
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Whether macroautophagy/autophagy is physiologically relevant to regulate mitochondrial function for a rapid and dynamic adaptation of yeast cells to respiratory growth was not fully understood until recently. May et al. (2020. Nat Commun) report that bulk autophagy provides serine as a one-carbon (1C) metabolite that controls respiratory growth onset by initiating mitochondrial initiator tRNAMet modification and mitochondrial translation linking autophagy mechanistically to mitochondrial function. We discuss the mechanistic interplay between autophagy, one-carbon-metabolism, and mitochondrial function and the possible implications in neurodegeneration, aging, and carcinogenesis.
Abbreviations: 10-formyl-THF: 10-formyl-tetrahydrofolate; OXPHOS: oxidative phosphorylation; PAS: phagophore assembly site
Autophagy is an evolutionarily conserved multi-step process that allows eukaryotic cells to recycle cellular components in response to nutrient limitation or various stress conditions. Bulk autophagy, e.g. under starvation conditions, is considered to be a rather nonselective process, whereas selective autophagy relies on the presence of cargo-specific receptors and/or specific ubiquitination of the cargo mediating the regulated degradation of, e.g. damaged proteins or organelles. Autophagy is initiated by the Atg1-kinase complex assembly at the phagophore assembly site (PAS) upon dephosphorylation of Atg13. The PAS expands for de novo biogenesis of the double-membrane phagophore that sequesters autophagy substrates and matures into an autophagosome. The autophagosome’s outer membrane subsequently fuses with the vacuole (lysosome in mammals) and releases an autophagic body (inner autophagosomal membrane) into the vacuolar lumen for substrate degradation [Citation1,Citation2]. Autophagy is induced by nutrient depletion or cellular stress and provides new metabolites (e.g. amino acids) as building blocks to fuel cellular energy and metabolic pathways [Citation1,Citation2]. However, despite the conservation of the autophagy machinery among eukaryotes, the physiological importance of autophagy and the fate of recycled metabolites in cellular homeostasis is poorly understood. In Nat. Commun., Ohsumi and colleagues [Citation1] reported that autophagy maintains the respiratory growth onset of the budding yeast Saccharomyces cerevisiae by regulating mitochondrial function and preserving energy metabolism.
Saccharomyces cerevisiae grown on the fermentable carbon source glucose favors glycolysis or fermentation for producing energy (ATP) and biomass. This is carried out by turning off genes of mitochondrial respiration (i.e. glucose-repressed genes) via a process known as glucose repression. However, this repression is relieved when glucose is limited, or cells are rapidly shifted from glucose to non-fermentable carbon sources, such as ethanol or glycerol. Yeast cells rapidly reprogram gene expression to switch bioenergetics from glycolysis to mitochondrial respiration for adaptation to non-fermentable (respiratory) substrates. Mitochondrial oxidative phosphorylation (OXPHOS) is a major energy conversion process and is achieved by multi-protein complexes within the mitochondria [Citation3]. On respiratory media, both nuclear- and mitochondrial-encoded OXPHOS subunits are induced by the synchronized cytosolic and mitochondrial translation program [Citation3]. Thus, the synchronized translation program orchestrates the synthesis of OXPHOS complexes to provide energy and to ensure mitochondrial biogenesis.
Mitochondria contain 1,000 proteins where 99% of mitochondrial proteins are nuclear-encoded and imported into mitochondria by mitochondrial import pathways. Approximately 1% of mitochondrial proteins are mitochondrial DNA (mtDNA)-encoded and synthesized by the mitoribosomes in the mitochondrial matrix. In S. cerevisiae, of the entire OXPHOS system, only seven subunits are encoded in mtDNA, one subunit of the respiratory chain complex III (Cob/cytochrome b), three subunits of complex IV (cytochrome c oxidase; Cox1, Cox2, Cox3) and three subunits of complex V (F1FO ATP synthase; Atp6, Atp8, Oli1/Atp9) [Citation3].
The mitochondrial translation is largely reminiscent of prokaryotic translation that usually requires formylated fMet-tRNAMet in addition to mitoribosomes, mRNA, and two mitochondrial initiation factors, Ifm1/mtIf2 and Aim23/mtIf3 [Citation4]. In mitochondria, the formylation of mitochondrial initiator Met-tRNAMet is catalyzed by Fmt1/formyltransferase (MTFMT in mammals) [Citation1,Citation5]. The formylated fMet-tRNAMet has a high affinity for Ifm1/mitochondrial translation initiation factor 2 that is recruited to the peptidyl site (P-site) of the ribosome to initiate efficient mitochondrial translation and function [Citation1,Citation5]. Interestingly, Fmt1-independent mitochondrial translation initiation pathways exist in yeast that are less efficient and depend on redundant genes RSM28, MSC6, or AEP3 [Citation1]. Thus, mitochondrial translation initiation is blocked when genetic redundancy is abolished by co-deletion of any redundant gene, RSM28, MSC6, or AEP3, in fmt1Δ cells. Translation initiation is the rate-limiting and highly regulated phase of translation. When the fidelity of mitochondrial translation initiation is compromised due to lack of formylated fMet-tRNAMet or the loss of Fmt1, the steady-state level of OXPHOS subunits is reduced, leading to respiratory growth defect. Thus, it is essential to maintain a sufficient pool of formylated fMet-tRNAMet within mitochondria, especially during diauxic shift when glucose is replaced by ethanol as a carbon source. How does autophagy control a pool of fMet-tRNAMet within mitochondria for translation initiation?
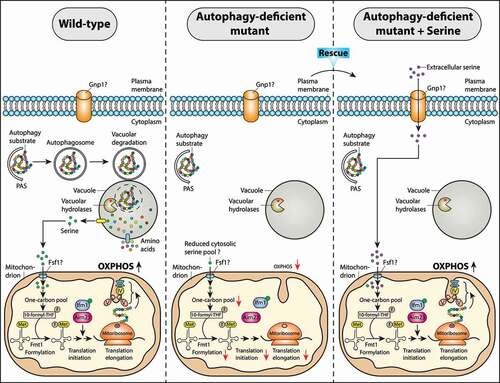
Here, May et al describe a highly relevant and novel molecular mechanism by which autophagy can promote yeast cells to adapt to non-fermentable substrates [Citation1]. The authors show that bulk autophagy (assessed by GFP-Atg8 cleavage, PAS formation, prApe1 maturation, and Atg13 dephosphorylation) is activated during diauxic shift (i.e. after glucose depletion) and the glucose-to-ethanol switch for supporting respiratory growth. This is maintained by trafficking of autophagy-derived serine into mitochondria via a yet unidentified mitochondrial serine transporter when yeast cells are grown on respiratory media. Mitochondrial serine subsequently enters in a series of reactions comprising one-carbon metabolism that generate 10-formyl-tetrahydrofolate (10-formyl-THF) as a formyl (-CHO) donor [Citation6]. Mitochondrial 10-formyl-THF provides a one-carbon (1 C) unit (formyl group) for Met-tRNAMet formylation and mitochondrial translation initiation within the organelle.
Furthermore, yeast cells lacking core autophagosome biogenesis genes ATG1 or ATG2 are compromised to adapt to respiratory growth conditions [Citation1]. Yeast atg2Δ mutant cells grown on ethanol show a prolongation by 50% in the lag phase (adaptive) duration of growth (tlag) compared to wild-type cells without affecting the log-phase growth rate (μlog). In addition, Met-tRNAMet formylation and mitochondrial translation initiation are impaired/delayed in atg2Δ cells, leading to respiration (OXPHOS) deficiency [Citation1]. Interestingly, L-serine supplementation to media can rescue the Met-tRNAMet formylation and mitochondrial translation defects, allowing atg2Δ cells to adapt to respiratory media comparable to wild-type cells (). Thus, the delayed respiratory growth onset in the atg2Δ mutant occurs due to the lack of precursor serine released from the vacuole via autophagy. Extracellular serine may be imported into the cells by a newly identified major plasma membrane serine transporter, Gnp1 [Citation7]. It should be noted that Met-tRNAMet formylation still can occur in atg2Δ cells, but with a delayed lag phase of growth (i.e. increased tlag) [Citation1]. This raises the possibility that other proteolytic processes such as the ubiquitin-proteasome system (UPS) may also contribute to generate metabolites such as serine for maintaining one-carbon metabolism within the organelle and regulate the fidelity of mitochondrial translation initiation on respiratory media. However, it remains undetermined if the cytosolic serine pool is indeed reduced below a critical threshold concentration in autophagy-deficient cells on respiratory media and thus became limiting for translation initiation and ATP synthesis in mitochondria. Are there additional metabolic pathways (e.g. lipid biosynthesis or maintenance of redox homeostasis) where autophagy-derived serine can be rapidly utilized promoting cellular homeostasis under changing environmental conditions such as respiratory growth? This is a challenging question for future work.
Figure 1. Model for autophagy‐mediated regulation of mitochondrial function via supplying serine during a metabolic switch from fermentation to respiration. When yeast S. cerevisiae cells are grown on non-fermentable carbon sources (e.g. glycerol), autophagy is activated and provides serine, a one-carbon donor for efficient Met-tRNAMet formylation and mitochondrial translation initiation, thereby promoting mitochondrial respiration. This allows wild-type cells to adapt to respiratory media. When autophagy is compromised as in the atg2Δ mutant, Met-tRNAMet formylation and mitochondrial translation are impaired causing respiration deficiency and the delayed respiratory growth onset phenotypes. L-serine supplementation in an atg2Δ mutant can rescue the Met-tRNAMet formylation and mitochondrial translation defects, allowing the mutant to resume respiratory growth with wild-type kinetics. The mitochondrial serine transporter that supplies the autophagy-derived serine into the mitochondria has not yet been identified (?). Abbreviations: PAS, phagophore assembly site; 10-formyl-THF, 10-formyl-tetrahydrofolate; OXPHOS, oxidative phosphorylation; III, IV, V are OXPHOS complexes III, IV, and V, respectively
Martin Graef and Jodi Nunnari showed previously that under amino‐acid starvation, mitochondrial function is crucial for the recruitment of the Atg1-Atg13 kinase complex to the PAS site and autophagy induction [Citation8]. The current study revealed that autophagy is crucial for the yeast to adapt to respiratory growth by maintaining mitochondrial respiration via mitochondrial one-carbon metabolism [Citation1]. Thus, it is likely that mechanisms of bi-directional cross-talk between autophagy and mitochondrial function exist in yeast. Furthermore, it remains an open question whether such a regulatory network exists in higher eukaryotes and is modulated under different stress conditions or between different cell types.
Does autophagy support mitochondrial respiration in more complex eukaryotes, similar to the budding yeast S. cerevisiae? It would be exciting to know if such a pathway is evolutionarily conserved and can be implemented for targeting therapeutics against cancer. It has been shown that autophagy and mitochondrial translation is upregulated in a subset of human cancer cells for their survival and growth [Citation2,Citation9]. Interestingly, several cancer cells are highly dependent on serine that is a one-carbon donor to the folate cycle, and contributes to methylation reactions, nucleotide synthesis, NADPH, and glutathione (GSH) synthesis for antioxidant defense [Citation6]. Thus, serine supports several metabolic pathways that are crucial for cancer cell survival and proliferation. A key question for future investigations is whether autophagy can provide metabolites such as serine via the recently discovered mitochondrial serine transporter SFXN1 (Fsf1 is a possible yeast homolog with 37% protein sequence similarity) to initiate mitochondrial translation in cancer cells [Citation10]. Thus, either blocking autophagy or inhibiting the transport of autophagy-derived metabolites (e.g. serine) into mitochondria for one-carbon metabolism (e.g. mitochondrial translation) may provide a potential rationale to develop SFXN1-targeted drugs as anticancer therapies.
Taken together, this study reveals that autophagy plays a crucial role in regulating cellular energy demand under respiratory growth conditions. Under these conditions, autophagy releases metabolites including serine to support Met-tRNAMet formylation, mitochondrial translation initiation, and respiration via one-carbon metabolism. This is exciting because formylated fMet-tRNAMet species are crucial for efficient intramitochondrial protein synthesis in humans and depend on serine catabolism. Defective mitochondrial tRNA modifications are observed in patients with mitochondrial disease [Citation11]. A better understanding about the interplay between autophagy and mitochondrial function could provide new therapeutic approaches for neurodegeneration, aging, and carcinogenesis where autophagy and mitochondrial function are perturbed.
Acknowledgments
We sincerely apologize to all colleagues and scientists whose relevant contribution is not cited due to space restrictions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- May AI, Prescott M, Ohsumi Y. Autophagy facilitates adaptation of budding yeast to respiratory growth by recycling serine for one-carbon metabolism. Nat Commun. 2020 Oct 7;11(1):5052.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017 Sep;17(9):528–542.
- Couvillion MT, Soto IC, Shipkovenska G, et al. Synchronized mitochondrial and cytosolic translation programs. Nature. 2016 May 26;533(7604):499–503.
- Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol Rev. 1983 Mar;47(1):1–45.
- Tucker EJ, Hershman SG, Kohrer C, et al. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011 Sep 7;14(3):428–434.
- Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016 Oct;16(10):650–662.
- Esch BM, Limar S, Bogdanowski A, et al. Uptake of exogenous serine is important to maintain sphingolipid homeostasis in Saccharomyces cerevisiae. PLoS Genet. 2020 Aug;16(8):e1008745.
- Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. Embo J. 2011 Jun 1;30(11):2101–2114.
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017 Jan;14(1):11–31.
- Kory N, Wyant GA, Prakash G, et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science. 2018 Nov 16;362(6416). DOI:https://doi.org/10.1126/science.aat9528
- Wei FY, Zhou B, Suzuki T, et al. Cdk5rap1-mediated 2-methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab. 2015 Mar 3;21(3):428–442.