
ABSTRACT
Spiral ganglion neurons (SGNs) are auditory neurons that relay sound signals from the inner ear to the brainstem. The ototoxic drug cisplatin can damage SGNs and thus lead to sensorineural hearing loss (SNHL), and there are currently no methods for preventing or treating this. Macroautophagy/autophagy plays a critical role in SGN development, but the effect of autophagy on cisplatin-induced SGN injury is unclear. Here, we first found that autophagic flux was activated in SGNs after cisplatin damage. The SGN apoptosis and related hearing loss induced by cisplatin were alleviated after co-treatment with the autophagy activator rapamycin, whereas these were exacerbated by the autophagy inhibitor 3-methyladenine, indicating that instead of inducing SGN death, autophagy played a neuroprotective role in SGNs treated with cisplatin both in vitro and in vivo. We further demonstrated that autophagy attenuated reactive oxygen species (ROS) accumulation and alleviated cisplatin-induced oxidative stress in SGNs to mediate its protective effects. Notably, the role of the antioxidant enzyme PRDX1 (peroxiredoxin 1) in modulating autophagy in SGNs was first identified. Deficiency in PRDX1 suppressed autophagy and increased SGN loss after cisplatin exposure, while upregulating PRDX1 pharmacologically or by adeno-associated virus activated autophagy and thus inhibited ROS accumulation and apoptosis and attenuated SGN loss induced by cisplatin. Finally, we showed that the underlying mechanism through which PRDX1 triggers autophagy in SGNs was, at least partially, through activation of the PTEN-AKT signaling pathway. These findings suggest potential therapeutic targets for the amelioration of drug-induced SNHL through autophagy activation.
Abbreviations: 3-MA: 3-methyladenine; AAV : adeno-associated virus; ABR: auditory brainstem responses; AKT/protein kinase B: thymoma viral proto-oncogene; Baf: bafilomycin A1; CAP: compound action potential; COX4I1: cytochrome c oxidase subunit 4I1; Cys: cysteine; ER: endoplasmic reticulum; H2O2: hydrogen peroxide; HC: hair cell; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; NAC: N-acetylcysteine; PRDX1: peroxiredoxin 1; PTEN: phosphatase and tensin homolog; RAP: rapamycin; ROS: reactive oxygen species; SGNs: spiral ganglion neurons; SNHL: sensorineural hearing loss; SQSTM1/p62: sequestosome 1; TOMM20: translocase of outer mitochondrial membrane 20; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end-labeling; WT: wild type.
Introduction
Sensorineural hearing loss (SNHL) is a common sensory deficit worldwide and has severe impacts on patients’ quality of life and is a burden on society. Although the disorder is multifactorial, SNHL can be associated, in large part, with the impairment of spiral ganglion neurons (SGNs) [Citation1,Citation2]. SGNs are the inner-ear auditory neurons that transmit sound signals transduced by hair cells (HCs) to the cochlear nuclei in the brainstem [Citation3]. SGNs are indispensable for hearing, and loss of SGNs contributes to irreversible SNHL. Moreover, a required number of intact SGNs is necessary for the function of cochlear implants, which are currently the only effective tool for restoring hearing in patients with severe SNHL [Citation4]. Therefore, a better understanding of the molecular mechanisms that mediate the damage and restoration of SGNs could provide novel treatment and prevention strategies.
Cisplatin is a chemotherapeutic agent widely used against many tumor types [Citation5]. However, cisplatin has ototoxic side effects in about 62% of chemotherapy patients [Citation6–8], and it causes bilateral, progressive, and irreversible SNHL [Citation9] by directly damaging the HCs, the stria vascularis, and SGNs [Citation10,Citation11]. Studies have demonstrated that cisplatin interferes with SGN function and causes SGN loss, resulting in elevation of compound action potential (CAP) and auditory brainstem response (ABR) thresholds [Citation8,Citation10]. It has been suggested that reactive oxygen species (ROS) production plays an important role in the pathogenesis of cisplatin-induced ototoxicity [Citation8,Citation9,Citation12], and recent data show that cisplatin increases ROS generation in cochlear cells via several signaling molecules, including enhancing the activity of NADPH oxidase [Citation13,Citation14] and XDH/xanthine oxidase [Citation15], interfering with antioxidant defense systems [Citation16,Citation17], and stimulating inflammation [Citation18,Citation19], which is one of the first events triggered by exposure to cisplatin. ROS might then cause auditory cell damage and eventually cell death via lipid peroxidation [Citation20], protein nitration [Citation21,Citation22], DNA damage [Citation23], and the amplification of inflammatory processes. Nevertheless, the mechanism of cisplatin-induced ototoxicity has not been fully clarified yet, and methods for hearing preservation during cisplatin-based chemotherapy in patients are lacking.
Autophagy is essential for cellular survival by eliminating dysfunctional organelles, protein aggregates, and damaged macromolecules and by recycling the breakdown products [Citation24,Citation25]. Basal levels of autophagy occur in all eukaryotic cells for quality control of cytoplasmic components and to maintain cell homeostasis, and autophagy is rapidly upregulated under many pathological conditions such as starvation, oxidative stress, etc [Citation25]. In the auditory system, autophagy has been shown to be essential for the development of the cochlea and for the maintenance of hearing [Citation26,Citation27], as well as for SGN function [Citation28]. However, limited research on autophagy in the hearing field has been reported, and most such studies have focused on utilizing autophagic mechanisms to treat SNHL caused by HC damage, and the role of autophagy in SGN injury induced by cisplatin as well as the underlying molecular mechanisms remain unclear.
PRDX1 (peroxiredoxin 1) is a typical 2-cysteine (Cys) peroxiredoxin that catalyzes reduction reactions, converting hydrogen peroxide (H2O2) to water, and works as a multifunctional anti-oxidant [Citation29]. The upregulation of PRDX1 in cells and tissues under oxidative stress is believed to be one of the cellular recovery reactions after oxidative damage [Citation29–31]. Although a previous report showed that PRDX1 is universally expressed in the cochlea, including the HCs, the lateral wall, and the spiral ganglion region, little information exists regarding the function of PRDX1 in the cochlea [Citation32], and direct evidence that PRDX1 plays a role in SGN damage has been lacking.
In this study, we first measured the autophagic flux in SGNs after cisplatin treatment, and we then conducted a full investigation of the neuroprotective effects of autophagy against cisplatin-induced SGN damage both in vitro and in vivo. Finally, we identified the role of the specific antioxidant enzyme PRDX1 in modulating autophagy in SGNs after cisplatin injury. We found that PRDX1 enhanced autophagy by activating the PTEN (phosphatase and tensin homolog)-AKT/protein kinase B signaling pathway in SGNs that were incubated with cisplatin, and the activation of autophagy protected SGNs against cisplatin-induced damage by reducing oxidative stress and apoptosis in SGNs.
Results
Cisplatin injury increases autophagy and activates autophagic flux in the cultured cochlear SGNs
We first determined changes in autophagy in SGNs after cisplatin treatment. The middle-turn cochlear explants from postnatal day (P) 3 wild-type (WT) C57BL/6 mice were cultured and treated with 50 μM cisplatin for 48 h and harvested for further analysis. The treatment was based on our previously published data [Citation33], which shows significant apoptosis and SGN loss under this condition. Representative images of transmission electron microscopy (TEM) analysis in showed that the endoplasmic reticulum (ER) and Golgi apparatus took on a circular structure that enveloped the targeted cytoplasmic constituents and formed double-membrane vesicles, i.e. autophagosomes (red arrows), that eventually fused with lysosomes and formed autolysosomes (yellow arrows) in which the contents were degraded and recycled. Statistical analysis demonstrated that there were significantly more autophagosomes and autolysosomes in the SGNs after cisplatin treatment compared with the controls (). The expression of the autophagy marker MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) in SGNs in response to cisplatin injury was examined, and the LC3B puncta in the damaged SGNs were significantly increased (). The protein expression of LC3B-II () was significantly enhanced in SGNs after cisplatin administration compared to control cultures. Together, these results showed that autophagy was increased in SGNs after cisplatin treatment.
Figure 1. Increased autophagy in cochlear SGNs after cisplatin treatment in vitro. The middle-turn cochleae and SGNs from P3 C57BL/6 WT mice were cultured and incubated with cisplatin for 48 h. (A) TEM analysis evaluated the autophagy in SGNs. Representative images showed that the ER and Golgi apparatus took on a circular structure, which enveloped the targeted cytoplasmic constituents and formed double membrane vesicles, i.e. autophagosomes (red arrows), that eventually fused with lysosomes and formed autolysosomes (yellow arrows) in which the contents are degraded and recycled by lysosome enzymes, n = 4. Scale bars: 2 μm. (B) Statistical analysis demonstrated that there were significantly more autophagosomes and autolysosomes in the SGNs after cisplatin treatment compared with the controls. (C) Immunostaining demonstrated that the LC3B puncta (LC3B-labeling, red) in the damaged SGNs (TUBB3-labeling, green) were significantly increased after cisplatin administration, n = 6. Scale bars: 5 μm. (D) Quantification of the LC3B fluorescent puncta. (E) The protein expression of LC3B-II in SGNs treated with cisplatin was significantly increased compared to the control group, n = 6. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
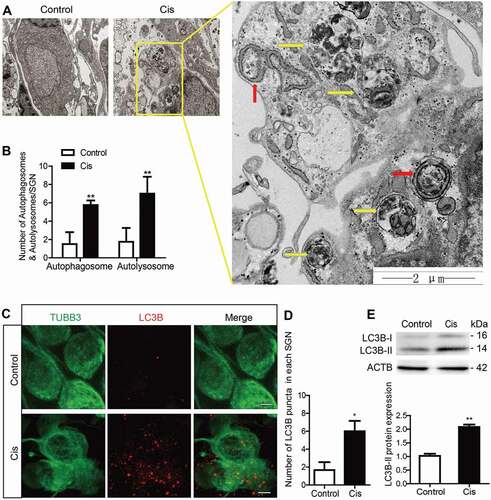
Next, given that autophagy is a dynamic process with multiple steps, we used several approaches to detect the autophagic flux in SGNs after cisplatin treatment. Bafilomycin A1 (Baf) treatment is a well-established assay for monitoring autophagosome biogenesis [Citation34,Citation35]. Baf inhibits the fusion of autophagosomes with lysosomes, thus blocking the degradation of autophagosomes. When cells are treated with Baf, the content of LC3B-II will be further increased if the autophagic flux is activated. Because the change in LC3B-II protein level or LC3B puncta reflects both autophagosome biogenesis and degradation, we used Baf treatment in SGNs to detect the effect of cisplatin on autophagosome biogenesis upon inhibition of autophagosomal degradation by Baf. As illustrated in , LC3B puncta were more abundant and the protein expression of LC3B-II was much higher in the Cis + Baf group than in the Baf-alone group or cisplatin-alone group (), suggesting that autophagosome synthesis in SGNs was enhanced by cisplatin injury even when the fusion of autophagosomes and lysosomes was blocked by Baf.
Figure 2. Cisplatin injury activates autophagic flux in the cultured cochlear SGNs. The middle-turn cochlear and SGN explants were cultured in vitro and treated with cisplatin (50 μM) alone or together with Baf (100 nM) for 48 h. (A) Immunostaining showed significantly more LC3B puncta in the Cis + Baf group compared with the Baf-alone group or cisplatin-alone group. Scale bars: 5 μm. (B) Quantification of the LC3B puncta in SGNs. (C) The expression of LC3B-II was significantly increased in the Cis + Baf group compared with Baf alone or cisplatin alone. (D) The protein levels of SQSTM1 were significantly decreased in cisplatin-treated SGNs and increased after Baf treatment compared with control SGNs. (E) Immunostaining showed anti-LC3B (green) and anti-SQSTM1 (red) labeling in SGNs. The colocalization puncta (white arrows) appeared after cisplatin treatment. Scale bars: 5 μm. (F) The cultured cochlear SGNs were incubated with Ad-mRFP-GFP-LC3B for 24 h, and then the medium was replaced with normal medium and the cultured SGNs were further treated with 50 μM cisplatin for 48 h. Immunostaining analysis showed that both the numbers of autophagosomes (yellow puncta, yellow arrows) and autolysosomes (red puncta, red arrows) were significantly increased in cisplatin-treated SGNs compared with the control group, n = 6 for each subtype. Scale bars: 5 μm. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01, *** P < 0.001
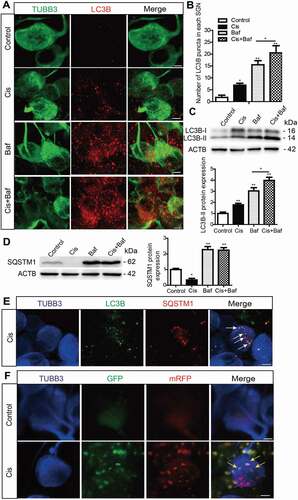
The cargo receptor protein SQSTM1/p62 (sequestosome 1), which interacts with ubiquitin and triggers the degradation of proteins in the proteasome or lysosome [Citation36], is an indicator of the autophagic degradation level. The protein expression of SQSTM1 was significantly decreased in cisplatin-treated SGNs compared with control SGNs (), confirming that the degradation of SQSTM1 was intensified in SGNs treated with cisplatin. However, the protein level of SQSTM1 was not significantly changed in SGNs co-treated with cisplatin and Baf compared with SGNs treated with Baf alone, which might be because the fusion of autophagosomes with lysosomes was blocked by Baf (). The co-localized LC3B and SQSTM1 puncta were found in the cytoplasm of SGNs after cisplatin treatment, indicating that the fusion of autophagosome-lysosome complexes was enhanced in SGNs after cisplatin injury ().
mRFP-GFP-LC3B is a dual-fluorescence fusion protein used to measure the level of autophagic flux in cells, and the yellow signal (the overlapping of red and green fluorescence) indicates autophagosomes that have not fused with lysosomes, while the red signal corresponds to autolysosomes. In this experiment, SGNs were transfected with the mRFP-GFP-LC3B adenovirus (Ad-mRFP-GFP-LC3B), and the numbers of both yellow puncta and red puncta were significantly increased in cisplatin-treated SGNs compared with the control group (), suggesting that both autophagosomes and autolysosomes were significantly increased in SGNs after cisplatin treatment. Taken together, all of the above results demonstrated that cisplatin could enhance both autophagosome synthesis and autophagosome-lysosome fusion, thus activating autophagic flux in SGNs.
Autophagy inhibits apoptosis and promotes the survival of SGNs after cisplatin injury in vitro
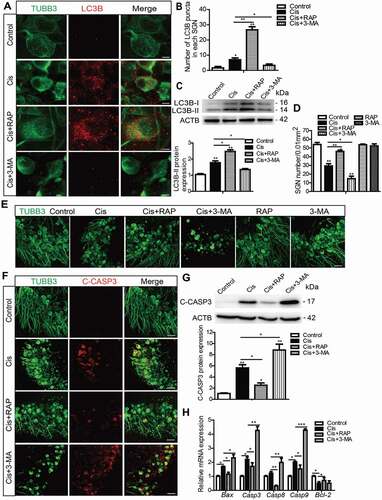
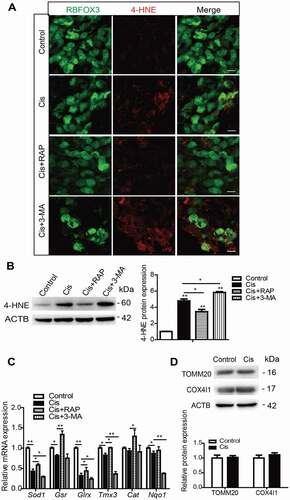
To examine the role of activated autophagy in the process of SGN damage induced by cisplatin, we performed experiments with upregulating or downregulating autophagy by the autophagy activator rapamycin (RAP) or the autophagy inhibitor 3-methyladenine (3-MA) in SGNs treated with cisplatin. The doses of RAP and 3-MA were determined according to our previous publication [Citation37] and our preliminary results of dose response (Figures S1A and S1B, Figures S1E and S1F), which showed that pretreatment of 0.1 μM RAP or 5 mM 3-MA effectively increased or decreased the number of surviving SGNs after cisplatin damage in vitro. First, we detected the effect of RAP and 3-MA on the expression of LC3B in SGNs exposed to cisplatin (, Figure S2A). The numbers of LC3B puncta and the LC3B-II protein expression level were both increased in the Cis + RAP group, while they were both decreased significantly in the Cis + 3-MA group compared with the cisplatin-only group (), validating that autophagosome formation was upregulated in SGNs treated with the autophagy activator and was downregulated with the autophagy inhibitor. The survival of SGNs was then determined, and it was shown that activating autophagy with RAP significantly increased the number of surviving SGNs, while inhibiting autophagy with 3-MA significantly decreased it after co-treatment with cisplatin (). However, the number of SGNs was not significantly changed when cells were treated with 0.1 μM RAP or 5 mM 3-MA alone without cisplatin (), demonstrating that the survival of SGNs was not affected by RAP or 3-MA alone at the concentration that we used in this study. We further investigated the effect of autophagy on regulating cisplatin-induced apoptosis of SGNs in vitro. After the drug treatment, cleaved-CASP3 (caspase 3) immunostaining showed that distinct cleaved-CASP3-positive SGNs were found in the cisplatin-treated group, while they were decreased in the Cis + RAP group but further increased in the Cis + 3-MA group compared to the cisplatin-only group (). Western blot assessment and statistical analysis of cleaved-CASP3 protein levels () were consistent with the immunostaining results, demonstrating that the CASP3-related apoptosis was reduced by RAP and increased by 3-MA in SGNs treated with cisplatin. Apoptosis analysis by TUNEL and flow cytometry assay were also similar to the results of cleaved-CASP3 detection, and the immunostaining signals of TUNEL in SGNs and the proportions of apoptotic cells were significantly reduced in the Cis + RAP group, while they were further increased in the Cis + 3-MA group (Figure S3A and S3B). The expression of apoptosis-related genes was also used to assess apoptosis in SGNs. The higher mRNA levels of the proapoptotic genes Casp3, Casp8, Casp9, and Bax induced by cisplatin were significantly decreased in the Cis + RAP group and increased in the Cis + 3-MA group compared with the cisplatin-only group, while the lower mRNA expression of the antiapoptotic gene Bcl2 was enhanced in the Cis + RAP group (). Taken together, these results suggest that the activation of autophagy promoted SGN survival and alleviated SGN apoptosis induced by cisplatin, while inhibition of autophagy attenuated SGN survival and enhanced cisplatin-induced SGN apoptosis.
Figure 3. Autophagy inhibits apoptosis and promotes the survival of SGNs after cisplatin injury in vitro. The cultured cochlear SGNs were pretreated with RAP (0.1 μM) or 3-MA (5 mM) for 6 h and then co-treated with 50 μM cisplatin for 48 h. (A and B) Immunostaining and quantification showed that the number of LC3B puncta (red) was significantly increased in the Cis + RAP group and decreased in the Cis + 3-MA group compared with cisplatin treatment alone. Scale bars: 5 μm. (C) The LC3B-II protein expression was significantly increased in the Cis + RAP group and decreased in the Cis + 3-MA group compared with cisplatin treatment alone. (D and E) SGN counting showed that RAP co-treatment promoted SGN survival compared with cisplatin exposure alone, while 3-MA accelerated SGN loss after cisplatin injury, and the number of SGNs was not significantly changed when cells were treated with 0.1 μM RAP or 5 mM 3-MA alone without cisplatin. Scale bars: 25 μm. (F and G) Fewer cleaved-CASP3-positive SGNs and lower protein levels of cleaved-CASP3were found in the Cis + RAP group, whereas the 3-MA co-treated SGNs had significantly more cleaved-CASP3-positive SGNs and higher cleaved-CASP3 protein expression compared with the cisplatin-only group. Scale bars: 25 μm. (H) The mRNA expression of Casp3, Casp8, Casp9, and Bax was significantly reduced in the Cis + RAP group and was significantly increased in the Cis + 3-MA SGNs compared to the cisplatin-only group, while the lower mRNA expression of the antiapoptotic gene Bcl-2 was enhanced in the Cis + RAP group. C-CASP3, cleaved-CASP3. n = 6 for each subgroup. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01, *** P < 0.001
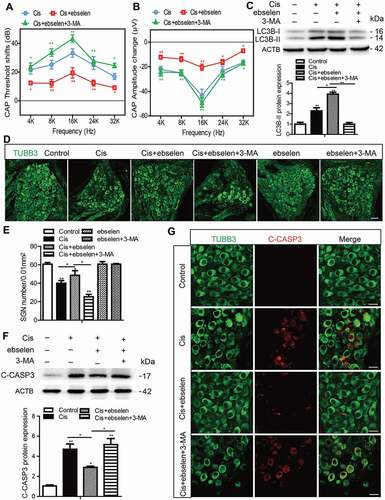
Autophagy inhibits apoptosis, promotes the survival of SGNs, and protects hearing function after cisplatin-induced hearing loss in mice
To determine whether autophagy can effectively protect SGNs and thus maintain hearing function after cisplatin-induced damage in vivo, RAP or 3-MA was co-injected with cisplatin in mice and the changes in hearing function, SGN survival, and SGN apoptosis were assessed. Before drug treatment, ABR tests were performed to make sure that all of the experimental mice had normal auditory function (Figure S4A). The cisplatin administration was performed according to our previous report [Citation33] showing that 3 mg/kg cisplatin intraperitoneal (i.p.) injection for 7 days leads to significant hearing loss and SGN loss in mice. The protocol for RAP treatment was based on our previous study [Citation38], and the method of 3-MA treatment was modified from the published report [Citation39]. First, the hearing function in mice after drug treatment was monitored by ABR and CAP. The ABR thresholds of all frequencies were increased in the cisplatin-treated group compared with the control group, and the shifts in thresholds were significantly lower in the Cis + RAP group, while they were much higher in the Cis + 3-MA group compared to those in the cisplatin-only mice (, Figure S4A). Treatment with RAP or 3-MA alone did not cause any significant ABR threshold shifts in the mice (Figure S4A). Given that the ABR tests the whole auditory transduction pathway, including the HCs, the auditory neurons, and the auditory cortex, we further measured the hearing function in mice after drug treatment by determining the CAP, which is the synchronized activity of the auditory nerve fibers. As illustrated in , a clear elevation in CAP threshold was observed across all tested frequencies after cisplatin administration, to some degree reflecting disturbed auditory nerve function by the ototoxic exposure. CAP input/output functions provide a more comprehensive view of neural activity at suprathreshold intensities, and shows the CAP amplitude measured at 90 dB SPL across frequencies demonstrating a significant reduction in the cisplatin group compared to the control group. Co-treatment with RAP significantly decreased the CAP threshold and increased the amplitudes compared with the cisplatin-only treatment in mice, whereas 3-MA administration had the opposite effect (). Together, these auditory function results suggest that the administration of RAP rescued the cisplatin-induced hearing loss, whereas 3-MA accelerated it in mice in vivo.
Figure 4. Autophagy inhibits apoptosis, promotes the survival of SGNs, and protects hearing function after cisplatin-induced damage in vivo. WT mice were injected i.p. with 3 mg/kg cisplatin daily for 7 days starting at P30 alone or co-treated with 1 mg/kg RAP i.p. every other day from P14 until P36, or with 15 mg/kg 3-MA i.p. every day from P14 until P36. (A) The ABR thresholds of all frequencies were increased in the cisplatin-treated group compared with the control group, and the shifts of thresholds were significantly lower in the Cis + RAP group, while they were much higher in the Cis + 3-MA group, compared to those in the cisplatin-only mice. (B and C) CAP threshold elevation (B) and amplitude reduction measured at 90 dB SPL (C) were observed across all tested frequencies. Co-treatment with RAP significantly decreased the CAP threshold and increased amplitudes compared with the cisplatin-only treatment in mice, whereas 3-MA administration had the opposite effect. (D and F) The LC3B puncta (D, red) and LC3B-II protein expression (F) in SGNs were increased after cisplatin administration, and RAP co-treatment further upregulated these, while 3-MA further downregulated them compared with the cisplatin-only group. Scale bars: 5 μm. (E and G) There were fewer cleaved-CASP3-positive SGNs and the protein level of cleaved-CASP3 was reduced in the Cis + RAP group compared with the cisplatin-only controls, while they were both increased in the Cis + 3-MA group. Scale bars: 12.5 μm. (H and I) Pretreatment with RAP protected SGN survival compared with the cisplatin-only group, while 3-MA accelerated SGN loss after cisplatin injury, and RAP or 3-MA treatment alone did not cause any significant differences in SGN numbers compared to the control group in the absence of cisplatin damage. Scale bars: 25 μm. n = 6 for each group. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
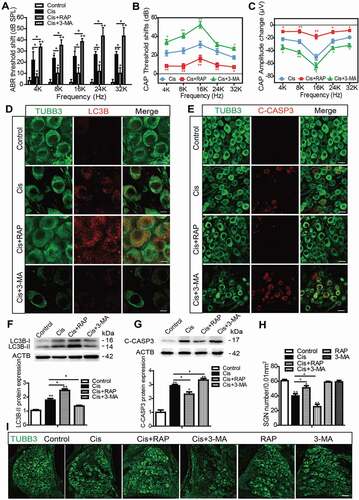
After physiological investigations, the cochleae were processed for immunostaining or western blot. The numbers of LC3B puncta and the LC3B-II protein expression level were increased significantly after cisplatin administration in SGNs compared to the control group (), indicating that cisplatin treatment also triggered autophagy activation in SGNs in vivo. In addition, both the numbers of LC3B puncta and the LC3B-II protein expression levels were significantly upregulated in the Cis + RAP group, whereas both were significantly downregulated in the Cis + 3-MA group compared with the cisplatin-only group (). Similar to the results in the in vitro experiment, cell counting showed that there were significantly more surviving SGNs in the Cis + RAP group but significantly fewer SGNs in the Cis + 3-MA group compared with the cisplatin-only group (). Treatment with RAP or 3-MA alone did not cause any significant differences in SGN numbers compared to the control group in the absence of cisplatin damage (). In addition, the cleaved-CASP3-positive SGNs and the protein level of cleaved-CASP3 were both reduced in the Cis + RAP group compared with the cisplatin-only controls, while they were both increased in the Cis + 3-MA group (). Together, these results demonstrated that activation of autophagy prevented SGN apoptosis, promoted SGN survival, and protected hearing function in vivo, while inhibition of autophagy made SGNs more vulnerable to the ototoxicity induced by cisplatin.
In addition, a previous study demonstrates that RAP treatment alleviates cisplatin-induced ototoxicity in rats in vivo [Citation40], and considering that cisplatin is well known to cause partial or complete death of the outer hair cells (OHCs), and to a lesser extent damage SGNs [Citation8], we therefore also analyzed the OHC loss after drug treatments in the mice to further evaluate the effect of autophagy on OHC survival after cisplatin damage in vivo. Because the OHCs in the basal turn are the most vulnerable to cisplatin injury [Citation6], the basal turn cochlear HCs were separated and stained with MYO7A/myosin 7a to measure the OHC loss in the different groups. Cisplatin treatment induced significant OHC loss in the basal turn of the cochlea, which was largely reduced after the RAP co-treatment, whereas it was significantly increased after co-treatment with 3-MA compared with the cisplatin-only group (Figure S4C). These results suggested that autophagy also plays a protective role against HC damage induced by cisplatin in vivo.
Autophagy attenuates cisplatin-induced oxidative stress in SGNs
Recent studies of hearing loss reveal that autophagy can serve in an anti-oxidative capacity by eliminating ROS in auditory HCs [Citation37,Citation41,Citation42]. In this study, we measured the changes in 4-HNE expression in order to evaluate the oxidative stress levels in SGNs. Compared with the cisplatin-only group, the immunostaining of 4-HNE () and the protein level of 4-HNE in SGNs () were significantly reduced after RAP cotreatment, while they were further increased in the cisplatin + 3-MA group, indicating that the level of oxidative stress was decreased in damaged SGNs by upregulating autophagy and was increased by inhibiting autophagy after cisplatin exposure. The accumulation of ROS was also demonstrated by the intensified immunostaining signals of MitoSOX Red in SGNs after cisplatin treatment (Figure S5A), and the immunostaining of MitoSOX Red was significantly reduced after RAP cotreatment, while it was increased in the Cis + 3-MA group. We also found that RAP significantly enhanced the mRNA expression of four antioxidant genes (Sod1, Gsr, Glrx, and Cat), while 3-MA significantly downregulated the expression of five antioxidant genes (Sod1, Gsr, Glrx, Tmx3, and Nqo1) compared with the cisplatin-only group (). Because autophagy can reduce oxidative damage and ROS levels through the removal of protein aggregates and damaged organelles such as mitochondria [Citation43], we also determined the efficiency of autophagy in removing and degrading damaged mitochondria in SGNs after cisplatin treatment. However, the protein expression of mitochondrial proteins, including the TOMM20 (translocase of outer mitochondrial membrane 20), which is an outer mitochondrial membrane protein, and the COX4I1/COX IV (cytochrome c oxidase subunit 4I1), which is an inner mitochondrial membrane protein, were not statistically changed in our experiments (), suggesting that the mitochondria are not eliminated by autophagy (mitophagy) in SGNs after cisplatin treatment.
Figure 5. Autophagy attenuates cisplatin-induced oxidative stress in SGNs. The cultured cochlear SGNs were pretreated with RAP (0.1 μM) or 3-MA (5 mM) for 6 h and then co-treated with 50 μM cisplatin for 48 h. (A and B) Compared with the cisplatin group, the immunostaining signals (A) and the protein levels of 4-HNE (B) in SGNs were significantly reduced after RAP cotreatment, while they were further increased in the Cis + 3-MA group. Scale bars: 12.5 μm. (C) RAP co-treatment significantly upregulated the mRNA expressions of Sod1, Gsr, Glrx, and Cat, while 3-MA co-treatment significantly downregulated the mRNA expressions of Sod1, Gsr, Glrx, Tmx3, and Nqo1 compared with the cisplatin-only group. (D) There were no statistically significant changes of mitochondrial protein levels (TOMM20 and COX4I1/COX IV) in our experiments. n = 6 for each group. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
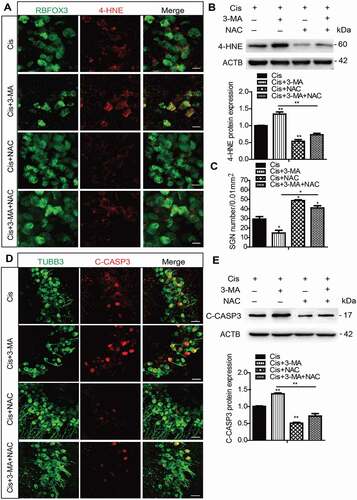
Next, a rescue experiment was performed using the antioxidant N-acetylcysteine (NAC) to further confirm the effect of autophagy on suppressing ROS accumulation in SGNs after cisplatin treatment. The dose of NAC was chosen according to our published study [Citation33] which shows that 2 mM NAC treatment successfully rescues the SGN loss from cisplatin damage in vitro. After NAC treatment, the immunofluorescence of 4-HNE () was weaker in the Cis + NAC group compared to the cisplatin-only group, and it was also lower in the Cis + 3-MA + NAC group compared to the Cis + 3-MA group (). Statistical analysis of the protein expression levels of 4-HNE in was consistent with the immunostaining results. The results of the MitoSOX assay also showed that the ROS levels were reduced in the Cis + NAC group compared to the cisplatin-only group, and they were decreased in the Cis + 3-MA + NAC group compared to the Cis + 3-MA group (Figure S5B). Accordingly, we found that the NAC treatment significantly increased the number of surviving SGNs () and decreased the expression of cleaved-CASP3 () in the Cis + NAC group compared to the cisplatin-only group, as well as in the Cis + 3-MA + NAC group versus the Cis + 3-MA group. Apoptosis analysis by TUNEL and flow cytometry assay also revealed that the immunofluorescence signals of TUNEL and the apoptotic cell percentages were reduced in the Cis + NAC group compared to the cisplatin-only group, and they were also decreased in the Cis + 3-MA + NAC group compared to the Cis + 3-MA group (Figure S3C and S3D). The NAC treatment alone without cisplatin did not cause significant differences in 4-HNE expression or SGN number (Figure S2B and S2C). Collectively, these observations suggest that antioxidant treatment successfully rescued the exacerbated apoptosis and SGN loss induced by autophagy inhibition after cisplatin injury.
Figure 6. Antioxidant treatment successfully rescued the increased SGN loss induced by autophagy inhibition after cisplatin injury. The cultured middle turn cochleae were treated with cisplatin (50 μM) alone, cotreated with NAC (2 mM), or pretreated with 3-MA (5 mM) for 6 h then cotreated with NAC for 48 h. (A and B) Immunofluorescence signals (A) and the protein expression levels (B) of 4-HNE were lower in the Cis + NAC group compared to the cisplatin-only group, and they were also reduced in the Cis + 3-MA + NAC group compared to the Cis + 3-MA group. Scale bars: 12.5 μm. (C-E) The NAC treatment significantly increased the number of surviving SGNs (C) and decreased the expression of cleaved-CASP3 (D and E) in the Cis + NAC group compared to the cisplatin-only group, as well as in Cis + 3-MA + NAC group versus the Cis + 3-MA group. Scale bars: 25 μm. n = 6 for each subgroup. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
PRDX1 is activated and regulates the autophagy activity in SGNs after cisplatin injury
To understand whether PRDX1 plays a role in SGNs after cisplatin damage, we investigated the expression changes of PRDX1 in cisplatin-treated SGNs. PRDX1 expression in cultured SGNs from P3 WT mice was significantly enhanced, even though the surviving SGN number was decreased after cisplatin treatment (). This result confirmed that PRDX1 signaling was activated in response to cisplatin-induced SGN damage. The experiments in prdx1−/− mice, in which PRDX1 expression was absent (Figures S6A and S6B), showed that there was no difference in the SGN numbers in prdx1−/− mice compared to the WT control mice without cisplatin damage, but the number of surviving SGNs was significantly reduced in the prdx1−/− mice compared to WT mice after cisplatin injury (), suggesting that PRDX1 deficiency aggravated SGN loss after cisplatin injury in vitro. In addition, a previous study reported that PRDX1 is also highly expressed in cochlear HCs, so we also measured the effect of PRDX1 on HC damage induced by cisplatin. However, our results suggest that PRDX1 was not effective in preventing HC damage-induced by cisplatin as the immunolabeling intensity of PRDX1 in cultured HCs of P3 WT mice exposed to cisplatin was similar to that in control HCs (Figure S6C), and there was no significant difference in HC loss between the prdx1−/− mice and WT mice after cisplatin treatment (Figures S6D and S6E).
Figure 7. PRDX1 is activated and regulates the autophagy activity in SGNs after cisplatin injury. The middle-turn cochleae and SGNs from P3 C57BL/6 WT mice or prdx1−/− mice were cultured and incubated with cisplatin for 48 h. (A and B) Immunostaining and western blot revealed that the expression of PRDX1 was significantly increased in SGNs after cisplatin administration. Scale bars: 12.5 μm. (C and D) The number of surviving SGNs was significantly reduced in prdx1−/− mice after cisplatin administration, while there was no difference in the SGN numbers in prdx1−/− mice compared to the WT control mice without cisplatin damage. Scale bars: 25 μm. (E-G) The puncta numbers of LC3B (E and F) and the protein expression of LC3B-II (G) were downregulated significantly in the Cis + prdx1−/− group compared with the cisplatin-only group. Scale bars: 5 μm. n = 6 for each subgroup. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
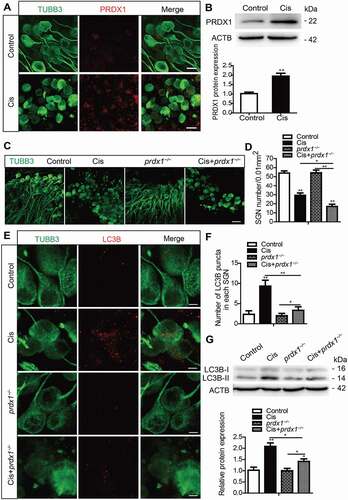
We then studied the role of PRDX1-dificiency in regulating autophagy in SGNs after cisplatin injury. Without cisplatin damage, there were no significant differences in either the numbers of LC3B puncta or in the protein expression level of LC3B-II in prdx1−/− mice compared to the control mice, but both of them were significantly decreased after cisplatin treatment in the Cis + prdx1−/− group compared with the cisplatin-treated WT group (), thus validating that deficiency in PRDX1 effectively suppressed autophagy in cisplatin-damaged SGNs, and indicating that PRDX1 might be involved in activating autophagy in SGNs.
The 2-Cys PRDX mimic ebselen and overexpression of PRDX1 protect against cisplatin-induced oxidative stress and apoptosis and reduce SGN loss by activating autophagy in SGNs
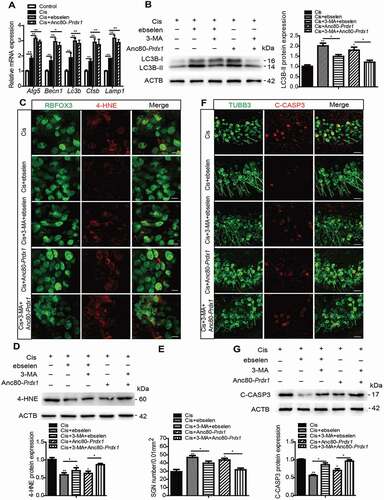
To further investigate the effect of PRDX1 on autophagy as well as subsequent neuroprotection in SGNs, we used two different methods to overexpress PRDX1 in SGNs treated with cisplatin. One way was to use the 2-Cys PRDX-mimic ebselen, which blocks intracellular H2O2 through 2-Cys PRDX-like mechanisms so as to mimic the effect of PRDX1. The other way was to use the adeno-associated virus (AAV) Anc80L65, which is reported to efficiently infect SGNs [Citation44], to construct the AAV virus Anc80L65-mNeonGreen-Prdx1-HA (Anc80-Prdx1) that overexpresses PRDX1 in SGNs after virus transfection. The efficiencies of virus transfection showed that about 65% of SGNs were transfected as indicated by the mNeonGreen reporter (Figure S6F), and the expression of PRDX1 was increased significantly in SGNs after transfection with Anc80-Prdx1 compared with the control group (Figure S6G). The dose of ebselen was determined according to a previous publication [Citation45] and to our preliminary dose response results (Figure S1C and S1G), which showed that pretreatment with 30 μM ebselen for 1 h significantly increased the number of surviving SGNs after cisplatin damage in vitro. It also showed that neither the number of SGNs nor the LC3B expression were significantly changed after SGNs were treated with 30 μM ebselen or Anc80-Prdx1 alone without cisplatin (Figure S2D and S2E), thus validating that the survival and autophagy of SGNs was not affected by ebselen or Anc80-Prdx1 alone at the dose that we used in this study. Next, the effect of PRDX1 on autophagy in SGNs after cisplatin damage was tested. RT-PCR results showed that the mRNA expressions of the autophagy-related genes Atg5, Becn1, and Lc3b and the lysosome-related genes Ctsb and Lamp1 were all upregulated in SGNs induced by cisplatin, and the overexpression of PRDX1 further enhanced the above gene transcription compared to the cisplatin-only group (). These above results together indicated that the upregulation of PRDX1 in SGNs after cisplatin exposure results in further activation of autophagic flux in SGNs. Moreover, ebselen and Anc80-Prdx1 both significantly increased the protein expression of LC3B-II in SGNs after cisplatin damage compared to the cisplatin-only group (), whereas the inhibition of autophagy by 3-MA effectively prevented the upregulation of LC3B-II expression induced by ebselen or Anc80-Prdx1. We further found that the expression of both 4-HNE () and cleaved-CASP3 () were significantly decreased and the number of surviving SGNs () was significantly increased in the Cis + ebselen group and Cis + Anc80-Prdx1 group compared to the cisplatin-only group, while co-treatment with 3-MA significantly reduced the effects of both ebselen and Anc80-Prdx1 on SGN ROS production, survival, and apoptosis. Collectively, these results show that both ebselen and PRDX1 overexpression could effectively activate autophagy, which further reduced oxidative stress and apoptosis in SGNs and increased the survival of SGNs after cisplatin injury.
Figure 8. Ebselen and overexpression of PRDX1 protect against cisplatin-induced ROS accumulation and apoptosis and reduce SGN loss by activating autophagy in SGNs. The cultured cochleae from WT mice were treated with cisplatin (50 μM) alone, cotreated with ebselen (30 μM, pretreated for 1 h), cotreated with Anc80-Prdx1 (4 × 1010 GC/mL, preincubation for 24 h), or cotreated with 3-MA (5 mM, pretreated for 6 h) for 48 h. (A) RT-PCR results showed that the mRNA expression of genes associated with autophagic flux, including Atg5, Becn1, Lc3b, Ctsb, and Lamp1, was upregulated in SGNs induced by cisplatin, and the overexpression of PRDX1 by ebselen or Anc80-Prdx1 further enhanced the above gene transcriptions compared to the cisplatin-only group. (B) Ebselen and Anc80-Prdx1 both significantly increased the protein expression of LC3B-II compared to the cisplatin-only group, whereas the co-treatment with 3-MA effectively prevented the upregulation of LC3B-II expression induced by ebselen or Anc80-Prdx1. (C-G) The expressions of 4-HNE (C and D) and cleaved-CASP3 (F and G) were significantly decreased, and the number of surviving SGNs (E) was significantly increased in the Cis + ebselen group and in the Cis + Anc80- Prdx1 group compared to the cisplatin-only group, while co-treatment with 3-MA significantly reduced the effects of both ebselen and Anc80-Prdx1 on SGN ROS production, cell survival, and apoptosis. Scale bars: 12.5 μm in (C). Scale bars: 25 μm in (F). n = 6 for each subgroup. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
Ebselen improves hearing function, promotes SGN survival, and inhibits SGN apoptosis after cisplatin-induced damage in vivo by activating autophagy
To verify the role of PRDX1 in activating autophagy in SGNs and consequently protecting SGNs against cisplatin damage in vivo, we further examined the role of ebselen in SGNs and hearing function in adult mice. The method of ebselen treatment in vivo was modified from the published study [Citation46], and the treatment of ebselen alone or ebselen together with 3-MA did not cause significant differences in ABR thresholds in mice without cisplatin administration (Figure S4D). We first compared the auditory neuron function by CAP assessments, and the CAP threshold shifts and amplitude changes of all frequencies were both lower in the Cis + ebselen group than in the cisplatin-only mice, while the CAP threshold shifts and amplitude changes were significantly increased in the Cis + ebselen + 3-MA group compared with the Cis + ebselen group (). Consistent with the in vitro data, ebselen administration significantly increased the protein expression of LC3B-II in SGNs compared to the cisplatin-only group, while 3-MA treatment inhibited the upregulation of LC3B in the Cis + ebselen + 3-MA group compared to the Cis + ebselen group (), indicating that ebselen treatment also effectively induced autophagy activation in SGNs in vivo. Accordingly, the mean density of middle-turn SGNs was increased in mice after co-treatment with ebselen compared to the cisplatin-only mice, whereas it decreased significantly in the Cis + ebselen + 3-MA group compared with the Cis + ebselen group (), but both the number of SGNs and the LC3B expression were not significantly changed after mice were treated with ebselen and 3-MA alone without cisplatin injury (, Figure S4E). Furthermore, the expression of cleaved-CASP3 was reduced in the Cis + ebselen mice compared to the cisplatin-only mice, while it was upregulated significantly in the Cis + ebselen + 3-MA group compared with the Cis + ebselen group (). Therefore, these results suggest that ebselen could effectively improve hearing function, promote SGN survival, and inhibit apoptosis by inducing autophagy in SGNs after cisplatin-induced damage in vivo.
Figure 9. Ebselen improves hearing function, promotes SGN survival, and inhibits SGN apoptosis after cisplatin-induced damage in vivo by activating autophagy. WT mice were injected i.p. with 3 mg/kg cisplatin alone daily for 7 days starting at P30 or with 15 mg/kg 3-MA i.p. every day from P14 until P36 or were co-injected i.p. with 15 mg/kg ebselen daily from P30 for 7 days. (A and B) The CAP threshold shifts (A) and amplitude changes (B) of all frequencies were both lower in the Cis + ebselen group than in the cisplatin-only mice, while in the Cis + ebselen + 3-MA group the CAP threshold shifts and amplitude changes were significantly increased compared with the Cis + ebselen group. (C) Ebselen administration significantly increased the protein expression of LC3B-II in SGNs compared to the cisplatin-only group, while 3-MA treatment inhibited the upregulation of LC3B in the Cis + ebselen + 3-MA group compared to the Cis + ebselen group. (D and E) The mean density of SGNs was increased after co-treatment with ebselen compared to the cisplatin-only mice, but decreased in the Cis + ebselen + 3-MA group compared with the Cis + ebselen group, and the number of SGNs was not significantly changed after mice were treated with ebselen or 3-MA alone without cisplatin injury. Scale bars: 25 μm. (F and G) The expression of cleaved-CASP3 was decreased in mice after co-treatment with ebselen compared to the cisplatin-only mice, whereas it decreased significantly in the Cis + ebselen + 3-MA group compared with the Cis + ebselen group, n = 6 for each group. Scale bars: 12.5 μm. All data are presented as the mean ± SEM, *P < 0.05, **P < 0.01
PRDX1 regulates the PTEN-AKT signaling pathway to activate autophagy in SGNs after cisplatin treatment
The mechanisms by which PRDX1 is involved in the regulation of autophagy remain unclear. Recent studies demonstrate that the PTEN-AKT signaling pathway plays a critical role in the regulation of autophagy [Citation18,Citation47,Citation48], and PTEN is highly expressed in auditory sensory neurons from embryonic day (E)10.5 to P7 [Citation49]. In addition, conditional depletion of PTEN leads to defects in nerve innervation and SGN survival during the development of the inner ear [Citation50]. Interestingly, it is showed that PRDX1 can bind to PTEN to form a dimer that inhibits PTEN’s phosphatase activity and thus maintains the activity of the PTEN-AKT signaling pathway [Citation51,Citation52]. Therefore, we hypothesized that PRDX1 can activate autophagy by regulating the PTEN-AKT signaling pathway. To test our hypothesis, we first measured the effect of PRDX1 on PTEN-AKT signaling in SGNs. The protein expression of PTEN was significantly increased and the ratio of phosphorylated (p)-AKT:AKT was remarkably decreased in cultured SGNs after cisplatin injury, and treatment with ebselen caused a higher expression of PTEN and a lower ratio of p-AKT:AKT in SGNs compared to the cisplatin-only group, while the lack of PRDX1 in SGNs from prdx1−/− mice led to an opposite pattern (), indicating that PRDX1 might upregulate PTEN-AKT signaling in SGNs. SF1670, a highly potent and specific inhibitor of PTEN, was used to further determine the upstream regulatory effect of PRDX1 on the PTEN-AKT signaling pathway. The dose of SF1670 was chosen based on our preliminary results of dose response, which showed that 15 μM SF1670 significantly decreased the number of surviving SGNs after cisplatin damage in vitro (Figure S1D and S1H), and treatment with SF1670 alone did not cause significant differences in SGN numbers or LC3B expression in SGNs without cisplatin damage (Figures S2F and S2G). Our results showed that SF1670 effectively inhibited the expression of PTEN and enhanced the phosphorylation of AKT in SGNs after cisplatin treatment, while co-treatment with ebselen reversed the effect of SF1670 as the expression of PTEN was increased and the ratio of p-AKT:AKT was decreased in the Cis + ebselen + SF1670 group compared to the Cis + SF1670 group (). In contrast, treatment with SF1670 did not change the expression of PRDX1 in SGNs after cisplatin incubation (). Taken together, these results suggest that PRDX1 stimulated the upstream PTEN-AKT signaling pathway in SGNs after cisplatin injury.
Figure 10. PRDX1 regulates the PTEN-AKT signaling pathway to activate autophagy in SGNs after cisplatin treatment. (A and B) The cultured cochleae and SGNs from WT mice or prdx1−/− mice were incubated with cisplatin (50 μM) for 48 h or cotreated with ebselen (30 μM, pretreated for 1 h) and cisplatin (50 μM) for 48 h. The protein expression of PTEN was significantly increased and the ratio of p-AKT:AKT was significantly decreased in cultured SGNs after cisplatin injury. Treatment with ebselen caused increased expression of PTEN and a lower ratio of p-AKT:AKT in SGNs compared to the cisplatin-only group, while the lack of PRDX1 in SGNs from prdx1−/− mice led to an opposite changing pattern. (C and D) The cultured cochleae and SGNs from WT mice were incubated with cisplatin (50 μM) alone, cotreated with ebselen (30 μM, pretreated for 1 h), or cotreated with SF1670 (15 μM) for 48 h. SF1670 effectively inhibited the expression of PTEN and enhanced the phosphorylation of AKT in SGNs after cisplatin treatment, and co-treatment with ebselen reversed the effect of SF1670 as the expression of PTEN was increased and the ratio of p-AKT:AKT was decreased in the Cis + ebselen + SF1670 group compared to the Cis + SF1670 group. In contrast, treatment with SF1670 did not change the expression of PRDX1 in SGNs after cisplatin incubation. (E-I) The inhibition of PTEN by SF1670 significantly decreased the expression of LC3B-II (E and F), increased cleaved-CASP3 expression (G and H), and reduced the number of surviving SGNs (I) compared to the cisplatin-only group, whereas co-treatment with ebselen rescued the above effect of SF1670 in SGNs. Scale bars: 25 μm. n = 6 for each group. All data are presented as the mean ± SEM, * P < 0.05, ** P < 0.01
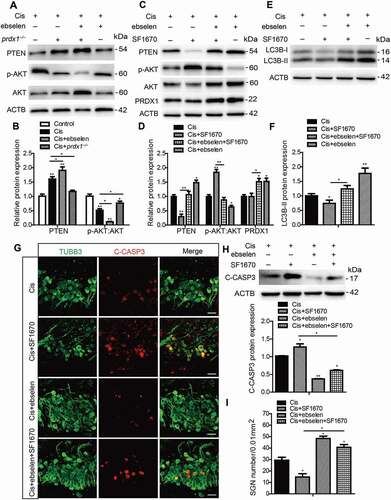
To verify the role of PTEN-AKT in activating autophagy and thus protecting SGNs against cisplatin damage, we further analyzed the level of LC3B-II, SGN apoptosis, and survival after inhibiting PTEN in SGNs. Inhibition of PTEN by SF1670 significantly decreased the expression of LC3B-II (), increased cleaved-CASP3 expression (), and reduced the number of surviving SGNs () compared to the cisplatin-only group, whereas co-treatment with ebselen rescued the above effect of SF1670 in SGNs. Collectively, these data indicated that PRDX1 activated autophagy and thus protected SGNs against cisplatin damage by upregulating the PTEN-AKT signaling pathway.
Discussion
Autophagy is a critical process involved in cochlear development and functional maturation [Citation26–28]. In the cochlea, the labeling of the autophagy marker LC3B is primarily localized in SGNs but not in glia cells, and the expression of LC3B increases intensely from E18.5 to P90 in SGNs, suggesting that autophagy might be a housekeeping mechanism necessary for SGN activity [Citation28]. In addition, several studies have reported that autophagy is enhanced in injured SGNs treated with aminoglycosides or lipopolysaccharide [Citation53–55], indicating that autophagy might also participate in auditory neuron damage. In the present study, we show for the first time that autophagy was significantly increased in the cochlear SGNs after cisplatin damage both in vitro and in vivo ( and ). Moreover, we demonstrated that the autophagic flux was effectively activated in SGNs treated with cisplatin, as evidenced by increased autophagosome synthesis and autophagosome-lysosome fusion as well as the intensified degradation of the SQSTM1 protein (). Our results also suggested that treatment with Baf alone increased the number of LC3B puncta more than cisplatin alone. We reason that Baf treatment can inhibit autophagosomal degradation, which in turn will lead to significant accumulation of autophagosomes in SGNs (as indicated by the huge increases in the number of LC3B puncta and in the LC3B-II protein level). Therefore, Baf and cisplatin appear to affect LC3B through different mechanisms, i.e. inhibition of macroautophagy (Baf alone treatment) that results in autophagosome accumulation is differentiated from stimuli (cisplatin alone treatment) that result in effectively activated autophagic flux. The state of autophagic flux is important for revealing autophagy’s functions and for identifying autophagy-related diseases. In inner ear disorders, He et al. reported that activated autophagic flux is seen in HCs after neomycin damage and that autophagy plays a protective role against neomycin injury [Citation37]. However, in another mouse model treated with kanamycin and furosemide, impaired autophagic flux in SGNs, based on the accumulated lipofuscin and higher level of SQSTM1 protein, leads to SGN degeneration, and restoring autophagic flux attenuated the degeneration of SGNs [Citation55]. Our study is the first to report that autophagic flux was activated during SGN injury induced by cisplatin and suggests that autophagy might play a role in the SGN damage process.
The role of autophagy can be paradoxical and dual in suppressing and promoting cell survival, that is, autophagy primarily acts as a protective mechanism that might prevent cell death by providing energy and removing damaged mitochondria and misfolded proteins under stress conditions, but uncontrolled upregulation of autophagy can lead to excessive “self-digestion” and result in cell death [Citation24,Citation25,Citation56]. In fact, the “self-cleaning” mechanism of autophagy seems vital for SGN survival and for maintaining physiological functions because the regeneration of SGNs is impossible in vivo. However, to date it is still not established as to whether autophagy plays a protective role [Citation40,Citation57] or destructive role [Citation58,Citation59] in cisplatin ototoxicity. In the current work, by manipulating autophagy in SGNs using an autophagy activator and inhibitor, we discovered that the further increase in autophagy significantly alleviated SGN damage and attenuated hearing loss in mice after cisplatin injury, while the inhibition of autophagy had the opposite effect ( and ). Therefore, although the activated autophagy induced by cisplatin in SGNs was accompanied by significant SGN impairment, we conclude that the specific contribution of autophagy functions as a pro-survival mechanism that protects SGNs against cisplatin-induced ototoxicity rather than being responsible for inducing apoptotic cell death in SGNs. Our results are also consistent with reports that the apoptosis of H460 cells induced by cisplatin is augmented by 3-MA treatment, possibly mediated by TP53/p53 activation [Citation60], that the inhibition of autophagy promotes cisplatin-induced apoptotic cell death through ATG5 and BECN1 (beclin 1) in A549 cells [Citation61], and that PINK1 protects HCs and SGNs from cisplatin-induced ototoxicity by inducing autophagy [Citation62], all of which prove that autophagy induced by cisplatin plays a protective role in cells against cisplatin damage, although through different regulatory mechanisms.
There are close interactions between ROS and autophagy as reflected in the fact that ROS can initiate autophagy by acting as cellular signaling molecules [Citation16,Citation63–65] and that autophagy in turn attenuates oxidative damage by engulfing and degrading oxidized substances [Citation66,Citation67]. The current studies tend to support that the excessive production of ROS could increase cell damage, while the role of autophagy could exhibit duality – cell damage or cell survival – depending on the activation level of autophagy. In terms of cisplatin-related results, inhibiting ROS production limits cisplatin-induced apoptosis and autophagy in HEI-OC1 cells, but apoptosis is unaffected when autophagy is inhibited [Citation58]. In this study, we found that the increased autophagy suppressed the ROS accumulation in SGNs induced by cisplatin, while blocking autophagy by 3-MA significantly reduced the ROS generation, and the NAC rescue experiments further confirmed the antioxidative role of autophagy in protecting against cisplatin-induced SGN damage (). Our findings are consistent with previous reports about hearing loss showing that autophagy is capable of protecting HCs and hearing by reducing oxidative stress after aminoglycoside injury [Citation37] or noise exposure [Citation68]. However, our results suggest that autophagy may attenuate cisplatin-induced oxidative stress in SGNs by regulating oxidative stress-related genes (), but not by directly degrading the damaged mitochondria (). Autophagy has been shown to reduce levels of ROS through pathways such as chaperone-mediated autophagy pathway [Citation69], the SQSTM1 delivery pathway [Citation70], etc., and the key step in how autophagy attenuates oxidative stress in SGNs after cisplatin injury needs to be further explored.
PRDX1 is the most abundant and ubiquitously distributed member of the peroxiredoxin family, the members of which are considered to be the primary cellular guardians against oxidative stress in all living organisms [Citation71,Citation72]. Studies indicate that PRDX1 might be a biomarker of different kinds of cell damage induced by cisplatin because cisplatin induces a dramatic increase in PRDX1 expression in primary hepatocytes [Citation73] and because deficiency of PRDX1 in murine embryonic fibroblasts makes them more susceptible to cisplatin-induced cytotoxicity [Citation74]. In our study, as expected, we found that PRDX1 expression was significantly increased in SGNs after cisplatin treatment, and deficiency of PRDX1 aggravated SGN loss after cisplatin injury in vitro (), indicating that PRDX1 might play a protective role in SGNs against cisplatin injury. However, we did not observe upregulation of PRDX1 protein expression in HCs after cisplatin treatment or a statistical difference in HC loss between the prdx1−/− and WT mice (Figure S6). This finding for HCs is supported by the study of Le et al., who show that deficiency of PRDX1 is ineffective in protecting HCs against cisplatin-induced injury [Citation32]. Chen et al. show that peroxiredoxin 3, another subtype in the peroxiredoxin family, regulates cochlear HC survival in response to noise trauma and aminoglycoside treatment. Reports indicate that PRDX1 is strongly expressed in the central nervous system, controls neuronal differentiation [Citation75], and provides neuroprotection [Citation76]. Thus, our finding is interesting in that it indicates that the difference in cochlear cell origin leads to different roles for the PRDX subtypes, and PRDX1 is important for protection of auditory neurons in cisplatin ototoxicity. Although autophagy also plays a protective role against HC damage induced by cisplatin in vivo (Figure S4), we speculate that it is not related to the cell protection and autophagy regulation effects of PRDX1, and the molecular mechanisms need to be further studied.
Notably, we also observed a novel mechanism underlying the neuroprotective role of PRDX1 in which PRDX1 activates autophagy and subsequently alleviates ROS accumulation and apoptosis in SGNs as well as preserves SGN survival after cisplatin injury. Only a few studies have reported the role of PRDX1 in regulating autophagy so far, which remains largely elusive and poorly understood. One study reported that PRDX1 deficiency induces excessive oxidative stress and impaired autophagic flux in macrophages after treatment with oxidized LDL [Citation77], thereby promoting atherosclerosis in mice. In addition, PRDX1 mimics could rescue the impaired lipophagic efflux induced by excessive oxidative stress. Consistent with these observations, our findings validate that deficiency of PRDX1 suppresses autophagy in cisplatin-damaged SGNs () and that the upregulation of PRDX1 in SGNs after cisplatin exposure results in further activation of autophagic flux in SGNs, which protects SGNs from cisplatin injury (). In contrast, however, another study showed that PRDX1 inhibits autophagy activation through inhibition of TRAF6-mediated BECN1 ubiquitination in macrophages treated with lipopolysaccharide [Citation78]. Therefore, the relationship between PRDX1 and autophagy appears to be complex and can be different under various conditions, and thus it would be important to further elucidate the effects as well as the underlying mechanisms of PRDX1 on regulating autophagy in other disorders. In addition, the pharmacological agent ebselen, which reacts with the thioredoxin system and has already been approved for clinical use by the US Food and Drug Administration, was found to effectively protect SGNs and improve hearing after cisplatin injury by activating autophagy ( and ). A clinical trial published in The Lancet [Citation79] showed that ebselen efficiently provides otoprotection against noise-induced hearing loss. Until now no clinical pharmacological therapies against SGN damage have been verified, and our study suggests that ebselen might be a potential candidate, with a novel mechanism of activating autophagy in SGNs.
Finally, and most importantly, we investigated the possible signaling pathways that might be regulated by PRDX1 when activating autophagy in SGNs. PTEN is a lipid phosphatase that negatively controls phosphoinositide 3 kinase/PI3K-AKT signaling and contributes to cellular processes such as proliferation, differentiation, migration [Citation19,Citation80], and autophagy [Citation18,Citation47,Citation48]. In our study, we found that the PTEN-AKT cascade participates in autophagy activation in SGNs, which has not been reported before, and this further supports that PTEN might be essential for auditory neuronal maintenance and survival and for accurate nerve innervations of HCs as previously reported [Citation50]. In addition, we confirmed the hypothesis that PRDX1 activates autophagy and thus protects SGNs against cisplatin damage, at least partially by regulating the PTEN-AKT signaling pathway. Considering that both PRDX1 and PTEN are universally expressed in various cells and tissues, our findings suggest that the regulation of autophagy by the PRDX1-PTEN-AKT signaling cascade might also play a role in other cell types, especially under conditions of oxidative stress, and this is worthy of further investigation.
In summary, we showed in this study that autophagy was activated in SGNs after cisplatin injury, and the activation of autophagy inhibited ROS accumulation and oxidative stress-induced apoptosis and thereby protected SGNs against cisplatin-induced injury. In contrast, the suppression of autophagy in SGNs made them more vulnerable to cisplatin damage. We demonstrated that PRDX1 could activate autophagy and thus inhibit ROS accumulation and apoptosis and attenuate the SGN damage induced by cisplatin, and the underlying mechanism appears to be through activation of the PTEN-AKT signaling pathways in SGNs. Additionally, our results also indicate that the clinically approved drug ebselen might be beneficial for treating SGN injury and SNHL caused by ototoxic drugs. Our findings provide new insights into the interplay between autophagy and cisplatin-induced oxidative stress in SGNs and suggest potential therapeutic strategies for the amelioration of drug-induced ototoxicity through autophagy activation.
Materials and methods
Experimental animals
C57BL/6 J WT mice were purchased from the Animal Center of Shandong University (Jinan, China). The Prdx1 knockout mice (prdx1−/−) in the C57BL/6 background were constructed by Beijing Biocytogen Co., Ltd. (Beijing, China). The design for the prdx1−/− mice was based on the report by Neumann et al. [Citation29]. The genotyping for prdx1−/− mice with PCR was performed using genomic DNA from tail tips incubated in 70 μL 50 mM NaOH at 98°C for 1 h, followed by neutralization in 7 μL 1 M HCl. The genotyping primers are listed in Table S1. All animals were bred and housed in a climate-controlled room with an ambient temperature of 23 ± 2°C and a 12/12-h light/dark cycle.
All animal procedures were performed according to protocols approved by the Animal Care Committee of Shandong University, China (No. ECAESDUSM 20,123,011) and were consistent with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and to prevent their suffering.
Organotypic culture of neonatal mouse cochlear SGNs and drug treatments in vitro
To ensure consistency, only the middle turns of P3 mouse cochleae were dissected out and cultured for all in vitro experiments. Tissue dissection and culture procedures were performed as described in published reports [Citation33]. Briefly, WT mice and prdx1−/− mice were decapitated at P3, and the cochlear capsule was removed from the temporal bones and the stria vascularis was removed. The middle turns of the cochlear explants containing the SGNs were then placed onto 10-mm coverslips (Fisher Scientific, PA) pre-coated with CellTaK (Corning, 354,241) and incubated in Dulbecco’s Modified Eagle Medium/F12 (DMEM/F12; Gibco, 11,330,032) supplemented with 10% fetal bovine serum (FBS; Gibco, 10,099,133 C) and ampicillin (50 mg/ml; Sigma-Aldrich, A5354) at 37°C in a 5% CO2 atmosphere.
The next day, samples were changed into fresh culture media containing 50 μM cisplatin (Sigma-Aldrich, P4394) alone or with the following drugs for 48 h as described in the text: Baf (100 nM; Abcam, ab120497); pretreated with RAP (0.1 μM; Abcam, ab120224) or 3-MA (5 mM; Sigma-Aldrich, M9281) for 6 h; NAC (2 mM; Sigma-Aldrich, A7250); pretreated with ebselen (30 μM; Sigma-Aldrich, E3520) for 1 h; SF1670 (15 μM; MedChemExpress, HY-15,842).
In vivo drug treatments
All experiments were performed using age and sex-matched C57BL/6 WT mice. Control mice were administered 0.9% physiological saline (0.6 ml/100 g body weight by i.p. injection) for 7 consecutive days starting on P30. Cisplatin (3 mg/kg) was i.p. injected to mice from P30 for 7 consecutive days alone or with the following drugs treatments as described in the text: 1 mg/kg RAP (Sigma-Aldrich, V900930) i.p. every other day from P14 until P36; 15 mg/kg 3-MA i.p. every day from P14 until P36; 15 mg/kg ebselen daily from P30 for 7 days.
TEM analysis
The cultured cochleae were fixed after drug treatment in a mixture of 2.5% glutaraldehyde (Sigma-Aldrich, G5882) and 2% paraformaldehyde (Servicebio, G1101). This was followed by rinsing with PBS (Thermo Fisher Scientific, 10,010,023), fixing in 1% osmic acid for 2 h, rinsing again with distilled water, dehydration with graded ethanol, and soaking and embedding in epon812 (Sigma-Aldrich, 45,345). Semi and ultra-thin radial sections were cut from the middle turns and stained with lead citrate and uranyl acetate. Finally, the sections were observed using a transmission electron microscope (JEOL-1200EX) at JiNan WeiYa Bio-Technology Co., Ltd., (Jinan, China).
Western blot
After the drug treatment, for in vitro groups the total protein was obtained from cultured SGNs using radio-immunoprecipitation assay lysis buffer (Sigma-Aldrich, R0278). For the adult cochlear SGNs in the in vivo groups, to ensure that the extracts were mainly from SGNs the basilar membrane and stria vascularis as well as other tissues were removed and only the modioli of the cochlea were collected from 6 mice in each subgroup in pre-chilled 0.1 M PBS. A grinding machine (Jingxin JXFSTPRP-48, Shanghai, China) was used to triturate the modioli with bone in the radio-immunoprecipitation assay lysis buffer, the mixture was centrifuged at 14,200 × g, and the supernatant was collected.
A total of 30 μg of each protein sample was separated on 12% SDS-PAGE gels. The primary antibodies were anti-LC3B (1:1,000 dilution; Cell Signaling Technology, 3868), anti-SQSTM1/p62 (1:500 dilution; Abcam, ab91526), anti-TOMM20 (1:1,000 dilution; Proteintech, 11,802-1-AP), anti-COX4I1 (1:1,000 dilution; Abcam, ab16056), anti-4-HNE (1:1000; Abcam, ab46545), anti-PRDX1 (1:1,000 dilution; GeneTex, GTX101705), anti-PTEN (1:1,000 dilution; Cell Signaling Technology, 9559), anti-p-AKT (1:1,000 dilution; Cell Signaling Technology, 4060), anti-AKT (1:1,000 dilution; Abcam, ab179463), and anti-ACTB/β-actin (1:2,000 dilution; ZSGB-BIO, TA-09). Semi-quantification of the western blot results was performed using ImageJ software to measure the intensities of the bands. The band densities were normalized to background, and then the relative optical density ratio was calculated by comparison to ACTB. Each experiment was repeated at least three times.
Auditory functional tests
The auditory function was measured in a soundproof chamber. Prior to the recording, mice were anesthetized with ketamine (50 mg/kg, intramuscular injection) and xylazine (6 mg/kg, intramuscular injection). During the recording, the body temperature of the mice was maintained at 38°C with a thermal static heating device (FHC; USA). To record ABRs, the non-inverting electrode was inserted at the vertex of the skull, and the reference electrode and the grounding electrode were inserted in the neck posterior to the auditory bullas of both sides. The TDT System III (Tucker-Davis Technologies, USA) hardware and software were used to generate stimuli and record the responses, with 1,024 stimulus repetitions per recording. The acoustic stimuli were 10 ms tone bursts and were played through a broadband speaker (MF1; TDT) that was placed 10 cm in front of the head of the animal. The ABR thresholds were tested at 4, 8, 16, 24, and 32 kHz. At each frequency, the test was started at 90 dB SPL and tracked in 5 dB steps before the response disappeared. The thresholds of all three groups were judged by the same person. To record CAPs, a silver ball electrode was placed at the round window membrane via a small hole drilled in the auditory bulla posterior-inferior to the external ear canal. Tone bursts (4, 8, 16, 24, and 32 kHz, 10 ms duration, 0.5 ms rise/fall time) were generated using Tucker Davis Technologies (TDT) System III (Tucker-Davis Technologies, USA) hardware. Cochlear amplification (500 ×) was achieved via a preamplifier (RA16PA, TDT), averaged 512 times, and filtered (cutoff frequency 3.5 kHz) to display the CAP of the auditory nerve. Stimulus intensity was decreased in 5 dB steps from 90 to 0 dB SPL. The focuses were the CAP amplitude defined as the voltage difference between the first negative peak (N1) and the following positive peak (P1) and plotted as the stimulus intensity level to generate I/O functions at each frequency, and the CAP threshold was defined as the sound intensity that elicited a 3 µV CAP amplitude.
Immunostaining
After culturing or cryosectioning, the cochlear explants or tissue sections were fixed with 4% paraformaldehyde, permeabilized with 1% Triton X-100 (Sigma-Aldrich, T8787) in PBS (Thermo Fisher Scientific, 10,010,023), and immersed in blocking solution (0.1% Triton X-100, 8% donkey serum (Sigma-Aldrich, D9663), 1% bovine serum albumin (Sigma-Aldrich, A1933), 0.02% sodium azide in PBS) at room temperature for 1 h. After blocking, the samples were incubated with primary antibodies against TUBB3/Tuj1 (1:1,000 dilution; Neuromics, MO15013), LC3B (1:400 dilution; Cell Signaling Technology, 3868), SQSTM1 (1:500 dilution; Abcam, ab91526), and RBFOX3/NeuN (1:500; Cell Signaling Technology,12,943), 4-HNE (1:500; Abcam, ab48506), cleaved-CASP3 (1:400 dilution; Cell Signaling Technology, 9664), PRDX1 (1:500 dilution; GeneTex, GTX101705), or MYO7A (1:800 dilution; Proteus Biosciences, 25–6790) diluted in blocking solution at 4°C overnight. The next day, cells were incubated with secondary antibodies (Invitrogen, A21202, A21206, A31571, A31573, A10040) along with DAPI (Sigma-Aldrich, D9542) at room temperature for 1 h. Coverslips were then mounted and the samples were observed under a laser scanning confocal microscope (Leica SP8; Leica, Germany).
Cryosectioning
Cochleae from adult mice were removed and fixed with 4% PFA in PBS at 4°C overnight. Tissues were then incubated in 10%, 20%, and 30% sucrose (Sigma-Aldrich, V900116) in 1× PBS, embedded in O.C.T compound (Tissue-Tek; Sakura Finetek, 4583), snap frozen on dry ice, and stored frozen at −80°C. Frozen sections were then cut into 7-μm sections using a cryostat (Leica CM 1850; Leica, Germany).
Real time- polymerase chain reaction (RT-PCR)
TRIzol (Life Technologies, 15,596,026) was used to obtain the total RNA following the manufacturer’s instructions. The cDNA was synthesized by reverse transcription using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1622) following the manufacturer’s protocol. Quantitative RT-PCR was performed using SYBR Premix Ex Taq (TaKaRa Bio, RR420A) with Gapdh as the housekeeping gene. All data were analyzed using an Eppendorf Realplex 2. PCR primers for the genes are listed in Table S1.
Virus administration
The Ad-mCherry-GFP-LC3 was purchased from Shanghai Genechem Co., Ltd. (GCD0165968). For adenovirus transduction, the cultured cochlear SGNs were incubated with 5 × 107 PFU/mL Ad-mRFP-GFP-LC3B for 24 h. The medium was then replaced with normal medium, and the cultured SGNs were further treated with 50 μM cisplatin and cultured for 48 h. Samples were then prepared for immunostaining and imaged with a confocal microscope.
The PRDX1-overexpressing AAV was constructed with the AAV vector Anc80L65 (Addgene, 92,307) containing the mouse Prdx1 gene. Briefly, the single-stranded AAV Anc80L65 carrying the coding sequence of Prdx1 driven by a Cag promoter was generated using a virus-free helper system and named Anc80L65-mNeonGreen-Prdx1-HA (Anc80-Prdx1). Anc80-Prdx1 was purified using an iodixanol (BioVision, M1248-250) step with a titer of 3 × 1013 GC/ml [Citation81]. Titers were calculated by qPCR with WPRE primers listed in Table S1. Virus aliquots were stored at −80 °C. The efficiencies of virus transfection and PRDX1 expression were evaluated first. Cochlear samples from P3 C57BL/6 J WT mice were cultured and treated with 4 × 1010 GC/ml Anc80-Prdx1. After incubation for 24 h, the medium was replaced with normal medium and the cells were further cultured for 48 h at 37°C in 5% CO2, and samples were then prepared for immunostaining or western blotting. Then, to test the effect of PRDX1 on autophagy, the cultured middle turn cochlea was first incubated with 4 × 1010 GC/ml Anc80-Prdx1 for 24 h, then the medium was replaced with normal medium and SGN explants were further treated with cisplatin (50 μM) or cisplatin (50 μM) + 3-MA (5 mM, pretreatment for 6 h) for 48 h at 37°C and 5% CO2. Samples were then prepared for western blot.
SGN counting
Six cochlear samples from each subgroup were used for SGN counting. As we previously reported [Citation33] with slight modification, SGNs were immunolabeled with the TUBB3 antibody and the cultured middle-turn cochlear explants were imaged using a confocal microscope using a 40 × oil-immersion lens. The acquisition mode was XYZ, and in the SGN somas 20–40 image planes were typically acquired. SGNs in which the nucleus comprised 40% of the soma area were counted in each optical section using the NIH ImageJ software. The total number of SGNs in each spiral ganglion explant was obtained by summing the SGN counts in all consecutive sections. The in vivo SGN counting was quantified from midmodiolar sections of cochleae dissected out of P30 mice. The number of SGNs in each section was divided by the area of Rosenthal’s canal, and the total number of SGNs was counted using NIH ImageJ software. Finally, the SGN density per unit area (0.01 mm2) was calculated.
Quantification of the autophagic structures and LC3B immunofluorescence signals
NIH ImageJ software was used for quantitative analysis. The autophagosomes and autolysosomes in each SGN were quantified from the original TEM images under high magnification. Autophagosomes are the double membrane vacuoles formed by the ER, Golgi apparatus, etc., and these took on a circular structure that enveloped the targeted cytoplasmic constituents like fragments of ribosomes or mitochondria. Autolysosomes are monolayer membrane structures that arise from the fusion of autophagosomes with lysosomes and contain undecomposed ER, mitochondria, and Golgi complexes or lipids, glycogen, etc. Four independent cochleae from each subgroup were used, and at least 10 SGNs were counted to get an average number of autophagic vacuoles and autolysosomes per SGN for each cochlea. The criteria for counting LC3B puncta were as previously reported [Citation37] with modifications. LC3B fluorescent puncta were quantified from the original confocal images taken under a confocal microscope using a 100 × oil-immersion lens, 4 × zoom, and a 0.5 μm distance between each optical section. We counted the LC3 fluorescent puncta in each SGN, which were labeled by TUBB3 antibody, and at least 50 SGNs were counted to get an average number of LC3B fluorescent puncta per SGN for each cochlea. Six independent cochlear cultures from each subgroup were counted.
Statistical analysis
Statistical analyses were conducted using Microsoft Excel and GraphPad Prism software. Data are presented as the mean ± SEM (standard error of the mean). All experiments were repeated at least three times, and n represents the number of independent cochlear samples from each subgroup. Two-tailed, unpaired Student’s t-tests were used to determine statistical significance when comparing two groups. Two-way ANOVA followed by a post-hoc Student-Newman-Keuls test was used for comparing the effects of more than one treatment as well as for functional hearing assessments. A value of P < 0.05 was considered to be statistically significant.
Supplemental Material
Download Zip (16.5 MB)Disclosure statement
There were no potential conflicts of interest to be disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Liu W, Wang X, Wang M, et al. Protection of Spiral Ganglion Neurons and Prevention of Auditory Neuropathy. Adv Exp Med Biol. 2019;1130:93–107.
- Géléoc GS, Holt JR. Sound strategies for hearing restoration. Science (New York, NY). 2014;344(6184):1241062.
- Nayagam BA, Muniak MA, Ryugo DK. The spiral ganglion: connecting the peripheral and central auditory systems. Hear Res. 2011;278(1–2):2–20.
- O’Donoghue G. Cochlear Implants — science, Serendipity, and Success. N Engl J Med. 2013;369(13):1190–1193.
- Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378.
- Schellens JH, Planting AS, Ma J, et al. Adaptive intrapatient dose escalation of cisplatin in patients with advanced head and neck cancer. Anticancer Drugs. 2001;12(8):667–675. .
- Marshak T, Steiner M, Kaminer M, et al. Prevention of cisplatin-induced hearing loss by intratympanic dexamethasone: a randomized controlled study. Otolaryngol Head Neck Surg. 2014;150(6):983–990. .
- Gentilin E, Simoni E, Candito M, et al. Cisplatin-Induced ototoxicity: updates on molecular targets. Trends Mol Med. 2019;25(12):1123–1132. .
- Rybak LP. Mechanisms of cisplatin ototoxicity and progress in otoprotection. Curr Opin Otolaryngol Head Neck Surg. 2007;15(5):364–369.
- Van Ruijven MW, De Groot JC, Klis SF, et al. The cochlear targets of cisplatin: an electrophysiological and morphological time-sequence study. Hear Res. 2005;205(1–2):241–248.
- Van Ruijven MW, De Groot JC, Smoorenburg GF. Time sequence of degeneration pattern in the guinea pig cochlea during cisplatin administration. A quantitative histological study. Hear Res. 2004;197(1–2):44–54.
- Langer T, Am Zehnhoff-dinnesen A, Radtke S, et al. Understanding platinum-induced ototoxicity. Trends Pharmacol Sci. 2013;34(8):458–469.
- Borse V, Rfh AA, Sheehan K, et al. Epigallocatechin-3-gallate, a prototypic chemopreventative agent for protection against cisplatin-based ototoxicity. Cell Death Dis. 2017;8(7):e2921.
- Mukherjea D, Jajoo S, Kaur T, et al. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid Redox Signal. 2010;13(5):589–598.
- Sheth S, Mukherjea D, Rybak LP, et al. Mechanisms of cisplatin-induced ototoxicity and otoprotection. Front Cell Neurosci. 2017;11:338.
- Dammeyer P, Hellberg V, Wallin I, et al. Cisplatin and oxaliplatin are toxic to cochlear outer hair cells and both target thioredoxin reductase in organ of Corti cultures. Acta Otolaryngol. 2014;134(5):448–454.
- Ravi R, Somani SM, Rybak LP. Mechanism of cisplatin ototoxicity: antioxidant system. Toxicol Pharmacol. 1995;76(6):386–394.
- Kaur T, Mukherjea D, Sheehan K, et al. Short interfering RNA against STAT1 attenuates cisplatin-induced ototoxicity in the rat by suppressing inflammation. Cell Death Dis. 2011;2(7):e180.
- Previati M, Lanzoni I, Astolfi L, et al. Cisplatin cytotoxicity in organ of Corti-derived immortalized cells. J Cell Biochem. 2007;101(5):1185–1197.
- Kilic K, Sakat MS, Akdemir FNE, et al. Protective effect of gallic acid against cisplatin-induced ototoxicity in rats. Braz J Otorhinolaryngol. 2019;85(3):267–274.
- Jamesdaniel S, Rathinam R, Neumann WL. Targeting nitrative stress for attenuating cisplatin-induced downregulation of cochlear LIM domain only 4 and ototoxicity. Redox Biol. 2016;10:257–265.
- Rathinam R, Ghosh S, Neumann WL, et al. Cisplatin-induced apoptosis in auditory, renal, and neuronal cells is associated with nitration and downregulation of LMO4. Cell Death Discov. 2015;1:15052.
- Van Houten B, Santa-Gonzalez GA, Camargo M. DNA repair after oxidative stress: current challenges. Curr Opin Toxicol. 2018;7:9–16.
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845–1846.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Fujimoto C, Iwasaki S, Urata S, et al. Autophagy is essential for hearing in mice. Cell Death Dis. 2017;8(5):e2780.
- Magarinos M, Pulido S, Aburto MR, et al. Autophagy in the vertebrate inner ear. Front Cell Dev Biol. 2017;5:56.
- De Iriarte Rodriguez R, Pulido S, Rodriguez-de La Rosa L, et al. Age-regulated function of autophagy in the mouse inner ear. Hear Res. 2015;330:39–50. Pt A
- Neumann CA, Krause DS, Carman CV, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565.
- Butterfield LH, Merino A, Golub SH, et al. From cytoprotection to tumor suppression: the multifactorial role of peroxiredoxins. Antioxid Redox Signal. 1999;1(4):385–402.
- Ledgerwood EC, Marshall JW, Weijman JF. The role of peroxiredoxin 1 in redox sensing and transducing. Arch Biochem Biophys. 2017;617:60–67.
- Le Q, Tabuchi K, Warabi E, et al. The role of peroxiredoxin I in cisplatin-induced ototoxicity. Auris Nasus Larynx. 2017;44(2):205–212.
- Liu W, Xu X, Fan Z, et al. Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects Against Cisplatin-Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea. Antioxid Redox Signal. 2019;30(11):1389–1410.
- Rubinsztein DC, Cuervo AM, Ravikumar B, et al. In search of an “autophagomometer”. Autophagy. 2009;5(5):585–589.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145.
- He Z, Guo L, Shu Y, et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy. 2017;13(11):1884–1904.
- Fu X, Sun X, Zhang L, et al. Tuberous sclerosis complex-mediated mTORC1 overactivation promotes age-related hearing loss. J Clin Invest. 2018;128(11):4938–4955.
- Wu X, He L, Chen F, et al. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PloS One. 2014;9(11):e112891.
- Fang B, Xiao H. Rapamycin alleviates cisplatin-induced ototoxicity in vivo. Biochem Biophys Res Commun. 2014;448(4):443–447.
- Menardo J, Tang Y, Ladrech S, et al. Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse Cochlea. Antioxid Redox Signal. 2012;16(3):263–274.
- Ye B, Fan C, Shen Y, et al. The Antioxidative Role of Autophagy in Hearing Loss. Front Neurosci. 2018;12:1010.
- Ureshino RP, Rocha KK, Lopes GS, et al. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid Redox Signal. 2014;21(1):123–137.
- Duarte MJ, Kanumuri VV, Landegger LD, et al. Ancestral Adeno-Associated Virus Vector Delivery of Opsins to Spiral Ganglion Neurons: implications for Optogenetic Cochlear Implants. Mol Ther. 2018;26(8):1931–1939.
- Kim SJ, Park C, Han AL, et al. Ebselen attenuates cisplatin-induced ROS generation through Nrf2 activation in auditory cells. Hear Res. 2009;251(1–2):70–82.
- Soyman Z, Uzun H, Bayindir N, et al. Can ebselen prevent cisplatin-induced ovarian damage? Arch Gynecol Obstet. 2018;297(6):1549–1555.
- Kim JH, Choi TG, Park S, et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 2018;25(11):1921–1937.
- Pang J, Fuller ND, Hu N, et al. Alcohol Dehydrogenase Protects against Endoplasmic Reticulum Stress-Induced Myocardial Contractile Dysfunction via Attenuation of Oxidative Stress and Autophagy: role of PTEN-Akt-mTOR Signaling. PLoS One. 2016;11(1):e0147322.
- Dong Y, Sui L, Yamaguchi F, et al. Role of phosphatase and tensin homolog in the development of the mammalian auditory system. Neuroreport. 2010;21(10):731–735.
- Kim HJ, Woo HM, Ryu J, et al. Conditional deletion of pten leads to defects in nerve innervation and neuronal survival in inner ear development. PLoS One. 2013;8(2):e55609.
- Cao J, Schulte J, Knight A, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. Embo J. 2009;28(10):1505–1517.
- Verrastro I, Tveen-Jensen K, Woscholski R, et al. Reversible oxidation of phosphatase and tensin homolog (PTEN) alters its interactions with signaling and regulatory proteins. Free Radic Biol Med. 2016;90:24–34.
- Guo S, Xu N, Chen P, et al. Rapamycin Protects Spiral Ganglion Neurons from Gentamicin-Induced Degeneration In Vitro. J Assoc Res Otolaryngology. 2019;20(5):475–487.
- Zuo WQ, Hu YJ, Yang Y, et al. Sensitivity of spiral ganglion neurons to damage caused by mobile phone electromagnetic radiation will increase in lipopolysaccharide-induced inflammation in vitro model. J Neuroinflammation. 2015;12:105.
- Ye B, Wang Q, Hu H, et al. Restoring autophagic flux attenuates cochlear spiral ganglion neuron degeneration by promoting TFEB nuclear translocation via inhibiting MTOR. Autophagy. 2019;15(6):998–1016.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075.
- Liu T, Zong S, Luo P, et al. Enhancing autophagy by down-regulating GSK-3β alleviates cisplatin-induced ototoxicity in vivo and in vitro. Toxicol Lett. 2019;313:11–18.
- Yin H, Yang Q, Cao Z, et al. Activation of NLRX1-mediated autophagy accelerates the ototoxic potential of cisplatin in auditory cells. Toxicol Appl Pharmacol. 2018;343:16–28.
- Xu F, Yan W, Cheng Y. Pou4f3 gene mutation promotes autophagy and apoptosis of cochlear hair cells in cisplatin-induced deafness mice. Arch Biochem Biophys. 2020;680:108224.
- Cho KH, Park JH, Kwon KB, et al. Autophagy induction by low-dose cisplatin: the role of p53 in autophagy. Oncol Rep. 2014;31(1):248–254.
- Chen J, Zhang L, Zhou H, et al. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol Med Rep. 2018;17(5):6859–6865.
- Yang Q, Sun G, Yin H, et al. PINK1 Protects Auditory Hair Cells and Spiral Ganglion Neurons from Cisplatin-induced Ototoxicity via Inducing Autophagy and Inhibiting JNK Signaling Pathway. Free Radic Biol Med. 2018;120:342–355.
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26(7):1749–1760.
- Chen Y, McMillan-Ward E, Kong J, et al. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15(1):171–182.
- Djavaheri-Mergny M, Amelotti M, Mathieu J, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281(41):30373–30382.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388.
- Scherz-Shouval R, Shvets E, Elazar Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy. 2007;3(4):371–373.
- Yuan H, Wang X, Hill K, et al. Autophagy attenuates noise-induced hearing loss by reducing oxidative stress. Antioxid Redox Signal. 2015;22(15):1308–1324.
- Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27(5–6):444–454.
- Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591.
- Ishii T, Warabi E, Yanagawa T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J Clin Biochem Nutr. 2012;50(2):91–105.
- Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid Redox Signal. 2005;7(5–6):768–777.
- Cho YE, Singh TS, Lee HC, et al. In-depth identification of pathways related to cisplatin-induced hepatotoxicity through an integrative method based on an informatics-assisted label-free protein quantitation and microarray gene expression approach. Mol Cell Proteomics. 2012;11(1):M111 010884.
- Ma D, Warabi E, Yanagawa T, et al. Peroxiredoxin I plays a protective role against cisplatin cytotoxicity through mitogen activated kinase signals. Oral Oncol. 2009;45(12):1037–1043.
- Yan Y, Sabharwal P, Rao M, et al. The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell. 2009;138(6):1209–1221.
- Lu Y, Zhang XS, Zhou XM, et al. Peroxiredoxin 1/2 protects brain against H(2)O(2)-induced apoptosis after subarachnoid hemorrhage. Faseb J. 2019;33(2):3051–3062.
- Jeong SJ, Kim S, Park JG, et al. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy. 2018;14(1):120–133.
- Min Y, Kim MJ, Lee S, et al. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy. 2018;14(8):1347–1358.
- Kil J, Lobarinas E, Spankovich C, et al. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2017;390(10098):969–979.
- Georgescu MM, Tumor Suppressor PTEN. Network in PI3K-Akt Pathway Control. Genes Cancer. 2010;1(12):1170–1177.
- Fripont S, Marneffe C, Marino M, et al. Production, Purification, and Quality Control for Adeno-associated Virus-based Vectors. J Vis Exp. 2019;143. DOI:https://doi.org/10.3791/58960.