
ABSTRACT
Selective recognition and elimination of misfolded polypeptides are crucial for protein homeostasis. When the ubiquitin-proteasome system is impaired, misfolded polypeptides tend to form small cytosolic aggregates and are transported to the aggresome and eventually eliminated by the autophagy pathway. Despite the importance of this process, the regulation of aggresome formation remains poorly understood. Here, we identify TRIM28/TIF1β/KAP1 (tripartite motif containing 28) as a negative regulator of aggresome formation. Direct interaction between TRIM28 and CTIF (cap binding complex dependent translation initiation factor) leads to inefficient aggresomal targeting of misfolded polypeptides. We also find that either treatment of cells with poly I:C or infection of the cells by influenza A viruses triggers the phosphorylation of TRIM28 at S473 in a way that depends on double-stranded RNA-activated protein kinase. The phosphorylation promotes association of TRIM28 with CTIF, inhibits aggresome formation, and consequently suppresses viral proliferation. Collectively, our data provide compelling evidence that TRIM28 is a negative regulator of aggresome formation.
Abbreviations: BAG3: BCL2-associated athanogene 3; CTIF: CBC-dependent translation initiation factor; CED: CTIF-EEF1A1-DCTN1; DCTN1: dynactin subunit 1; EEF1A1: eukaryotic translation elongation factor 1 alpha 1; EIF2AK2: eukaryotic translation initiation factor 2 alpha kinase 2; HDAC6: histone deacetylase 6; IAV: influenza A virus; IP: immunoprecipitation; PLA: proximity ligation assay; polypeptidyl-puro: polypeptidyl-puromycin; qRT-PCR: quantitative reverse-transcription PCR; siRNA: small interfering RNA
Introduction
To maintain proper cellular processes, misfolded polypeptides are largely degraded by the ubiquitin–proteasome system or autophagy pathway [Citation1-7]. When the ubiquitin-proteasome system is impaired or compromised, some misfolded polypeptides tend to form small cytosolic aggregates that are selectively transported to the aggresome, a perinuclear inclusion, via dynein motor protein-mediated and microtubule-mediated retrograde transport [Citation8,Citation9]. Misfolded polypeptides accumulated in the aggresome are eventually removed from the cell through aggrephagy, a subtype of autophagy [Citation10].
There are several cellular machineries involved in the selective recognition and aggresomal targeting of misfolded polypeptides: HDAC6 (histone deacetylase 6), BAG3 (BAG cochaperone 3), and the CED (CTIF-EEF1A1-DCTN1) complex. HDAC6 recognizes and transports misfolded polypeptides containing polyubiquitin chains to the aggresome [Citation11]. BAG3 acts as a cochaperone to regulate the activity of HSPA/HSP70 and promotes aggresomal targeting of misfolded polypeptides in either a polyubiquitin-dependent or polyubiquitin–independent manner [Citation12]. Finally, the CED complex along with UPF1 promotes the formation of an aggresome containing polyubiquitinated misfolded polypeptides [Citation13-15]. In this complex, EEF1A1 directly binds to the misfolded polypeptides [Citation15] and generates an aggresome formation signal [Citation16]. DCTN1 interacts with the dynein motor protein and promotes the retrograde transport of misfolded polypeptides along microtubules to the aggresome [Citation15,Citation17,Citation18]. CTIF functions as a scaffold protein linking EEF1A1 with DCTN1 through its N–terminal region spanning amino acid residues (aa) 12–53 [Citation15].
It is well known that the aggresome is associated with a variety of biological and physiological events [Citation1-7]. For instance, inefficient aggresome formation causes the accumulation of small and dispersed cytosolic aggregates of misfolded polypeptides, thereby inducing proteotoxic stress [Citation1,Citation2]. In neurons, aggregates or inclusions containing misfolded polypeptides are a representative feature of many neurodegenerative diseases: amyloid fibrils in Alzheimer disease and SNCA/α–synuclein Lewy bodies in Parkinson disease [Citation3–6,Citation8,Citation19-22]. It should be noted that inclusion bodies in neurodegenerative diseases are biochemically and morphologically similar to the aggresome observed in cultured cells [Citation15,Citation21]. Efficient proliferation or replication of several DNA or RNA viruses also involves an aggresome or aggresome–like structure [Citation23-26]. IAVs (influenza A viruses) exploit host cellular factors involved in aggresome formation and disassembly for capsid disassembly during viral entry [Citation27]. For efficient viral replication and packaging, the adenovirus also uses the cellular aggresome response to inactivate the MRE11-RAD50-NBS1 complex [Citation28]. In the case of herpes simplex virus, artificial disruption of the aggresome (or an aggresome-like structure) is known to cause a decrease in the production of intracellular infectious particles [Citation29]. Furthermore, viral factories produced by African swine fever virus are structurally and biochemically similar to aggresomes, suggesting that an aggresome is a cellular site for viruses to concentrate structural proteins for efficient virion assembly [Citation30].
TRIM28/TIF1β/KAP1 (tripartite motif containing 28), a member of the TRIM family of proteins, was first identified as a cofactor for Krüppel-associated box zinc finger protein transcription factors [Citation31]. Many subsequent studies have revealed that TRIM28 participates in various molecular or biological processes, including transcriptional regulation, the DNA damage response, regulation of retrotransposons, epithelial–mesenchymal transition, antiviral response, and autophagy [Citation32–39]. As for autophagy, it is likely that the role of TRIM28 in this process is somewhat dependent on pathophysiological conditions or the tissue/organ system. In glioblastoma multiforme, TRIM28 markedly correlates with the tumor grade and activates autophagy [Citation40]. TRIM28 triggers SUMOylation and the activity of PIK3C3/VPS34, inducing autophagosome formation [Citation33]. In contrast, TRIM28 is involved in the ubiquitination and degradation of AMPK, consequently suppressing autophagy [Citation41].
In this study, we demonstrate an inhibitory role of TRIM28 in the formation of the aggresome containing misfolded polypeptides. The inhibitory function of TRIM28 is mediated by a direct interaction between TRIM28 and CTIF. In particular, under the conditions where inflammatory responses are activated via either treatment with poly I:C or viral infection, a EIF2AK2/PKR (eukaryotic translation initiation factor 2 alpha kinase 2) triggers the phosphorylation of TRIM28 at S473 and promotes association between TRIM28 and CTIF. The increased interaction inhibits aggresome formation, consequently suppressing viral proliferation.
Results
TRIM28 directly interacts with CTIF, a component of an aggresome formation complex
To understand the molecular mechanism determining how aggresome formation via the CED complex [Citation13] is regulated, we searched for cellular factors interacting with a CTIF fragment, either CTIF(1–53) or CTIF(12-53), harboring a minimal region of CTIF required for CED complex formation and aggresomal targeting [Citation15]. To this end, we carried out a GST (glutathione S-transferase) affinity-isolation assay followed by LC-MS/MS (liquid chromatography-tandem mass spectrometry; ). To set up conditions favorable for aggresome formation, HEK293T cells were treated with MG132, a potent proteasome inhibitor [Citation9]. The specificity of the affinity-isolation of MYC-CTIF(1-53)-GST or MYC-CTIF(12-53)-GST was confirmed by western blotting with an anti-GST antibody (Fig. S1A). Through the LC-MS/MS analysis, we identified seven putative CTIF-interacting proteins (Table S1). Among them, two proteins have been implicated in aggresome formation or autophagy: EEF1A1P5 (a pseudogene product homologous to EEF1A1, a CED component) [Citation15,Citation16] and TRIM28 [Citation33,Citation40,Citation41].
Figure 1. TRIM28 competes with EEF1A1 and DCTN1 for the binding to CTIF. (A) Silver staining of the GST affinity isolation of MYC-GST, MYC-CTIF(1-53)-GST, and MYC-CTIF(12-53)-GST. HEK293T cells were transiently transfected with one of the plasmids. Two days later, the cells were treated with MG132 for 12 h before cell harvest. The bands analyzed via LC-MS/MS are indicated by numbers. The proteins identified in each band are listed in Table S1. The arrowhead marks the position of an immunoglobulin heavy chain. (B) In vitro GST affinity-isolation assay using recombinant proteins. The extracts of E. coli expressing GST or a GST-fused protein were mixed with the extracts of E. coli expressing 6× His-TRIM28. The mixtures were subjected to GST affinity-isolation experiments. Representative images obtained from two biological replicates (n = 2) are presented. (C) Immunoprecipitation (IP) of endogenous TRIM28. HEK293T cells were either treated or not treated with MG132. After cell lysis, the cell extracts were digested with RNase A and subjected to IP with the anti-TRIM28 antibody; n = 3. (D) Proximity ligation assay (PLA) involving either FLAG or FLAG-TRIM28 and either MYC or MYC-CTIF. The PLA was performed on HeLa cells transiently expressing the indicated proteins. Nuclei were stained with DAPI (blue). Representative images obtained from three biological replicates (n = 3) are presented. Scale bar: 10 μm. (E) IP of FLAG-CTIF using the extracts of cells depleted of endogenous TRIM28. HEK293T cells transiently expressing FLAG-CTIF and depleted of endogenous TRIM28 were treated with MG132 before cell harvest. The cell extracts digested with RNase A were subjected to IP with FLAG M2 affinity gel. After western blotting with the indicated antibodies, the band intensities were quantitated using the ImageJ software. The intensities of co-IPed proteins were normalized to those of immunoprecipitated FLAG-CTIF. The normalized levels from the undepleted cells were arbitrarily set to 1.0. The average values of normalized intensities from three biological replicates are presented at the bottom of each image. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values. These values of each blot are provided in Table S2; n = 3. (F) IP of FLAG-CTIF in the extracts of cells depleted of either EEF1A1 or DCTN1. As performed in panel (E), except that the cells were depleted of either endogenous EEF1A1 or endogenous DCTN1; n = 3
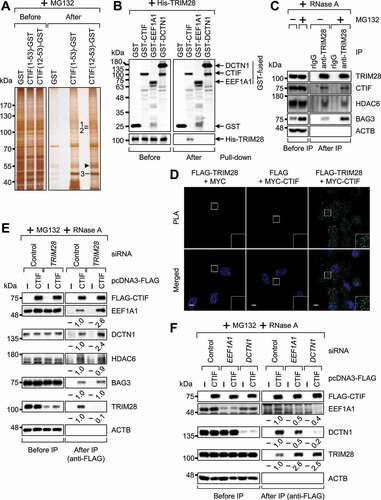
We next validated the interaction between TRIM28 and CTIF using several experimental approaches. First, the GST affinity-isolation experiments revealed that CED components (EEF1A1 and DCTN1) and TRIM28 were enriched in the affinity-isolation of CTIF(12-53)-GST but not that of GST (Fig. S1B). In addition, immunoprecipitation (IP) experiments revealed that DCTN1 and TRIM28 were preferentially enriched in the IP of FLAG-CTIF, relative to the IP of FLAG–CTIF(54-598) lacking the N-terminal aa 1–53 (Fig. S1C). Second, an in vitro GST affinity-isolation assay with recombinant proteins showed that His-TRIM28 selectively interacted with GST-CTIF but not with GST-EEF1A1 or GST-DCTN1 (), indicating a direct interaction between TRIM28 and CTIF. Third, endogenous CTIF co–IPed (coimmunoprecipitated) with TRIM28 in a way that was independent of treatment with MG132 and RNase A (). Of note, HDAC6 and BAG3, other known adaptors involved in aggresome formation [Citation11-13], also turned out to be enriched in the IP of TRIM28, thus pointing to a possible role of TRIM28 in HDAC6-mediated or BAG3-mediated aggresome formation. Fourth, an in situ PLA (proximity ligation assay) performed on HeLa cells revealed that FLAG-TRIM28 and MYC-CTIF yielded specific interaction signals (), suggesting a specific interaction between TRIM28 and CTIF within the cell. Finally, endogenous CTIF was found to be enriched in the IPs of full-length TRIM28 and of its C-terminal half, not its N-terminal half (Fig. S1D). All our in vivo and in vitro data indicate that the C-terminal half of TRIM28 interacts with the N-terminal aa 12–53 of CTIF.
TRIM28 hinders CED complex formation through competition with EEF1A1 and DCTN1 for binding to CTIF
Efficient transport of misfolded polypeptides into the aggresome requires the stable association of CTIF, EEF1A1, and DCTN1 [Citation15]. Because EEF1A1 and DCTN1 interact with the minimal domain (aa 12–53) of CTIF required for CED complex formation [Citation15], we hypothesized that TRIM28 may compete with EEF1A1 or DCTN1 for the binding to CTIF. Indeed, downregulation of TRIM28 by a specific small interfering RNA (siRNA) increased the amounts of co-IPed EEF1A1 and DCTN1 by 2.6- and 2.4-fold, respectively, in the IP of FLAG-CTIF without significantly affecting the amounts of co-IPed HDAC6 and BAG3 (). Conversely, overexpression of TRIM28 decreased the amounts of co-IPed EEF1A1 and DCTN1 (by approximately 3- and 5-fold, respectively), but not HDAC6 and BAG3, in the IP of FLAG-CTIF (Fig. S1E). In addition, the downregulation of either EEF1A1 or DCTN1 increased the amount of co-IPed TRIM28 by approximately 2.5-fold in the IP of FLAG-CTIF (). All these results support our conclusion that TRIM28 competes with EEF1A1 and DCTN1 for the binding to CTIF.
Next, we investigated a possible role of TRIM28 in the ability of the CED complex to associate with misfolded polypeptides via the above-mentioned competition. To this end, we assessed the levels of co-IPed polypeptidyl-puros (polypeptidyl-puromycins; Fig. S1F), which are prematurely terminated translation products (mostly misfolded polypeptides) generated after transient treatment with puromycin and are targeted into the aggresome [Citation14,Citation15,Citation42,Citation43]. IP experiments with FLAG-CTIF showed that TRIM28 downregulation increased the amount of co-IPed polypeptidyl-puro by 1.9-fold (Fig. S1F), suggesting that TRIM28 disrupts the CED complex and consequently inhibits the ability of this complex to associate with misfolded polypeptides.
TRIM28 inhibits the CED-driven aggresomal formation
To understand the biological consequences of the TRIM28-mediated inhibition of CED complex formation, we first tested whether the downregulation of TRIM28 affects aggresomal targeting of CTIF. It is known that the size, but not the number, of CTIF-containing aggresomes increases when the ubiquitin-proteasome system is impaired [Citation15]. In agreement with this notion, we observed a significant increase in the size, but not the number, of CTIF-containing aggresomes when cells were treated with MG132 (–C). Of note, TRIM28 downregulation further increased the aggresome size before or after MG132 treatment, with only a marginal effect on the number of aggresomes per cell (–C). In addition, TRIM28 downregulation increased the size of the aggresomes containing endogenous ubiquitin (Fig. S2A), which is known to colocalize with the aggresome during MG132 treatment [Citation15,Citation16].
Figure 2. Downregulation of TRIM28 enhances the formation of aggresomes. (A–C) The effect of TRIM28 downregulation on the formation of aggresomes containing MYC-CTIF. HeLa cells either undepleted or depleted of TRIM28 were transiently transfected with a plasmid expressing MYC-CTIF. The cells were treated with either DMSO or MG132 before fixation. (A) Immunostaining of MYC-CTIF (red). Nuclei were stained with DAPI (blue). Scale bar: 10 μm; n = 3. (B) The diameter of an aggresome containing MYC-CTIF. Immunostained images in panel A were quantitated. More than 30 cells were analyzed from each of the three biological replicates. Statistical analysis was performed by one-way ANOVA with post hoc Tukey’s honestly significant difference test; ***, P < 0.00001. (C) The number of aggresomes containing MYC-CTIF per cell in panel A. Two-tailed, equal-sample variance Student’s t test was conducted to calculate the P values; #, not significant. (D–F) The effect of TRIM28 or CTIF downregulation on the formation of aggresomes containing polypeptidyl-puro. As performed in panels (A–C), except that HeLa cells depleted of TRIM28, CTIF, or both were treated with MG132 for 12 h and puromycin for 1 h before fixation. (D) Western blotting confirming specific downregulation of endogenous proteins. (E) Immunostaining of polypeptidyl-puro (green). Scale bar: 10 μm; n = 3. (F) Relative percentages of cells containing either an aggresome or dispersed aggregates of polypeptidyl-puro presented in panel (E). Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values; #, not significant; **, P < 0.01
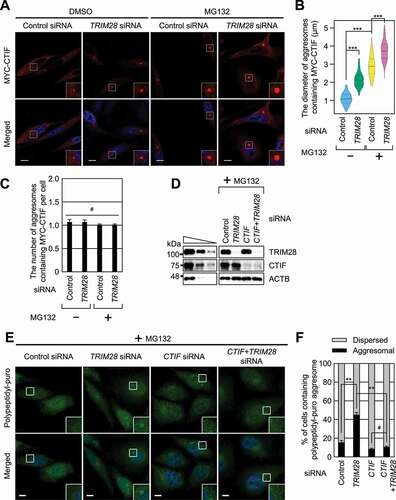
Next, we investigated the influence of TRIM28 downregulation on the formation of the aggresome containing polypeptidyl-puro, a type of misfolded polypeptides (–F). To this end, HeLa cells were treated with puromycin at a low concentration, after which the aggresome containing polypeptidyl-puro was hardly detectable (). In this condition, the TRIM28 downregulation significantly increased the proportion of cells containing the polypeptidyl-puro aggresome (). Notably, the increase in aggresome formation after the TRIM28 downregulation was found to be almost completely reversed when CTIF was simultaneously downregulated (), suggesting that the formation of aggresomes upon TRIM28 downregulation is largely mediated by the CED complex.
We corroborated the above results by showing that the overexpression of TRIM28 inhibited the aggresomal targeting and formation of various misfolded polypeptides or aggresomal components (Fig. S2B,C), including (i) a GFP-CFTR-ΔF508 (green fluorescent protein-fused mutant version of cystic fibrosis transmembrane conductance regulator), which harbors a disease-associated mutation found in cystic fibrosis and is targeted to the aggresome [Citation9,Citation11]; (ii) RFP-Ub (red fluorescent protein-fused ubiquitin) [Citation15,Citation16]; (iii) polypeptidyl-puro; and (iv) a Parkinson disease-associated protein, SYN1 (synphilin 1)-GFP. It is known that SYN1 interacts with SNCA and is enriched in cytoplasmic inclusion bodies in neuronal tissues [Citation44,Citation45]. All these data suggest that TRIM28 acts as a negative regulator of CED-mediated aggresome formation.
TRIM28 targets a step in aggresome formation
Our observation that the increase in aggresome diameter after TRIM28 downregulation may be due to either promoted aggresome formation or inefficient removal of the aggresome possibly caused by autophagy inhibition by TRIM28 downregulation. To clarify the direct or indirect effect of TRIM28 downregulation on aggresome formation, we carried out a single-particle analysis to directly visualize the motion of GFP-CTIF particles in live MEFs (mouse embryonic fibroblasts) by line-scan confocal microscopy [Citation14]. We counted the GFP-CTIF particles that reached the aggresome within a span of 30 s. On average, approximately 10.6 and 14.4 independent GFP-CTIF particles reached the aggresome during the 30 s in the control cells and TRIM28-depleted MEFs, respectively ( and Video S1). These data indicate that TRIM28 directly affects the frequency of GFP-CTIF particles (and possibly misfolded polypeptides) that move toward the aggresome.
Figure 3. TRIM28 inhibits a step in aggresome formation. (A) The frequency of GFP-CTIF particles that reach the aggresome. The number of GFP-CTIF particles (aggregates) per cell that reached the aggresome during a span of 30 s was determined. In total, 56 undepleted and 56 TRIM28-depleted cells from three independent experiments were examined. Statistical analysis was performed by one-way ANOVA with post hoc Tukey’s honestly significant difference test. (B–E) The influence of autophagy inhibition on TRIM28-mediated inhibition of aggresome formation. HeLa cells stably expressing GFP-LC3B were depleted of TRIM28, ATG5, or both. One day later, the cells were transiently transfected with a plasmid expressing MYC-CTIF. The cells were treated with MG132 before fixation; n = 3. (B) Western blotting showing specific downregulation of the tested proteins. (C) The number of GFP-LC3B puncta per cell. Immunostained images in panel (D) were quantitated. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values. *, P < 0.05; **, P < 0.01. (D) Immunostaining of MYC-CTIF (red) and GFP-LC3B (green). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (E) The diameter of an aggresome containing MYC-CTIF. Immunostained images in panel (D) were quantitated. Statistical analysis was performed by one-way ANOVA with post hoc Tukey’s honestly significant difference test; ***, P < 0.00001
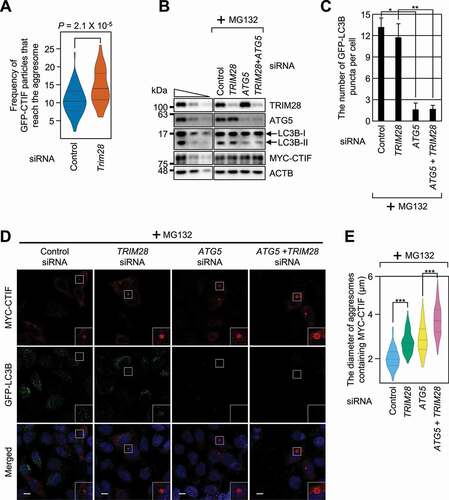
We next investigated whether TRIM28-mediated inhibition of aggresome formation is linked to autophagy. For this purpose, the cells were either undepleted or depleted of TRIM28, ATG5 (a central protein for autophagosome formation), or both (). Under our conditions, downregulation of ATG5 significantly reduced the conversion of LC3B-I to LC3B-II () and the number of LC3B puncta (; both are hallmarks of autophagy) and increased the diameter of the aggresome containing MYC-CTIF, suggesting that the aggresome removal depends on autophagy. In contrast, downregulation of TRIM28 only marginally affected the number of LC3B puncta but significantly increased the aggresome diameter even in the cells depleted of ATG5. In agreement with these observations, an increase in the diameter of the aggresome after TRIM28 downregulation was observed in the cells treated with a potent autophagy inhibitor, bafilomycin A1 (Fig. S3). All these data provide supporting evidence that TRIM28 specifically targets a step in aggresome formation.
The S473 residue is important for the TRIM28-mediated inhibition of aggresome formation
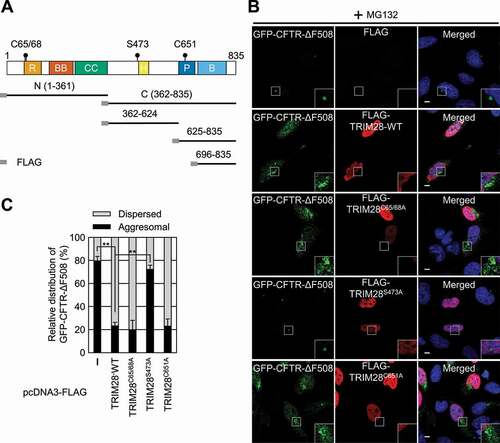
TRIM28 is a multidomain protein. The N-terminal half contains a RING domain, followed by two B-box domains and a coiled-coil region. The RING domain has an intrinsic E3 ubiquitin ligase activity. The C-terminal half of TRIM28 contains an HP1 protein-binding domain, a PHD domain (which has an intrinsic E3 SUMO ligase activity), and bromodomain. We were curious about which region of TRIM28 contributes to its inhibitory role in aggresome formation. For this purpose, we first delineated a minimal domain of TRIM28 required for CTIF binding. Using a series of deletion variants of TRIM28, we determined a region spanning aa 362–624 (containing the HP1-binding domain) as the minimal region for the binding of TRIM28 to CTIF (Fig. S4A).
As an alternative approach, we constructed three TRIM28 variants that harbor amino acid substitution(s) at C65 and C68 (critical for the ubiquitin ligase activity promoted by MAGE proteins) [Citation46], S473 (specifically phosphorylated in response to viral RNA in a EIF2AK2-dependent manner) [Citation39], or C651 (critical for SUMO ligase activity) [Citation47] (). We observed that approximately 80% of the cells showed a distinct aggresome containing GFP-CFTR-ΔF508 under MG132 treatment conditions ( and Fig. S4B). In contrast, when FLAG-TRIM28-WT, FLAG-TRIM28C65,68A, or FLAG-TRIM28C651A was overexpressed, approximately 20% of the cells contained the GFP-CFTR-ΔF508 aggresome. Moreover, overexpression of FLAG-TRIM28S473A did not significantly affect the relative level of the GFP-CFTR-ΔF508 aggresome, indicating that the S473 residue is essential for TRIM28-mediated inhibition of aggresome formation.
Figure 4. The S473 residue is responsible for TRIM28-mediated inhibition of aggresome formation. (A) A schematic diagram of the TRIM28 protein. The mutation sites are indicated by solid circles. Deletion variants of TRIM28 with the N-terminal FLAG-tag are also indicated. R, RING domain; BB, two B-box-type zinc finger domains; CC, coiled-coil motif; H, HP1-binding domain; P, PHD domain; and B, bromodomain. (B) Immunostaining of GFP-CFTR-ΔF508 and FLAG-TRIM28, either WT or its variant. HeLa cells stably expressing GFP-CFTR-ΔF508 were transiently transfected with a plasmid expressing FLAG, FLAG-TRIM28-WT, or its variant. Two days later, the cells were treated with MG132 for 12 h before fixation. The cells were stained with the anti-GFP antibody (green) and an anti-FLAG antibody (red). Nuclei were stained with DAPI (blue). Scale bar: 10 μm; n = 3. (C) Relative distribution of GFP-CFTR-ΔF508. Relative percentages were determined by counting the cells containing either the aggresome or the dispersed aggregates of GFP-CFTR-ΔF508 in the immunostaining images. To accurately assess the effect of exogenously expressed FLAG-TRIM28, only the cells expressing both GFP-CFTR-ΔF508 and exogenous FLAG-TRIM28 were counted. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values. **, P < 0.01
EIF2AK2-mediated S473 phosphorylation of TRIM28 is critical for the inhibition of aggresome formation
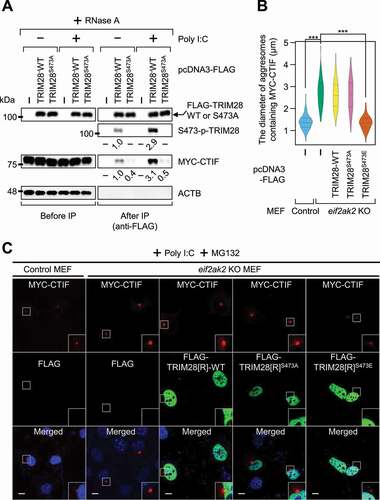
TRIM28 is phosphorylated at the S473 residue after either viral infection or poly I:C treatment in a EIF2AK2-dependent manner [Citation39]. Poly I:C is a widely used synthetic double-stranded RNA that helps to recapitulate the conditions of virus-infected cells. As expected, poly I:C treatment increased the phosphorylation of endogenous TRIM28 by approximately 3-fold in the cells either untreated or treated with MG132 (Fig. S5A). To investigate a possible role of S473 phosphorylation in the interaction between TRIM28 and CTIF, we next carried out IP experiments using the extracts of cells either treated or not treated with poly I:C ( and Fig. S5B). Poly I:C treatment resulted in an approximately 3-fold increase in both the level of phosphorylation of TRIM28 and the amount of co-IPed CTIF in the IPs of TRIM28. In contrast, the S473A substitution in TRIM28 decreased by approximately 2-fold the amount of co-IPed CTIF regardless of poly I:C treatment (). By contrast, the S473E substitution in TRIM28 (a phospho-mimetic form) increased the amount of co-IPed CTIF by approximately 3-fold even in the cells not treated with poly I:C (Fig. S5B), suggesting that the phosphorylation of S473 promotes an interaction between TRIM28 and CTIF. Furthermore, treatment of MEFs with poly I:C caused a reduction in the size of aggresomes containing MYC-CTIF (Fig. S5C,D). On the contrary, the size of the aggresomes was not significantly affected by the treatment with poly I:C in eif2ak2 KO (knockout) MEFs (Fig. S5C,D). Under these conditions, overexpression of FLAG-TRIM28S473E, but not FLAG-TRIM28-WT or FLAG-TRIM28S473A, significantly reduced the size of the aggresome containing MYC-CTIF (). The above data indicate that activated EIF2AK2 (upon poly I:C treatment) triggers TRIM28 phosphorylation at S473, promotes the interaction between TRIM28 and CTIF, and consequently leads to inefficient aggresome formation.
Figure 5. EIF2AK2-mediated S473 phosphorylation of TRIM28 inhibits the formation of aggresomes containing MYC-CTIF. (A) IP of FLAG-TRIM28-WT or FLAG-TRIM28S473A. HEK293T cells transiently expressing MYC-CTIF and FLAG, FLAG-TRIM28-WT, or FLAG-TRIM28S473A were either transfected or not transfected with poly I:C. The cell extracts were digested with RNase A and subjected to IP with FLAG M2 affinity gel; n = 3. (B and C) The impact of EIF2AK2 on aggresome formation. Either WT or eif2ak2 KO MEFs transiently expressing MYC-CTIF and FLAG, FLAG-TRIM28-WT, or its variant were transfected with poly I:C. The cells were treated with MG132 for 12 h before cell fixation; n = 3. (B) The diameter of aggresomes containing MYC-CTIF. Statistical analysis was performed by one-way ANOVA with post hoc Tukey’s honestly significant difference test; ***, P < 0.00001. (C) Immunostaining of MYC-CTIF (red) and FLAG-TRIM28 or its variant (green). Nuclei were stained with DAPI (blue). Scale bar: 10 μm
To corroborate the above conclusion, we carried out complementation experiments using TRIM28 siRNA and siRNA-resistant (R) FLAG-TRIM28[R]-WT, FLAG-TRIM28[R]S473A, or FLAG-TRIM28[R]S473E. As expected, all tested aggresomes containing GFP-CFTR-ΔF508, MYC-CTIF, or RFP-Ub were dispersed or reduced in size during treatment with poly I:C under MG132 treatment conditions ( and Fig. S6). Under the same conditions, downregulation of TRIM28 abrogated or reversed the observed dispersion or reduction in the size of aggresomes ( and Fig. S6). Notably, the expression of FLAG-TRIM28[R]-WT or FLAG-TRIM28[R]S473E, but not FLAG-TRIM28[R]S473A, at a level comparable to that of endogenous TRIM28 in TRIM28-depleted cells caused a poly I:C-induced dispersion of the aggresome containing GFP-CFTR-ΔF508 (). All these findings support our notion that S473 phosphorylation of TRIM28 is crucial for TRIM28–mediated inhibition of aggresome formation.
Figure 6. Poly I:C–induced TRIM28 phosphorylation hinders the formation of aggresomes containing GFP-CFTR-ΔF508. HeLa cells stably expressing GFP-CFTR-ΔF508 were treated with a control siRNA or TRIM28 siRNA. One day later, the cells were transfected with a plasmid expressing siRNA-resistant FLAG-TRIM28[R] (WT, S473A, or S473E) or a plasmid expressing only FLAG, which served as a negative control. One day later, the cells were either mock-transfected or transfected with poly I:C. The cells were treated with MG132 for 12 h before cell fixation; n = 3. (A) Western blotting validating specific downregulation of endogenous TRIM28 by means of siRNA and the expression of FLAG-TRIM28[R]-WT, FLAG-TRIM28[R]S473A, or FLAG-TRIM28[R]S473E at a level comparable to that of endogenous TRIM28. (B) Immunostaining of GFP-CFTR-ΔF508 (green) and exogenously expressed FLAG-TRIM28[R]-WT, FLAG-TRIM28[R]S473A, or FLAG-TRIM28[R]S473E (red). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (C) Relative percentages of cells containing either aggresomal or dispersed GFP-CFTR-ΔF508. To accurately assess the effect of exogenously expressed FLAG-TRIM28, only the cells expressing both GFP-CFTR-ΔF508 and exogenous FLAG-TRIM28 were counted. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values. *, P < 0.05; **, P < 0.01
![Figure 6. Poly I:C–induced TRIM28 phosphorylation hinders the formation of aggresomes containing GFP-CFTR-ΔF508. HeLa cells stably expressing GFP-CFTR-ΔF508 were treated with a control siRNA or TRIM28 siRNA. One day later, the cells were transfected with a plasmid expressing siRNA-resistant FLAG-TRIM28[R] (WT, S473A, or S473E) or a plasmid expressing only FLAG, which served as a negative control. One day later, the cells were either mock-transfected or transfected with poly I:C. The cells were treated with MG132 for 12 h before cell fixation; n = 3. (A) Western blotting validating specific downregulation of endogenous TRIM28 by means of siRNA and the expression of FLAG-TRIM28[R]-WT, FLAG-TRIM28[R]S473A, or FLAG-TRIM28[R]S473E at a level comparable to that of endogenous TRIM28. (B) Immunostaining of GFP-CFTR-ΔF508 (green) and exogenously expressed FLAG-TRIM28[R]-WT, FLAG-TRIM28[R]S473A, or FLAG-TRIM28[R]S473E (red). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (C) Relative percentages of cells containing either aggresomal or dispersed GFP-CFTR-ΔF508. To accurately assess the effect of exogenously expressed FLAG-TRIM28, only the cells expressing both GFP-CFTR-ΔF508 and exogenous FLAG-TRIM28 were counted. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values. *, P < 0.05; **, P < 0.01](/cms/asset/f601636d-6024-41c7-97ac-58ad4b85f4d4/kaup_a_1909835_f0006_oc.jpg)
TRIM28 suppresses proliferation of influenza A viruses by regulating aggresome formation
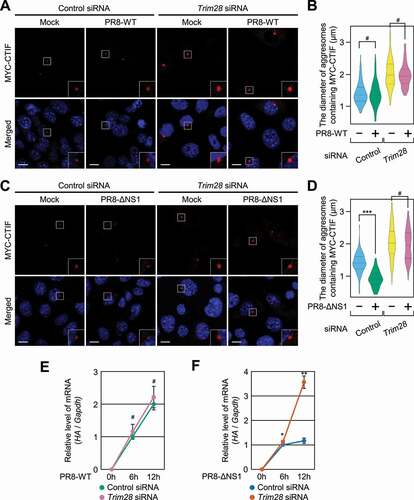
Considering that (i) IAVs take advantage of the aggresome machinery for efficient capsid disassembly [Citation27], (ii) NS1 (nonstructural protein 1) of IAV directly binds to EIF2AK2 and inhibits EIF2AK2 activation [Citation48-52], and (iii) TRIM28-mediated inhibition of aggresome formation depends on S473 phosphorylation of TRIM28 via EIF2AK2 (), we investigated a possible interplay among IAV infection, EIF2AK2 activation, and TRIM28-mediated inhibition of aggresome formation. To this end, we employed one of two IAVs, either strain PR8-WT (wild-type A/Puerto Rico/8/34, which blocks EIF2AK2 activation) or strain PR8-ΔNS1 (which lacks NS1 and thereby activates EIF2AK2), and compared the proliferation of these two viruses in the cells either undepleted or depleted of TRIM28 (). Infection with PR8-ΔNS1 caused a significant increase in TRIM28 phosphorylation and the level of phosphorylation of endogenous EIF2S1/EIF2α (which is a previously well-known substrate for EIF2AK2 and therefore served as an indicator of EIF2AK2 activity) as compared to infection with PR8-WT (Fig. S7), proving that PR8-ΔNS1 efficiently activated EIF2AK2 under our conditions. Accordingly, infection with strain PR8-ΔNS1 significantly reduced the diameter of aggresomes containing MYC-CTIF in a manner that was reversed by TRIM28 downregulation (). By contrast, infection with PR8-WT had no significant influence on the diameter of the aggresomes (). Of note, in agreement with a report that efficient entry of IAVs into host cells involves aggresome machinery [Citation27], PR8-WT proliferation was not significantly affected by TRIM28 downregulation (), whereas the proliferation of PR8-ΔNS1 was found to be significantly increased when TRIM28 was downregulated (). All these data indicate that TRIM28-mediated inhibition of aggresome formation suppresses viral proliferation.
Figure 7. Proliferation of the influenza A virus (IAV) lacking NS1 depends on TRIM28. (A and B) The effect of infection with IAV strain PR8-WT on aggresomes containing MYC-CTIF. MEFs either undepleted or depleted of TRIM28 were either mock-infected or infected with PR8-WT for 12 h at a multiplicity of infection of 2; n = 3. (A) Immunostaining of MYC-CTIF (red). Scale bar: 10 μm. (B) The diameter of an aggresome containing MYC-CTIF. Statistical analysis was performed by one-way ANOVA with post hoc Tukey’s honestly significant difference test; ***, P < 0.00001. (C and D) The effect of PR8-ΔNS1 infection on aggresomes containing MYC-CTIF. As performed in panels A and B, except that the cells were infected with strain PR8-ΔNS1. (C) Immunostaining of MYC-CTIF (red). Scale bar: 10 μm. (D) The diameter of an aggresome containing MYC-CTIF. ***, P < 0.00001. (E) The time course of relative expression levels of IAV HA mRNA in the cells infected with strain PR8-WT. MEFs either undepleted or depleted of TRIM28 were infected with PR8-WT at a multiplicity of infection of 2 and harvested at the indicated time points after infection. The HA mRNA level was normalized to that of Gapdh mRNA at each time point. Two-tailed, equal-sample variance Student’s t test was carried out to calculate the P values; n = 3; #, not significant. (F) The time course of relative expression levels of IAV HA mRNA in the cells infected with PR8-ΔNS1; n = 3; *, P < 0.05; **, P < 0.01
Discussion
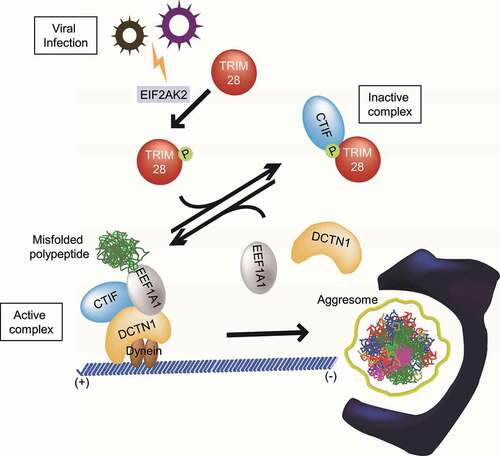
The aggresome-autophagy pathway is one of the major cellular mechanisms contributing to protein homeostasis in the cell. Some viruses often employ aggresome machinery for their efficient proliferation and replication. In this study, we demonstrate an inhibitory role of TRIM28 in the formation of the aggresome containing misfolded polypeptides (). This inhibitory function of TRIM28 is mediated by a direct interaction between TRIM28 and CTIF, which is promoted by TRIM28 phosphorylation by EIF2AK2. This interaction disrupts the CED complex and consequently hinders the movement of misfolded polypeptides toward the aggresome. We also demonstrate that the EIF2AK2-TRIM28-CTIF-aggresome axis contributes to the suppression of IAV proliferation, highlighting the biological importance of this axis. Considering that a variety of mammalian viruses associates with aggresomes or aggresome–like structures during their life cycle [Citation23-26], our data open up a new possibility that this axis could function as a host defensive mechanism against viral infection.
Figure 8. The proposed model for the regulation of aggresome formation mediated by TRIM28. Depending on relative amounts of TRIM28 and the extent of EIF2AK2-induced S473 phosphorylation of TRIM28, CTIF may form one of two alternative complexes: CTIF-TRIM28 (a complex inactive in terms of aggresome formation) and the CED complex (a complex active in terms of aggresome formation). See the Discussion section for details
It is well known that TRIM28 mainly acts as a transcriptional cofactor in the nucleus. Consistently with this notion, we also observed the nuclear localization of TRIM28 throughout this study. Nevertheless, our PLA results showed that PLA signals between TRIM28 and CTIF were largely present in the cytoplasm (). Because the PLA technique allows to determine an intracellular site where a protein-protein interaction takes place, most of the TRIM28-CTIF interaction may occur in the cytoplasm. Indeed, we observed that a small proportion of TRIM28 was present in the cytoplasm (Fig. S8). In addition, Yang et al. has proposed that TRIM28 accumulates in the cytoplasm after cells are treated with a pan-HDAC inhibitor and that cytoplasmic TRIM28 interacts with acetylated HSPA/HSP70 for modulating autophagopore formation [Citation33]. Therefore, it is most likely that a small amount of TRIM28 present in the cytoplasm is sufficient for TRIM28–mediated inhibition of aggresome formation.
Several reports imply that aside from the viral life cycle, TRIM28 has a potential function in the regulation of the expression of SNCA and MAPT/Tau (microtubule associated protein tau) proteins enriched in inclusion bodies that are found in neuronal cells affected by various neurodegenerative diseases [Citation53-56]. In general, TRIM28 is upregulated in the tissues of patients with neurodegenerative disease [Citation54,Citation56]. The upregulated TRIM28 increases the levels of SNCA and MAPT [Citation54]. Furthermore, a reduction in the amount of TRIM28 in adult mice causes a decrease in the levels of SNCA and MAPT proteins and relieves the adverse effects of these proteins [Citation54]. Although it remains unclear whether a change in TRIM28 abundance is a cause or consequence of neurodegenerative diseases, it should also be noted that (i) TRIM28 functions as a negative regulator of aggresome formation (according to our study), (ii) the aggresome observed in cultured cells is biochemically and morphologically similar to the inclusion bodies found in cells affected by various neurodegenerative diseases [Citation21], and (iii) the distribution of CTIF overlaps with that of SNCA in the cerebellar molecular layer of patients with Parkinson disease [Citation15]. Therefore, it is most likely that TRIM28 participates in the formation of inclusion bodies found in the cells of patients with neurodegenerative diseases. In this regard, future studies should address the possible biological and pathological roles of TRIM28 in various neurodegenerative diseases.
Materials and methods
Cell culture
HEK293T cells (fetal; ATCC, CRL-3216), HeLa cells (female; ATCC, CCL-2), HeLa cells stably expressing GFP-LC3B, and MEFs (a gift from Dr. Byung-Yoon Ahn, Korea University, Seoul, Korea) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, D6429) containing 10% fetal bovine serum (Sigma, TMS-013-BKR) and 1% penicillin-streptomycin solution (Sigma, P4333). HeLa cells stably expressing GFP-CFTR-ΔF508 [Citation15] were cultured in media supplemented with 0.4 mg/ml G418.
To induce aggresome formation, the cells were treated with MG132 (5 μM; Calbiochem, 474,790) predissolved in DMSO (BioShop, DMS666.50) for 12 h. To specifically label newly synthesized misfolded polypeptides, the cells were treated with puromycin (10 μg/ml for IPs and 0.1 or 0.025 μg/ml for the confocal microscopy assay) for 1 h. Where indicated, the cells were treated with bafilomycin A1 (100 nM; Calbiochem, 196,000) to inhibit autophagy. To trigger cellular stress induced by double-stranded RNAs, the cells were transfected with poly I:C (1 μg/ml; InvivoGen, tlrl-picw) 12 h before cell harvest.
Two IAVs – PR8-WT (wild-type A/Puerto Rico/8/34) and PR8-ΔNS1– were propagated in embryonated chicken eggs. MEFs seeded in 60-mm dishes were either mock-infected with PBS (phosphate-buffered saline; [8.1 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4]) or infected with PR8-WT, or PR8-ΔNS1 at a MOI (multiplicity of infection) of 2 for 1 h. The viral inoculum was washed with PBS three times and then maintained in DMEM supplemented with 2% of fetal bovine serum and 1% of penicillin-streptomycin. The infected cells were collected at 6 or 12 hours post infection (hpi) or fixed with 3.65–3.80% formaldehyde (Sigma, F8775) at 12 hpi. The collected cells were subjected to western blotting or qRT-PCR (quantitative reverse-transcription PCR), whereas the fixed cells were immunostained.
Plasmid construction
The following constructs have been reported previously or purchased from the indicated company: pcDNA3-FLAG, pcDNA3-FLAG-CTIF, and pCMV-MYC-CTIF [Citation57]; pCMV-MYC-GST, pCMV-MYC-CTIF(1-53)-GST, pCMV-MYC-CTIF(12-53)-GST, and pcDNA3-FLAG–CTIF(54-598) [Citation15]; pGEX-6p-1 [Citation58]; pGEX-6p-1-CTIF, pGEX-6p-1-EEF1A1, and pGEX-6p-1-DCTN1 [Citation14]; pCXbsr-mRFP-Ub [Citation16]; pEGFP-N1-SYN1 [Citation44]; pCNS-TRIM28 (KU026684; 21 C Frontier Human Gene Bank, KRIBB, Korea); pCMV-MYC (Clontech, 635,689); and pRSET A (Invitrogen, V35120).
To construct pCMV-MYC-TRIM28, the cDNA (complementary DNA) of TRIM28 sequences was amplified by PCR (polymerase chain reaction) using pCNS-TRIM28 as a template and specific oligonucleotides: 5′-CGGAATTATTATGGCGGCCTCCGCGGCGGCAGCCTCGGCA-3′ (sense) and 5′-CCGCTCGAGTCAGGGGCCATCACCAGGGCCACCAGACAG-3′ (antisense), where the underlined nucleotides specify EcoRΙ and XhoΙ sites, respectively. The PCR-amplified fragment was digested with EcoRΙ and XhoΙ and ligated to EcoRΙ/XhoΙ-digested pCMV-MYC.
To construct the pcDNA3-FLAG-TRIM28 plasmid, pcDNA3-FLAG-Dcp1a [Citation59] was digested with EcoRΙ and NotΙ and then ligated with two fragments: (i) an EcoRΙ/BamHI fragment of the PCR-amplified 5′ half of TRIM28 cDNA and (ii) a BamHΙ/NotΙ fragment of pCMV-MYC-TRIM28 corresponding to the 3′ half of TRIM28 cDNA. The EcoRΙ/BamHI fragment was amplified by PCR using pCNS-TRIM28 as a template and two oligonucleotides: 5′-CGGAATTCATGGCGGCCTCCGCGGCGGCAGCCTCGGC-3′ (sense) and 5′-GCTCAGGGGCCCTGGGGCTCTTGGAGGGGC-3′ (antisense), where the underlined nucleotides denote EcoRI sites.
To generate plasmids pcDNA3-FLAG–TRIM28(1-361) and pcDNA3-FLAG–TRIM28(362-835), an EcoRI/XhoI fragment of pcDNA3-FLAG-TRIM28 was ligated to a PCR-amplified EcoRI/XhoI fragment encoding the aa 1–361 or 362–835 region of TRIM28, respectively. The fragment encoding the aa 1–361 region of TRIM28 was amplified by PCR using pcDNA3-FLAG-TRIM28 as a template and specific oligonucleotides: 5′-GGCTAACTAGAGAACCCACTGCTTACTGGC-3′ (sense) and 5′-CCGCTCGAGTCAAAGGGCTGTGTTGTTGTCACTCTCCAGAGC-3′ (antisense), where the underlined nucleotides specify the XhoI site. The fragment encoding the aa 362–835 region of TRIM28 was amplified by PCR using pcDNA3-FLAG-TRIM28 as a template and specific oligonucleotides: 5′-CCGGAATTCTTGCTTTCTAAGAAGTTGATCTACTTCCAG-3′ (sense) and 5′-CAAACAACAGATGGCTGGCAACTAGAAGGC-3′ (antisense), where the underlined nucleotides specify the EcoRI site.
To generate plasmids pcDNA3-FLAG–TRIM28(362-624), pcDNA3-FLAG–TRIM28(625-835), and pcDNA3-FLAG–TRIM28(696-835), an EcoRI/XhoI fragment of pcDNA3-FLAG–TRIM28(362-835) was ligated to a PCR-amplified EcoRI/XhoI fragment encoding the aa 362–624, 625–835, or 696–835 region of TRIM28. The fragment encoding the aa 362–624, 625–835, or 696–835 region of TRIM28 was amplified by PCR using pcDNA3-FLAG–TRIM28(362-835) as a template and specific oligonucleotides: (i) 5′-CCGGAATTCTTGCTTTCTAAGAAGTTGATCTACTTCCAG-3′ (sense) and 5′-ACCTCGAGTCAACTGTCATCCAGGGTTCCCGGGCCACCACC-3′ (antisense) for aa 362–624, (ii) 5′-CCGAATTCGCCACCATTTGCCGTGTCTGCCAG-3′ (sense) and 5′-CCGCTCGAGTCAGGGGCCATCACCAGGGCCAC-3′ (antisense) for aa 625–835, and (iii) 5′-CCGAATTCCTCTCACCAGCCAACCAGCGGAAATG-3′ (sense) and 5′- CCGCTCGAGTCAGGGGCCATCACCAGGGCCAC −3′ (antisense) for aa 696–835, where the underlined nucleotides specify the EcoRI and XhoI site, respectively.
pcDNA3-FLAG-TRIM28[R]-WT was constructed by two-step PCR. First, the 5′ fragment encoding the N-terminal half of TRIM28[R]-WT and the 3′ fragment encoding the C-terminal half of TRIM28[R]-WT were amplified by PCR using pcDNA3-FLAG-TRIM28 as a template with specific oligonucleotides: (i) 5′-ACTATAGGGAGACCCAAGCTGCCAC-3′ (sense) and 5′- CTCTCACAAAAGAGGACTAGCGGCTCGTGTTTGTGTACGTTGCAATAG-3′ (antisense) for the amplification of the 5′ fragment and (ii) 5′- ACGTACACAAACACGAGCCGCTAGTCCTCTTTTGTGAGAGCTGTG-3′ (sense) and 5′-GAGGTCCCACTGAAACTTCATCTCG-3′ (antisense) for the 3′ fragment (mutated sequences are italicized). Next, the two PCR-amplified fragments were mixed and reamplified by PCR with the sense oligonucleotide used for the amplification of the 5′ fragment and the antisense oligonucleotide used for the amplification of the 3′ fragment. The resulting PCR-amplified fragment was digested with BamHI and then ligated to a BamHI fragment of pcDNA3-FLAG-TRIM28.
Variants expressing FLAG-TRIM28[R]C65,68A, FLAG-TRIM28[R]S473A, FLAG-TRIM28[R]S473E, or FLAG-TRIM28[R]C651A were constructed by means of a PCR-based replacement of the corresponding nucleotide sequences with an alanine or glutamate codon in pcDNA3-FLAG-TRIM28[R].
A plasmid expressing His–tagged TRIM28 was generated by inserting TRIM28 cDNA into pRSET A.
DNA or siRNA transfection
For IP experiments, HEK293T cells were transiently transfected with a plasmid by the calcium phosphate method, as described previously [Citation60-62]. For confocal microscopy experiments, cells were transiently transfected with the various plasmids using Lipofectamine 2000 (HeLa cells; Invitrogen, 11,668,019) or a Neon™ Transfection System (for MEFs; Invitrogen, MPK5000S).
To downregulate endogenous proteins, the cells were transfected with 100 or 200 nM in vitro-synthesized siRNA (Gene Pharma) via Lipofectamine 3000 (Invitrogen, L3000150) or Oligofectamine (Life Technologies, 12,252,011). The following siRNA sequences were employed in this study: 5′-r(GAAGUGGAGAUCGCACACA)d(TT)-3′ for human CTIF, 5′-r(GCAUGAACCCCUUGUGCUG)d(TT)-3′ for human TRIM28, 5′-r(CCAAAGACAUCGUGGAGAATT)d(TT)-3′ for mouse Trim28, 5′-r(CCCAGGACACAGAGACUUU)d(TT)-3′ for human EEF1A1, 5′-r(GGAUGCAAUUGAAGCUCAU)d(TT)-3′ for human ATG5, and 5′-r(CAGAGAAGGCAGAACUAAA)d(TT)-3′ for human DCTN1. The siRNA sequences for the control have been described elsewhere [Citation58]. Cells were harvested or fixed 2 days after DNA transfection or 3 days after siRNA transfection.
Antibodies
Primary antibodies against the following proteins were used in this study: TRIM28 (KAP1; Novus, NB500–158), TRIM28 pS473 (BioLegend, 654,102), CTIF [Citation57], DCTN1/p150glued (BD Biosciences, 610,474), EEF1A1 (Merck Millipore, 05-235), HDAC6 (Bethyl Laboratories, A301-342A), BAG3 (Proteintech, 10,599-1-AP), puromycin (12D10; Merck Millipore, MABE343), GFP (Santa Cruz Biotechnology, sc-9996), ACTB/β-actin (Sigma, A5441), GST (Bethyl Laboratories, A190–122A), His (GE Healthcare, 27-4710-01), FLAG (DYKDDDDK; Cell Signaling Technology, 14,793; Sigma, F1804; or Sigma, A8592), ubiquitin (Lys63–specific; Merck Millipore, 05-1308), LC3B (Cell Signaling Technology, 3868), ATG5 (Cell Signaling Technology, 12,994), EIF2A/EIF2S1 (EIF2α; Cell Signaling Technology, 9722), phospho-EIF2S1 (Ser51; Cell Signaling Technology, 9721), SNRNP70/U1 snRNP70 (Santa Cruz Biotechnology, sc-390,899), GAPDH (Ab Frontier, LF-PA0212), and MYC (9E10; Calbiochem, OP10L; or Cell Signaling Technology, 2272).
The following secondary antibodies against the primary antibodies were applied for western blotting and immunostaining: a horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Merck Millipore, AP124P), horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Merck Millipore, AP132P), Alexa Fluor® 488-conjugated goat anti-mouse IgG antibody (Invitrogen, A11017), and a rhodamine-conjugated goat anti-rabbit IgG antibody (Invitrogen, 31,670).
Western blotting
Whole-cell extracts or IP samples were incubated with 2× sample buffer (10% β-mercaptoethanol, 4% SDS [sodium dodecyl sulfate], 100 mM Tris-HCl, pH 6.8, 15% glycerol, and 0.008% bromophenol blue) and boiled for 5 min at 95°C. The samples were analyzed by SDS polyacrylamide gel electrophoresis. After that, the samples were transferred to Protran Premium nitrocellulose (Amersham, 10,600,021). The primary and secondary antibodies described above were used to detect the specific proteins. After western blotting, the band intensities of the obtained images were quantitated using the ImageJ software (version 1.52a).
Quantitative reverse-transcription PCR (qRT-PCR)
Total-RNA samples were purified with the TRIzol Reagent (Thermo Scientific, 15,596,018) and were utilized to synthesize cDNA using RevertAid Reverse Transcriptase (Thermo Scientific, EP0442). qRT-PCRs were performed with gene-specific oligonucleotides and the Light Cycler 480 SYBR Green I Master Mix (Roche, 04887352001) on a Light Cycler 480 II machine (Roche). The following gene-specific oligonucleotides for the amplification of mRNAs were used in this study: 5′-GGCCCAACCACAACACAAAC-3′ (sense) and 5′-AGCCCTCCTTCTCCGTCAGC-3′ (antisense) for IAV HA mRNAs and 5′-TGGCAAATTCCATGGCACC-3′ (sense) and 5′-AGAGATGATGACCCTTTTG–3′ (antisense) for Gapdh mRNAs.
Immunoprecipitation
IP experiments were performed on HEK293T cells as described elsewhere [Citation60-62]. Briefly, HEK293T cells were harvested by centrifugation at 3,000 × g for 10 min at 4°C. The pellet was resuspended in NET-2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM PMSF [phenylmethylsulfonyl fluoride; Sigma, P7626], 2 mM benzamidine hydrochloride [Sigma, B6506], 0.05% NP-40 [IGEPAL® CA–630; Sigma, I8896], 10 mM sodium fluoride [Sigma, 201,154], and 0.25 mM sodium orthovanadate [Sigma, 13,721-39–6]). Next, the suspended cells were sonicated and centrifuged. For preclearing, the cell extracts were mixed with either protein G agarose 4B beads (Incospharm, 1105–3) or protein A agarose 4B beads (Incospharm, 1106-3) and incubated for 1 h at 4°C. The precleared samples were incubated for 3 h at 4°C with antibody-conjugated beads or FLAG M2 affinity gel (Sigma, A2220). The beads were washed five times with NET-2 buffer and subjected to elution with 2× sample buffer. The samples before and after IP were analyzed by western blotting.
Where indicated, the supernatant after sonication was treated with RNase A to rule out a possible RNA-mediated indirect protein-protein interaction during IP.
Immunostaining
HeLa cells were fixed with 3.65–3.80% formaldehyde for 30 min and permeabilized with 0.5% Triton X-100 (Sigma, T8787) for 10 min. The fixed cells were treated with 1.5% bovine serum albumin (BSA; Bovogen Biologicals, BSAS0.1) for 1 h for blocking nonspecific binding sites. After that, the cells were probed with a primary antibody diluted in 0.5% BSA (incubation for 1 h). Then, the primary antibody was detected by incubation with an appropriate secondary antibody conjugated to either Alexa Fluor 488 or rhodamine and diluted in 0.5% BSA. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Biotium, 40,011). Immunostained cells were examined under an LSM 510 Meta, LSM 700, or LSM 800 microscope (Carl Zeiss).
The in situ proximity ligation assay
This experiment was performed using the In Situ Proximity Ligation Assay (Merck Millipore, DUO92001, DUO92005, and DUO92014). Briefly, cultured cells were fixed with formaldehyde and permeabilized with Triton X-100. The fixed cells were sequentially incubated in blocking buffer for 1 h at 37°C, in antibody diluent buffer containing primary antibodies for 1 h at 37°C, and in antibody diluent buffer containing secondary antibodies conjugated with either a PLUS or MINUS PLA probe. After that, the samples were incubated in ligation buffer with ligase for 30 min at 37°C. Next, the samples were incubated with a polymerase mixture and Amplification Green buffer for 100 min at 37°C for rolling circle amplification. The resultant samples were stained with DAPI and visualized using the LSM 800 Carl Zeiss confocal microscope.
The in vivo GST affinity-isolation assay
The GST affinity–isolation assay was performed as described previously [Citation60-62]. Briefly, HEK293T cells transiently expressing a GST-tagged protein were pelleted and washed with ice-cold PBS. The pellets were resuspended in NET–2 buffer and sonicated on ice. After centrifugation, the supernatant was incubated with protein G agarose 4B beads for 1 h at 4°C for preclearing. The precleared supernatant was mixed with the glutathione Sepharose 4B (GE Healthcare, 17-5279-01) in NET-2 buffer for 3 h incubation at 4°C. The resin-bound samples were washed three times with ice-cold NET-2 buffer and were subjected to elution with 2× sample buffer.
The in vitro GST affinity-isolation assay
This procedure was conducted using recombinant GST, GST-CTIF, GST-EEF1A1, GST-DCTN1, or 6× His-TRIM28. Escherichia coli BL21(DE3)pLysS was transformed with a plasmid encoding a recombinant protein. The recombinant proteins were induced by the addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside followed by 3 h incubation at 4°C. After that, the cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Triton X-100, 1 mM dithiothreitol, 10% [v:v] glycerol, 1 mM PMSF, and 2 mM benzamidine hydrochloride [Sigma, B6506]) and sonicated. The lysate of cells expressing a GST-fused protein was mixed with the lysate of cells expressing a 6× His-fused protein and kept in incubation buffer (10 mM HEPES pH 7.4, 1.5 mM magnesium acetate, 150 mM potassium acetate, 2.5 mM dithiothreitol, and 0.05% NP-40) for 30 min at 4°C. Next, the glutathione Sepharose 4B resin was added to the mixture and incubated for 2 h at 4°C. The resin-bound proteins were washed four times using incubation buffer and eluted with 2× sample buffer.
LC-MS/MS
SDS-PAGE and silver staining were performed as described previously [Citation60]. In-gel digestion and LC-MS/MS were conducted by Proteomtech (Korea).
Optical microscopy and image acquisition
Single-molecule imaging in live cells was performed by highly inclined and laminated optical sheet (HILO) microscopy [Citation63]. GFP-CTIF was excited by a 488-nm laser (Cobolt MLDTM 488 nm). A high-numerical-aperture (NA) objective (Olympus UPLXAPO100xOPH, NA = 1.45, oil immersion) in an inverted microscope (Olympus IX71) was used to focus the excitation lasers and gather the emitted photons. The emitted light was filtered through emission filter sets (Semrock FF01-600/37-25 and FF01-520/35-25 for GFP) using a DV2 multichannel imaging system (Photometrics) that was placed in front of the EMCCD (electron-multiplying charge-coupled device) camera (Andor iXon 897). All images were recorded by the SOLIS imaging software (Andor) every 100 ms. To maintain the viability of cells during the fluorescence imaging, the imaging was performed in a CO2 incubation chamber at 37°C (Live Cell Instrument).
Nucleocytoplasmic fractionation
HeLa cells were lysed with hypotonic buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 0.1% Triton X-100, 10 mM EDTA, 1 mM PMSF, and 2 mM benzamidine hydrochloride) and incubated for 10 min on ice. After centrifugation, the supernatant was collected for the cytoplasmic fraction. The pellet was washed three times with NET-2 buffer and resuspended in hypotonic buffer. The lysates were sonicated and centrifuged. After that, the supernatant was collected as the nuclear fraction. Western blotting using antibodies against SNRNP70 (a marker for the nuclear fraction) and GAPDH (a marker for the cytoplasmic fraction) was performed to confirm proper nucleocytoplasmic fractionation.
Statistical analysis
For statistical comparisons, two-tailed and equal-variance Student’s t test was performed with significance defined as a P value < 0.05 or < 0.01. In most cases, data obtained from at least three independently performed biological replicates were analyzed, unless indicated otherwise in the figure legends. Data in all graphs are presented as the mean ± standard deviation. In the case of IP experiments, we presented average values of quantification obtained from three biological replicates. The corresponding P values for each quantitation are summarized in Table S2.
To determine the diameter of CTIF-containing aggresomes per cell, more than 30 individual cells were visualized in each experiment by means of LSM 510 Meta or LSM 800. The size of CTIF bodies was measured using Zeiss LSM Image Browser. One-way ANOVA followed by Tukey’s honestly significant difference test was carried out to calculate the statistical significance of the results. The results obtained from at least three biological replicates were counted independently.
To determine the frequency of GFP-CTIF aggregates moving into the aggresome (), we counted the GFP-CTIF particles entering the aggresome during 300 frames in ImageJ (version 1.51 j). One-way ANOVA with post hoc Tukey’s honestly significant difference test was carried out to compute the statistical significance.
For quantification of aggresomes or dispersed aggregates in cells, more than 50 cells were imaged in each experiment with three biological replicates. A two-tailed equal-variance Student’s t test was performed for statistical analysis. Cell counting was performed by two experienced independent investigators in a blinded manner.
Supplemental Material
Download Zip (7.1 MB)Acknowledgments
We thank Dr. Byung-Yoon Ahn for providing the MEFs
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298.
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366.
- Chin LS, Olzmann JA, Li L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem Soc Trans. 2010;38:144–149.
- Richter-Landsberg C, Leyk J. Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol. 2013;126:793–807.
- Takalo M, Salminen A, Soininen H, et al. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am J Neurodegener Dis. 2013;2:1–14.
- Yan J. Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014;33:567–580.
- Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol. 2013;4:1–26.
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530.
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898.
- Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905.
- Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738.
- Gamerdinger M, Kaya AM, Wolfrum U, et al. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12:149–156.
- Park Y, Park J, Kim YK. Crosstalk between translation and the aggresome-autophagy pathway. Autophagy. 2018;14:1079–1081.
- Park Y, Park J, Hwang HJ, et al. Nonsense-mediated mRNA decay factor UPF1 promotes aggresome formation. Nat Commun. 2020;11:3106.
- Park J, Park Y, Ryu I, et al. Misfolded polypeptides are selectively recognized and transported toward aggresomes by a CED complex. Nat Commun. 2017;8:15730.
- Meriin AB, Zaarur N, Sherman MY. Association of translation factor eEF1A with defective ribosomal products generates a signal for aggresome formation. J Cell Sci. 2012;125:2665–2674.
- Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacol Ther. 2011;130:348–363.
- Urnavicius L, Zhang K, Diamant AG, et al. The structure of the dynactin complex and its interaction with dynein. Science (New York, NY). 2015;347:1441–1446.
- Benzinger TL, Gregory DM, Burkoth TS, et al. Propagating structure of Alzheimer’s beta-amyloid(10-35) is parallel beta-sheet with residues in exact register. Proc Natl Acad Sci U S A. 1998;95:13407–13412.
- Der-Sarkissian A, Jao CC, Chen J, et al. Structural organization of alpha-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530–37535.
- McNaught KS, Shashidharan P, Perl DP, et al. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148.
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17.
- Wileman T. Aggresomes and autophagy generate sites for virus replication. Science (New York, NY). 2006;312:875–878.
- Gaete-Argel A, Márquez CL, Barriga GP, et al. Strategies for success. Viral infections and membraneless organelles. Front Cell Infect Microbiol. 2019;9:336.
- Olasunkanmi OI, Chen S, Mageto J, et al. Virus-induced cytoplasmic aggregates and inclusions are critical cellular regulatory and antiviral factors. Viruses. 2020:12. doi: https://doi.org/10.3390/v12040399.
- Zheng K, Jiang Y, He Z, et al. Cellular defence or viral assist: the dilemma of HDAC6. J Gen Virol. 2017;98:322–337.
- Banerjee I, Miyake Y, Nobs SP, et al. Influenza A virus uses the aggresome processing machinery for host cell entry. Science (New York, NY). 2014;346:473–477.
- Liu Y, Shevchenko A, Shevchenko A, et al. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J Virol. 2005;79:14004–14016.
- Nozawa N, Yamauchi Y, Ohtsuka K, et al. Formation of aggresome-like structures in herpes simplex virus type 2-infected cells and a potential role in virus assembly. Exp Cell Res. 2004;299:486–497.
- Heath CM, Windsor M, Wileman T. Aggresomes resemble sites specialized for virus assembly. J Cell Biol. 2001;153:449–455.
- Friedman JR, Fredericks WJ, Jensen DE, et al. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10:2067–2078.
- Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci. 2017;42:297–311.
- Yang Y, Fiskus W, Yong B, et al. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A. 2013;110:6841–6846.
- van Gent M, Sparrer KMJ, Gack MU, et al. Their roles in antiviral host defenses. Annu Rev Virol. 2018;5:385–405.
- Jaworska AM, Wlodarczyk NA, Mackiewicz A, et al. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal: concise review. Stem Cells. 2019. DOI: https://doi.org/10.1002/stem.3109.
- Hage A, Rajsbaum R. To TRIM or not to TRIM: the balance of host-virus interactions mediated by the ubiquitin system. J Gen Virol. 2019. DOI:https://doi.org/10.1099/jgv.0.001341
- Patil G, Li S. Tripartite motif proteins: an emerging antiviral protein family. Future Virol. 2019;14:107–122.
- Czerwinska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci. 2017;24:63.
- Krischuns T, Günl F, Henschel L, et al. Phosphorylation of TRIM28 enhances the expression of IFN-β and proinflammatory cytokines during HPAIV infection of human lung epithelial cells. Front Immunol. 2018;9:2229.
- Peng Y, Zhang M, Jiang Z, et al. TRIM28 activates autophagy and promotes cell proliferation in glioblastoma. Onco Targets Ther. 2019;12:397–404.
- Pineda CT, Ramanathan S, Fon Tacer K, et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160:715–728.
- Lelouard H, Ferrand V, Marguet D, et al. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol. 2004;164:667–675.
- Szeto J, Kaniuk NA, Canadien V, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189–199.
- Zaarur N, Meriin AB, Gabai VL, et al. Triggering aggresome formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J Biol Chem. 2008;283:27575–27584.
- Engelender S, Kaminsky Z, Guo X, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114.
- Doyle JM, Gao J, Wang J, et al. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010;39:963–974.
- Ivanov AV, Peng H, Yurchenko V, et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–837.
- Li S, Min JY, Krug RM, et al. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology. 2006;349:13–21.
- Bergmann M, Garcia-Sastre A, Carnero E, et al. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol. 2000;74:6203–6206.
- Kochs G, García-Sastre A, Martínez-Sobrido L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol. 2007;81:7011–7021.
- Sharma K, Tripathi S, Ranjan P, et al. Influenza A virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS One. 2011;6:e20215.
- García-Sastre A, Egorov A, Matassov D, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330.
- Nenasheva VV, Tarantul VZ. Many faces of TRIM proteins on the road from pluripotency to neurogenesis. Stem Cells Dev. 2019. DOI:https://doi.org/10.1089/scd.2019.0152
- Rousseaux MW, Revelli JP, Vazquez-Velez GE, et al. Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. eLife. 2018:7. doi: https://doi.org/10.7554/eLife.36768.
- Watanabe M, Hatakeyama S. TRIM proteins and diseases. J Biochem. 2017;161:135–144.
- Rousseaux MW, De Haro M, Lasagna-Reeves CA, et al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife. 2016:5. doi: https://doi.org/10.7554/eLife.19809.
- Kim KM, Cho H, Choi K, et al. A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 2009;23:2033–2045.
- Kim YK, Furic L, Desgroseillers L, et al. Mammalian Staufen1 recruits Upf1 to specific mRNA 3ʹUTRs so as to elicit mRNA decay. Cell. 2005;120:195–208.
- Fenger-Gron M, Fillman C, Norrild B, et al. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915.
- Ryu I, Won YS, Ha H, et al. eIF4A3 phosphorylation by CDKs affects NMD during the cell cycle. Cell Rep. 2019;26:2126–2139 e2129.
- Park OH, Ha H, Lee Y, et al. Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol Cell. 2019;74:494–507 e498.
- Jeong K, Ryu I, Park J, et al. Staufen1 and UPF1 exert opposite actions on the replacement of the nuclear cap-binding complex by eIF4E at the 5ʹ end of mRNAs. Nucleic Acids Res. 2019;47:9313–9328.
- Tokunaga M, Imamoto N, Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods. 2008;5:159–161.