a high protein purity of cytosolic or membranous fractions under different induction conditions. GAPDH was used as cytosolic reference marker and ATP1A1/Na,K-ATPase was used as membranous reference marker. n = 3 repeats. Data were presented as mean ± SEM. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. (C and D) Stimulated emission depletion (STED) microscope images showed that after the hypoxia induction, membranous CLDN5 (red arrowheads in i and ii) delocalized, aggregated and was surrounded by LC3 in the cytosol (yellow arrowheads in ii). iii are high magnification images of the cytosolic proteins in ii. The numbers of LC3-surrounded CLDN5 clusters per image were counted and quantified. (E and F) Immuno-electronmicroscope (IEM) images showed CLDN5 (black arrows) was packaged in autophagosome-like vesicles in bEnd.3 cells after CoCl2-induced hypoxia treatment. i and ii, normoxia control; iii and iv, hypoxia-treated group. ii and iv are high magnification scans of the red line-marked regions in i and iii respectively. The numbers of gold particles in autophagosome-like vesicles were quantified. P value indicates two-tailed unpaired t test. MF: membranous fraction; CF: cytosolic fraction. M, mitochondria; N, nucleus, AP, autophagosome; CQ: Chloroquine, 30 μmol/L; Rapa: rapamycin, 50 nmol/L. Data were presented as mean ± SEM. n = 3. *, P < 0.05; **, P < 0.01. N.S: no significance. Scale bars: 500 nm
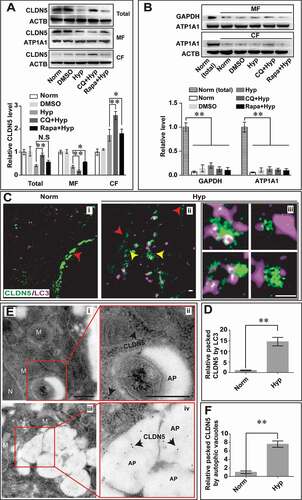
Next, long-term effect of autophagy on CLDN5 subcellular distribution and expression under hypoxia was additionally analyzed in bEnd.3 cells. The results showed that after 200 μmol/L CoCl2-induced hypoxic treatment for a serial time (0, 6, 12, 18, 24, 48 h), the membranous CLDN5 gradually decreased and redistributed into the cytosol, and colocalized with cytosolic LC3 till 48 h (red arrows, Fig. S6A). Quantitative analyses of protein immunoblotting showed a gradual degradation of total endothelial CLDN5, accompanying with the decreased expression of autophagic marker of SQSTM1/p62 and increased expression of LAMP1, ATG5 and LC3II:I (Fig. S6B).
Based on above findings, we next questioned whether other TJ components of TJP1 or OCLN were also degraded by autophagic pathway under hypoxic treatment. The immunofluorescent staining showed that TJP1, but not OCLN, colocalized with autophagic marker of LC3 in endothelial cytosol after 24 h hypoxia treatment (white arrows and circles, Fig. S7).
Besides autophagosome-lysosome pathway, other mechanisms such as ubiquitin-proteasome system (UPS) is also responsible for intracellular protein degradation in eukaryotic cells. Therefore, the possibility of UPS in the degradation of aggregated CLDN5 was investigated in addition. As shown in Figure S8, the treatment of proteasome inhibitor MG-132 could not block the degradation of CLDN5 in bEnd.3 cells under hypoxia treatment. In contrast, blockage of autophagy by CQ significantly inhibited the degradation of endothelial CLDN5 (Fig. S8). This indicates that the degradation of CLDN5 under hypoxia is independent of the UPS.
We next explored the direct evidences confirming the participation of autophagy in the endothelial CLDN5 degradation under hypoxia. To this end, stimulated emission depletion (STED) microscopy and immunoelectron microscopy (IEM) scanning was applied to visualize the autophagosomes and the cytosolic localization of CLDN5. The results showed that under normoxic condition, CLDN5 localized on the membrane of bEnd.3 cells (red arrowhead, Ci). After hypoxic stimulus, CLDN5 redistributed and was surrounded by LC3 in the cytosol (yellow arrowheads, Cii,iii,D). IEM results further revealed that the immunogold-labeled CLDN5 was packaged in the autophagosome-like vesicles in bEnd.3 cells post hypoxic treatment (black arrows, ). These data reveal that autophagy functions on the degradation of cytosolic CLDN5 in endothelial cells under hypoxia.
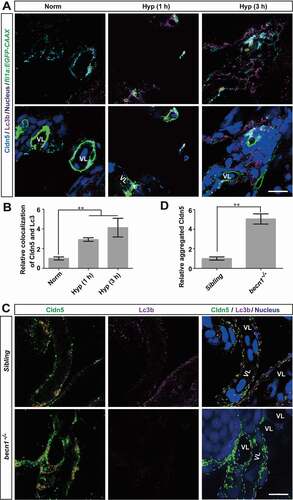
To verify our findings on the role of endothelial autophagy by in vivo evidence, we applied immunofluorescent staining to assess the localization of Cldn5 and Lc3b in zebrafish cerebrovascular ECs after hypoxia treatment for 1 or 3 h respectively. As shown in , a cytosolic accumulation of Cldn5 which colocalized with upregulated Lc3b in the BMECs of zebrafish larvae, especially in the perinuclear space, was clearly observed (yellow circles, ). To further confirm the specificity of autophagy on the clearing of accumulated Cldn5 in the cytosol caused by hypoxia, the becn1/beclin 1 mutated zebrafish was applied, in which autophagic process is specifically and efficiently blocked [Citation32]. The results revealed that hypoxia treatment on homozygous becn1 mutants caused a severe cytosolic aggregation of Cldn5 in zebrafish BMECs (). In comparison with that in the siblings, the abnormally accumulated Cldn5 in the BMECs of becn1 mutants could not be degraded timely after exposure to hypoxia for 1 h ().
Figure 6. Autophagy suppresses the redistribution of membranous Cldn5 in zebrafish cerebrovascular endothelial cells. (A and B) Colocalization of Lc3b and Cldn5 (yellow circles) was observed in the cytosol of zebrafish BMECs after N2-induced hypoxia for 1 or 3 h respectively. Endothelial eGFP-specific transgenic Tg(fli1a:EGFP-CAAX) zebrafish (5 dpf) were used. The integrated optical density (IOD) of colocalized Lc3b and Cldn5 in the cytosol of zebrafish cerebrovascular endothelial cells (in yellow circles) was quantified. n = 4 fishes per group. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. (C and D) Autophagy deficiency caused a cytosolic accumulation of Cldn5 (red circles) in the BMECs of homozygous becn1 mutated zebrafish, especially in the perinuclear space, after N2-induced hypoxia treatment for 1 h. IOD of colocalized Lc3b and Cldn5 in the cytosol of zebrafish cerebrovascular endothelial cells (in red circles) was quantified. n = 4 fishes for each group. P value indicates two-tailed unpaired t test. **, P < 0.01. Norm: normoxia; Hyp: hypoxia; VL: cerebrovascular lumen. LC3: microtubule-associated protein 1 light chain 3; 3-MA: 3-methyladenine, 10 mmol/L; CQ: chloroquine, 30 μmol/L; Rapa: rapamycin, 50 nmol/L. Yellow circles indicate the colocalization of LC3 and Cldn5 in the cytosol of BMECs and white dotted lines indicate the boundary of BMECs of zebrafish. Scale bars: 5 μm
Above results indicate that autophagy could alleviate the breakdown of BBB integrity caused by hypoxia by participating in the redistribution and degradation of membranous CLDN5 in cerebral ECs.
An involvement of caveolar endocytosis in hypoxia-induced redistribution of endothelial CLDN5
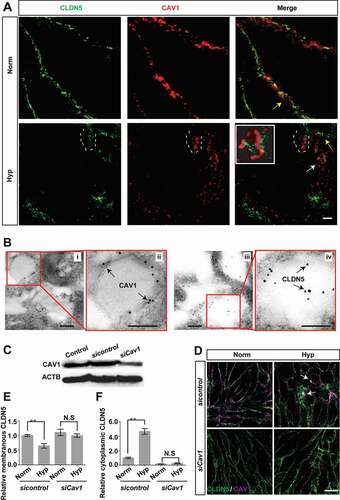
We have shown in above results that autophagy is involved in the degradation of CLDN5. However, the underlying mechanisms promoting the delocalization of membranous CLDN5 into cytosol under hypoxia is still not fully understood. Previous studies have shown that an increased expression of CAV1 (caveolin 1), a marker of caveolae-mediated endocytosis, precedes the decrease in CLDN5 levels [Citation33] and knockdown of Cav1 with siRNA could block the CLDN5 redistribution [Citation34]. Hence, we next asked whether CAV1 or caveolae-mediated endocytosis is responsible for the redistribution of endothelial CLDN5 post hypoxia induction. To this end, ultrastructural details of CLDN5 and CAV1 were scanned by STED microscopy and IEM respectively. CLDN5 and CAV1 in bEnd.3 were captured to colocalize on the endothelial cell membrane under normoxic conditions (yellow arrows, ). However, after hypoxic stimulus, CLDN5 and CAV1 redistributed from the cell membrane into the cytosol from round 3 h-hypoxic induction on (white dotted lines, ; Fig. S9A). Meanwhile, CLDN5 was shown to be packaged into CAV1-labeled vesicles (white arrows and enlarged image in white square, ). IEM scans revealed that immunogold-labeled CAV1 proteins were imaged on the membrane of caveolae-like vesicles and CLDN5 was packaged inside (black arrows, ). Moreover, when Cav1 was specifically knocked down (), the redistribution of CLDN5 from the endothelial membrane into the cytosol was partially inhibited under hypoxia treatment (). These suggest that the hypoxia-induced redistribution of CLDN5 is mediated by caveolae-dependent endocytosis.
Figure 7. Caveolae-mediated endocytosis is involved in the redistribution of endothelial CLDN5 after hypoxia induction. (A) The localization of CLDN5 and CAV1 in bEnd.3 cells under CoCl2-induced hypoxia treatment was imaged by stimulated emission depletion (STED) microscope. White dotted lines-labeled region showed a surrounding of CLDN5 by CAV1 beneath the endothelial cell membrane (yellow arrows). Caveolae-liked vesicle packaging the aggregated CLDN5 (white arrow) was captured and the higher magnification image was shown in the white square. Scale bar: 1 μm. (B) CAV1 and CLDN5 in bEnd.3 cells under CoCl2-induced hypoxia treatment was imaged by immuno-electronmicroscope (IEM). Immunogold-labeled CAV1 was captured and found on the membrane of caveolae-liked vesicle while immunogold-labeled CLDN5 localized inside of the caveolae-liked vesicle. i and ii, normoxia control; iii and iv, hypoxia-treated group. ii and iv are high magnification scans of the red line-marked regions in i and iii respectively. Scale bars: 100 nm. (C-F) After knock-down of Cav1, the redistribution of membranous CLDN5 into the cytosol of bEnd.3 cells (white arrows in D) was suppressed under CoCl2-induced hypoxic conditions. The integrated optical density (IOD) of membranous or cytosolic CLDN5 was quantified. CoCl2: 200 μmol/L, treated for 12 h; Norm: normoxia; Hyp: hypoxia. sicontrol: monolayer of bEnd.3 was transiently transfected with scrambled negative control siRNA. siCav1: monolayer of bEnd.3 was transiently transfected with Cav1 siRNA. n = 3 images for each experiment. Data were presented as mean ± SEM. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. N.S: no significance. Scale bar: 10 μm
Some studies have reported that CAV1 could suppress the activation of autophagy [Citation35], while other studies show that autophagy induced by hypoxia mediates the degradation of CAV1 by binding to and activating EGFR in a ligand-independent manner [Citation36]. Since we have shown that caveolae-mediated endocytosis of CLDN5 under hypoxia is a main cause of the impairment of BBB and autophagy could partially alleviate this impairment, we therefore questioned whether this alleviation of barrier dysfunction is due to the blockade of caveolae-mediated endocytosis. Hereby, the effect of autophagy on the degradation of CAV1 was first analyzed. The results showed that hypoxia treatment caused a significant degradation of CAV1 (Fig. S9A; ). This loss could be efficiently blocked by the autophagy inhibitor of CQ, but was further enhanced by the autophagy activator of Rapa (). In addition, the IEM scanning presented a direct evidence of immunogold-labeled CAV1 in autophagosome-like vesicles in bEnd.3 cells after hypoxia induction (black arrows, ). To additionally check whether the loss of CAV1 itself will increase the BBB permeability, infiltration of FITC-dextran (10 kDa) through endothelial cell layer was measured after knockdown of Cav1. The result showed no impairment of endothelial barrier function in siCav1-transfected bEnd.3 monolayer (). Together, these data indicate that autophagy could mediate the degradation of Cav1, therefore affecting the endocytosis of membranous CLDN5 and maintaining of BBB function ().
Figure 8. Autophagy mediates the degradation of endocytosed CAV1 under hypoxia. (A and B) CoCl2-induced hypoxia caused degradation of CAV1 in monolayer bEnd.3 cells. Blocking of autophagy by CQ significantly inhibited the degradation of CAV1 while enhancing of autophagy by Rapa promoted its degradation. n = 5. P value indicates one-way ANOVA with Dunnett’s multiple comparisons test. (C and D) Immunogold-labeled CAV1 (black arrows) was imaged by immuno-electronmicroscope (IEM) and was found in autophagosome-like vesicles of bEnd.3 cells after CoCl2-induced hypoxia treatment. The right panel is a high magnification scan of the red line-marked region in the left panel. The numbers of gold particles which represent packaged CAV1 in caveolae per image were counted for quantification analyses. n = 6 images analyzed. Scale bars: 200 nm. (E) knock-down of Cav1 itself in bEnd.3 cells showed no effect on the paracellular permeability of cell monolayer under normoxic condition. n = 3. CoCl2: 200 μmol/L, treated for 12 h. Norm: normoxia; Hyp: hypoxia; sicontrol: monolayer of bEnd.3 was transiently transfected with scrambled negative control siRNA. siCav1: monolayer of bEnd.3 was transiently transfected with Cav1 siRNA. CQ: chloroquine. Rapa: rapamycin. FITC-dextran: Fluorescein-labeled dextran. Papp: apparent permeability coefficient. LC3: microtubule-associated protein 1 light chain 3. Data were presented as mean ± SEM. P value indicates two-tailed unpaired t test. *, P < 0.05, **, P < 0.01. N.S: no significance
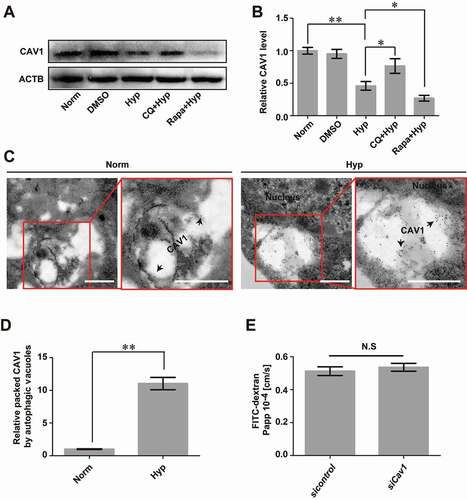
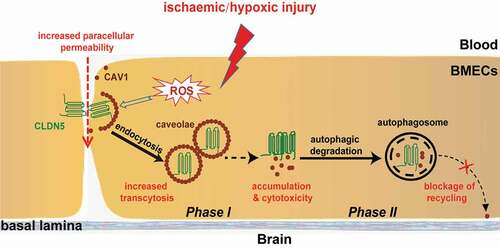
Figure 9. A proposed model of the role of autophagy on protecting the integrity of BBB under hypoxia. Hypoxia treatment in cerebrovascular endothelial cells induces a redistribution of membranous CLDN5 which is further endocytosed by CAV1-composed caveolae, impairing the integrity and permeability of BBB. Meanwhile, endothelial autophagy mediates the clearance of aggregated CLDN5 and CAV1 in the cytosol to reduce cytotoxicity and to block the recycling of CAV1 back to cell membrane, suppressing further redistribution of membranous CLDN5 and preventing BBB from fast disruption. control (CLDN5/LC3B/Nucleus)
Clathrin-mediated transcytosis is another important transcytotic pathway regulating BBB function. To explore whether clathrin-mediated transcytosis is responsible for the redistribution of membranous CLDN5 or is affected by autophagy under hypoxic injury, immunofluorescent staining of clathrin with LC3 or CLDN5 was performed in bEnd.3 cells after hypoxia treatment. The results indicate that clathrin is not responsible for the endocytosis of delocalized membranous CLDN5 (Fig. S9B). Moreover, clathrin showed no colocalization with autophagic marker of LC3 at all (Fig. S9C). This suggests that the cytosolic internalization of CLDN5 was mainly mediated by caveolae-mediated transcytosis at the early stages of hypoxia induction.
In summary, our findings revealed a protective role of hypoxia-induced autophagy on maintaining BBB integrity. Autophagy affects the redistribution of membranous CLDN5 by degrading CAV1 and regulating the caveolae-mediated endocytosis under hypoxia treatment (). Meanwhile, autophagy participates in the degradation of CLDN5, therefore reducing the cytotoxicity caused by ROS and aggregated proteins in the cytosol.
Discussion
The major finding of this study is that autophagy alleviates the hypoxia-induced BBB injury via regulating the redistribution and degradation of CLDN5. Under early hypoxic induction, ROS is generated and may attack tight junction structure and proteins including CLDN5 [Citation25,Citation37–39], leading to an increase of the paracellular permeability and impairment of the endothelial barrier. Subsequently, CAV1-composed caveolae packaged delocalized membranous CLDN5 and mediated its endocytosis, further contributing to the impairment of endothelial barrier by increased transcytosis. Moreover, with the redistribution and abnormal accumulation of CLDN5 and CAV1 in the cytosol, severe cytotoxicity may promote the endothelial apoptosis and cause further endothelial injury and BBB disruption (). In contrast, autophagy was activated in response to the impairment to degrade accumulated cytosolic proteins and to reduce cytotoxicity. Meanwhile, autophagic degradation of CAV1 may block its recycling back to the plasma membrane [Citation40,Citation41], preventing further endocytosis of membranous CLDN5 and CAV1-mediated transcytosis (). Besides, the internalized CLDN5 could follow a recycling rather than a degradation pathway to re-assemble the tight junction complex during barrier recovery [Citation6,Citation42]. These all together, attenuate the endothelial injury and prevent BBB from fast disruption after early hypoxic induction ().
As an essential physiological barrier between peripheral blood and brain, BBB functions on the maintenance of the homeostasis of the CNS. BBB collapse is a common event in the clinical manifestation of CNS diseases including stroke, as well as a cause of secondary damage to brain parenchyma. Clinical studies including contrast-enhanced MRI inspections has already revealed a breakdown of BBB permeability in the brain of stroke patients [Citation43]. However, the role of BBB in the pathological process of stroke has rarely been studied. During the pathological process of stroke, a hypoxic microenvironment is usually induced in the cerebrovascular endothelium of the lesion, which further impairs the cell-cell junctions and leads to infiltration of peripheral blood cells into brain, resulting in CNS damage. In our study, the hypoxia-induced BBB dysfunction was verified by sets of in vitro and in vivo investigations where decreased TEER values or enhanced infiltration of dye penetrant was observed.
Autophagy, a lysosomal-dependent cellular process, has been reported to be involved in multiple diseases [Citation44]. Recent studies have suggested that hypoxia can activate autophagy [Citation45], and functions on clearing the aggregated proteins during degenerative diseases and aging [Citation46]. In this study, the relationship between autophagy and the impairment of BBB was first explored in brain tissues from patients with ICH stroke and MCAO/R mice. The colocalization of upregulated LC3B and aggregated CLDN5 indicates that autophagy might be activated and involved in stroke-caused BBB impairment. In the clinical studies of stroke, sex and age are considered as essential biological variables, our investigated samples from both male and female patients of different ages, and female and male mice all showed consistent findings of autophagy involvement. To further confirm the effects of autophagy on BBB, we applied multiple brain endothelial cell lines and zebrafish models to explore the role of autophagy on the hypoxia-induced BBB impairment. Our results obtained by EM analyses, and by the fusion events of GFP-tagged LC3 puncta displayed that autophagy was activated in bEnd.3 cells and zebrafish BMECs in response to hypoxia stimuli. Furthermore, the upregulated expression of Lc3b and its colocalization with Cldn5 in the cytosol of zebrafish BMECs upon hypoxic stimulation were confirmed. These results indicate that endothelial autophagy is involved in hypoxia-induced BBB impairment.
To confirm the role of autophagy on the pathologic process of BBB dysfunction, we analyzed the change of BBB permeability and the cellular distribution of CLDN5 by pharmacological inhibitors or enhancer of autophagy post hypoxic induction. The paracellular permeability was significantly increased by the treatment with a pharmacological inhibitor of 3-MA, as seen by the decreased TEER and intensified infiltration of dyes through the endothelial barrier. In contrast, the increased permeability was prevented following the autophagy enhancer of Rapa treatment. Moreover, inhibition of autophagy by CQ or 3-MA impaired the tightness of zebrafish BBB. These results suggest autophagy plays an essential role in alleviating BBB impairment upon hypoxic stress. Upon these findings, we next demonstrated that autophagy was conducive for the stability of membranous CLDN5 in bEnd.3 cells under hypoxia treatment (by CoCl2 or 1% O2 induction). Inhibition of autophagy promoted the redistribution and accumulation of CLDN5, resulting in severe impairment of BBB function. Besides CoCl2- and 1% O2-induced hypoxic injury models, we also applied OGD-induced model on bEnd.3 cells to better mimic ischemic stroke in vitro. The findings confirmed that autophagy was activated in endothelial cells on OGD-induced model, and autophagy affected the cytosolic internalization of membranous CLDN5 and monolayer permeability (Fig. S5). In the in vivo studies, the accumulation of Cldn5 in BMECs of the autophagy-deficient zebrafish (becn1 mutants) was also imaged after exposure to hypoxia. Although artificial enhancement of autophagy did not significantly inhibit the loss of CLDN5 from the cell membrane, it might be beneficial to keep the stability of membrane CLDN5 and to maintain the membrane integrity of endothelial cells via indirect functions. In this study, the pharmacological inhibitors of 3-MA or CQ was applied to block the autophagic pathway. These unspecific chemicals may have side effects since they have been reported to affect other cellular pathways. Therefore, to generate and to use stable genetic endothelial cell lines or model animals related to autophagic pathway factors will help to investigate specific autophagic effects on of the protection of BBB under stroke in further studies.
In this study, we sought the underlying mechanism by which autophagy regulates the redistribution of CLDN5 under hypoxia. It was reported that the delocalization of TJ proteins under hypoxia induction from membrane to cytosol might be affected by endocytosis [Citation47]. Nevertheless, there is still lack of direct and convincing data supporting this. In the current study, STED and IEM analyses were performed to show that CLDN5 was packed by CAV1 and entered the cytoplasm in caveolae-like vesicles. Thereafter, autophagy was activated to degrade the endocytosed caveolae (vesicles containing CLDN5 and CAV1) through downstream autophagosomes/autolysosome (). During the endocytosis of CLDN5, inhibiting the expression or function of Cav1 caused a delayed redistribution of CLDN5 from endothelial membrane (). These outcomes directly prove that caveolae-mediated endocytosis is responsible for redistribution of CLDN5 under hypoxia. Our data also confirm that autophagy is involved in the degradation of CAV1 to suppress endocytosis of CLDN5, which was consistent with other findings that autophagy degrades CAV1 through binding to and activation of epidermal growth factor receptor in a ligand-independent manner [Citation48]. Additionally, loss of CAV1 itself showed no significant effect on paracellular permeability. Our data also suggest that autophagy may have a negative feedback effect on CAV1, thereby preventing the redistribution of CLDN5. The degradation of CAV1 by autophagosomes/autolysosome suppressed the redistribution of CLDN5 from cell membrane to a certain extent, further protecting BBB from fast disruption.
There are also reports indicating that endocytosis and endosomes are usually involved in the protein transport [Citation49]. In our study, we also explored whether clathrin-mediated transcytosis is responsible for the redistribution of membranous CLDN5 or is affected by autophagy under hypoxic injury. The results illustrate that hypoxia-induced redistribution of CLDN5 is mainly dependent on caveolae-mediated endocytosis, but less via clathrin-mediated transcytosis, thereby exacerbating BBB impairment.
In addition to the autophagy-lysosomal pathway, the UPS is also responsible for intracellular protein degradation in eukaryotic cells. Therefore, the role of UPS in the degradation of aggregated CLDN5 was additionally checked. The results indicated that the degradation of endothelial CLDN5 caused by hypoxia was independent of the UPS. Taken together, our results demonstrate that autophagy is involved in the degradation of cytosolic CLDN5 induced by hypoxia treatment. Another TJ component of TJP1, but not OCLN, was also found to colocalize with LC3 in the cytosol under long-term hypoxia induction, indicating a presence of selective protein degradation of autophagy [Citation50,Citation51]. The degradation of OCLN in bEnd.3 cells may depend on the UPS, since OCLN has been reported as a target of the E3 ubiquitin-protein ligase [Citation52].
Under short-term hypoxic induction, autophagy might be activated and function as a protective mechanism by which cells defend against external/internal stress. We showed in this study that during the early period of hypoxic (ischemic) injury, autophagy degrades and removes the accumulated proteins in the cytosol rapidly. These accumulated proteins might be not only the delocalized membranous proteins like CLDN5, but also other cytosolic proteins. If these proteins could not be cleared timely, it may cause severe cytotoxicity to induce endothelial apoptosis, which will further accelerate the disruption of BBB. In summary, our study highlights CLDN5 as sensitive marker of hypoxic injury, and provides the possibility that modulation of endothelial autophagy could be beneficial in clinical therapy of CNS disease related to BBB breakdown.
Materials and methods
Brain tissues from patients
Clinical research approval for this study (PJ2017074) was issued by the Medical Ethics Committee of the Affiliated Hospital of Guangdong Medical University. Brain tissue samples were obtained from patients with hemorrhagic stroke (n = 14 with 8 males and 6 females, aged from 37–59 years old). Control brain tissue samples were taken from non-stroke patients with primary glioblastoma, which was away from the lesion site and had to be removed for the surgery (n = 12 with 6 males and 6 females, aged from 37–57 years old).
Focal ischemic stroke model
Mice (C57BL/6 J, male and female, 22–25 g; Charles River Labs) MCAO/R surgery was performed following a modified protocol based on the previous description [Citation53]. In brief, mice were anesthetized with an intraperitoneal injection of chloral hydrate (300 mg/kg, i.p.). The right common carotid artery (CCA) was exposed through a midline neck incision. Next, the external carotid artery (ECA) and internal carotid artery (ICA) were carefully dissected. A 6–0 nylon monofilament coated with silicon resin (ϕ 0.21 ± 0.02 mm; Doccol Corporation, 602123PK10) was pushed into the ICA till the tip meets a slight resistance at the origin of the middle cerebral artery (MCA). After 90 min of MCAO, the monofilament was removed and reperfusion was performed for indicated time (0, 12, 24 h respectively). Mice in the sham group underwent identical operations with experimental groups except for ligation of the arteries and insertion of the nylon monofilaments. Mice were then sacrificed and the cortical and hippocampal sections were obtained using a freezing microtome for the subsequent experiments. Sample sizes of male mice: sham, n = 12; MCAO, n = 9; MCAO and 12 h reperfusion, n = 8; MCAO and 24 h reperfusion, n = 8. Sample sizes of female mice (> 16-month old): sham, n = 6; MCAO, n = 7; MCAO and 12 h reperfusion, n = 6; MCAO and 24 h reperfusion, n = 6.
Hypoxia-induced BBB impairment model in zebrafish and the autophagy analysis in vivo
To generate the hypoxia-induced BBB impairment model in zebrafish, wild-type AB and the endothelial specific transgenic zebrafish lines of Tg(kdrl:ras-cherry)s916 and Tg(fli1a:EGFP-CAAX) were used. The model was generated according to a modified version of previously described technique [Citation54]. In general, zebrafish (5 dpf, n = 12) in a 5 mL embryo medium-filled well plate were transferred to the modular incubator chamber (Billups-Rothenberg, MIC-101) filled with N2 and placed in an incubator at 28.5°C. For controls, the larvae were treated using the same process under normoxia condition. The dissolved oxygen value in embryo medium was measured and the HIF1A level of the larvae was analyzed to determine the hypoxia index. The survival curve of zebrafish was also monitored in parallel. The brain sections (150 μm thick) from zebrafish larvae after fixation in 4% paraformaldehyde (PFA) were obtained using a vibrating microtome (Leica VT1000S; Leica Microsystem, Germany).
For autophagy analyses, 30 μmol/L chloroquine (CQ; Sigma-Aldrich, C6628), 10 mmol/L 3-methyladenine (3-MA; Sigma-Aldrich, M9281), 20 μmol/L MG-132 (Sigma-Aldrich, M7449) or 50 nmol/L Rapa (Cell Signaling Technology, 9904) was used to incubate the larvae for 1 h under normoxic conditions respectively, followed by incubation in the modular incubator chamber filled with N2 to ensure hypoxia. Besides, homozygous becn1 mutated zebrafish was generated by using the CRISPR-Cas9 system [Citation55] with targeting site (sequence: 5ʹ-GTCTCAGCTTCCAGAAGAAG-3ʹ) located at the exon7 of becn1, which finally resulted in a translation truncation [Citation32].
All animal experiments were performed in accordance with the guidelines for animal care of Guangdong State Regulations on Laboratory Animal Management.
Cell culture and treatment
Brain microvascular endothelial cells (bEnd.3 or hCMEC/D3, Bioleaf Biotech) were used in this study. After formation of a confluent monolayer, the cells were exposed to CoCl2 (Sigma-Aldrich, C8661) or low oxygen containing gas (94% N2, 5% CO2, and 1% O2) as previously reported [Citation56]. For OGD induction, bEnd.3 cells were incubated in glucose-free medium (Procell, PM 150,443) with low oxygen containing gas for 6 h. Cell viability was evaluated by methyl thiazolyl tetrazolium (MTT) assay. For autophagy analysis, medium containing 10% fetal bovine serum (FBS), 50 nmol/L Rapa, 30 μmol/L CQ, or 10 mmol/L 3-MA was used to incubate the monolayer cells for 2 h respectively. Then the cells were exposed to hypoxia and cultured under the consistent concentrations of Rapa, CQ, or 3-MA, respectively.
Measurements of BBB permeability
The TEER value and paracellular permeability were measured to reflect the barrier property of the endothelial monolayer in vitro. For TEER measurement, bEnd.3 cells were seeded on collagen-coated semipermeable membrane filters (Millipore, USA), and their monolayer electrical resistance was monitored by the CellZscope®-System (NanoaAnalytics GmbH, Muenster, Germany) with 2 or 4 h-interval [Citation12]. After formation of the endothelial monolayer of bEnd.3 cells on membrane filters, the paracellular permeability was measured according to the amount of infiltrated fluorescein-labeled dextran (10 kDa, 0.5 mg/L) through the endothelial monolayer [Citation57].
To analyze the tightness of BBB in vivo, the dye diffusion assay was applied in transgenic Tg(kdrl:ras-Cherry)s916 zebrafish larvae (5 dpf) as previously described [Citation4]. Briefly, fluorescein sodium (375 Da, 5 nl, 1 mg/ml) or FITC-dextran (10 kDa, 5 nl, 0.5 mg/ml) was injected via the primary head sinus of the larvae. The larvae were immediately mounted in 1% low-melting agarose (LMA). Subsequently, 3D images of the cerebrovasculature were captured on a FV3000 confocal laser scanning microscope (Olympus, Japan). The relative fluorescence intensity of the dyes was measured and analyzed by Image J software.
Immunostaining, immunoblotting and data analysis
Monolayer of bEnd.3 or hCMEC/D3 cultured on coverslips were fixed with acetone for 10 min on ice. After blocking, the cells were incubated with primary antibodies including CLDN5 (Invitrogen, 35–2500), TJP1/ZO-1 (Invitrogen, 40–2200), OCLN/occludin (Invitrogen, 33–1500), LC3A/B (Cell Signaling Technology, 4108), CHC/clathrin heavy chain (Santa Cruz Biotechnology, sc-12,734) and CAV1/caveolin-1 (Santa Cruz Biotechnology, sc-894), followed by Alex Fluor 488-conjugated goat anti-mouse IgG (1:200; ThermoFisher Scientific, A-11,029), Alex Fluor 647-conjugated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch, 111–605-003), and DAPI (1:1000; Sigma-Aldrich, D9542) staining. To visualize the proteins at ultrastructural level, the localization of CLDN5, LC3, and CAV1 was imaged using a Leica TCS SP8 STED 3X microscope (Leica, Solms, Germany). The numbers of LC3-surrounded CLDN5 clusters per image were counted and quantified. For tissue immunostaining, the brain sections were incubated with primary antibodies of CLDN5 (Invitrogen, 35–2500), LC3B (Sigma-Aldrich, L7543), and secondary antibodies of Alex Fluro 488-conjugated goat anti-mouse IgG, Alex Fluro 647-conjugated goat anti-rabbit IgG, and DAPI. Images were obtained by FV3000 confocal laser scanning microscope.
For the fluorescent signal evaluation, the stained signals in the cytoplasm or membrane were first selected from at least 3 representative images. The mean fluorescence intensity in each compartment was then analyzed by Image-Pro Plus software and the integrated optical density (IOD) was calculated as (IOD = area × average density). For the evaluation of the expression of TJ proteins, the stained signals in endothelial membrane or cytosol were selected and quantified respectively. To evaluated the autophagic levels, the total LC3 intensity was quantified. For the quantification of LC3-CLDN5 colocalization in the cytosol, the colocalized cytosolic LC3-CLDN5 signals were first selected. Then, the IOD values from the images of the hypoxic groups were normalized to that of the control groups and the relative ratios were presented in the graphs.
For immunoblotting, the total cell lysate and subcellular fractions of membrane or cytosol was obtained by using RIPA buffer (Beyotime, China) or the membrane and cytosol protein extraction kit (Beyotime, China) respectively. In the process of subcellular fractionation, cells were harvested and lysed on ice with extraction buffer I, with protease inhibitor complex (Beibo, China) added additionally. For zebrafish Hif1a detection, the brain part of the larvae was cut and lysed. The lysate was then centrifuged at 700 × g for 10 min at 4°C to remove the nuclei and unbroken cells. The supernatant was collected and centrifuged again at 14,000 × g for 30 min at 4°C to separate cytosol proteins (supernatant) and membrane fragment (precipitate). The precipitate was then lysed in extraction buffer II and centrifuged at 14,000 × g for 5 min at 4°C to obtain the membrane proteins (supernatant). Next, the levels of CLDN5, TJP1, OCLN, CAV1, LC3A/B, LAMP1, ATG5, SQSTM1/p62 protein, and ATP1A1/Na,K-ATPase were analyzed by western blotting analyses. In brief, protein samples were separated by 8%-15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, ISEQ00010). The membranes were then incubated with antibodies of LAMP1 (Santa Cruz Biotechnology, AB24170), ATG5 (Cell Signaling Technology, 12,994), SQSTM1/p62 protein (Cell Signaling Technology, 39,749), and ATP1A1/Na,K-ATPase (Abbkine, Abp51894). Western blot bands were analyzed by adding ECL advance western blotting detection reagents (Millipore, WBKLS0100) and imaged using SmartChemi-500 imaging system (Sage Creation Science, Beijing, China). The immunoblot bands were normalized to the loading controls before quantification analyses.
Transmission electron microscopy (TEM)
Autophagosomes in bEnd.3 cells were imaged by electron microscopy as previously described [Citation12]. For analysis of the autophagosomes in BMECs of zebrafish larvae (AB, 5 dpf), the larvae were fixed with 2.5% glutaraldehyde and 1% osmium tetroxide in 0.2 mol/L sodium phosphate buffer for 12 h. The head of the zebrafish was dissected and dehydrated in graded ethanol (50, 75, 95%). Next, the mid-/hindbrain was cut into 70 nm-thick sections and stained with uranyl acetate and lead citrate. The samples were imaged on a JEM-1400 electron microscope (JEM, Tokyo, Japan), and the autophagic vesicles in the BMECs of zebrafish were quantified.
To determine whether autophagy was involved in the degradation of CLDN5, the autophagosome-like vesicles and the subcellular localization of CLDN5 were imaged by immunoelectron microscopy (IEM). Briefly, cells were fixed in a mixture of 4% paraformaldehyde (w:v) and 2% glutaraldehyde (v:v) in 0.2 mol/L sodium phosphate buffer. After washing with PBS, the cells were dehydrated in a graded series of ethanol (50, 70, 85, 95%) and permeabilized with increasing concentrations of LR-White resin (50, 70, 100% LR-White; London Resin, 14,381-UC) on ice. Ultrathin sections (70 nm-thick) were incubated with monoclonal mouse anti-CLDN5 or rabbit anti-CAV1 antibodies (1:50) overnight at 4°C and then incubated with IgG conjugated to 10 nm gold particles (Sigma-Aldrich, G7652 for CLDN5, G7402 for CAV1) for 2 h at room temperature. Finally, the region with gold particles were imaged by JEM-1400 electron microscope (JEM, Tokyo, Japan). For quantification analyses, the numbers of gold particles which represent packaged CLDN5 or CAV1 in autophagosome-like vesicles or caveolae per image were counted respectively.
Flow cytometry analysis
For apoptosis analysis, cells were harvested and stained with annexin V-FITC and propidium iodide (PI; Roche, 1,348,639). The early apoptotic cells (annexin V-positive and PI-negative) and late apoptotic cells (annexin V-positive and PI-positive) were analyzed using a FACS Calibur flow cytometer (BD Biosciences, USA). To quantify the level of ROS, bEnd.3 cells were collected and incubated with 2ʹ,7ʹ-Dichlorodihydrofluorescein diacetate (DCFH-DA, 10 μmol/L; Sigma-Aldrich, D6883) in serum-free medium at 37°C in a 5% CO2 incubator for 20 min. After washing with PBS, the cells were examined using a FACS Calibur flow cytometer (BD Biosciences, USA).
Transfection of plasmid DNA and small interfering RNAs
Endothelial bEnd.3 cells at 80% confluence were transfected with GFP-LC3 or with a control empty vector using Lipofectamine-3000 reagent (Invitrogen, L3000-015), in accordance with the manufacturer’s instructions. After transfection for 24 h, the cells were treated with 200 μmol/L CoCl2 for 12 h. Then, autophagy flux was lively tracked by Leica microscope to evaluate the number of GFP-LC3 puncta and its fusion ability.
For small interfering RNA transfection, the Cav1 siRNA reagent kit (Origene, SR404266) or scrambled negative control siRNA (Origene, SR30004) was used to transfect the bEnd.3 cells according to the manufacturer’s instructions. After incubation in 10% FBS medium for 48 h, the cells were treated with 200 μmol/L CoCl2 for 12 h. The level of CLDN5 or CAV1 was evaluated by immunostaining.
Statistical analysis
The results are presented as means ± standard error (mean ± SEM). All the experiments were conducted for at least 3 times. Two-tailed Student’s t-test (for two groups) or one-way analysis of variance (ANOVA followed by Tukey’s test for more than two groups) was applied to determine statistical significance. P < 0.05 and P < 0.01 were indicated by * and ** respectively.
Supplemental Material
Download Zip (2.4 MB)Acknowledgments
We thank Dr. Honghui Huang (Southwest University, China) for kindly providing the becn1 mutant zebrafish line and Dr. Dong Liu (Nantong University, China) for providing Tg(fli1a:EGFP-CAAX) transgenic zebrafish line. For critical reading of and comments on the manuscript, we are indebted to Dr. Canhua Huang, Dr. Yun Zhao and Dr. Jincai Luo.
Disclosure statement
The authors declare no conflict of interests.
Supplemenary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Abbott N, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53.
- Flierl MA, Stahel PF, Rittirsch D, et al. Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit Care. 2009;13(1):R12.
- Jingjing Z, J?Rg P, Hartwig W, et al. Establishment of a neuroepithelial barrier by Claudin5a is essential for zebrafish brain ventricular lumen expansion. Proc Natl Acad Sci USA. 2010;107(4):1425–1430.
- Liao Z, Yang Z, Piontek A, et al. Specific binding of a mutated fragment of Clostridium perfringens enterotoxin to endothelial claudin-5 and its modulation of cerebral vascular permeability. Neuroscience. 2016;327:53–63.
- Liu J, Weaver J, Jin X, et al. Nitric oxide interacts with caveolin-1 to facilitate autophagy-lysosome-mediated claudin-5 degradation in oxygen-glucose deprivation-treated endothelial cells. Mol Neurobiol. 2015;53(9):1–13.
- Stamatovic SM, Keep RF, Wang MM, et al. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009 Jul 10;284(28):19053–19066. DOI:https://doi.org/10.1074/jbc.M109.000521. PubMed PMID: 19423710; PubMed Central PMCID: PMC2707189.
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51.
- Ishida Y, Nagata K. Autophagy eliminates a specific species of misfolded procollagen and plays a protective role in cell survival against ER stress. Autophagy. 2009;5(8):1217–1219.
- De Meyer GR, Grootaert MO, Michiels CF, et al. Autophagy in vascular disease. Circ Res. 2015;116(3):468–479.
- Varga M, Fodor E, Vellai T. Autophagy in zebrafish. Methods. 2015;75:172–180.
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25(6):354–363.
- Yang Z, Huang C, Wu Y, et al. Autophagy protects the blood-brain barrier through regulating the dynamic of claudin-5 in short-term starvation. Front Physiol. 2019;10:2. PubMed PMID: 30713499; PubMed Central PMCID: PMCPMC6345697. eng.
- Marchi S, Corricelli M, Trapani E, et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med. 2015;7(11):1403–1417.
- Xia P, Wang S, Du Y, et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. Embo J. 2014;32(20):2685–2696.
- Matsuda J, Namba T, Takabatake Y, et al. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy. 2018;14(1):53–65.
- Bhattacharya A, Wei Q, Shin JN, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. 2015;12(11):1731–1739.
- Kim SS, Im SH, Yang JY, et al. Zebrafish as a screening model for testing the permeability of blood-brain barrier to small molecules. Zebrafish. 2017;14(4):322–330.
- Eunmyong L, Yeon K, Aylwin N, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10(4):572–587.
- Saera-Vila A, Kish PE, Louie KW, et al. Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy. 2016 Oct 2;12(10):1864–1875. PubMed PMID: 27467399; PubMed Central PMCID: PMC5066936.
- Hosseini R, Lamers GE, Hodzic Z, et al. Correlative light and electron microscopy imaging of autophagy in a zebrafish infection model. Autophagy. 2014;10(10):1844–1857.
- Kim W, Russell C. Mitophagy and neurodegeneration: the zebrafish model system. Autophagy. 2013;9(11):1693–1709.
- Markus R, Reutens DS, Read S, et al. Hypoxic tissue in ischaemic stroke: persistence and clinical consequences of spontaneous survival. Brain. 2004;127(Pt 6):1427–1436.
- Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54.
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009 Apr;43(4):348–364. PubMed PMID: 19241241.
- Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. Faseb J. 2007;21(13):3666–3676.
- Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Eur J Cancer Suppl. 2009;29(6):2570–2581.
- Li H, Wu J, Shen H, et al. Autophagy in hemorrhagic stroke: mechanisms and clinical implications. Prog Neurobiol. 2017;163-164:79–97.
- McMahon J, Huang X, Yang J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci Off J Soc Neurosci. 2012;32(45):15704–15714.
- Martínez A, Portero-Otin M, Pamplona R, et al. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010;20(2):281–297.
- Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15(5):767–776.
- Cutting AS, Del Rosario Y, Mu R, et al. The role of autophagy during group B Streptococcus infection of blood-brain barrier endothelium. J Biol Chem. 2014;289(52):35711–35723.
- Dong G, Zhang Z, Duan K, et al. Beclin 1 deficiency causes hepatic cell apoptosis via endoplasmic reticulum stress in zebrafish larvae. FEBS Lett. 2020 Apr;594(7):1155–1165. PubMed PMID: 31823348.
- Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114(5):459–469.
- Liu J, Jin X, Liu KJ, et al. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci Off J Soc Neurosci. 2012;32(9):3044–3057.
- Chen Z-H, Cao J-F, Zhou J-S, et al. Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;306(11):1016–1025.
- Chen Z, Nie SD, Qu ML, et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018;9(7):771.
- Ferrero E, Fulgenzi A, Belloni D, et al. Cellfood improves respiratory metabolism of endothelial cells and inhibits hypoxia-induced reactive oxygen species (ros) generation. J Physiol Pharmacol. 2011 Jun;62(3):287–293. PubMed PMID: 21893688.
- Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014 Jan 30;123(5):625–631. PubMed PMID: 24300855; PubMed Central PMCID: PMC3907751.
- Chen S, Wang Y, Zhang H, et al. The antioxidant MitoQ protects against CSE-induced endothelial barrier injury and inflammation by inhibiting ROS and autophagy in human umbilical vein endothelial cells. Int J Biol Sci. 2019;15(7):1440–1451. PubMed PMID: 31337974; PubMed Central PMCID: PMC6643142.
- Hertzog M, Monteiro P, Le Dez G, et al. Exo70 subunit of the exocyst complex is involved in adhesion-dependent trafficking of caveolin-1. PLoS One. 2012;7(12):e52627. PubMed PMID: 23300727; PubMed Central PMCID: PMC3531403.
- Pelkmans L, Zerial M. Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae. Nature. 2005 Jul 7;436(7047):128–133. PubMed PMID: 16001074.
- Jiang X, Andjelkovic AV, Zhu L, et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018 Apr - May;163-164:144–171. PubMed PMID: 28987927; PubMed Central PMCID: PMC5886838.
- Topakian R, Barrick TR, Howe FA, et al. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 2010;81(2):192–197.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Cosin-Roger J, Simmen S, Melhem H, et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017;8(1):98.
- Crippa V, Carra S, Rusmini P, et al. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy. 2010;6(7):958–960.
- Stamatovic SM, Johnson AM, Sladojevic N, et al. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. 2017;1397(1):54–65.
- Chen Y, Henson ES, Xiao W, et al. Tyrosine kinase receptor EGFR regulates the switch in cancer cells between cell survival and cell death induced by autophagy in hypoxia. Autophagy. 2016;12(6):1029–1046.
- Babst M, Katzmann DJ, Estepa-Sabal EJ, et al. ESCRT-III: an endosome-associated heterooligomeric protein complex required for MVB sorting. Dev Cell. 2002;3(2):271–282.
- Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987.
- Liang RJ, Lingeman E, Ahmed S, et al. Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol. 2018;217(10):3354–3367.
- Andreas T, Deyu F, Yun-Cai L, et al. The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch. J Biol Chem. 2002;277(12):10201–10208.
- Stubbe T, Ebner F, Richter D, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33(1):37–47.
- Braga MM, Silva ES, Moraes TB, et al. Brain zinc chelation by diethyldithiocarbamate increased the behavioral and mitochondrial damages in zebrafish subjected to hypoxia. Sci Rep. 2016;6:20279.
- Hwang WY, Fu Y, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–229.
- Wu W, Lin C, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. Embo J. 2016;35(13):1368–1384.
- Gaillard PJ, Voorwinden LH, Nielsen JL, et al. Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci. 2001;12(3):215–222.