
ABSTRACT
The WDR45 gene is localized on the X-chromosome and variants in this gene are linked to six different neurodegenerative disorders, i.e., ß-propeller protein associated neurodegeneration, Rett-like syndrome, intellectual disability, and epileptic encephalopathies including developmental and epileptic encephalopathy, early-onset epileptic encephalopathy and West syndrome and potentially also specific malignancies. WDR45/WIPI4 is a WD-repeat β-propeller protein that belongs to the WIPI (WD repeat domain, phosphoinositide interacting) family. The precise cellular function of WDR45 is still largely unknown, but deletions or conventional variants in WDR45 can lead to macroautophagy/autophagy defects, malfunctioning mitochondria, endoplasmic reticulum stress and unbalanced iron homeostasis, suggesting that this protein functions in one or more pathways regulating directly or indirectly those processes. As a result, the underlying cause of the WDR45-associated disorders remains unknown. In this review, we summarize the current knowledge about the cellular and physiological functions of WDR45 and highlight how genetic variants in its encoding gene may contribute to the pathophysiology of the associated diseases. In particular, we connect clinical manifestations of the disorders with their potential cellular origin of malfunctioning and critically discuss whether it is possible that one of the most prominent shared features, i.e., brain iron accumulation, is the primary cause for those disorders.
Abbreviations: ATG/Atg: autophagy related; BPAN: ß-propeller protein associated neurodegeneration; CNS: central nervous system; DEE: developmental and epileptic encephalopathy; EEG: electroencephalograph; ENO2/neuron-specific enolase, enolase 2; EOEE: early-onset epileptic encephalopathy; ER: endoplasmic reticulum; ID: intellectual disability; IDR: intrinsically disordered region; MRI: magnetic resonance imaging; NBIA: neurodegeneration with brain iron accumulation; NCOA4: nuclear receptor coactivator 4; PtdIns3P: phosphatidylinositol-3-phosphate; RLS: Rett-like syndrome; WDR45: WD repeat domain 45; WIPI: WD repeat domain, phosphoinositide interacting
WDR45/WIPI4
The human WDR45 gene is located on the X-chromosome [Citation1] and is ubiquitously expressed in all tissues [Citation2,Citation3]. Spatial expression profiling in mice brains revealed that WDR45 is more abundantly expressed in the hippocampus, a region involved in learning and memory, compared to the rest of the brain [Citation4]. Human WDR45 encodes for a protein of 360 amino acids, which is composed of a N-terminal β-propeller followed by a short intrinsically disordered region (IDR) (Figure S1A). β-propellers are structural motifs that are present in numerous proteins and are mostly involved in protein-protein interactions [Citation5,Citation6]. They are built of four to eight blades/β-sheets, consisting of a four-stranded antiparallel β-strands. These blades are packed in a circular, ring-like array. β-propellers are often composed by the so-called WD-repeats, although not all of them contain this amino acid sequence motif [Citation7,Citation8]. The WD-repeat was originally defined as a core unit of approximately 40 amino acids, ending with tryptophan (W) and aspartic acid (D) residues [Citation9]. WD-repeat-containing proteins form a large protein family that is present in all eukaryotes. They are involved in a variety of functions, including transcription regulation, signal transduction, cell cycle control, autophagy and apoptosis [Citation7]. WDR45 is one of the WD-repeat proteins [Citation5,Citation10]. The WD-repeats of WDR45 are bracketed by glycine-histidine and tryptophan-aspartic acid pairs, and are predicted to fold into a 7-bladed β-propeller (Figure S1A-B) [Citation5]. Although the crystal structure of WDR45 is not resolved yet, based on the structure of some of its human and yeast homologs (see below) that have been solved [Citation11–14], the overall conformation of WDR45 can be predicted with accuracy (Figure S1B).
WDR45 is highly conserved in mammals, and the amino acid sequences of the human, mouse, pig, bovine and horse protein have more than 97% identity [Citation15]. Together with WIPI1, WIPI2 and WIPI3/WDR45B, it forms the WIPI (WD repeat domain, phosphoinositide interacting) protein family [Citation9]. This is why WDR45 is also known as WIPI4. Like WDR45, all WIPI proteins have 7 WD repeats and can be grouped into two clades according to their amino acid sequence identity. One clade contains WIPI1 and WIPI2, and the other WDR45B and WDR45 (Figure S1C) [Citation15]. The first blade and the C-terminal IDR are the regions that are most divergent between the members from the two clades (Figure S1D). Consequently, the amino acid sequence identity and similarity between the members of the different clades are both less than 31%, while the sequence identity and similarity between human WDR45 and WDR45B is 46.0% and 59.8%, respectively, and between WIPI1 and WIPI2 is 53.8% and 68.1%, respectively. These characteristics also explains the redundant roles in autophagy between the members of the same clade. A common characteristic of all WIPI proteins is the F/LRRG motif that allows them to bind to phosphoinositides, and therefore WIPI proteins are also known as β-propellers that bind polyphosphoinositides (PROPPINs) [Citation16]. This motif is localized in blade 6 (Figure S1D) and mediates their specific binding to phosphatidylinositol-3-phosphate (PtdIns3P) and phosphatidylinositol-3,5-biphosphate [Citation3,Citation15,Citation17].
WIPI proteins have homologs in all eukaryotic species and their number varies in between organisms [Citation3,Citation18]. For example, yeast Saccharomyces cerevisiae possesses 3 WIPI proteins (Atg18, Atg21 and Hsv2), while Caenorhabditis elegans has 2 (APG-18 and EPG-6) [Citation19–21]. Phylogenic analyses have shown that the majority of the WIPI protein homologs fall in one of the two claves described above [Citation3,Citation18], probably reflecting different conserved molecular functions.
The role of WDR45 and the other WIPI proteins in autophagy
WIPI proteins are PtdIns3P-binding effectors that are part of the ATG machinery [Citation10]. Autophagy is an evolutionary conserved process essential to maintain cellular homeostasis, in which cell components such as dysfunctional and/or aggregated proteins, damaged or redundant organelles, and other cytoplasmic material, are degraded in lysosomes for the subsequent recycling of the resulting metabolites () [Citation22,Citation23]. Autophagy is maintained at a basal level in most cells and is induced by nutrient deprivation and other stresses [Citation22,Citation23]. This process is characterized by the maturation of a transient membranous structure known as the phagophore, into a double-membraned vesicle, called the autophagosome, which then fuses with lysosomes to release its cytoplasmic cargo into the hydrolytic interior of this organelle [Citation22,Citation23]. This process is regulated by ATG (autophagy-related) proteins, which are organized in functional complexes [Citation22,Citation23]. One of them, is the class III phosphatidylinositol 3-kinase complex, which comprises BECN1, ATG14, PIK3R4/VPS15, PIK3C3/VPS34 and NRBF2, and catalyzes the local synthesis of PtdIns3P on the phagophore membrane [Citation23–25]. PtdIns3P has an important function in the formation of autophagosomes as it acts as a scaffold for the recruitment of specific components of the ATG machinery [Citation23,Citation24]. The expansion and eventual closure of the phagophore to form an autophagosome depends on conjugation of members of the MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) protein family to phosphatidylethanolamine, a process that involves two ubiquitin-like conjugation systems [Citation23]. First, the E1 enzyme-like ATG7 and the E2 enzyme-like ATG10 lead to the formation of the ATG12–ATG5-ATG16L1 complex. Second, the E1 enzyme-like ATG7 and the E2 enzyme-like ATG3, guided by the ATG12–ATG5-ATG16L1 complex, promote the formation of the conjugates between LC3 proteins and phosphatidylethanolamine on the phagophore membrane [Citation23,Citation26].
Figure 1. The autophagy pathway and the role of the WIPI proteins. Autophagy is an evolutionary conserved process essential to maintain cellular homeostasis. Autophagy is characterized by the development of a transient membranous cisterna known as the phagophore, into a double-membraned vesicle, called the autophagosome, which then fuses with lysosomes to release its cargo into the hydrolytic interior of this organelle. The process of autophagosome formation and maturation is regulated by autophagy-related (ATG) proteins. While WIPI1 and WIPI2 play a key role during the early stages of the biogenesis of an autophagosome, WDR45 and WDR45B have an upstream function, later during the autophagosome formation
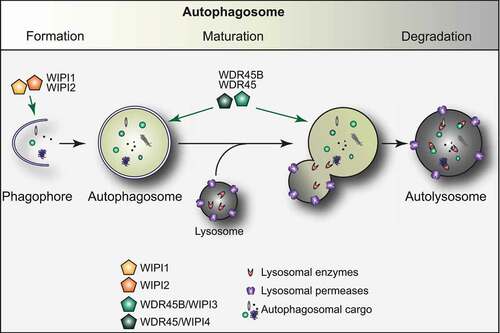
WIPI proteins participate to both the biogenesis and maturation of autophagosomes () [Citation10]. The members of the two different evolutionary clades, i.e., WIPI1/WIPI2 and WDR45B/WDR45, function differently in these two steps of autophagy [Citation15,Citation21,Citation27]. WIPI2 has been shown to connect PtdIns3P production with the recruitment of the ATG12–ATG5-ATG16L1 complex for subsequent LC3 lipidation at the phagophore [Citation15], with ATG16L1 directly binding to WIPI2 [Citation28]. In addition, WIPI2 interacts with RAB11A at early stages of autophagosome formation [Citation29]. WIPI1 also acts at early steps of autophagy [Citation30] and interacts with WIPI2, possibly supporting WIPI2 in ATG16L1 recruitment for LC3 protein lipidation [Citation27,Citation31]. The notion that WIPI1 and WIPI2 have a function different from WDR45 (and WDR45B), i.e., act at a different step of autophagosome formation, is supported by the observation that depletion of WIPI1 or WIPI2 suppresses the formation of LC3 puncta, while downregulation of WDR45B or WDR45 enhances their number. This suggests that WIPI1 and WIPI2 are mainly involved in the formation of phagophores while WDR45B and WDR45 are principally participating in phagophore expansion and/or closure, or even autophagosome maturation [Citation15,Citation21,Citation27]. Thus, WDR45 and WDR45B, mainly function downstream of WIPI1 and WIPI2, and possibly control the size and maturation of nascent autophagosomes [Citation27]. In C. elegans, mutants of atg-18, the WIPI1 and WIPI2 homolog, and epg-6, the WDR45B and WDR45 counterpart, show distinct phenotypes in autophagy with ATG-18 functions upstream of EPG-6 [Citation21], confirming a different function of those proteins in autophagy.
Genetic studies in mice have revealed that the central nervous system (CNS)-specific wdr45−/–deficient mice display an impaired autophagy flux with accumulation of SQSTM1/p62- and ubiquitin-positive protein aggregates in neurons () [Citation2]. The autophagy defect in wdr45-/- mice, however, is weaker compared to mice lacking core Atg genes, which may indicate that the autophagy impairment is restricted to neurons [Citation2]. Similar to the conditional wdr45-/- animals, full body wdr45b-/- mice also show an autophagy defect [Citation4], however, the affected brain regions are not overlapping, i.e., defects were most obvious in thalamus, cortex and hippocampus for wdr45-/- animals while the cerebellum was mostly affected in the wdr45b-/- mice, indicating that these genes are required for autophagy in different brain regions. Interestingly, wdr45b-/- wdr45-/- double deficient mice exhibit a more severe autophagy defect in the brain than single knockout animals and moreover, die within a day after birth similar to mice lacking core Atg genes [Citation4]. These observations also support the notion that WDR45B and WDR45 function redundantly, and that these two proteins are critical for neural homeostasis.
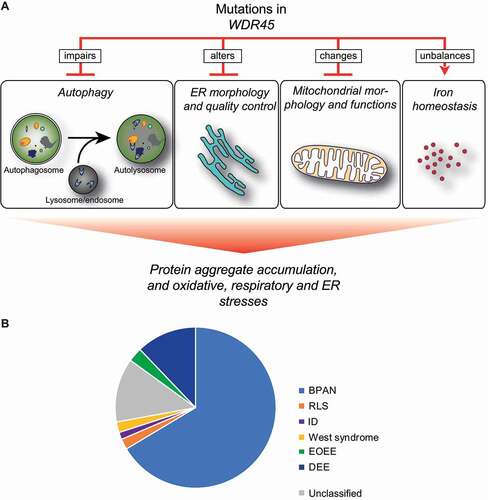
Figure 2. Cellular functions that are altered by mutations in WDR45 and WDR45 variant distribution in between diseases. (A) Effects of WDR45 mutations on cellular functions. At cellular level, WDR45 mutations cause autophagy impairment, alterations of ER morphology and quality control, perturbation of mitochondria morphology and function, and unbalances in iron homeostasis. These impairments result in protein aggregates accumulation, and enhanced oxidative, respiratory and ER stresses. (B) WDR45 variants found in BPAN, RLS, ID, EOEE, DEE and West syndrome patients, but also that have not been associated to a specific disease
Although there are not too many functional lines of evidence to explain the involvement of WDR45 in autophagy, it has been very well established that this protein interacts with ATG2A and ATG2B [Citation27,Citation32–36], stronger than do other WIPI proteins [Citation32]. ATG2A and ATG2B have redundant functions that are essential for autophagy progression [Citation37]. Binding between ATG2A and WDR45 has been suggested to be mediated by Asp1376, Glu1378 and Lys1539 of ATG2A, and Lys89 and Lys134 of WDR45 [Citation32,Citation34] and via a WIPI-interacting-region (WIR)-motif present in ATG2A [Citation38]. Mechanistically, this latter motif unwinds and wraps around WDR45 (or other WIPI proteins) and binds to three sites on three different propeller blades [Citation38]. Two other residues in WDR45, Asn15 and Asp17, may also mediate WDR45 binding to ATG2A [Citation27]. Crucially, it has been shown that ATG2 proteins are able to transfer phospholipids between adjacent liposomes [Citation35,Citation36,Citation39] and binding of WDR45 to ATG2A or ATG2B accelerate this lipid transfer activity [Citation36,Citation40]. Since the ATG2A-WDR45 complex tethers membranes in vitro and probably in vivo [Citation34,Citation41], it might well be that it is involved in the transport of lipids from the ER to one or more other subcellular compartments, including nascent autophagosomes.
The functional relevance in autophagy of the ATG2A-WDR45 and ATG2B-WDR45 complexes remains to be fully established, especially because WDR45 appears not to play a similar major role in autophagy as the ATG2 proteins [Citation42]. Although, ATG2A and ATG2B are required for phagophore formation during autophagy [Citation37], their interaction with WDR45 is dispensable for phagophore formation and therefore not required for early autophagy events [Citation42]. However, mutations in the WIR-motif in ATG2A could not rescue the autophagy defect phenotype of ATG2A-depleted cells, indicating that this complex is required for autophagy [Citation38].
WDR45 also forms a larger complex that contains ATG2A, ULK1 and 5ʹ AMP-activated protein kinase (AMPK) under nutrient-rich conditions. When autophagy is induced or AMPK is specifically activated, WDR45 and ATG2A dissociate from this complex and more WDR45-positive punctate structures are formed [Citation27,Citation31]. Thus, autophagy induction leads to the activation and translocation of ATG2A-WDR45 possibly onto phagophore membranes, contributing to autophagosome formation [Citation31]. Importantly, under autophagy-induced conditions, WDR45 localizes with WDR45B, which is also found on lysosomal structures, indicating that WDR45-ATG2A might also have a role in autophagosome maturation [Citation27]. It remains unclear whether the ATG2B-WDR45 is regulated in a similar manner.
WDR45 functions in other cellular pathways
Increasing evidence shows that WDR45 also functions in other important cellular pathways, such as endoplasmic reticulum (ER) homeostasis or mitochondria organization (). In full body wdr45-/- mice, large amounts of ER proteins significantly accumulate in the brain. This accumulation is caused by a reduced ability in degrading ER proteins via the proteasome and lysosome, resulting in ER expansion, elevated ER stress and eventually cell death [Citation43]. By treating wdr45-/- neurons or WDR45-depleted HeLa cells with the autophagy inducer rapamycin or by reducing ER stress, those cells were rescued form undergoing apoptosis, indicating that autophagy induction can compensate to a certain degree the cellular insults caused by WDR45 depletion. Although, this result suggests that WDR45 is involved in ER quality control, the ER stress in wdr45-/- mice was only observed in old animals, whereas the autophagy defect was already detected in young animals, indicating that impaired autophagy might also contribute to the enhanced ER stress.
Loss of WDR45 leads to a variety of morphological alterations in mitochondria as well. An abnormal accumulation of mitochondrial proteins and enlarged mitochondria are observed in wdr45−/–deficient mice brain and WDR45-/- HeLa cells, respectively [Citation43,Citation44]. These data highlight that WDR45 loss in neuronal and non-neuronal cells cause mitochondrial damage and oxidative stress, pointing to a possible autophagy-independent function of WDR45. Notably, two mass spectrometry analyses have uncovered interactors of WDR45 that have not been connected to autophagy yet. This observation strengths the notion of a possible autophagy-independent function of WDR45.
WDR45-associated disorders
Gene mutations in WDR45 are associated with several neurodevelopmental and neurodegenerative disorders and have been shown to can occur sporadically. The phenotypic spectrum of patients with WDR45 variants is broad, ranging from severe early-onset epileptic encephalopathies in males to mild cognitive impairment in females. The differences in severity between male and female patients is very likely because WDR45 is X-chromosome linked. The course of WDR45-associated disorders is mostly progressive and patients with mild and relative static cognitive issues in childhood, can develop dystonia and dementia later in adulthood. Six different, albeit overlapping disorders, i.e., ß-propeller protein associated neurodegeneration (BPAN), Rett-like syndrome (RLS), intellectual disability (ID), developmental and epileptic encephalopathy (DEE), early-onset epileptic encephalopathy (EOEE) and West syndrome, have been associated with variants in WDR45. The vast majority of the WDR45 variants (81%) are de novo nucleotide changes, but there are 9 cases, including BPAN and West syndrome clinical manifestations, where the variant has been inherited (). The origin of the variants has not been determined in 12% of the cases ().
Table 1. Reported WDR45 variants associated with different diseases
To get an overview of all the documented patients that harbor a WDR45 variant (independently of the origin of the variant), we performed a systematic literature search. This search revealed that to date (November 2020), 140 WDR45 variants have been identified and associated to a pathology (). In a recent report, limited to 123 patients [Citation45] 17 new cases were described, but not conclusively categorized into any diseases. We therefore, listed those cases as “unclassified” in our analysis (, and 5). Altogether, our analysis showed that WDR45 variants have led to a BPAN, a RLS, an ID a DEE a EOEE and a West syndrome diagnosis in 66.4%, 2.1%, 1.4%, 12.1%, 2.9% and 2.1% of the cases respectively (). 12.9% of the cases are unclassified (). This analysis further revealed that nine different WDR45 variants can lead to different clinical manifestations thereby causing different disorders ().
Table 2. Similar WDR45 variants causing different diseases
ß-propeller protein associated neurodegeneration (BPAN)
Variants in WDR45 have been identified in numerous patients suffering from neurodegeneration with brain iron accumulation (NBIA) (). NBIA is characterized by iron accumulation generally observed in the globus pallidus and substantia nigra, and occasionally in the cortex and cerebellum [Citation44,Citation46,Citation47]. So far 10 different genes have been identified and associated with the different NBIA subtypes [Citation48].
In 2012, two laboratories discovered that variants in WDR45 were responsible a particular subset of idiopathic NBIA patients [Citation46,Citation49,Citation50]. These patients were characterized by a global developmental delay in early childhood, intellectual disability, impairment of motor skills, speech and seizures. The difference between these patients and the ones suffering from other NBIA pathologies, was that symptoms of the disease remained non-progressive until early adulthood or even later before they started to exhibit signs of parkinsonism, dystonia and dementia [Citation49]. This type of NBIA was originally described as static encephalopathy of childhood with neurodegeneration in adulthood (SENDA) [Citation50], but subsequently replaced with BPAN [Citation46,Citation49]. It is estimated that BPAN accounts for 35–40% of all cases of NBIA [Citation51].
The pathognomonic sign of BPAN is the presence of a hyperintense ‘halo’ on T1-weighted MRI images at the level of substantia nigra and the cerebral peduncle, but less in globus pallidus.This is a unique imaging characteristic, in addition to the diagnosis by gene sequencing, that permits to distinguish BPAN from other forms of NBIA [Citation44,Citation46]. Currently, there are no specific biomarkers for BPAN, making clinical diagnosis difficult especially in early infancy. In one case report, however, a persistent elevation of the ENO2/NSE (enolase 2) in both serum and cerebrospinal fluid was found in a patient still in childhood age. ENO2 is an enzyme that is exclusively expressed in neuronal cells and has been shown to be a reliable marker protein for neural injury [Citation52]. This finding therefore indicates that neuronal damage might already start in infancy but also that ENO2 might be a suitable biomarker for an early diagnosis of BPAN [Citation53]. Another retrospective study that summarized clinical characteristics of ten BPAN children found that a calculated ratio of plasma:serum concentration of soluble (s) TFRC (transferrin receptor) divided by logarithm (log) of plasma:serum concentration of FT (ferritin; sTFRC/logFT) might be another possible biomarker to detect BPAN. Other biomarkers connected to iron metabolism, including iron, transferrin, ferritin levels in the serum, however, were normal. Interestingly, 5 out of 6 patients that were tested for ENO2 also showed an increased serum level of ENO2 [Citation54]. Despite these encouraging observation, analyses of additional patients are necessary to confirm whether ENO2 levels or sTFRC/logFT in the serum could indeed be used as a biomarker for an early diagnosis [Citation46,Citation55].
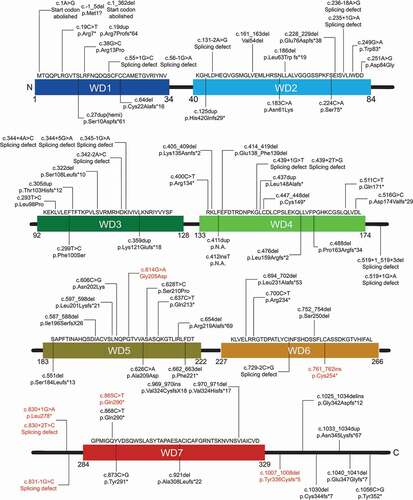
BPAN-associated genetic changes are quite different in nature, ranging from missense to nonsense variants, frame shifts, splicing defects and deletions (). Moreover, mapping all BPAN-associated variants over the entire WDR45 sequence, did not results in specific domains that could be associated to BPAN (). So far, 93 patients carrying a WDR45 variant have been conclusively associated to BPAN, and 84 of them are females while only 9 are male (). Notably, male cases that have somatic mosaic variants or germline variants, albeit rare, in WDR45 can survive but are associated with a more severe phenotype than those observed in heterozygous females [Citation45,Citation56–58]. This gives rise to the assumption that the majority of germline WDR45 variants are likely lethal for males probably because this gene is located on the X chromosome.
Figure 3. Schematic distribution of the documented WDR45 variants causing BPAN over the entire protein sequence. 93 BPAN-associated WDR45 variants have been reported. Of the patients, 84 are women, 9 are males. Those variants also found in patients affected by another disorder are highlighted in red. N.A., not available
Rett-like syndrome (RLS)
RLS is characterized by seizures, motor abnormalities, abnormal hand movements, problems with language, features of autism and the tendency to lose skills throughout adulthood [Citation59]. These symptoms start between 6 and 18 months of age [Citation59]. The most common genetic cause for RLS has been attributed to MECP2 variants, but in recent years several other genes, including WDR45, CDKL5 and FOXG1, have been linked to RLS as well [Citation59]. We found in total seven diagnosed RLS cases with WDR45 variants that are all female (). As for BPAN, the variants associated with RLS are distributed over the entire WDR45 amino acid sequence (). Overlapping clinical features exist between early stages of BPAN and RLS, e.g., developmental delay, stereotypical hand movements, seizures, sleep disorder and spasticity. Therefore, in four out of the seven cases (), patients that were initially diagnosed with RLS or displayed RLS-like symptoms, were later reevaluated to have BPAN, based on iron accumulation that only manifested at an older age [Citation60–63]. Although careful examination of brain MRI scans in retrospect might reveal signs of iron accumulation [Citation62,Citation63], it is often very difficult to detect this at early stages of BPAN [Citation46]. Therefore, to avoid misdiagnosis and as proposed already in 2013 by Hayflick and colleagues [Citation46], genetic testing for WDR45 variants of putative RLS patients, especially with no variants in common RLS-associated genes, should be performed or even included into the gene testing panel for RLS (https://www.ncbi.nlm.nih.gov/gtr/tests/500160/).
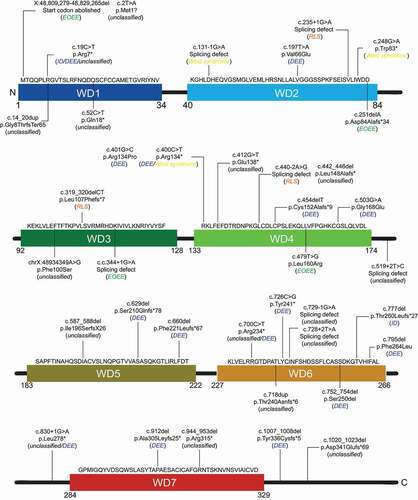
Figure 4. Schematic distribution of the documented WDR45 variants causing RLS, ID, EOEE, DEE and West syndrome, and that have not been associated to a specific disease. Disorder caused by the indicated variants are in between brackets, in italic. Thirty variants in WDR45 cause disorder other than BPAN with twenty-two female and eight male patients. The diagnosis is not conclusive in 18 cases; therefore, annotated as “unclassified”
Although cases are diagnosed with RLS [Citation55,Citation64,Citation65], longitudinal follow-up of those patients will show whether they will evolve BPAN pathology (e.g., signs of iron accumulation in the brain). Since the overall frequency of WDR45 variants in RLS cases is rather low compared to the overall numbers of RLS patients, more studies/patient follow-ups are required to confirm whether a RLS diagnosis could reflect an early symptom of BPAN in patients with WDR45 variations.
Intellectual disability (ID)
ID is defined by significant impairment of cognitive and adaptive functions with the disease onset occurring before 18 years of age. ID has an estimated worldwide prevalence of 1–3%, affecting up to 0.5% of the population in Western countries with moderate or severe forms (IQ <50) [Citation66]. Disease-associated variants have been so far associated with multiple genes, including WDR45B and WDR45 [Citation67,Citation68]. Genetic alterations in ID ranges from cytogenetic abnormalities to single nucleotide variants, in rare cases even epigenetic defects in methylations has been identified [Citation69]. Yet the genetic cause of ID remains elusive in over 50% of the cases [Citation70].
So far, only two reported cases, both female () with ID are caused by a variant in WDR45 () [Citation67,Citation71]. One 3-year-old ID patient with a nonsense variant in the first WD-repeat of WDR45, and one 29-year-old ID patient with a frameshift variant in the sixth WD repeat () [Citation67,Citation71], showing that these ID patients do not carry a similar type of variant in a specific domain of WDR45. Moreover, no iron accumulation has been reported in the brain of these two patients. In light of the knowledge acquired from WDR45-associated RLS diagnosed patients, it would be interesting to determine on a long term whether these two patients will develop a brain iron accumulation later in life.
Epileptic encephalopathies
a) DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY (DEE)DEE is characterized by refractory seizures, electroencephalographic (EEG) abnormalities and frequent epileptiform activity that contribute to developmental slowing and regression in the childhood that can lead to an early death [Citation72,Citation73].
There are 17 reported DEE patients carrying a variant in WDR45 (, ). As depicted in , the DEE-associated WDR45 variants are distributed over the entire protein, without concentrating to a specific region. So far, there are four male cases within DEE patients (). Interestingly, Khoury and coworkers described a 10-year-old boy with profound developmental delay, spastic quadriparesis, and intractable epilepsy with tonic and atypical absence seizures [Citation74]. This patient was first noted to have a development delay at 9 months, showed diffusely decreased tone at 17 months and seizures started at the age of two [Citation74]. Only at the age of six, MRI brain scans showed iron accumulation in globus pallidus interna and externa, and signs of iron accumulation in substantia nigra [Citation74]. This late onset of brain iron accumulation suggests that iron deposition is not one of the first symptoms of WDR45-associated DEE, similar to what is observed in BPAN.
b) EARLY-ONSET EPILEPTIC ENCEPHALOPATHIES (EOEE)
Another disorder that is linked with WDR45 is EOEE which is characterized by severe epilepsies and neurodevelopmental disorders [Citation56]. Further clinical signs of EOEE include stormy seizures associated with an altered interictal EEG pattern. The disease-related epileptic disorders are highly heterogeneous in terms of etiology as they can have symptomatic causes (e.g,, cortical malformation or strokes), or can have metabolic or genetic origin [Citation75]. In fact, variants in over 100 genes, including KCNQ2, STXBP1, SCN2A, PNPO, PIGA, SEPSECS and WDR45, were shown or suggested to cause EOEE [Citation76]. Based on the interictal/ictal EEG pattern, type of seizures and age at onset, EOEE is further sub-classified into Ohtahara syndrome, early myoclonic encephalopathy, Dravet syndrome and Lennox-Gastaut syndrome, Landau-Kleffner syndrome and West syndrome [Citation77]. In contrast to DEE where first symptoms occur later, symptoms of EOEE, such as severe epilepsies, are already occurring in the first 3 months of life [Citation56,Citation74].
There are only four cases of EOEE patients, one of them a male, associated with a variant in WDR45 (). These variants include a deletion, a splicing defect and a missense variant in WDR45 and, as for the other disorders, those variants are not concentrated at a specific region of the protein (, ). As for RLS, none of the four EOEE patients display signs of brain iron accumulation, at the time when they were tested which was at the early stage of EOEE [Citation56,Citation78].
c) WEST SYNDROME
West syndrome is the most common subtype of EOEE with an incidence of 2–5 per 10,000 births [Citation79], and it principally manifests at 3–12 months of age, predominantly affecting males, i.e., in 60–70% of the cases [Citation80]. This disease represents as a specific epileptic encephalopathy characterized by unique type of attacks, called infantile spasms, hypsarrhythmia and delays in the psychomotoric development [Citation81,Citation82]. West syndrome can also be part of more complex disorders, where some may manifest with a developmental delay, like for example in DEE [Citation83]. Neurodevelopmental delay and/or regression have interchangeably been part of the definition of West syndrome, and are currently considered as part of the triad of symptoms characterizing this disease, i.e., infantile spasms, hypsarrhythmia, and retardation.
So far only 3 cases of West syndrome patients carrying a variant in WDR45 have been documented (), making the West syndrome frequency with WDR45-associated diseases as low as the one of ID or EOEE (). As for all WDR45-associated diseases, the documented WDR45 variants causing West syndrome are also distributed throughout the whole protein. The 3 patients are all male, which might indicate a gender-specificity for WDR45-associated West syndrome cases. These individuals showed early-onset intractable seizures, profound intellectual disability and developmental delay, and the MRI brain scans showed cerebral atrophy [Citation84,Citation85]. Interestingly, like DEE and EOEE, West syndrome patients showed no brain iron accumulation and altogether, these three WDR45-associated disorders are also the ones that show most frequently male patients. This could imply that diseases progressions that eventually lead to brain iron accumulation cannot or hardly be tolerated by male.
Cancer
Almost all of the documented WDR45-related diseases as of today fall into the neurodevelopmental and neurodegenerative disorders category described above, however, in a metadata analysis study by Lebovitz and colleagues, WDR45 was identified as a potential target for specific cancers. In particular, they analyzed DNA sequences and RNA expression levels of ATG genes from cancer patients using datasets from The Cancer Genome Atlas consortium (http://cancergenome.nih.gov/). They found that WDR45 is one of the ATG genes that is frequently genetically altered in uterine corpus endometrial carcinoma patients [Citation86], indicating that WDR45 variants might also contribute to cancer development. A further indication that WDR45 may play a role in cancer comes from a bioinformatics study that showed that WDR45 expression is affected by OTUD5, a deubiquitinase. Reduced expression of OTUD5 correlates with a poor prognosis in cervical cancer and when OTUD5 is depleted, WDR45 expression is reduced, suggesting a possible connection between WDR45 function and cervical cancer [Citation87].
Potential connections between WDR45 molecular functions and the pathogenicity associated with its variants
Mouse models are often key to relate consequences of a gene mutation to the pathophysiology of the disease it is causing. So far, three different wdr45-/- mouse models have been described [Citation2,Citation4,Citation43]. The first was a CNS-specific wdr45-/- mouse [Citation2]. On a cellular level, these mouse brains show axonal swelling, and accumulation of ubiquitin- and SQSTM1/p62-positive aggregates, but little neuronal damage [Citation2]. These mice also display deficits in cognitive functions such as learning and memory, however, only older animals show motor dysfunction [Citation2]. The others mouse models are constitutive, full-body wdr45-/- mice [Citation4,Citation43] and display similar phenotypes as the CNS-specific wdr45-/- animals with older mice display neuronal loss and the appearance of seizures [Citation43]. However, none of the mice models accumulated iron in the brain. Notably, all wdr45-/- mice are born normally and survive the post-natal starvation period, which is in contrast to mice lacking essential ATG proteins [Citation43]. Therefore, although a mild autophagy defect was observed in brain cells of those animals and some cognitive dysfunction might be connected with this alteration, it seems that Wdr45 is not an essential ATG gene, in agreement with most of the cellular studies (see section The role of WDR45 and the other WIPI proteins in autophagy).
Another approach to connect molecular dysfunctions with disease pathophysiology is by using patient material. The first study that investigated WDR45-associated disease patient material, in particular BPAN patient material, showed that WDR45 levels were significantly reduced in lymphoblastoid cells derived from 5 BPAN patients [Citation50]. Moreover, compared to cells from healthy family members, those cells displayed a reduction in autophagic flux and a augmentation of lipidated LC3 and LC3-positive labeled structures, which is indicative of an accumulation of autophagosomes and/or autophagosomal intermediates [Citation50]. More recently, Seibler and colleagues investigated fibroblasts derived from one BPAN patient in more detail [Citation88] and also observed a defect in autophagy as others did [Citation2,Citation24,Citation43,Citation50]. The examined cells also showed drastic alterations in mitochondrial membrane potential and network organization, resulting in elevated mitochondrial stress [Citation88]. Mitochondrial dysfunction is a reoccurring phenotype in WDR45-/- cell lines, including HeLa [Citation43] and SH-SY5Y [Citation88] cells, but it is also observed in swollen axons of the wdr45-/- mice [Citation2]. The altered mitochondria morphology and function is of particular interest since this phenotype is associated with many other neurodegenerative diseases, e.g., Huntington and Parkinson diseases, Rett syndrome, etc [Citation89]., but also other NBIA disorders, including PKAN, PLAN, CoPAN and MPAN [Citation90]. Thus, mitochondrial dysfunction might be a crucial trait underlying the neurodegeneration that characterizes WDR45-associated diseases.
The most striking brain phenotype of WDR45-related patients is, however, an iron deposition in the basal ganglia region. Interestingly and like mitochondrial dysfunction, brain iron accumulation is not only observed in WDR45-associated patients, but also in individuals affected many other neurodegenerative diseases, including Huntington’s, Parkinson’s and Alzheimer’s diseases. Those patients exhibit a dysfunction in iron metabolism, elevated free iron and iron deposition at the sites of neurodegeneration [Citation91]. As iron reacts with hydrogen peroxide and catalyzes the generation of highly reactive hydroxyl radicals, high levels of iron can increase the cellular oxidative stress and even trigger an iron-dependent form of cell death, known as ferroptosis [Citation92,Citation93]. Consistently, increased levels in oxidative stress-related marker proteins and an elevated susceptibility to iron-induced stress has been observed in BPAN patient derived fibroblasts [Citation88]. Interestingly, two studies could show that BPAN patient-derived cells, i.e., fibroblast and dopaminergic neurons, display a defect in iron homeostasis and an increase in iron level, which are accompanied by lower amounts of ferritin [Citation88,Citation94]. The observed iron overload and the reduced ferritin levels could be restored by inducing autophagy in BPAN patient-derived dopaminergic neurons [Citation88], indicating that some phenotypical alterations in WDR45-depleted cells can be overcome by enhancing autophagy. Mechanistically, iron overload in the cells could be due to an overexpression of the SLC11A2/DMT1 (solute carrier family 11 member 2), which is upregulated in BPAN patient cells [Citation94] as well as in cells lacking PLA2G6, the gene than when mutated causes another NBIA disorder, PLAN [Citation95]. In contrast, another iron cell importer, the transferrin receptor, is downregulated in BPAN patient-derived cells [Citation94]. Transferrin receptor is required for both the endocytosis of extracellular transferrin-bound iron and the storage of iron in ferritin cages and its subsequent controlled release [Citation96,Citation97]. Therefore, misregulation of this receptor likely also contribute to the observed cellular iron overload.
Although the number of studies that have investigated the WDR45 dysfunction and the resulting pathology are limited, they concurrently show that WDR45 mutations leads to an alteration of autophagy progression, mitochondrial morphology and function, ER stress and iron homeostasis (). However, it remains to be determined whether WDR45 dysfunction affects multiple cellular processes simultaneously, or whether the inhibition of a single pathway has pleiotropic negative effects on several other cellular processes.
Conclusions
Six different WDR45-associated neurodegenerative disorders have been identified so far. Amongst those, BPAN is the only one that appears to be exclusively caused by variants in WDR45 while the other disorders can also be caused by other gene variants or environmental factors. Because of WDR45’s role in autophagy (), most of the investigations have tried to connect autophagy dysfunction with the onset and pathophysiology of those disorders. WDR45 depletion, however, also causes cellular alterations that are unrelated to autophagy because of its probable function in other cellular pathways (), similar to other ATG genes [Citation98–100]. The best characterized molecular function of WDR45 is promoting lipid transfer together with ATG2 proteins between adjacent membranes. This function at the phagophore-ER membrane contact sites is important for autophagy [Citation34–41,Citation101]. It cannot be excluded a priori that these proteins also participate in transferring lipids at other membrane contact sites and that a disruption caused by WDR45 mutations could contribute to the pathogenicity of WDR45-associated diseases. In particular, misregulated ER-mitochondria membrane contact sites could explain the morphological and functional alterations of mitochondria and ER in cells lacking WDR45 [Citation102,Citation103]. The most studied cellular function of WDR45 is the one in autophagy, however, the very partial autophagy inhibition [Citation2,Citation4,Citation27,Citation50,Citation88] and the absence of developmental defects in mice lacking Wdr45 [Citation2,Citation4,Citation43], suggest that WDR45 is not a core ATG gene. Interestingly, when depleted together with WDR45B, the severity of the autophagy and developmental defects in mice drastically increase [Citation4], similarly to animals lacking core ATG genes [Citation104,Citation105]. This notion indicates that WDR45 and WDR45B might function redundantly at least in the brain. Therefore, it is tempting to speculate that WDR45B expression levels might influence the severity of WDR45-associated diseases, i.e., lower WDR45B expression levels lead to a severer pathology. This notion and a reciprocal causative connection between these two genes are also evoked by the fact that variants in WDR45B can lead to ID [Citation66,Citation67].
Besides general bulk autophagy, it might be that WDR45 is required for selective autophagy that target for example mitochondria (i.e, mitophagy) or ferritin (i.e, ferritinophagy) [Citation106]. The role of WDR45 in those pathways has not been studied, yet it could provide important insights why WDR45 depletion is causing the observed phenotypical and functional alterations; i.e., ferritinophagy plays a crucial role in iron homeostasis, something that is misregulated in many WDR45-associated disorders. During ferritinophagy, the NCOA4 (nuclear receptor coactivator 4) interacts with ferritin and is required for the delivery of iron complexed with ferritin to lysosomes via autophagosomes [Citation107,Citation108]. Interestingly, mice lacking NCOA4 show increased levels of ferritin and iron in several tissues [Citation109,Citation110], something reminiscent with BPAN patients. Thus, the potential connection between WDR45 and ferritinophagy could be an explanation why WDR45-associated disorders are often accompanied with iron accumulation.
It is still unknown which cellular alterations, present in patient material, are the primary cause of these pathologies, and which are consequences. In this context, it is important to note that some BPAN patients did not show iron accumulation at their first diagnosis, but were detected in their brains with age [Citation55]. Moreover, patients suffering of other specific WDR45-associated disorders, i.e, ID, EOEE and West syndrome, do not show iron accumulation at all. Interestingly, in those disorders the frequency of male patients is much higher than in those that develop iron accumulation (). This may indicate that iron accumulation is a consequence of one or more WDR45-associated cellular dysregulations and not a primary feature.
Despite the variety of clinical manifestations in patients that harbor WDR45 variants, it appears that there is not a phenotype-genotype correlation () [Citation45]. Genetic variants are diverse and are distributed over the entire amino acids sequence of WDR45 (, ) and in nine cases the variants are even causing different diseases (). Thus, it is possible that because of the complex and compact structure of WDR45, variants in different parts all lead to the disruption of the -propeller structure (Figure S1B), resulting in a complete loss-of-function [Citation50]. It cannot be excluded, however, that some variants are leading to gain-of-function proteins.
Although there is overlap in the spectrum of clinical symptoms and genetic and environmental factors are likely playing roles in the clinical manifestations of WDR45-associated disorders, it remains still unclear, why the disease severity varies so much. Fundamentally, it is crucial to determine the primary cellular defects caused by WDR45 variants and try to correlate the consequences with the appearance of biomarkers in patients that may appear at different disease stages. This knowledge will be vital to design effective therapies that specifically counteract the causes of WDR45-associated disorders rather than the consequences.
Added in proof
A manuscript providing additional mechanistic insights on the function of WDR45 has very recently been published [Citation111]. This study reveals the redundant function of WDR45 and WDR45B, in particular in the maturation of autophagosomes into autolysosomes in neurons. Mechanistically, WDR45 and WDR45B interact with the tether protein EPG5 and facilitate its localization to late endosomes and lysosomes. Thus, in the absence of WDR45 and WDR45B, the correct assembly of the EPG5-depepndent SNARE complexes that are required for autophagosome-lysosome fusion is compromised. An additional important observation in this study is that although autophagosomes can still be formed in the absence of WDR45 and WDR45B, their size is greatly reduced. This might be due to insufficient lipid transfer during autophagosome formation because it could be partially rescued by ATG2A overexpression. Of note, the autophagosome maturation defects were not observed in other non-neuronal cells or when any of the genes was deleted singularly in neurons.
Supplemental Material
Download Zip (1.8 MB)Acknowledgments
The authors thank Rubén Gómez-Sánchez for the critical reading of the manuscript. F.R. is supported by ZonMW TOP (91217002), ALW Open Programme (ALWOP.310), Open Competition ENW-KLEIN (OCENW.KLEIN.118), Marie Skłodowska-Curie Cofund (713660) and Marie Skłodowska Curie ETN (765912) grants. M.M. has been supported by NBIADA. F. R. and M.M. are also supported by the Million Dollar Bike Ride (MDBR) grant program. The authors also thank the Stichting Ijzersterk (https://www.stichtingijzersterk.nl/) for their support and generous financial contribution.
Disclosure statement
No conflict of interest was declared by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. Handb Clin Neurol. 2018;147:293–305.
- Zhao YG, Sun L, Miao G, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015;11:881–890.
- Proikas-Cezanne T, Waddell S, Gaugel A, et al. WIPI-1a (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004;23(58):9314–9325.
- Ji C, Zhao H, Li D, et al. Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 2020;16(4):615–625.
- Hor CHH, Tang BL. Beta-propeller protein-associated neurodegeneration (BPAN) as a genetically simple model of multifaceted neuropathology resulting from defects in autophagy. Rev Neurosci. 2019;30:261–277.
- Pons T, Gomez R, Chinea G, et al. Beta-propellers: associated functions and their role in human diseases. Curr Med Chem. 2003;10(6):505–524.
- Jain BP, Pandey S. WD40 Repeat proteins: signalling scaffold with diverse functions. Protein J. 2018;37(5):391–406.
- Smith TF, Gaitatzes C, Saxena K, et al. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci. 1999;24(5):181–185.
- Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58(14):2085–2097.
- Proikas-Cezanne T, Takacs Z, Donnes P, et al. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207–217.
- Scacioc A, Schmidt C, Hofmann T, et al. Structure based biophysical characterization of the PROPPIN Atg18 shows Atg18 oligomerization upon membrane binding. Sci Rep. 2017;7(1):14008.
- Watanabe Y, Kobayashi T, Yamamoto H, et al. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem. 2012;287(38):31681–31690.
- Liang R, Ren J, Zhang Y, et al. Structural conservation of the two phosphoinositide-binding sites in WIPI Proteins. J Mol Biol. 2019;431(7):1494–1505.
- Lei Y, Tang D, Liao G, et al. The crystal structure of Atg18 reveals a new binding site for Atg2 in Saccharomyces cerevisiae. Cell Mol Life Sci. 2020. in press. https://doi.org/10.1007/s00018-020-03621-9.
- Polson HE, De Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010;6(4):506–522.
- Dove SK, Piper RC, McEwen RK, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. Embo J. 2004;23(9):1922–1933.
- Krick R, Tolstrup J, Appelles A, et al. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett. 2006;580(19):4632–4638.
- Rieter E, Vinke F, Bakula D, et al. Atg18 function in autophagy is regulated by specific sites within its beta-propeller. J Cell Sci. 2013;126(2):593–604.
- Krick R, Henke S, Tolstrup J, et al. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy 2008;4(7):896–910.
- Zhang H, Wu F, Wang X, et al. The two C. elegans ATG-16 homologs have partially redundant functions in the basal autophagy pathway. Autophagy. 2013;9(12):1965–1974.
- Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21(2):343–357.
- Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26(8):624–635.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364.
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186(6):773–782.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750.
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16(8):461–472.
- Bakula D, Muller AJ, Zuleger T, et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. 2017;8(1):15637.
- Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55(2):238–252.
- Puri C, Vicinanza M, Ashkenazi A, et al. The RAB11A-positive compartment Is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P-RAB11A. Dev Cell. 2018;45(1):114–131.
- Mauthe M, Jacob A, Freiberger S, et al. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation [Research Support, Non-U.S. Gov’t]. Autophagy. 2011;7(12):1448–1461.
- Bakula D, Mueller AJ, Proikas-Cezanne T. WIPI beta-propellers function as scaffolds for STK11/LKB1-AMPK and AMPK-related kinase signaling in autophagy. Autophagy 2018;14(6):1082–1083.
- Zheng JX, Li Y, Ding YH, et al. Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy 2017;13(11):1870–1883.
- Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature 2010;466(7302):68–76.
- Chowdhury S, Otomo C, Leitner A, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci USA. 2018;115(42):E9792–E9801.
- Valverde DP, Yu S, Boggavarapu V, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. 2019;218(6):1787–1798.
- Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 2019;8:e45777.
- Velikkakath AK, Nishimura T, Oita E, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23(5):896–909.
- Ren J, Liang R, Wang W, et al. Multi-site-mediated entwining of the linear WIR-motif around WIPI beta-propellers for autophagy. Nat Commun. 2020;11(1):2702.
- Osawa T, Kotani T, Kawaoka T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26(4):281–288.
- Osawa T, Ishii Y, Noda NN. Human ATG2B possesses a lipid transfer activity which is accelerated by negatively charged lipids and WIPI4. Genes Cells. 2020;25(1):65–70.
- Gomez-Sanchez R, Rose J, Guimaraes R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 2018;217(8):2743–2763.
- Bozic M, Van Den Bekerom L, Milne BA, et al. A conserved ATG2-GABARAP family interaction is critical for phagophore formation. EMBO Rep. 2020;21(3):e48412.
- Wan H, Wang Q, Chen X, et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 2020;16(3):531–547.
- Kruer MC, Boddaert N. Neurodegeneration with brain iron accumulation: a diagnostic algorithm. Semin Pediatr Neurol. 2012;19(2):67–74.
- Adang LA, Pizzino A, Malhotra A, et al. Phenotypic and imaging spectrum associated with WDR45. Pediatr Neurol. 2020;109:56–62.
- Hayflick SJ, Kruer MC, Gregory A, et al. Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013;136(6):1708–1717.
- Hayflick SJ. Unraveling the Hallervorden-Spatz syndrome: pantothenate kinase-associated neurodegeneration is the name. Curr Opin Pediatr. 2003;15(6):572–577.
- Wang ZB, Liu JY, Xu XJ, et al. Neurodegeneration with brain iron accumulation: insights into the mitochondria dysregulation. Biomed Pharmacother. 2019;118:109068.
- Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91(6):1144–1149.
- Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45(4):445–449.
- Stige KE, Gjerde IO, Houge G, et al. Beta-propeller protein-associated neurodegeneration: a case report and review of the literature. Clin Case Rep. 2018;6(2):353–362.
- Haque A, Polcyn R, Matzelle D, et al. New Insights into the role of neuron-specific enolase in neuro-Inflammation, neurodegeneration, and neuroprotection. Brain Sci. 2018;8(2):33.
- Takano K, Shiba N, Wakui K, et al. Elevation of neuron specific enolase and brain iron deposition on susceptibility-weighted imaging as diagnostic clues for beta-propeller protein-associated neurodegeneration in early childhood: additional case report and review of the literature. Am J Med Genet A. 2016;170(2):322–328.
- Belohlavkova A, Sterbova K, Betzler C, et al. Clinical features and blood iron metabolism markers in children with beta-propeller protein associated neurodegeneration. Eur J Paediatr Neurol. 2020;28:81–88.
- Hoffjan S, Ibisler A, Tschentscher A, et al. WDR45 mutations in Rett (-like) syndrome and developmental delay: case report and an appraisal of the literature. Mol Cell Probes. 2016;30(1):44–49.
- Abidi A, Mignon-Ravix C, Cacciagli P, et al. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet. 2016;24(4):615–618.
- Zarate YA, Jones JR, Jones MA, et al. Lessons from a pair of siblings with BPAN. Eur J Hum Genet. 2016;24(7):1080–1083.
- Akcakaya NH, Salman B, Gormez Z, et al. A Novel and mosaic WDR45 nonsense variant causes beta-propeller protein-associated neurodegeneration identified through whole exome sequencing and X chromosome heterozygosity analysis. Neuromolecular Med. 2019;21(1):54–59.
- Ehrhart F, Sangani NB, Curfs LMG. Current developments in the genetics of Rett and Rett-like syndrome. Curr Opin Psychiatry. 2018;31(2):103–108.
- Chard M, Appendino JP, Bello-Espinosa LE, et al. Single-center experience with Beta-propeller protein-associated neurodegeneration (BPAN); expanding the phenotypic spectrum. Mol Genet Metab Rep. 2019;20:100483.
- Morisada N, Tsuneishi S, Taguchi K, et al. [A woman with beta-propeller protein-associated neurodegeneration identified by the WDR45 mutation presenting as Rett-like syndrome in childhood]. No To Hattatsu. 2016;48(3):209–212.
- Okamoto N, Ikeda T, Hasegawa T, et al. Early manifestations of BPAN in a pediatric patient. Am J Med Genet A. 2014;164(12):3095–3099.
- Ohba C, Nabatame S, Iijima Y, et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet. 2014;59(5):292–295.
- Kulikovskaja L, Sarajlija A, Savic-Pavicevic D, et al. WDR45 mutations may cause a MECP2 mutation-negative Rett syndrome phenotype. Neurol Genet. 2018;4(2):e227.
- Percy AK, Lane J, Annese F, et al. When Rett syndrome is due to genes other than MECP2. Translational science of rare diseases. 2018;3(1):49–53.
- Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11(1):161–187.
- Hamdan FF, Srour M, Capo-Chichi JM, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10(10):e1004772.
- Suleiman J, Allingham-Hawkins D, Hashem M, et al. WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: a consistent neurodevelopmental syndrome. Clin Genet. 2018;93(2):360–364.
- Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80(2):117–126.
- De Ligt J, Willemsen MH, Van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929.
- Fieremans N, Van Esch H, Holvoet M, et al. Identification of intellectual disability genes in female patients with a Skewed X-inactivation pattern. Hum Mutat. 2016;37(8):804–811.
- Carvill GL, Liu A, Mandelstam S, et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia 2018;59(1):e5–e13.
- Steward CA, Roovers J, Suner MM, et al. Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A. NPJ Genom Med. 2019;4(1):31.
- Khoury J, Kotagal P, Moosa ANV. Epileptic encephalopathy and brain iron accumulation due to WDR45 mutation. Seizure 2019;71:245–246.
- Hwang SK, Kwon S. Early-onset epileptic encephalopathies and the diagnostic approach to underlying causes. Korean J Pediatr. 2015;58(11):407–414.
- Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53(8):1387–1398.
- Von Deimling M, Helbig I, Marsh ED, et al. Epileptic encephalopathies-clinical syndromes and pathophysiological concepts. Curr Neurol Neurosci Rep. 2017;17(2):10.
- Russo C, Ardissone A, Freri E, et al. Substantia Nigra swelling and dentate nucleus T2 Hyperintensity may be early magnetic resonance imaging signs of beta-propeller protein-associated neurodegeneration. Mov Disord Clin Pract. 2019;6(1):51–56.
- Saemundsen E, Ludvigsson P, Rafnsson V. Risk of autism spectrum disorders after infantile spasms: a population-based study nested in a cohort with seizures in the first year of life. Epilepsia 2008;49(11):1865–1870.
- Zeka N, Gerguri A, Bejiqi R, et al. Compare of the west syndrome with other syndromes in the epileptic encephalopathy - kosovo experience. Open Access Maced J Med Sci. 2017;5(7):925–928.
- Osborne JP, Lux AL, Edwards SW, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom infantile spasms study (UKISS) on contemporary causes and their classification. Epilepsia 2010;51(10):2168–2174.
- Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58(4):512–521.
- Salar S, Moshe SL, Galanopoulou AS. Metabolic etiologies in West syndrome. Epilepsia Open. 2018;3:134–166.
- Krey I, Krois-Neudenberger J, Hentschel J, et al. Genotype-phenotype correlation on 45 individuals with West syndrome. Eur J Paediatr Neurol. 2020;25:134–138.
- Nakashima M, Takano K, Tsuyusaki Y, et al. WDR45 mutations in three male patients with West syndrome. J Hum Genet. 2016;61(7):653–661. .
- Lebovitz CB, Robertson AG, Goya R, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015;11(9):1668–1687.
- Bai M, Che Y, Lu K, et al. Analysis of deubiquitinase OTUD5 as a biomarker and therapeutic target for cervical cancer by bioinformatic analysis. PeerJ 2020;8:e9146.
- Seibler P, Burbulla LF, Dulovic M, et al. Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 2018;141(10):3052–3064.
- Dard L, Blanchard W, Hubert C, et al. Mitochondrial functions and rare diseases. Mol Aspects Med. 2020;71:100842.
- Aoun M, Tiranti V. Mitochondria: a crossroads for lipid metabolism defect in neurodegeneration with brain iron accumulation diseases. Int J Biochem Cell Biol. 2015;63:25–31.
- Li K, Reichmann H. Role of iron in neurodegenerative diseases. J Neural Transm (Vienna). 2016;123(4):389–399.
- Ward RJ, Zucca FA, Duyn JH, et al. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13(10):1045–1060.
- Kim GH, Kim JE, Rhie SJ, et al. The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol. 2015;24(4):325–340.
- Ingrassia R, Memo M, Garavaglia B. Ferrous iron up-regulation in fibroblasts of patients with beta propeller protein-associated neurodegeneration (BPAN). Front Genet. 2017;8:18.
- Beck G, Shinzawa K, Hayakawa H, et al. Deficiency of calcium-independent phospholipase A2 beta induces brain iron accumulation through upregulation of divalent metal transporter 1. PLoS One. 2015;10(10):e0141629.
- Recalcati S, Gammella E, Buratti P, et al. Molecular regulation of cellular iron balance. IUBMB Life. 2017;69(6):389–398.
- Arosio P, Elia L, Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69(6):414–422.
- Bestebroer J, V’Kovski P, Mauthe M, et al. Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 2013;14(10):1029–1041.
- Mauthe M, Reggiori F. ATG proteins: are we always looking at autophagy? Autophagy 2016;12(12):2502–2503.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell 2019;177(7):1682–1699.
- Hollenstein DM, Kraft C. Autophagosomes are formed at a distinct cellular structure. Curr Opin Cell Biol. 2020;65:50–57.
- Lee KS, Huh S, Lee S, et al. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci USA. 2018;115(38):E8844–E8853.
- Tubbs E, Rieusset J. Metabolic signaling functions of ER-mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol. 2017;58(2):R87–R106.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434.
- Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature 2004;432(7020):1032–1036.
- Zhang Y, Mikhael M, Xu D, et al. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid Redox Signal. 2010;13(7):999–1009.
- Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16(11):1069–1079.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509(7498):105–109.
- Bellelli R, Federico G, Matte A, et al. NCOA4 deficiency impairs systemic iron homeostasis. Cell Rep. 2016;14(3):411–421.
- Santana-Codina N, Gableske S, Quiles Del Rey M, et al. NCOA4 maintains murine erythropoiesis via cell autonomous and non-autonomous mechanisms. Haematologica. 2019;104(7):1342–1354.
- Ji C, Zhao H, Chen D, Zhang H, Zhao YG. β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr Biol. 2021;S0960-9822(21):00146-9. doi:https://doi.org/10.1016/j.cub.2021.01.081
- Zitser J, Giladi N, Gurevich T. A Case with Beta-Propeller Protein Associated Neurodegeneration with Smooth Response to Levodopa Treatment. Mov Disord Clin Pract 2018; 5:327–9.
- Fonderico M, Laudisi M, Andreasi NG, Bigoni S, Lamperti C, Panteghini C, et al. Patient Affected by Beta-Propeller Protein-Associated Neurodegeneration: A Therapeutic Attempt with Iron Chelation Therapy. Front Neurol 2017; 8:385.
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015; 10:845–58.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–9.
- Ishiyama A, Kimura Y, Iida A, Saito Y, Miyamoto Y, Okada M, et al. Transient swelling in the globus pallidus and substantia nigra in childhood suggests SENDA/BPAN. Neurology 2018; 90:974–6.
- Willoughby J, Duff-Farrier C, Desurkar A, Kurian M, Raghavan A, Study DDD, et al. Functional mRNA analysis reveals aberrant splicing caused by novel intronic mutation in WDR45 in NBIA patient. Am J Med Genet A 2018; 176:1049–54.
- Lim SY, Tan AH, Ahmad-Annuar A, Schneider SA, Bee PC, Lim JL, et al. A Patient with Beta-Propeller Protein-Associated Neurodegeneration: Treatment with Iron Chelation Therapy. J Mov Disord 2018; 11:89–92.
- Long M, Abdeen N, Geraghty MT, Hogarth P, Hayflick S, Venkateswaran S. Novel WDR45 Mutation and Pathognomonic BPAN Imaging in a Young Female With Mild Cognitive Delay. Pediatrics 2015; 136:e714–7.
- Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, et al. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging 2015; 36:2004 e9- e15.
- Takeuchi J, Ito K, Takeda A, Saitsu H, Matsumoto N. A Male Case with β-Propeller Protein-Associated Neurodegeneration (BPAN) with Somatic Mosaic Mutation in WDR45. J Alzheimers Dis Parkinsonism 2016; 6:2161–0460.1000270.
- Ozawa T, Koide R, Nakata Y, Saitsu H, Matsumoto N, Takahashi K, et al. A novel WDR45 mutation in a patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Am J Med Genet A 2014; 164A:2388–90.
- Rathore GS, Schaaf CP, Stocco AJ. Novel mutation of the WDR45 gene causing beta‐propeller protein‐associated neurodegeneration. Movement Disorders 2014; 29:574–5.
- Ozgun N, Ozer L, Yaramis A. A rare cause of epileptic encephalopathy: a beta-propeller protein associated neurodegeneration case with a new mutation and literature review. Turk J Pediatr 2020; 62:109–13.
- Ryu SW, Kim JS, Lee SH. Beta-Propeller-Protein-Associated Neurodegeneration: A Case of Mutation in WDR45. J Clin Neurol 2015; 11:289–91.
- Yoganathan S, Arunachal G, Sudhakar SV, Rajaraman V, Thomas M, Danda S. Beta Propellar Protein-Associated Neurodegeneration: A Rare Cause of Infantile Autistic Regression and Intracranial Calcification. Neuropediatrics 2016; 47:123–7.
- Sato R, Koga M, Iwama K, Mizuguchi T, Matsumoto N, Kanda T. [A case of novel WDR45 mutation with beta-propeller protein-associated neurodegeneration (BPAN) presenting asymmetrical extrapyramidal signs]. Rinsho Shinkeigaku 2020.
- Rohani M, Fasano A, Akhoundi FH, Haeri G, Lang AE, Rahimi Bidgoli MM, et al. Beta-propeller protein associated neurodegeneration (BPAN); the first report of three patients from Iran with de novo novel mutations. Parkinsonism Relat Disord 2019; 61:231–3.
- Araujo R, Garabal A, Baptista M, Carvalho S, Pinho C, de Sa J, et al. Novel WDR45 mutation causing beta-propeller protein associated neurodegeneration (BPAN) in two monozygotic twins. J Neurol 2017; 264:1020–2.
- Van Goethem G, Livingston JH, Warren D, Oojageer AJ, Rice GI, Crow YJ. Basal ganglia calcification in a patient with beta-propeller protein-associated neurodegeneration. Pediatr Neurol 2014; 51:843–5.
- Ichinose Y, Miwa M, Onohara A, Obi K, Shindo K, Saitsu H, et al. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). Neurol Clin Pract 2014; 4:175–7.
- Tiedemann LM, Reed D, Joseph A, Yoo SH. Ocular and systemic manifestations of beta-propeller protein-associated neurodegeneration. J AAPOS 2018; 22:403–5.
- Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014; 76:473–83.
- Wynn DP, Pulst SM. A novel WDR45 mutation in a patient with beta-propeller protein-associated neurodegeneration. Neurol Genet 2017; 3:e124.
- Kim MK, Kim NY, Hong S, Ma HI, Kim YJ. Presynaptic Dopaminergic Degeneration in a Patient with Beta-Propeller Protein-Associated Neurodegeneration Documented by Dopamine Transporter Positron Emission Tomography Images: A Case Report. J Mov Disord 2017; 10:161–3.
- Tschentscher A, Dekomien G, Ross S, Cremer K, Kukuk GM, Epplen JT, et al. Analysis of the C19orf12 and WDR45 genes in patients with neurodegeneration with brain iron accumulation. J Neurol Sci 2015; 349:105–9.
- Verhoeven WM, Egger JI, Koolen DA, Yntema H, Olgiati S, Breedveld GJ, et al. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat Disord 2014; 20:332–6.
- Endo H, Uenaka T, Satake W, Suzuki Y, Tachibana H, Chihara N, et al. Japanese WDR45 de novo mutation diagnosed by exome analysis: A case report. Neurol Clin Neurosci 2017; 5:131–3.
- Hornemann F, Le Duc D, Roth C, Pfaffle R, Huhle D, Merkenschlager A. Childhood Dystonia-Parkinsonism Following Infantile Spasms-Clinical Clue to Diagnosis in Early Beta-Propeller Protein-Associated Neurodegeneration. Neuropediatrics 2020; 51:22–9.
- Kaleka G, McCormick ME, Krishnan A. Beta-Propeller Protein-Associated Neurodegeneration (BPAN) Detected in a Child with Epileptic Spasms. Cureus 2019; 11:e5404.
- Xiong Q, Li W, Li P, Zhao Z, Wu C, Xiao H. Functional evidence for a de novo mutation in WDR45 leading to BPAN in a Chinese girl. Mol Genet Genomic Med 2019; 7:e858.
- Uchino S, Saitsu H, Kumada S, Nakata Y, Matsumoto N. Stereotypic Hand Movements in beta-Propeller Protein-Associated Neurodegeneration: First Video Report. Mov Disord Clin Pract 2015; 2:190–1.
- Chen H, Qian Y, Yu S, Xiao D, Guo X, Wang Q, et al. Early onset developmental delay and epilepsy in pediatric patients with WDR45 variants. Eur J Med Genet 2019; 62:149–60.
- Xixis KI, Mikati MA. Epileptic spasms: a previously unreported manifestation of WDR45 gene mutation. Epileptic Disord 2015; 17:467–72.
- Redon S, Benech C, Schutz S, Despres A, Gueguen P, Le Berre P, et al. Intragenic deletion of the WDR45 gene in a male with encephalopathy, severe psychomotor disability, and epilepsy. Am J Med Genet A 2017; 173:1444–6.
- Burger BJ, Rose S, Bennuri SC, Gill PS, Tippett ML, Delhey L, et al. Autistic Siblings with Novel Mutations in Two Different Genes: Insight for Genetic Workups of Autistic Siblings and Connection to Mitochondrial Dysfunction. Front Pediatr 2017; 5:219.
- Takano K, Goto K, Motobayashi M, Wakui K, Kawamura R, Yamaguchi T, et al. Early manifestations of epileptic encephalopathy, brain atrophy, and elevation of serum neuron specific enolase in a boy with beta-propeller protein-associated neurodegeneration. Eur J Med Genet 2017; 60:521–6.
- Spiegel R, Shalev S, Bercovich D, Rabinovich D, Khayat M, Shaag A, et al. Severe infantile male encephalopathy is a result of early post-zygotic WDR45 somatic mutation. Clin Genet 2016; 90:560–2.
- Khalifa M, Naffaa L. Exome sequencing reveals a novel WDR45 frameshift mutation and inherited POLR3A heterozygous variants in a female with a complex phenotype and mixed brain MRI findings. Eur J Med Genet 2015; 58:381–6.