
ABSTRACT
RASAL2 (RAS protein activator like 2), a RASGTPase activating protein, can catalyze the hydrolysis of RAS-GTP into RAS-GDP to inactivate the RAS pathway in various types of cancer cells. However, the cellular function of RASAL2 remains elusive. Here we showed that RASAL2 can attenuate PRKAA/AMPKα phosphorylation by recruiting phosphatase PPM1B/pp2cβ, thus inhibiting the initiation of basal autophagy under normal conditions. In addition, we found that glucose starvation could induce dissociation of PPM1B from RASAL2 and then RASAL2 at S351 be phosphorylated by PRKAA, followed by the binding of phosphorylated-RASAL2 with to PIK3C3/VPS34-ATG14-BECN1/Beclin1 complex to increase PIK3C3 activity and autophagy. Furthermore, RASAL2 S351 phosphorylation facilitated breast tumor growth and correlated to poor clinical outcomes in breast cancer patients. Our study demonstrated that the phosphorylation status of RASAL2 S351 can function as a molecular switch to either suppress or promote AMPK-mediated autophagy. Inhibition of RASAL2 S351 phosphorylation might be a potential therapeutic strategy to overcome the resistance of AMPK-activation agents.
Abbreviations: AICAR: aminoimidazole carboxamide ribonucleotide; AMPK: adenosine 5‘-monophosphate (AMP)-activated protein kinase; ATG14: autophagy related 14; C.C: compound C; CQ: chloroquine; DKO: double-knockout; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4/VPS15: phosphoinositide-3-kinase regulatory subunit 4; PPM1B/pp2cβ: protein phosphatase, Mg2+/Mn2+ dependent 1B; PRKAA/AMPKα: protein kinase AMP-activated catalytic subunit alpha; PtdIns: phosphatidylinositol; PtdIns3P: phosphatidylinositol-3-phosphate; RASAL2: RAS protein activator like 2; RasGAPs: RasGTPase activating proteins; SQSTM1/p62: sequestosome 1; TNBC: triple-negative breast cancer.
KEYWORDS:
Introduction
Macroautophagy/autophagy is an evolutionarily conserved eukaryotic process that degrades misfolded proteins and damaged organelles to maintain cellular homeostasis in response to various stresses [Citation1–3]. During the initiation of autophagy, PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3), the only class III phosphatidylinositol 3-kinase (PtdIns3K) in mammals, interacts with BECN1/Beclin1, ATG14 (autophagy related 14), and PIK3R4/Vps150 (phosphoinositide-3-kinase regulatory subunit 4) at the phagophore to form functional complexes and generates phosphatidylinositol-3-phosphate (PtdIns3P) by phosphorylating the 3-position of PtdIns [Citation4]. The initiation of autophagy is regulated by several critical upstream molecules including adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK) and MTOR (mechanistictarget of rapamycin kinase) [Citation5–7]. AMPK can promote PIK3C3 activity and autophagy either through phosphorylation of TSC2 and RPTOR/Raptor to suppress MTOR activity or through direct phosphorylation and activation of ULK1 (unc-51 like autophagy activating kinase 1) and BECN1 [Citation8–11]. Although these known AMPK substrates have been well documented, the signaling pathways for AMPK to regulate the initiation of autophagy and other substrates of AMPK involved in these events remain to be further defined.
Recently, RASGTPase activating proteins (RASGAPs) have emerged as an expanding class of tumor suppressors that can catalyze the hydrolysis of RAS-GTP into RAS-GDP to inactivate RAS [Citation12]. As one of the RASGAPs, RASAL2 (RAS protein activator like 2) initially identified as a potential tumor suppressor in a functional cell-based screen and down-regulates in various types of cancers, such as luminal B breast cancer, lung cancer, ovarian cancer, bladder cancer and nasopharyngeal carcinoma [Citation13–19]. In addition, RASAL2 can function as a RHO-GAP and binds with ECT2 (epithelial cell transforming 2) to regulate RHO activity in human malignant astrocytoma cells [Citation20]. In contrast, RASAL2 is reported to be over-expressed in triple-negative breast cancer (TNBC) and plays an oncogenic role through interacting with ARHGAP24 (Rho GTPase activating protein 24), a RAC1 GAP protein, to activate small GTPase RAC1 [Citation21]. Moreover, RASAL2 is upregulated and targeted through the LATS2 (large tumor suppressor kinase 2)-YAP1 (Yes1 associated transcriptional regulator) axis in colorectal cancer [Citation22]. Thus, the role of RASAL2 in cancer seems to be context dependent. However, it is unknown whether RASAL2 has a universal role in the regulation of other important cellular activities.
In this study, we showed that glucose deprivation induces the binding of RASAL2 to the PIK3C3/VPS34-containing complex in a PRKAA-dependent manner; mechanistically, PRKAA directly phosphorylates RASAL2 at S351 to switch the function of RASAL2 from a suppressor to an activator in the formation of basal autophagy.
Results
RASAL2 negatively regulates autophagy under nutrient-rich condition
To determine whether RASAL2 was able to modulate autophagy, we first examined the effect of RASAL2 depletion on autophagosome formation in CRISPR/Cas9 system generated RASAL2-negative MCF-7 (KO) cells or MCF-7 cells (human breast adenocarcinoma cell line) with an empty CRISPR-cas9 plasmid as control (CT). We found increased accumulation of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-II and degradation of SQSTM1/p62 (sequestosome 1) in RASAL2-KO MCF7 (), MDA-MB-231 (human triple-negative breast cancer cell line) and A549 cells (human non-small-cell lung carcinoma cell lines) (Figure S1), while re-expression of RASAL2 in RASAL2-KO MCF7 cells restored the protein level of LC3-II and SQSTM1 to that of the RASAL2-CT MCF7 cells (). We further determined the regulatory function of RASAL2 on autophagy by using electron microscopy and found that depletion of RASAL2 resulted in increased autophagosome and autolysosome accumulation (). The increased LC3-II level observed might be a result of promotion of autophagic flux or of inhibition of LC3-II degradation. We thus used GFP-RFP-LC3, which was used to distinguish the yellow puncta autophagosomes (GFP-RFP-positive), and red puncta autolysosomes (RFP-positive and GFP-negative) whose GFP fluorescence was quenched in acidic pH [Citation23] to address this issue, and found a significant increase of red puncta and reduction of yellow puncta in RASAL2-KO MCF7 cells (). These observations were further confirmed by using CQ (chloroquine), a lysosome inhibitor, where we found that CQ-enhanced LC3-II accumulation was further increased in RASAL2-KO MCF7 cells, indicating that RASAL2 might directly affect autophagic flux ().
Figure 1. Silencing RASAL2 initiates basal autophagy. (A, B, E) Autophagy assays were measured in RASAL2-CT or RASAL2-KO MCF7 cells. (A) Immunoblotting detection of autophagy-related proteins in these cells, (B) transfected with or without MYC-RASAL2, or (E) in the presence or absence of 50 μM chloroquine (CQ) for 4 h. (C) TEM analysis of autophagosomes and autolysosomes (scale bars: 0.5 μm). (D) GFP-RFP-LC3 plasmids were transfected in these cells and monitored using a fluorescence microscope. White boxed regions in the panels are enlarged. Scale bars: 10 μm. Representative images of RFP-positive and GFP-negative LC3-labeled autolysosomes are shown. Data represent the mean ± SEM, n = 50, **p < 0.01). (F) RASAL2-KO MCF7 cells transfected with MYC-RASAL2 (WT), GAP activity–deficient mutant (MYC-RASAL2-GAP-mut), or GAP domain deletion mutant (MYC-RASAL2-ΔGAP) were used for immunoblotting analyses with indicated antibodies. The relative amount of LC3-II was semi-quantified by grayscale analysis
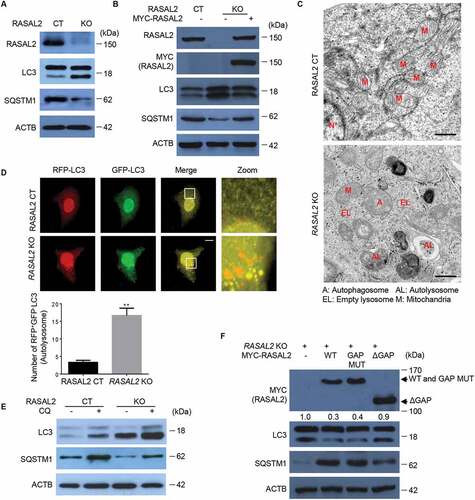
We next sought to test whether the role of RASAL2 in autophagy depends on its RASGAP activity. As it is known that K435E and K585E double mutations in RASAL2 result in the loss of RASGAP activity [Citation21], we therefore expressed the RASAL2 K435/K585E mutant and a GAP domain deletion mutant in RASAL2-KO MCF7 cells. We found that the RASAL2 RASGAP mutant retained its activity to inhibit autophagy while the GAP deletion mutant did not, suggesting that RASGAP activity is dispensable for RASAL2 function in autophagy, while RASAL2 still needs the GAP domain to exert its inhibitory role in autophagy (). Collectively, these findings suggested that RASAL2 inhibits the initiation of basal autophagy independently of its GAP activity.
RASAL2 attenuates PRKAA activity and autophagy through PPM1B
To explore the mechanism underlying RASAL2-regulated autophagy, we transfected Myc-RASAL2 into RASAL2-KO MCF7 cells, followed by immunoprecipitation and mass spectrometry analysis. The result showed that PRKAA was a potential RASAL2 interacting protein (Figure S2A and Table S1). We confirmed this physical interaction by co-expressing MYC-RASAL2 and FLAG-PRKAA in 293 T cells followed by co-IP assays () and further examined the endogenous interaction in MCF7 cells by using anti-RASAL2 antibody to immunoprecipitate endogenous PRKAA (Figure S2B).
Figure 2. RASAL2 attenuates PRKAA activity and autophagy through PPM1B. (A) 293 T cells co-transfected with FLAG-PRKAA and MYC-RASAL2 were used for immunoprecipitation by anti-FLAG (M2) beads. (B) Immunoblotting analysis of MCF7 cells transfected with increasing amount of MYC-RASAL2 as indicated. (C) Western blots of p-PRKAA and LC3 in RASAL2-CT and RASAL2-KO MCF7 cells treated with AICAR (0.5 mM, 2 h) or Compound C (C.C.) (20 μM, 4 h) as indicated. The relative amount of LC3-II and p-PRKAA was semi-quantified by grayscale analysis. (D) PRKAA control (CT) and PRKAA double-knockout (DKO) MCF7 cells transfected with or without RASAL2 si-RNA were used for immunoblotting analysis with indicated antibodies. (E) Endogenous PPM1B and RASAL2 were immunoprecipitated from MCF7 cells by using anti-RASAL2 or anti-PPM1B antibodies or IgG control. (F) Co-IP assays were performed by using PPM1B antibody or IgG control in RASAL2-CT and RASAL2-KO MCF7 cells. The relative amount of immunoprecipitated PRKAA was semi-quantified by grayscale analysis. (G) PPM1B de-phosphorylates PRKAA in vitro. 293 T cells transfected with FLAG-PRKAA were treated with AICAR (0.5 mM, 2 h). Then the activated FLAG-PRKAA protein were purified by using M2 beads and incubated with purified GST-PPM1B protein or its mutant in the present or absent of purified HIS-RASAL2 in kinase buffer at 30°C for 30 min. The relative amount of immunoprecipitated PPM1B and p-PRKAA was semi-quantified by grayscale analysis. (H) Western blots of the indicated proteins in RASAL2-CT, PPM1B-KO, RASAL2-KO and PPM1B/RASAL2 double-KOMCF7 cells. The relative amount of LC3-II and p-PRKAA was semi-quantified by grayscale analysis. (I) MCF7 cells generated as in (H) were transfected with GFP-RFP-LC3 and RFP-positive-GFP-negative LC3-labeled autolysosomes were qualified. White boxed regions in the panels are enlarged. Scale bars: 10 μm. Data represent the mean ± SEM, n = 50. *p < 0.05, **p < 0.01
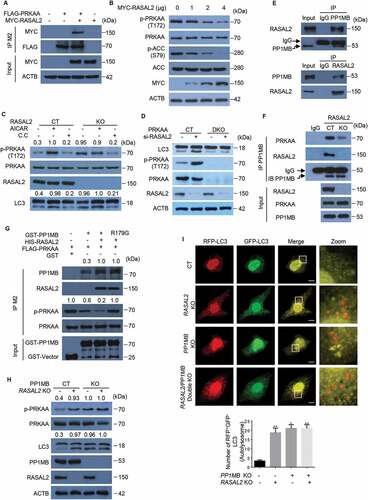
As AMPK is one of the key regulators in autophagy [Citation24], we therefore investigated whether RASAL2 regulates autophagy through PRKAA. We first transfected increased amounts of MYC-RASAL2 plasmid in MCF7 cells and found the phosphorylation level of PRKAA and ACC (Acetyl-CoA carboxylase), a substrate of PRKAA, were significantly decreased (). Moreover, MCF7 cells treated with the AMPK activator AICAR (aminoimidazole carboxamide ribonucleotide) resulted in the accumulation of LC3-II; while the increased accumulation of LC3-II triggered by RASAL2 knockout were attenuated after AMPK inhibitor compound C (C.C) was added in (). To further verify the role of RASAL2 in PRKAA phosphorylation, we transfected RASAL2 si-RNA in PRKAA-CT and PRKAA1/AMPKα1 and PRKAA2/AMPKα2 double-knockout (DKO) MCF7 cells (). As expected, the accumulation of LC3-II drastically decreased in PRKAA-DKO MCF7 cells. However, silencing RASAL2 could significantly increase the accumulation of LC3-II in PRKAA-CT, but not in PRKAA-DKO MCF7 cells. Collectively, these findings suggested that RASAL2 plays a pivotal role in attenuating PRKAA phosphorylation.
We next investigated the mechanisms by which RASAL2 regulates PRKAA phosphorylation. Sequence and structure analysis of RASAL2 did not implicate it as a putative phosphatase for PRKAA. Mass spectrometry results showed that PPM1B/pp2cβ (protein phosphatase, Mg2+/Mn2+ dependent 1B), a metal-dependent protein phosphatase that can dephosphorylate phosphoserine and phosphothreonine residues [Citation25], was also in the immunocomplex (Figure S2A and Table S1). We validated this association by a co-immunoprecipitation analysis showing the endogenous interaction between RASAL2 and PPM1B in MCF7 (), MDA-MB-231 and A549 cells (Figure S2C). As PPP2 (protein phosphatase 2) can bind and dephosphorylate PRKAA [Citation26], we therefore postulated that RASAL2 attenuates PRKAA phosphorylation might be dependent on PPM1B. As expected, endogenous co-immunoprecipitation analysis showed that the physical interaction of PPM1B with PRKAA was dramatically reduced in RASAL2-KO MCF7 cells as compared with RASAL2-CT MCF7 cells (). Moreover, we performed an in vitro experiment by mixing activated FLAG-PRKAA with GST-PPM1B or GST-PPM1BR179G kinase-dead mutant [Citation27] in the presence or absence of purified HIS-RASAL2 () and showed that incubation of PRKAA with PPM1B leads to a decrease in the amount of phosphorylated PRKAA. In addition, PPM1B complexed with RASAL2 showed enhanced interaction with PRKAA and significantly attenuated PRKAA phosphorylation. Remarkably, RASAL2 also enhanced PPM1BR179G-PRKAA interaction, although this interaction showed no PPM1B kinase activity. Together, these results suggested that RASAL2 is critical for PPM1B-PRKAA interaction and subsequent PRKAA dephosphorylation.
To further investigate the contribution of PPM1B to the negative effect of RASAL2 on autophagy, we generated PPM1B-KO, RASAL2-KO and PPM1B/RASAL2 DKO cells. PPM1B knockout led to an increase in PRKAA phosphorylation () and RFP+-GFP–LC3 puncta accumulation () in both RASAL2-CT and RASAL2-KO MCF7 cells. However, depletion of RASAL2 could significantly increase the phosphorylation level of PRKAA and the accumulation of LC3-II in PPM1B-CT MCF7 cells, but not in PPM1B-KO MCF7 cells. The same results were also observed in MDA-MB-231 cells (Figure S2D). Taken together, these data indicate that RASAL2 attenuates PRKAA phosphorylation and autophagy via PPM1B.
Glucose starvation disassociates PPM1B from RASAL2, enhances PRKAA-RASAL2 interaction
We then asked whether the RASAL2-PRKAA interaction is regulated by autophagy-promoting stimuli. As shown in , glucose deprivation, but not other autophagy stimuli such as amino acid starvation or Earle’s Balanced Salt Solution (EBSS), dramatically increased RASAL2-PRKAA interaction in 293 T cells. This observation was further confirmed on endogenous proteins in MCF7 cells and MDA-MB-231 cells (). As glucose deprivation-induced autophagy was mediated prevailingly by the activation of AMPK signaling pathway [Citation6,Citation24], we therefore tested whether PRKAA activation is required for this increased PRKAA-RASAL2 interaction. As expected, treatment with AICAR led to an up-regulation of PRKAA-RASAL2 interaction, while the enhanced RASAL2-PRKAA interaction triggered by glucose starvation was attenuated after C.C treatment (). Moreover, PRKAA-RASAL2 interaction was greatly elevated in PRKAAT172E mutation that mimicked PRKAA phosphorylation, while PRKAAT172A lost its ability to bind to RASAL2. These results suggested that PRKAA activation is required for its binding to RASAL2 ().
Figure 3. Glucose starvation disassociates PPM1B from RASAL2 and enhances PRKAA-RASAL2 interaction. (A) Glucose deprivation increases PRKAA-RASAL2 interaction. 293 T cells co-transfected with FLAG-PRKAA and MYC-RASAL2 were cultured with glucose starvation (4 h), amino acids deprivation (6 h), or EBSS (4 h) as indicated. The co-IP analyses were performed by using M2 beads. The relative amount of co-eluted MYC-RASAL2 was semi-quantified by grayscale analysis. (Band F) MCF7 or MDA-MB-231 cells were cultured with/without glucose starvation medium and immunoprecipitation were performed as indicated. (C) MCF7 cells treated with AICAR (0.5 mM, 2 h) or C.C (20 μM, 4 h) were incubated with or without glucose starvation medium for 4 h. The co-IP assay was performed by using PRKAA antibody. The relative amount of immunoprecipitated RASAL2 were semi-quantified by grayscale analysis. (D) 293 T cells co-transfected with MYC-RASAL2 and FLAG-PRKAA or PRKAA mutants were used for immunoprecipitation by using anti-FLAG (M2) beads. The relative amount of immunoprecipitated MYC-RASAL2 was semi-quantified by grayscale analysis. (E) 293 T cells co-transfected with FLAG-PPM1B and MYC-RASAL2 were incubated with or without glucose-starvation medium for 4 h, the cell lysates were used for immunoprecipitation by using anti-FLAG (M2) beads. (G) MCF7 cells transfected with increased amount of MYC-PPM1B in the present or absent of glucose starvation medium for 4 h. Immunoblotting analyses were performed. The relative amount of p-PRKAA was semi-quantified by grayscale analysis. (H) RASAL2-CT and RASAL2-KO MCF-7 or MDA-MB-231 cells were incubated with/without glucose starvation medium for 4 h. Immunoblotting analyses were performed. The relative amount of LC3-II was semi-quantified by grayscale analysis. (I) RASAL2-CT and RASAL2-KO MCF-7 cells transiently transfected with GFP-RFP-LC3 were incubated with/without glucose starvation medium for 4 h and monitored by using fluorescence microscopy. Representative images of RFP-positive-GFP-negative LC3-labeled autolysosomes were shown. White boxed regions in the panels are enlarged. Scale bars: 10 μm. Data represent the mean ± SEM. n = 50, **p < 0.01
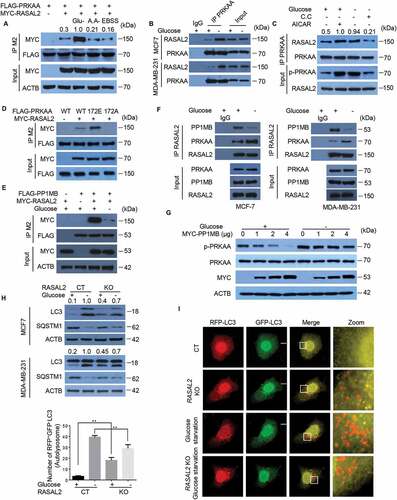
We further tested whether PPM1B-RASAL2 interaction is regulated by glucose starvation. After co-expression of FLAG-PPM1B and MYC-RASAL2 in 293 T cells, we found that glucose deprivation rapidly decreased the association of RASAL2 with PPM1B (). The dissociation of PPM1B from RASAL2 upon glucose starvation was further validated by endogenous co-immunoprecipitation assays in MCF7 cells and MDA-MB-231 cells (). Moreover, expression of ectopic PPM1B dramatically decreased the phosphorylation level of PRKAA under nutrient-rich but not glucose starvation conditions in MCF7 (). These results indicated that glucose deprivation could induce disassociation of PPM1B from RASAL2 and then promoted interaction of RASAL2 with phosphorylated-PRKAA.
We next sought to test the function of this enhanced RASAL2-PRKAA interaction in glucose-starvation induced autophagy. We treated RASAL2-CT and RASAL2-KO cells with or without glucose starvation medium for 4 h. As expected, RASAL2 depletion led to the accumulation of LC3-II and degradation of SQSTM1 under normal conditions. However, upon glucose starvation, the accumulation of LC3-II and degradation of SQSTM1 were significantly down regulated in RASAL2-KO as compared with RASAL2-CT in MCF-7 and MDA-MB-231 cells (). We further confirmed these observations by using GFP-RFP-LC3 plasmid and the results showed that upon glucose starvation, GFP–RFP+-LC3 puncta dramatically increased in RASAL2-CT cells while this response was greatly impaired in the RASAL2-KO cells (). Together, these results suggested that instead of binding with PPM1B and attenuating the basal level of PRKAA activity and autophagy under nutrient-rich conditions, RASAL2 enhances binding with PRKAA and might play a positive role in glucose-deprivation-induced autophagy.
AMPK phosphorylated RASAL2 at S351
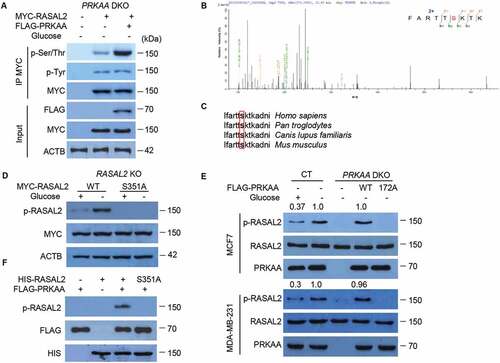
To further elucidate the mechanism by which activated PRKAA regulates the interaction with RASAL2 for glucose-starvation-induced autophagy, a simple proposal put forward was that PRKAA might directly phosphorylate RASAL2. To verify this hypothesis, we transfected MYC-RASAL2 with or without FLAG-PRKAA in PRKAA-DKO MCF7 cells and treated cells with glucose starvation medium for 4 h. After immunoprecipitation with MYC-beads, we found a dramatic increase in phosphorylation at serine/threonine but not tyrosine of RASAL2 in the cells co-transfected with MYC-RASAL2 and FLAG-PRKAA as compared with MYC-RASAL2 alone (), indicating that RASAL2 could be phosphorylated by PRKAA. In addition, mass-spectrometry analysis showed that RASAL2 was phosphorylated at S351 (), which matches the optimal AMPK substrate motif and is an evolutionarily conserved residue across different species (). Phosphorylation at RASAL2 S351 was further verified by a specific anti-phosphorylated RASAL2S351 antibody in RASAL2-KO MCF7 cells transfected with RASAL2 WT or RASAL2S351A mutant (mutation of S351 into alanine) and showed this glucose-starvation-induced phosphorylation was totally eliminated in S351A mutant of RASAL2, but not WT RASAL2 (). Moreover, the RASAL2 S351 phosphorylation was abrogated by PRKAA-DKO and could be restored by re-expression of WT PRKAA, but not the catalytically inactive PRKAAT172A mutant upon glucose starvation in MCF7 and MDA-MB-231 cells (). Finally, the in vitro phosphorylation assay showed that the activated PRKAA phosphorylated HIS-RASAL2at S351 but not RASAL2 S351A (). Together, these results indicated that PRKAA phosphorylates RASAL2 at S351 both in vitro and in vivo.
Figure 4. PRKAA phosphorylates RASAL2 at S351. (A) PRKAA-DKO MCF7 cells co-transfected MYC-RASAL2 with or without FLAG-PRKAA were cultured in the glucose-starvation medium for 4 h. The co-IP analyses were performed by using anti-MYC beads. (B) Identification of phosphorylation site(S) on RASAL2 by mass spectrometry. Immunoprecipitations of MYC-RASAL2 from the lysates of PRKAA-DKO MCF7 cells co-transfected MYC-RASAL2 with FLAG-PRKAA as generated in (A) were used for mass spectrometry analysis. The results showed a tryptic peptide (FARTTpSKTKAD) from FLAG-PRKAA treated RASAL2 protein with a mass shift of +79.9663 Da at the serine residue. The results suggested that S351 was phosphorylated. (C) S351 in human RASAL2 protein is evolutionally conserved in different species. S351 residues in the box are highlighted in bold. (D) RASAL2-KO MCF7 cells transfected with indicated RASAL2 constructs were incubated with or without glucose deprivation for 4 h. The cell lysates were used for immunoblotting as indicated. (E) MCF7 or MDA-MB-231 cells with or without PRKAA DKO and reconstituted expression of FLAG-PRKAA (WT or T172A) were cultured in the presence or absence of glucose starvation medium for 4 h. The cell lysates were used for immunoblotting as indicated. The relative amount of p-S351 RASAL2 was semi-quantified by grayscale analysis. (F) PRKAA phosphorylates RASAL2 in vitro. In vitro kinase assay carried out with purified HIS-RASAL2 or HIS-RASAL2S351A, purified activated-FLAG-PRKAA complex prepared from 293 T cells expressing FLAG-PRKAA after glucose starvation for 4 h and nonradioactive ATP, and mixed in the reaction buffer for 20 min at 30°C. Immunoblotting analyses were performed with the indicated antibodies
S351 phosphorylated RASAL2 binding with PIK3C3 complex and facilitates PIK3C3 activity and formation of basal autophagy
To understand the functional consequence of RASAL2 phosphorylation in autophagy initiation, we performed immunoprecipitation and mass spectrometry analysis to find the potential binding partners of RASAL2 before and after glucose starvation. The result showed that PIK3C3 was associated with RASAL2 upon glucose starvation (Figure S3A and Table S2). We confirmed this glucose-starvation-induced physical interaction endogenously by using RASAL2 antibody in MCF7 and MDA-MB-231 cells (). In addition, co-immunoprecipitation analyses of RASAL2-KO MCF7 cells transfected with MYC-RASAL2 (WT or S351A) showed that RASAL2S351A, in contrast to its WT counterpart, lost its ability to bind to PIK3C3 (Figure S3B). However, S351D mutant, mimicking S351-phosphorylated RASAL2, increased PIK3C3-RASAL2 association even in the glucose-sufficient condition (Figure S3C). In line with the finding that RASAL2 phosphorylation is required for the glucose-deprivation-induced interaction between RASAL2 and PIK3C3, PRKAA-DKO inhibited this glucose-starvation-induced association between RASAL2 and PIK3C3, and such an inhibition was overridden by re-expression of WTPRKAA, but not the catalytically inactive PRKAAT172A mutant in MCF7 cells () and MDA-MB-231 cells (Figure S3D).
Figure 5. S351 phosphorylates RASAL2 binding with PIK3C3 complex and facilitates PIK3C3 activity and formation of basal autophagy. (A) MCF7 or MDA-MB-231 cells were incubated with/without glucose starvation medium for 4 h. Immunoprecipitation of endogenous RASAL2 were performed. (B) MCF7 with or without PRKAA DKO and reconstituted expression of FLAG-PRKAA1 (WT or T172A) were transfected with MYC-RASAL2 and cultured in the presence or absence of glucose-deprived medium for 4 h, then immunoprecipitation of MYC-RASAL2 were performed. (C) MCF7 cells were incubated with/without glucose starvation medium. Immunoprecipitation of endogenous RASAL2 were performed. (D) RASAL2-KO MCF7 cells transfected with MYC-RASAL2 (WT or S351A) were incubated with/without glucose-deprived medium for 4 h. Cell lysate were used for immunoprecipitation by using anti-MYC beads. (E – I) RASAL2-CT andRASAL2-KO MCF7 cells stably expressing MYC-RASAL2 (WT, S351D, S351A). (E) The PIK3C3 complex were immunoprecipitated and PIK3C3 lipid kinase activity were measured with the Class III PtdIns3P ELISA Kit and the PtdIns3P level were normalized to the amount of ATG14 used in the assay; or (F) the PIK3C3 complex were immunoprecipitated and incubated with 3H-PtdIns. Results were reported as means ± SEM of three replicates. **p < 0.01, N.S., not significant. (G) GFP signals were observed in these cells transfected with GFP-FYVE plasmid and the statistics of GFP-positive puncta per cell were calculated. Scale bars, 10 μm. Data represent the mean ± SEM. n = 50. **p < 0.01, N.S., not significant. (H) Lysates from these cells were used for immunoprecipitation and immunoblotting with the antibodies as indicated. (I) GFP-RFP-LC3 plasmids were transfected in these cells and monitored using fluorescence microscope and RFP-positive and GFP-negative LC3-labeled autolysosomes were qualified. White boxed regions in the panels are enlarged. Scale bars, 10 μm. Representative images of RFP-positive and GFP-negative LC3-labeled autolysosomes are shown. Data represent the mean ± SEM, n = 50. **p < 0.01, N.S., not significant
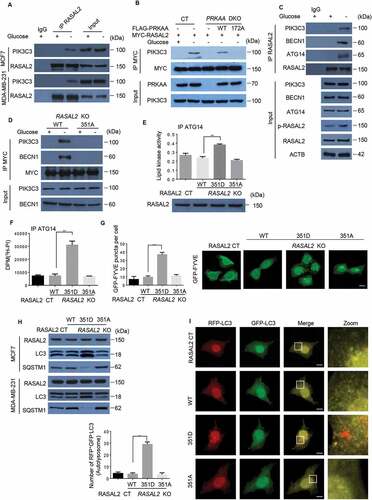
Next, we examined if S351 phosphorylated RASAL2 could bind with the components of PIK3C3 complex. As expected, glucose deprivation effectively resulted in the association of endogenous RASAL2 with PIK3C3, BECN1 and ATG14 () in MCF7 cells. In addition, co-immunoprecipitation analyses showed that RASAL2S351A, in contrast to its WT counterpart, lost its ability to bind to PIK3C3 complex under the condition of glucose starvation in MCF-7 () and MDA-MB-231 cells (Figure S3E). However, we could not detect the RASAL2-UVRAG interaction (Figure S3F), suggesting that S351 phosphorylated RASAL2 is not involved in UVRAG-associated cellular activity.
Knowing that S351 phosphorylation positively regulates the RASAL2-PIK3C3 interaction, we wondered whether this binding might regulate the kinase activity of PIK3C3. After transfection with FLAG-PIK3C3 in RASAL2-CT and RASAL2-KO MCF7 cells stably expressing MYC-RASAL2 (WT, S351D, S351A), the lipid kinase activity of PIK3C3 was greatly elevated in RASAL2S351D mutant (). The positive effect of RASAL2 on PIK3C3 lipid kinase activity could be due to two possible mechanisms: 1. RASAL2 phosphorylation might modulate PIK3C3 complex formation [Citation28]; or 2. RASAL2 phosphorylation might affect PIK3C3 binding to its substrates PtdIns [Citation5]. As expected, glucose starvation enhances PIK3C3-BECN1-ATG14 complex formation. However, this RASAL2 phosphorylation-enhanced PIK3C3 activity was not due to the alteration of PIK3C3-BECN1-ATG14 complex formation, because RASAL2 depletion did not affect the interactions among them (Figure S3G). To determine whether RASAL2 phosphorylation affects PIK3C3 binding to its substrates PtdIns, we immunoprecipitated ATG14-associated PIK3C3 complex from MCF7 cells with or without RASAL2 KO and reconstituted expression of MYC-RASAL2 (WT, S351D, S351A), and incubated with 3H-labeled PtdIns. The results showed that the PIK3C3 complex isolated from RASAL2S351D MCF7 cell lysates binds more PtdIns than that of the PIK3C3 complex isolated from WT RASAL2 or RASAL2S351A (), suggesting a positive regulation of PIK3C3-PtdIns binding via RASAL2S351 phosphorylation. In accordance with these findings, the number of PtdIns3P puncta (green) was largely induced in S351D but abrogated in WT or S351A mutant of RASAL2, as defined by GFP-FYVE (). These results show that RASAL2 phosphorylation at S351 is required for the activation of the PIK3C3 complex.
RASAL2S351 phosphorylation also promotes the formation of basal autophagy. This was supported by observation of the significant increase in the accumulation of LC3-II and degradation of SQSTM1 in RASAL2S351D mutant, alongside the drastic decrease in RASAL2S351A mutant in MCF7 and MDA-MB-231 cells (). Moreover, RFP+GFP−LC3 puncta were dramatically increased in S351D mutant in MCF-7 cells, indicating RASAL2 phosphorylation at S351 promotes autophagosome formation (). Collectively, these data strongly suggest that RASAL2 phosphorylation at S351 by PRKAA facilitates the formation of autophagy.
In addition to phosphorylation of RASAL2 at S351, PRKAA could also bind andphosphorylate BECN1 at S93/S96 to enhance PIK3C3 activity upon glucose starvation [Citation11]. We showed that RASAL2 S351 phosphorylation was not involved in the PRKAA-BECN1 signaling axis, because RASAL2 depletion did not affect PRKAA binding with BECN1 and subsequent BECN1 S93/S96 phosphorylation (Figure S3H). This finding was further supported by an in vitro experiment mixing FLAG-PRKAA and GST-BECN1 with or without MYC-RASAL2 in the presence or absence of glucose (Figure S3I). The results showed that glucose starvation led to a promotion in PRKAA-BECN1 interaction and BECN1 phosphorylation. However, both of these values remained unchanged after RASAL2 was added in.
RASAL2 S351 phosphorylation promotes breast tumorigenesis and correlates with a poor prognosis in breast cancer patients
Tumor cells induce autophagy to help cell survival in response to cellular stress like nutrient deprivation and chemotherapeutic agents [Citation1]. We therefore investigated whether the RASAL2S351 phosphorylation has an effect on cell physiological response to glucose starvation. As expected, RASAL2 knockout dramatically inhibited cell proliferation under glucose starvation condition, while reconstituted expression of WT RASAL2 or RASAL2S351D, but not RASAL2S351A, attenuated this RASAL2-deficiency-induced cell death in both MCF-7 and MDA-MB-231 cells (). However, cell proliferation was largely unaffected in these cells under normal conditions, which was consistent with previously reported finding that cell proliferation was not affected significantly by reconstituting or suppressing RASAL2 in breast cancer cells () [Citation17,Citation21]. These results suggested the role of RASAL2 S351 phosphorylation in supporting breast tumor cell survival under the condition of glucose starvation. We further investigated the biological importance of RASAL2 S351 phosphorylation in tumorigenesis by subcutaneously injecting MCF7 and MDA-MB-231 cells with or without RASAL2 KO and reconstituted expression of MYC-RASAL2 (WT, S351D, S351A) into nude mice. The results showed that expression of RASAL2S351D significantly promoted tumor growth with increased autophagy, while RASAL2S351A greatly suppressed tumor growth and autophagy in both MCF7 and MDA-MB-231 cells (). These results indicated that RASAL2 S351 phosphorylation and subsequent autophagy activation plays an instrument role in breast tumor growth.
Figure 6. RASAL2 S351 phosphorylation promotes breast cancer growth and indicates a poor prognosis in breast cancer patients. (A and B) RASAL2-CT or RASAL2-KO MCF7 and MDA-MB-231 cells with reconstituted expression of MYC-RASAL2 WT, MYC-RASAL2351D and MYC-RASAL2351A were cultured in the present (A) or absent (B) of glucose-deprived medium for 24 h, and cell viability was determined by MTT assays. Results are reported as means ± SEM of three replicates. **p < 0.01, N.S., not significant. (C – E) RASAL2-CT and RASAL2-KO MCF7 or MDA-MB-231 cells stably expressing MYC-RASAL2 (WT, S351D, S351A) were subcutaneously injected into nude mice (n = 5/group). (C) Tumor volumes were calculated, and (D) representative tumors were photographed. (E) Tumor tissues in (C) were used for immunoblotting as indicated. Data represent the mean ± SEM of five mice. *p < 0.05, **p < 0.01. (F and G) IHC staining of breast tissue microarray (TMA) with antibody against RASAL2 S351 phosphorylationand PRKAA-T172 phosphorylation were performed. Brown staining indicates positive immune reactivity. Chi-squareanalysis was performed depending on the staining scores. (H) Kaplan-Meier curves showing the overall survival rates of breast cancer patients with high or low expression of p-S351-RASAL2. Statistical significance was calculated by using a log-rank test
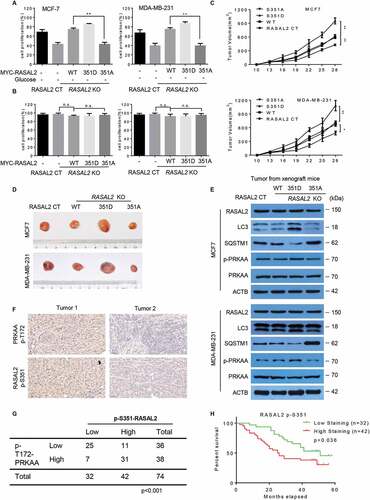
We next analyzed a breast tissue microarray (TMA) containing 74 human breast cancer specimens in various subtypes by using both PRKAAT172 and RASAL2 S351 antibodies and showed that the phosphorylation levels of PRKAA were statistically correlated with the phosphorylation levels of RASAL2 S351 (). Although RASAL2 expression level failed to display a significant correlation with overall survival in breast cancer patients [Citation21], we found that high RASAL2 S351 phosphorylation levels correlated with poor survival in these patients, with the median survival being 51 months for low p-S351-RASAL2 expression patients and 32.5 months for high p-S351-RASAL2 expression patients respectively (). Together, these results suggest that PRKAA-mediated RASAL2 S351 phosphorylation facilitates breast tumor growth and correlates with poor clinical outcomes in breast cancer patients.
Discussion
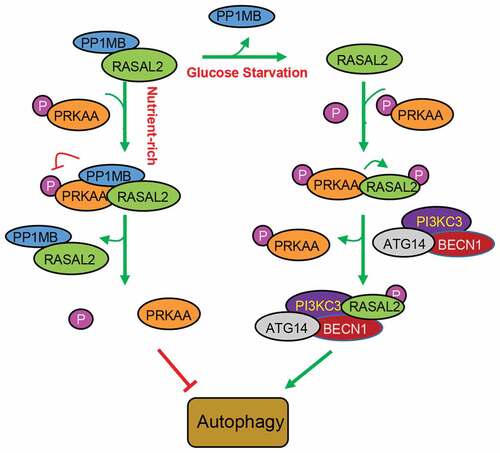
RASGAPs were recognized as tumor suppressors for their role in turning off RAS by catalyzing RAS-GTP hydrolysis, thus resulting in the inhibition of cell polarization, cell differentiation, cell migration and invasion of cancer cells [Citation12,Citation29,Citation30]. Recent studies showed that DAB2IP, a member of RAS GAPs, could inhibit autophagy initiation, indicating a critical role of RAS GAPs in the regulation of autophagy [Citation31,Citation32]. In this study, we demonstrated that RASAL2, one of the RASGAPs, could attenuate PRKAA-phosphorylation through PPM1B to keep phosphorylated AMPK at a low level, thus inhibiting the initiation of basal autophagy under the normal conditions. Additionally, RASAL2 is a direct substrate of PRKAA. Glucose deprivation could activate AMPK and released PPM1B from RASAL2. Activated PRKAA then phosphorylated RASAL2 at S351, which resulted in an increase in the binding of RASAL2 with PIK3C3 complex, as well as the interaction between PIK3C3 and its substrate PtdIns to increase PIK3C3 activity toward PtdIns3P production and autophagy (). These data provide strong evidence for the functional switch of RASAL2S351 phosphorylation, from suppressor to activator in AMPK-mediated autophagy.
Figure 7. Schematic model of RASAL2 regulation on autophagy. Under normal circumstances, the complex of RASAL2 andphosphorytase PP1MB (RASAL2/PP1MB) can bind to phosphorylated PRKAA (p-PRKAA), dephosphorylating PRKAA and thus keeping autophagy at a low level. Additionally, RASAL2 is a direct substrate of PRKAA. Glucose deprivation can activate AMPK and release PP1MB from RASAL2. Activated PRKAA then phosphorylates RASAL2 at S351, resulting in an increase in the binding of RASAL2 with PI3KC3 complex, as well as the interaction between PI3KC3 and its substrate PtdIns to increase PI3KC3 activity toward PtdIns3P production and autophagy
The role of RASAL2 in breast cancer seems to be context dependent. RASAL2 is an oncogenic protein and over-expressed in TNBC and ER-negative tumor cells, e.g. MDA-MB-231 cells, to induce mesenchymal invasion and metastasis. In contrast, the expression of RASAL2 is reduced and correlates with metastasis, recurrence and poor disease outcome in luminal B ER+ tumor cells like MCF-7 cells [Citation17,Citation21]. However, our results showed that the biological effects of RASAL2 on autophagy were the same in MCF7, MDA-MB-231 and lung cancer cell A549. In addition, glucose starvation dissociates PPM1B from RASAL2, leading to PRKAA phosphorylation of RASAL2 at S351 and subsequent binding with PIK3C3 core complex. Autophagy activation was also the same in MDA-MB-231 and MCF7 cells, suggesting a universal role of RASAL2 in autophagy.
The role of autophagy in cancer development is complicated [Citation33,Citation34]. Initially, autophagy was demonstrated to inhibit cancer development as depletion of autophagy-related genes in the mouse could result in the initiation of neoplasia [Citation35,Citation36]. In contrast, autophagy was found to be up regulated in the hypoxic region of tumors to promote tumor cell survival [Citation37]. Autophagy could also exert its cyto-protective role to help cancer cell survival from a variety of stresses induced by therapies such as chemotherapy and radiotherapy, and to develop therapeutic resistance [Citation38–40]. Elevated basal autophagy was demonstrated in various cancer types to promote tumor cell survival, metastasis and tumorigenesis, and the expression of autophagy markers in these cancers was associated with poor prognosis [Citation41–44]. In this study, we found that RASAL2-S351-phosphorylation-induced autophagy significantly promoted MCF-7 and MDA-MB-231 cell growth in xenograft mice and correlated with poor survival in breast cancer patients. These findings once again support the cyto-protective role of autophagy in connecting cellular energy state and cellular fate in response to stress. RASAL2 also has some non-autophagic functions like regulation of tumor angiogenesis and inhibition of cell invasion and migration [Citation14,Citation20,Citation45]. Therefore, it will be important to elucidate whether RASAL2 S351 phosphorylation can regulate these non-autophagic functions. To fully address these issues, RASAL2 S351A and S351D knock in mice would be a useful tool.
It is worth noting that the effect of RASAL2 as a tumor suppressive RASGAP protein is largely dependent on its functional RASGAP domain. However, as an oncogenic protein in TNBC and high-grade serious ovarian cancer, RASAL2 promoted tumor cell metastasis, which was irrelevant to its RASGAP activity [Citation17,Citation21]. In this study, we found that RASGAP activity is dispensable for RASAL2 function in autophagy, as the RASGAP activity–deficient RASAL2 mutant retained its activity to inhibit autophagy. This might be because the S351 phosphorylation site and RASGAP domain are far apart from each other. In this respect, the detailed structure of RASAL2 would be needed to fully understand this phosphorylation process.
AMPK functions as a metabolic tumor suppressor and can oppose tumor growth by disrupting gene expression involved in cellular metabolism and regulating several well-established tumor suppressors such as p53, LKB1 and TSC2 [Citation46]. Various AMPK-activation agents like metformin, aspirin and flavones have been developed for cancer treatment and prevention [Citation47]. However, these AMPK-activation agents might also induce autophagy, which exerts a cyto-protective role to help cancer cells develop resistance and cause undesired survival [Citation48,Citation49]. In this study, we showed the activation of PRKAA could phosphorylate RASAL2 at S351 to induce cyto-protective autophagy, which in turn promoted survival of these cells. These findings suggest that activation of AMPK while prevent RASAL2 S351 phosphorylation may be a potential therapeutic strategy to overcome the resistance of these AMPK-activation agents.
In summary, our study revealed the phosphorylation status could switch the function of RASAL2 from a suppressor to an activator in AMPK-mediated autophagy. We also highlighted that the importance of RASAL2 S351 phosphorylation in promoting breast tumorigenesis and correlation with a poor prognosis in breast cancer patients. An enhanced understanding of RASAL2 S351 phosphorylation might help establish a potential therapeutic strategy to overcome the resistance of AMPK-activation agents and lead to personalized treatment for breast cancer.
Materials and methods
Reagents and antibodies
Compound C (C.C; 171261), ATP (20–306), chloroquine (CQ) (C6628), AICAR (A9978), MYC-beads (A7470), M2 beads (M8823) and anti–HIS antibodies (SAB1306082) were bought from Sigma-Aldrich; glutathione-sepharose 4B beads (17075601) from GE Healthcare Life Sciences; and [3H]-phosphatidylinositol (ART0184) from American Radiolabeled Chemicals. Antibodies including LC3 (3868), SQSTM1/p62 (23214), PIK3C3/VPS34 (for WB) (4263), phospho-BECN1/Beclin1 S93 (14717), BECN1/Beclin1 (3495), phospho-PRKAA/AMPKαT172 (2535S), PRKAA/AMPKα (5831S) and anti-phospho-tyrosine (9411) were purchased from Cell Signaling Technology; anti-phospho-serine/threonine (PP2551) from ECM Biosciences; GST (sc-138), MYC (sc-40), ACTB/β-actin (sc-47778), protein A/G agarose (sc-2003), rabbit and mouse IgG (sc-2027 and sc-2025) from Santa Cruz Biotechnology; PPM1B/Pp2cβ (ab70804) from Abcam, RASAL2 antibody (NBP1-82579) from Novus Biologicals and ATG14 (PD026) antibody from MBL International Corporation. Anti-PIK3C3/VPS34 (Echelon, Z-R015) antibody was used for immunoprecipitation. RASAL2 S351 phospho-specific antibody was custom produced by Youke Biosciences Shanghai, China.
Cell culture, transfection, and treatment
293 T cells (HCL4517) were obtained from GE life sciences; and MCF7 (HTB-22), MDA-MB-231 (HTB-26) and A549 (CCL-185) cells from the American Type Culture Collection (ATCC). 293 T, MCF7, A549 cells and MDA-MB-231 were maintained in DMEM (Dulbecco’s modified eagle medium) (Gibco; 11965092) supplemented with 10% FBS (fetal calf serum) and 100 U/ml penicillin-streptomycin. The detailed methods of transfections were performed as described previously [Citation50]. Glucose-free DMEM (11966025), Hanks’ buffer (with calcium and glucose) (14025076), EBSS (Earle’s Balanced Salt Solution) (24010043), and dialyzed FBS (30067334) were purchased from Thermo Scientific. Transient transfections were performed using Lipofectamine 2000 (Invitrogen; 11668030) according to the manufacturer’s instructions. The following siRNA duplexes were used: RASAL2 siRNA-GCCUGAAAGGCUCCAUCAA; for glucose starvation, cells were cultured in the same medium mentioned above expect using glucose-free DMEM. EBSS treatment was prepared by replacing the medium to EBSS. Amino acid starvation was prepared by replacing the medium to Hanks’ buffer supplemented with 10% dialyzed FBS and 1% HEPES (Invitrogen,15630106). The chemicals were used as follow: AICAR, 0.5 mM, 2 h; compound C, 20 μM, 4 h and chloroquine, 50 μM, 4 h.
Plasmids and mutagenesis
Human RASAL2 (NM_170692.2), PRKAA/AMPKα (NM_006252.3), ATG14 (NM_014924.4) and PPM1B/pp2cβ (NM_002706.5) were generated by PCR and cloned into pcDNA3.1 N-flag (Addgene, 52535; deposited by Adam Antebi) or MYC-his-pcDNA3.1 vector (Invitrogen, V80020). GFP-RFP-LC3 was purchased from Addgene (21074; deposited by Tamotsu Yoshimori). FLAG-PIK3C3, MYC-BECN1, MYC-RASAL2-GAP-Mut and MYC-RASAL2-ΔGAP constructs were developed as previously described [Citation21,Citation51]. RASAL2, PRKAA and PPM1B point mutations were generated by PCR based site-directed mutagenesis. For prokaryotic expression and purification, RASAL2 and PPM1B were inserted into the bacterial expression construct (pGEX-5X) or (pET30a). Primers were list in Table S3.
CRISPR-Cas9-mediated knockout cell lines
Stable knockout cell lines were generated by retroviral infection of lentiCRISPRv2 plasmid or/and PX458 plasmid purchased from Addgene as reported previously [Citation52]. The following gRNA sequences were: RASAL2 gRNA 5ʹ-CCTGAGATACTCCGTCTACG-3ʹ, PPM1B gRNA 5ʹ- CCGAATCGGCAACGGCGCTA −3ʹ. The RASAL2/PPM1B double-knockout cell line or AMPKα1/α2 DKO cell line were generated by using a lentiCRISPRv2-sgRNA-RASAL2 plasmid (5′-CCTGAGATACTCCGTCTACG-3′) and a PX458-sgRNA-PPM1B plasmid (5′-CCGAATCGGCAACGGCGCTA-3′) or a lentiCRISPRv2-sgRNA-PRKAA1 plasmid (5′-GGCTGTCGCCATCTTTCTCC-3′) and a PX458-sgRNA-PRKAA2 plasmid (5′-GTCAGCCATCTTCGGCGCGC-3′) respectively. Stable cell lines were established by selection with puromycin or/and GFP.
GFP-RFP-LC3 and GFP-FYVE reporter assay
GFP-RFP-LC3 and GFP-FYVE (Addgene, 140047; deposited by Harald Stenmark) reporter assays were performed as described previously [Citation50]. Briefly, cells transfected with GFP-RFP-LC3 or GFP-FYVE plasmids were cultured on gelatinized coverslips for 48 h. The fluorescence of GFP-RFP-LC3 or GFP-FYVE puncta was obtained by using a FV3000 Olympus confocal microscope (Olympus, Tokyo, Japan). Autophagy was determined by counting the numbers of puncta in 50 cells with a 60× objective.
Transmission electronmicroscopy
Transmission electron microscopy (TEM) was performed as described previously [Citation53]. Briefly, RASAL2-CT and RASAL2-KO MCF7 cells were fixed in 0.1 M sodium cacodylate buffer (pH 7.2) containing 2.5% glutaraldehyde and 2% paraformaldehyde for 4 h and post-fixed with 1% osmium tetroxide for 1.5 h at room temperature. After processing a standard manner and embedding in EMbed 812 (Electron Microscopy Sciences,14900), the ultrathin sections (60 nm) were stained with 2% uranyl acetate and 0.3% lead citrate by standard methods [Citation54]. Stained grids were examined under a Philips CM-12 electron microscope (FEI) and photographed with a Gatan (4kX2.7k) digital camera (Gatan).
Co-immunoprecipitation and mass spectrometry analysis
Immunoprecipitation of FLAG-tag, HIS-tag, GST-tag, MYC-tag or endogenous proteins were performed as described [Citation51] for mass spectrometry analysis. To determine the RASAL2 binding proteins, immunoprecipitated MYC-RASAL2 was from RASAL2-KO MCF7 cells transfected with MYC-RASAL2 in the presence or absence of glucose deprived medium for 4 h. To determine the phosphorylation site of RASAL2, immunoprecipitated MYC-RASAL2 was from PRKAA1/PRKAA2-DKO MCF7 cells co-transfected MYC-RASAL2 with or without FLAG-PRKAA were cultured in the glucose-starvation medium for 4 h. All the immunoprecipitates were washed and separated on SDS-PAGE gels. Protein were than stained with silver or coomassie blue and distinct bands were excised from the gel and subjected to mass spectrometry analysis as described previously [Citation55].
In vitro PIK3C3 lipid kinase assay
The PIK3C3 lipid kinase assay was examined by using the class III ELISA kit according to the manufacture (Echelon Biosciences; K-3000). Briefly, ATG14 protein complex were enriched by using ATG14 antibody, and then mixed with PI (phosphatidylinositol) substrates in kinase reaction buffer for 2 h at 37°C. After quench the reactions by adding EDTA, products were diluted and incubated with PtdIns3P detector (provided by the kit) in PtdIns3P-coated microplate for competitive binding to the PtdIns3P detector. The amount of PtdIns3P detector protein bound to the plate was determined through absorbance at 450 nm on a plate reader. The concentration of PtdIns3P in the reaction mixture was calculated as reversed to the amount of PtdIns3P detector protein bound to the plate.
Purification of recombinant proteins
HIS-RASAL2, GST-PRKAA, GST-BECN1,GST-PPM1B and GST-PPM1BR179G were expressed in BL21 (DE3) E. coli cells (BioFeng, 8618) by induction with 0.1 mM isopropyl thiogalactoside (Sigma-Aldrich, I6758) for 20 h at 30°C and affinity purified with Ni-NTA resin (QIAGEN,30210) or glutathione-sepharose resin according to the manufacturer’s protocol. MYC-RASAL2 was prepared from the 293 T cells transfected with MYC-RASAL2 and treated with glucose starvation for 4 h and then eluted from the MYC beads by adding an excess amount of MYC peptide (Sigma-Aldrich, M2435).
In vitro AMPK phosphorylation assay
In vitro kinase assays were performed as described [Citation28,Citation56]. For phosphorylation of RASAL2:purified FLAG-PRKAA was prepared from the 293 T cells transfected with FLAG-PRKAA and incubated with glucose starvation for 4 h and eluted from the M2 beads by adding the excess amount of 3xFLAG peptide. One µg of purified FLAG-PRKAA complex and 0.5 µg purified HIS-RASAL2 or HIS-RASAL2S351A were mixed in the reaction buffer (40 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 0.1 µg/µl BSA (Sigma-Aldrich, V900933) and 0.2 mM AMP (Selleck, S9366) for 20 min at 30°C. The reaction was initiated by addition of non-radioactive ATP and terminated by addition of SDS sample loading buffer. The reaction mixture was loaded onto SDS-PAGE, and the phosphorylation state was analyzed by western blot using RASAL2 phospho-S351 antibody. For phosphorylation of BECN1:purified FLAG-PRKAA prepared from 293 T cells transfected with FLAG-PRKAA before treatment with or without glucose starvation for4h. After immunoprecipitation using anti-FLAG (M2) beads, FLAG-PRKAA proteins were washed and incubated with purified GST-BECN1 protein in the presence or absence of purified MYC-RASAL2 and nonradioactive ATP in the reaction buffer for 20 min at 30°C.
In vitro PPM1B dephosphorylation assay
Purified FLAG-PRKAA was prepared from 293 T cells transfected with FLAG-PRKAA before treated with AICAR for 2 h. After immunoprecipitation by using anti-FLAG (M2) beads, FLAG-PRKAA proteins were washed and incubated with GST-PPM1B or its mutation in the present or absent of HIS-RASAL2 in kinase buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 1 mg/ml BSA and complete protease inhibitors) at 30°C for 30 min [Citation57].
Phosphatidylinositol binding ability assay
PtdIns-PIK3C3 binding ability assay was performed as described previously [Citation58]. In brief, immunoprecipitated ATG14-containing PIK3C3 complex, immobilized on protein A/G-agarosebeads, was washed 3 times by using washing buffer (100 mM NaCl, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA). The beads were then incubated in 60 μl of reaction buffer containing 10 mM MnCl2, 10 μl of 440 μM ATP (Sigma-Aldrich, A6419), and 10 μl of 2 μg/μl phosphatidylinositol (Sigma-Aldrich, P6636) containing 1 μCi of [3H]-phosphatidylinositol (American Radiolabeled Chemicals, ART0184) for 30 min. The beads were then washed 3 times by washing buffer and detected via liquid scintillation counting.
Cell proliferation assay
Cell Proliferation Assay was performed as described previously [Citation50]. Briefly, 2 × 104 cells were seeded in 96-well plates and treated with or without glucose-starvation medium for 24 h. Then, 20 μl of Cell Titer-Blue reagent (Promega, G8081) was added to each well, shaken for 10s, and incubated at 37°C for 4 h. Fluorescence was measured at an excitation wavelength of 579 nm and emission of 584 nm.
Xenograft tumor growth
To asses tumor growth in vivo, female nude mice (six-week-old) were injected subcutaneously with 1.5 × 106 tumor cells (n = 5 for each group) as previously described [Citation50]. Tumor size was measured at regular intervals and tumor volume was determined as volume = (width)2 x length/2. All animal procedures and maintenance were conducted in accordance with the protocols approved by the Institutional Animal Care and Use Committees at Nantong University and Fudan University.
IHC for tissue microarrays
Breast cancer tissue microarray slides (ZL-BRCsur1221) were purchased from Shanghai Ouwei Biotech Co. Ltd. (SOBC, Shanghai, China). All patients gave consent for the use of their tissue samples and clinical data. This study was approved by the Institutional Review Board at Huashan Hospital, Fudan University in Shanghai, China. Immunohistochemical staining of tissue microarray was carried out previously described protocols [Citation59]. Briefly, TMA were deparaffinized and rehydrated and treated with 1% hydrogen peroxide to inactive peroxidase activity. After antigen retrieval and blocking, the samples were incubated with primary antibodies, biotinylated secondary antibody and avidin-biotin peroxidase complex solution and were developed with the 3-amino-9-ethylcarbazole solution. H score was determined according to the percentage of positive tumor cells and the staining intensity. The correlation between AMPKα phosphorylation and RASAL2S351 phosphorylation were determined by using the χ2 test. Overall survival curves were calculated by the Kaplan–Meier method and the rate were compared by using the log-rank test.
Statistical analyses
All results were expressed as means ± SEM of three independent experiments. Statistical analyses were performed using student’s t-test, one-way ANOVA or two-way ANOVA by Graphpad Prism 6.07 software. P < 0.05 was considered significant.
Availability of data and materials
All data generated or analyzed during this study are included either in this article or in the supplementary information files.
Ethics approval and consent to participate
The study was approved by the Institutional Animal Care and Use Committee of Nantong University in Nantong and Institutional Review Board of Huashan Hospital (NO. NS81874146) of Fudan University in Shanghai, China.
Supplemental Material
Download Zip (6.3 MB)Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410.
- Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. Bioessays. 2013;35(1):34–45.
- Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276.
- Matsunaga K, Morita E, Saitoh T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190(4):511–521.
- Funderburk SF, Wang QJ, The Beclin YZ. 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362.
- Kim J, Guan KL. AMPK connects energy stress to PIK3C3/VPS34 regulation. Autophagy. 2013;9(7):1110–1111.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750.
- Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226.
- Hardie DGAMP. K and autophagy get connected. Embo J. 2011;30(4):634–635.
- Kalender A, Selvaraj A, Kim SY, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401.
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303.
- Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul. 2014;55:1–14.
- Huang Y, Zhao M, Xu H, et al. RASAL2 down-regulation in ovarian cancer promotes epithelial-mesenchymal transition and metastasis. Oncotarget. 2014;5(16):6734–6745.
- Hui K, Gao Y, Huang J, et al. RASAL2, a RAS GTPase-activating protein, inhibits stemness and epithelial-mesenchymal transition via MAPK/SOX2 pathway in bladder cancer. Cell Death Dis. 2017;8(2):e2600.
- Jia Z, Liu W, Gong L, et al. Downregulation of RASAL2 promotes the proliferation, epithelial-mesenchymal transition and metastasis of colorectal cancer cells. Oncol Lett. 2017;13(3):1379–1385.
- Li N, Li S. RASAL2 promotes lung cancer metastasis through epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;455(3–4):358–362.
- McLaughlin SK, Olsen SN, Dake B, et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell. 2013;24(3):365–378.
- Min J, Zaslavsky A, Fedele G, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16(3):286–294.
- Wang Z, Wang J, Su Y, et al. RASAL2 inhibited the proliferation and metastasis capability of nasopharyngeal carcinoma. Int J Clin Exp Med. 2015;8(10):18765–18771.
- Weeks A, Okolowsky N, Golbourn B, et al. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am J Pathol. 2012;181(2):662–674.
- Feng M, Bao Y, Li Z, et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J Clin Invest. 2014;124(12):5291–5304.
- Pan Y, Tong JHM, Lung RWM, et al. RASAL2 promotes tumor progression through LATS2/YAP1 axis of hippo signaling pathway in colorectal cancer. Mol Cancer. 2018;17(1):102.
- Klionsky DJ. A human autophagy interaction network. Autophagy. 2012;8(4):439–441.
- Egan D, Kim J, Shaw RJ, et al. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–644.
- Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK activation: role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev. 2016;28:15–26.
- Moore F, Weekes J, Hardie DG. Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem. 1991;199(3):691–697.
- Kusuda K, Kobayashi T, Ikeda S, et al. Mutational analysis of the domain structure of mouse protein phosphatase 2Cbeta. Biochem J. 1998;332(Pt 1):243–250.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141.
- Fukata M, Kuroda S, Nakagawa M, et al. Cdc42 and Rac1 regulate the interaction of IQGAP1 with beta-catenin. J Biol Chem. 1999;274(37):26044–26050.
- Bourguignon LY, Gilad E, Rothman K, et al. Hyaluronan-CD44 interaction with IQGAP1 promotes Cdc42 and ERK signaling, leading to actin binding, Elk-1/estrogen receptor transcriptional activation, and ovarian cancer progression. J Biol Chem. 2005;280(12):11961–11972.
- Liao H, Xiao Y, Hu Y, et al. microRNA-32 induces radioresistance by targeting DAB2IP and regulating autophagy in prostate cancer cells. Oncol Lett. 2015;10(4):2055–2062.
- Yu L, Tumati V, Tseng SF, et al. DAB2IP regulates autophagy in prostate cancer in response to combined treatment of radiation and a DNA-PKcs inhibitor. Neoplasia. 2012;14(12):1203–1212.
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930.
- Nyfeler B, Eng CH. Revisiting autophagy addiction of tumor cells. Autophagy. 2016;12(7):1206–1207.
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820.
- Yue Z, Jin S, Yang C, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082.
- Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64.
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–666.
- Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1–11.
- Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838.
- Espina V, Mariani BD, Gallagher RI, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One. 2010;5(4):e10240.
- Galavotti S, Bartesaghi S, Faccenda D, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32(6):699–712.
- Li J, Yang B, Zhou Q, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34(6):1343–1351.
- Whelan KA, Chandramouleeswaran PM, Tanaka K, et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene. 2017;36(34):4843–4858.
- Hui K, Wu S, Yue Y, et al. RASAL2 inhibits tumor angiogenesis via p-AKT/ETS1 signaling in bladder cancer. Cell Signal. 2018;48:38–44.
- Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–37.
- Li W, Saud SM, Young MR, et al. Targeting AMPK for cancer prevention and treatment. Oncotarget. 2015;6(10):7365–7378.
- Wang Y, Yang Z, Zheng G, et al. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKα1 and nuclear AMPKα2 pathways. Life Sci. 2019;225:64–71.
- Wu H, Ding J, Li S, et al. Metformin promotes the survival of random-pattern skin flaps by inducing autophagy via the AMPK-mTOR-TFEB signaling pathway. Int J Biol Sci. 2019;15(2):325–340.
- Jiang X, Bao Y, Liu H, et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene. 2017;36(50):6850–6862.
- Yang Y, Fiskus W, Yong B, et al. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A. 2013;110(17):6841–6846.
- Imam S, Talley S, Nelson RS, et al. TRIM5α degradation via autophagy is not required for retroviral restriction. J Virol. 2016;90(7):3400–3410.
- Ylä-Anttila P, Vihinen H, Jokitalo E, et al. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–164.
- Bao Y, Ding Z, Zhao P, et al. Autophagy inhibition potentiates the anti-EMT effects of alteronol through TGF-β/Smad3 signaling in melanoma cells. Cell Death Dis. 2020;11(4):223.
- Chang C, Su H, Zhang D, et al. AMPK-dependent phosphorylation of GAPDH Triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell. 2015;60(6):930–940.
- Zhao Y, Wang Q, Qiu G, et al. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 2015;13(7):1407–1417.
- Tasdelen I, van Beekum O, Gorbenko O, et al. The serine/threonine phosphatase PPM1B (PP2Cβ) selectively modulates PPARγ activity. Biochem J. 2013;451(1):45–53.
- Qian X, Li X, Cai Q, et al. Phosphoglycerate kinase 1 phosphorylates beclin1 to induce autophagy. Mol Cell. 2017;65(5):917–931.e6.
- Su D, Fu X, Fan S, et al. Role of ERRF, a novel ER-related nuclear factor, in the growth control of ER-positive human breast cancer cells. Am J Pathol. 2012;180(3):1189–1201.