
ABSTRACT
ULK1 (unc-51 like autophagy activating kinase) has a central role in initiating macroautophagy/autophagy, a process that contributes to atherosclerosis and neointima hyperplasia, or excessive tissue growth that leads to vessel dysfunction. However, the role of ULK1 in neointima formation remains unclear. We aimed to determine how Ulk1 deletion affected neointima formation and to investigate the underlying mechanisms. We measured autophagy activity, vascular smooth muscle cell (VSMC) migration and neointima hyperplasia in cultured VSMCs and ligation-injured mouse carotid arteries from male wild-type (WT, C57BL/6 J) and VSMC-specific ulk1 knockout (ulk1 KO) mice. Carotid artery ligation in WT mice increased ULK1 protein expression, and concurrently increased autophagic flux and neointima formation. Treating human aortic smooth muscle cells (HASMCs) with PDGF (platelet derived growth factor) increased ULK1 expression, activated autophagy, and promoted cell migration. Further, smooth muscle cell-specific deletion of Ulk1 suppressed autophagy, inhibited VSMC migration, and impeded neointima hyperplasia. Mechanistically, Ulk1 deletion inhibited autophagic degradation of histone acetyltransferase protein KAT2A/GCN5 (K[lysine] acetyltransferase 2A), resulting in accumulation of KAT2A that directly acetylated TUBA/α-tubulin and subsequently increased protein levels of acetylated TUBA. The acetylation of TUBA increased microtubule stability and inhibited VSMC directional migration and neointima formation. Finally, local transfection of Kat2a siRNA decreased TUBA acetylation and prevented the attenuation of vascular injury-induced neointima formation in ulk1 KO mice. These findings suggest that Ulk1 deletion inhibits neointima formation by reducing autophagic degradation of KAT2A and increasing TUBA acetylation in VSMCs.
Abbreviations: ACTA2/α-SMA: actin, alpha 2, smooth muscle, aorta; ACTB: actin beta; ATAT1: alpha tubulin acetyltransferase 1; ATG: autophagy related; BECN1: beclin 1; BP: blood pressure; CAL: carotid artery ligation; CQ: chloroquine diphosphate; EC: endothelial cells; EEL: external elastic layer; FBS: fetal bovine serum; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HASMCs: human aortic smooth muscle cells; HAT1: histone acetyltransferase 1; HDAC: histone deacetylase; IEL: inner elastic layer; IP: immunoprecipitation; KAT2A/GCN5: K(lysine) acetyltransferase 2A; KAT8/hMOF: lysine acetyltransferase 8; MAP1LC3: microtubule associated protein 1 light chain 3; MYH11: myosin heavy chain 11; PBS: phosphate-buffered saline; PDGF: platelet derived growth factor; PECAM1/CD31: platelet and endothelial cell adhesion molecule 1; RAC3: Rac family small GTPase 3; SIRT2: sirtuin 2; SPP1/OPN: secreted phosphoprotein 1; SQSTM1/p62: sequestosome 1; TAGLN/SM22: transgelin; TUBA: tubulin alpha; ULK1: unc-51 like autophagy activating kinase; VSMC: vascular smooth muscle cell; VVG: Verhoeff Van Gieson; WT: wild type.
Introduction
Macroautophagy/autophagy is a catabolic process that maintains cellular homeostasis by turnover of damaged cytosolic material and is tightly regulated by more than 30 ATG (autophagy related) proteins [Citation1]. Although autophagy has generally been considered to be nonspecific degradation, it also degrades specific cargo, such as organelles and proteins [Citation2–4]. Basal autophagy is required for maintaining normal vascular function. Accumulating evidence suggests that dysregulated autophagy is implicated in cardiovascular diseases, including atherosclerosis [Citation5,Citation6], restenosis [Citation7], vascular aging [Citation8], and angiogenesis [Citation9]. However, it is still debated whether autophagy is a protective or harmful mechanism in vascular pathology. One possible reason for this discrepancy may be that the major results are obtained under either ATG5- or ATG7-deficient conditions, but mammalian autophagy occurs by at least two different pathways: a conventional ATG5-ATG7-dependent pathway and an alternative ATG5-ATG7-independent pathway [Citation10]. Thus, further investigation is required to determine the precise role of autophagy in vascular diseases using a mouse with vascular smooth muscle cell (VSMC)-specific deletion of Ulk1 (unc-51 like autophagy activating kinase), in which both ATG5-ATG7-dependent and -independent autophagy are inhibited. ULK1 is a serine/threonine kinase that activates both ATG5-ATG7-dependent and -independent autophagy pathways [Citation10], participates in the initiation of autophagy [Citation11], and regulates the maturation of autophagosomes [Citation12]. ULK1 not only plays a role in the activation of autophagy, but also regulates many other important physiological activities, including protein transportation, innate immune response, neuronal development, glucose and lipid metabolism [Citation13]. However, it is unknown whether or not ULK1 is involved in the development of neointima hyperplasia.
VSMCs are a major component of vascular media, and are vital for maintaining vessel homeostasis [Citation14]. VSMCs respond to vascular injury and pro-atherosclerotic factors by migrating to the intima, which is a key event in the development of many vascular diseases and a consequence of mechanical vascular injury. VSMC migration is initiated by multiple environmental cues that stimulate receptors on the cell surface, activating multiple signaling cascades that alter the cytoskeletal structure of the cell, increasing the migration capacity of VSMCs [Citation15]. Microtubules, one of the major components of the eukaryotic cytoskeleton, are composed of tubulin heterodimers made of TUBA/alpha-tubulin and TUBB/beta-tubulin. The TUBA is subjected to posttranslational modifications, including detyrosination, acetylation, and polyglutamylation, which affect the function and stability of microtubules [Citation16,Citation17]. Given that TUBA acetylation is an established marker of microtubule stability [Citation18], reversible acetylation on the ε-amino group of TUBA Lys40 stabilizes microtubule structures and contributes to regulating cell motility [Citation19]. Microtubule acetylation in mammalian cells is performed by ATAT1 (TUBA N-acetyltransferase 1) [Citation20] and the elongator protein complex [Citation21] and removed by HDAC (histone deacetylase) family member HDAC6 and SIRT2 (sirtuin 2) [Citation22,Citation23]. KAT2A/GCN5 (K[lysine] acetyltransferase 2A) is a conserved histone acetyltransferase involved in a broad range of cellular processes, including gene transcription, differentiation, DNA repair, nucleosome assembly, and cell cycle regulation [Citation24]. Our recent work showed that KAT2A contains a MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-interacting region (LIR) domain that binds to the autophagic effector protein MAP1LC3, mediating the degradation of KAT2A. Suppression of autophagy results in the accumulation of KAT2A, which acetylates TUBA and inhibits VSMC migration [Citation25]. However, it is unclear whether KAT2A regulates neointima formation in vivo. In this study, we used VSMC-specific ulk1 knockout (ulk1 KO) mice to determine the role of autophagy in VSMC migration and neointima formation. We concluded that Ulk1 deletion in VSMCs suppresses autophagy, results in accumulation of KAT2A, and subsequently inhibits the VSMC migration and neointima formation that follow vascular injury.
Results
ULK1 upregulation is associated with neointima hyperplasia in response to vascular injury
To determine the relationship between ULK1 expression and vascular remodeling, we performed carotid artery ligation in C57BL/6 J mice and measured endogenous ULK1 protein expression in carotid arteries 4 weeks after vascular injury. We found that ULK1 expression was more than double the immunohistochemical staining score compared with that of sham group, shown by immunohistochemical staining ( and ) and immunofluorescence staining (Fig. S1A and S1B). Then we detected protein expression of SQSTM1/p62 (sequestosome1), an autophagic flux marker. Results showed that SQSTM1 protein levels were significantly (P < 0.05) lower in the carotid artery ligation group than in the sham group (, , S1C and S1D), suggesting that carotid artery ligation increases autophagic flux.
Figure 1. ULK1 upregulation is associated with neointima formation in mice subjected to carotid artery ligation (CAL). (A-B) C57BL/6 J mice were subjected to CAL, ULK1 protein levels in carotid arteries were determined by immunohistochemistry 28 d after injury. Scale bar: 50 µm. (C-D) immunohistochemical staining for SQSTM1. Scale bar: 50 µm. (E) Representative images of Verhoeff Van Gieson (VVG) and hematoxylin and eosin (H&E) staining. White triangle indicates media area, black triangle indicates neointima area. Scale bar: 50 µm. IEL, internal elastic layer; EEL, external elastic layer. (F) Quantitative analysis of lumen area and lumen:media ratio in sham and CAL mice. **P< 0.01, n = 5 in each group. (G) Five days after injury, arteries were collected, opened longitudinally, spread onto an agar plate with the luminal surface facing upward, and immunostained for ACT2A/α-SMA (actin alpha 2), a specific marker of smooth muscle cells. Scale bar: 20 µm. n = 5 in each group
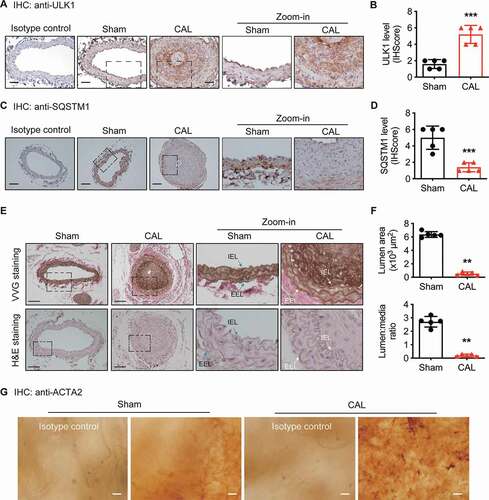
ULK1 protein upregulation was associated with neointima hyperplasia, as shown by a decrease in the lumen:media ratio determined by Verhoeff Van Gieson (VVG) and hematoxylin and eosin (H&E) staining ( and ). To directly examine whether autophagy influenced VSMC migration during vascular remodeling, we performed en face staining [Citation26,Citation27] 5 d after ligation. The result showed that vascular injury resulted in greater positive ACTA2/α-SMA (actin alpha 2) staining in intimal cells than a sham operation (). Because ACTA2-positive cells observed in the neointima on the fifth day after injury are reported to be migrating rather than proliferating cells [Citation28], these data suggest that ULK1 protein upregulation may enhance VSMC migration and promote neointima hyperplasia in response to vascular injury.
ULK1 deficiency inhibits cell migration in cultured HASMCs
Because ULK1 is critical for inducing autophagy, we assessed the role of ULK1 in regulating autophagy and VSMC migration in cultured HASMCs. PDGF (platelet derived growth factor) is important for vascular remodeling in response to vascular injury [Citation29]. We therefore treated HASMCs with PDGF-BB (20 ng/mL) and examined its effect on ULK1 expression and autophagy activity. PDGF-BB treatment increased ULK1 protein levels ( and ), promoted the conversion of nonlipidated LC3-I into phosphatidylethanolamine-conjugated LC3-II, and reduced SQSTM1 protein levels (, , and ), suggesting autophagy activation.
Figure 2. Gene silencing of ULK1 inhibits vascular smooth muscle cell migration in response to PDGF. (A-D) Human aortic smooth muscle cells (HASMCs) were treated with PDGF (platelet-derived growth factor BB, 20 ng/ml) for 12 and 24 h, and protein levels of ULK1, SQSTM1, and LC3 were measured by western blot and densitometry, *P< 0.05, n = 3–4. (E-F) GFP-LC3-expressing HASMCs were treated with PDGF, chloroquine (CQ, 3 µM), or PDGF + CQ for 24 h. (E), Representative images of GFP-LC3 staining. Scale bar: 20 µm. (F) Quantitation of autophagic vacuoles (AV) per cell. n = 3–4, *P< 0.05 vs. Ctrl (control); #P< 0.05 vs. PDGF or CQ. (G-J) Western blot analysis of ULK1, SQSTM1, and LC3 in HASMCs transfected with siCtrl (control siRNA) or siULK1 (ULK1 siRNA). n = 3–4, **P< 0.01, vs. siCtrl. (K-L) HASMCs were transfected with siCtrl or siULK1 for 48 h and treated with PDGF (20 ng/ml) for 24 h. (K) Cell migration was determined by transwell migration assays. Scale bar: 20 µm. (L) Migrated cells were quantified. n = 3–4, *P< 0.05 vs. siCtrl. #P< 0.05 vs. PDGF/siCtrl
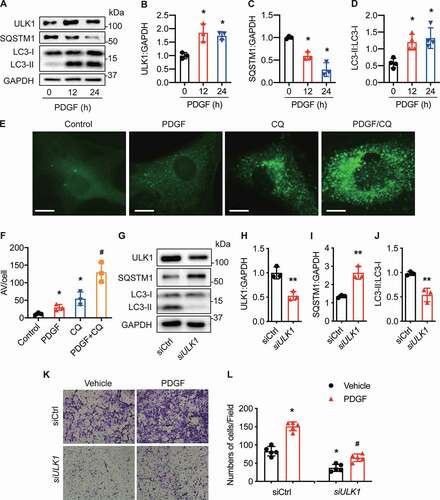
To study the effects of PDGF-BB on autophagic flux, we treated HASMCs with PDGF-BB in the presence or absence of chloroquine (CQ), a lysosome and autophagosome fusion inhibitor [Citation30]. In the absence of CQ, PDGF-BB treatment enhanced the number and distribution of GFP-LC3 punctate structures. Addition of CQ further increased autophagosome formation in cells treated with PDGF-BB ( and 2F). These results indicate that PDGF-BB acts by inducing autophagosome formation rather than by disrupting maturation into autolysosomes.
We further studied whether autophagy suppression by gene silencing of ULK1 would inhibit VSMC migration in cultured HASMCs. ULK1 siRNA abolished ULK1 protein expression and concomitantly suppressed autophagic flux, as evidenced by both reduced conversion of LC3-I to LC3-II and increased SQSTM1 levels (). The transwell assay indicated that PDGF-BB treatment significantly (P< 0.05) promoted cell migration. However, ULK1 knockdown inhibited HASMC migration in both basal and PDGF-BB-treated conditions, as indicated by lower numbers of migrating cells ( and ). Collectively, these results demonstrate that inhibiting autophagy by silencing ULK1 inhibits VSMC migration in cultured cells.
Ulk1 deletion inhibits neointima formation
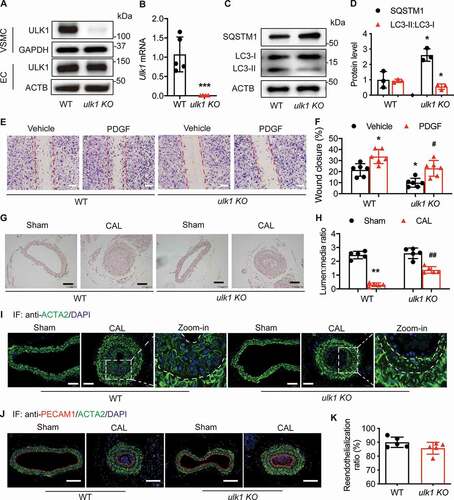
To investigate the role of ULK1 in neointima formation in vivo, we developed VSMC ulk1 conditional knockout (ulk1 KO) mice and performed sham or carotid artery ligation surgery in these mice. As shown in , ULK1 protein and mRNA expression were abolished in VSMCs isolated from ulk1 KO mouse aortas ( and ). However, Ulk1 gene deletion in VSMCs did not affect ULK1 protein expression in endothelial cells (EC) isolated from ulk1 KO mouse aortas (), indicating successful creation of VSMC ulk1 conditional knockout mice. To determine if Ulk1 gene deletion inhibited autophagy, we detected protein levels of SQSTM1 and LC3. In ulk1 KO mouse VSMCs, SQSTM1 protein levels were significantly (P< 0.05) increased whereas LC3-II levels were lower than in WT VSMCs ( and ), indicating flux suppression. We further examined the effect of deletion of Ulk1 on autophagic flux by treating WT and ulk1 KO VSMCs with CQ, and found that administration of CQ in ulk1 KO VSMCs induced LC3-II accumulation and further increase in SQSTM1 levels, indicating that deletion of Ulk1 reduces autophagic flux by inhibiting autophagosome formation rather than by disrupting maturation into autophagolysosomes (Fig. S1E-G).
Figure 3. Deletion of Ulk1 in VSMCs inhibits autophagic flux and attenuates neointimal formation. (A) Western blot analysis of ULK1 levels in vascular smooth muscle cells (VSMCs) and endothelial cells (EC) from wild-type (WT) and ulk1 KO mouse aortas. (B) Messenger RNA expression in VSMCs isolated from WT and ulk1 KO mice was analyzed by RT-PCR. (C-D) Protein levels of SQSTM1 and LC3 in VSMCs from WT and ulk1 KO mouse aortas. n = 5, *P< 0.05 vs. WT. (E-F) WT and ulk1 KO VSMCs were treated with or without PDGF (20 ng/ml) for 24 h. Cell migration was measured by scratch wound assay. (E) Representative images of VSMC migration in scratch wound assay. Scale bar: 1 mm. (F) Quantification of wound closure of VSMCs. n = 5, *P< 0.05 vs. WT. #P< 0.05 vs. WT PDGF. (G-H) Carotid artery sections from WT and ulk1 KO mice subjected to sham or carotid artery ligation (CAL) were collected and stained with hematoxylin and eosin to evaluate neointima formation. (G) Representative images of WT and ulk1 KO mice subjected to sham operation and carotid artery ligation. The white triangle indicates vascular media area, the black triangle indicates neointima area. Scale bar: 50 µm. (H) Quantitative analysis of lumen:media ratio in WT and ulk1 KO mice. **P< 0.01, n = 5. ##P< 0.01 vs. WT CAL. (I), Representative images of immunostaining of ACTA2. (J-K) Seven days after CAL, carotid arteries were collected and immunostained to evaluate reendothelialization. (J) Representative immunostaining images for PECAM1 (red, EC marker) and ACTA2 (green, smooth muscle cell marker). Scale bar: 100 µm. (K) Reendothelialization was calculated as the ratio of the surface covered by PECAM1-positive cells to the total luminal surface, n = 5 in each group
As BECN1/Beclin 1 silencing inhibits autophagy and promotes neointima formation [Citation31], we checked whether VSMC-specific deletion of Ulk1 affected BECN1 expression in vascular cells. We found that primary VSMCs and ECs in WT mice had similar BECN1 levels as in ulk1 KO mice (Fig. S1H-K). In primary ECs, the expression of SQSTM1 and LC3-II in WT mice was not different from that of ulk KO mice (Fig. S1L-N), suggesting that Ulk1 deletion in VSMCs did not affect autophagic flux in ECs. We also examined if deletion of Ulk1 affected the expression of other ATG proteins, including ULK2, ATG5, and ATG7, and we did not find that deletion of Ulk1 affected the expression of these proteins (Fig. S1O and S1P).
We next evaluated the effects of VSMC-specific Ulk1 deletion on vascular functions and found that ulk1 KO mice had lower blood pressure than WT mice (Fig. S2A-C). Resistant mesenteric arteries of WT and ulk1 KO had no difference in constriction induced by U46619, a TBXA2R (thromboxane A2 receptor) agonist (Fig. S2D). Endothelium-mediated mesenteric artery relaxation induced by acetylcholine (Ach) was similar between ulk1 KO mice and WT littermates (Fig. S2E). In the presence of L-NAME (NOS3 [nitric oxide synthase 3, endothelial cell] inhibitor), we found no difference in sodium nitroprusside (SNP)-induced vessel relaxation between ulk1 KO mice and WT mice (Fig. S2F).
We then tested whether Ulk1 gene deletion inhibited VSMC migration. Consistent with the previous findings in HASMCs, lack of ULK1 in mouse VSMCs significantly (P< 0.05) inhibited cell migration into the wound area with or without PDGF ( and ).
To determine the effects of Ulk1 deletion on in vivo vascular remodeling, we analyzed neointima formation in ulk1 KO mice. Ulk1 deletion did not visibly alter vessel structure. After injury, the lumen:media ratio significantly (P< 0.01) decreased in WT mice, indicating increased neointima formation. Whereas Ulk1 gene deletion significantly (P< 0.01) prevented ligation-enhanced neointima area ( and ). Next, we immunostained vessel sections for ACTA2 (a VSMC marker), and found that VSMCs were the predominant cell type in the neointimas ().
Early luminal reendothelialization in response to mechanical arterial injury is an important factor determining neointima size [Citation32]. To evaluate the reendothelialization rate, we collected sham and ligated arteries 7 d after ligation and immunostained the vessel sections with an antibody against PECAM1/CD31 (platelet and endothelial cell adhesion molecule 1). The results showed that the endothelial layer in ligated arteries completely recovered at this time point, compared to arteries subjected to a sham operation. Specific deletion of Ulk1 in VSMCs did not affect the reendothelialization rate ( and ) and cell apoptosis (Fig. S2G). Taken together, our data suggest that inhibiting autophagy by deleting Ulk1 in VSMCs postpones VSMC migration and neointima formation.
Ulk1 deletion inhibits VSMC migration ex vivo
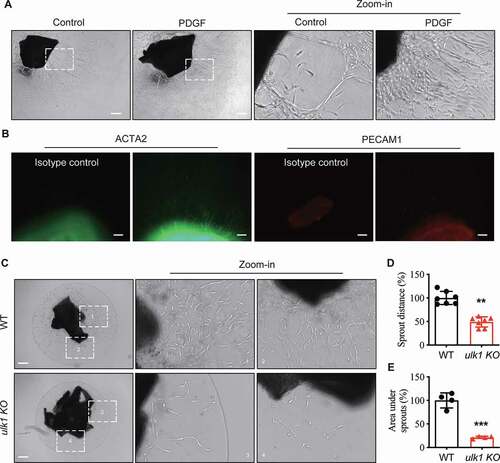
We also examined the effects of Ulk1 deletion on ex vivo VSMC migration using an aortic ring assay, as previously described [Citation28]. Firstly, we determined the identity of migrating cells using WT aortic rings. The identities of migrating cells in neovessel structures were determined by whole mount immunostaining with antibodies for PECAM1 (a marker of ECs) or ACTA2 (a marker of VSMC). PDGF-BB-treated aortic rings had more neovessel formation than control rings (). Whole mount staining for PECAM1 and ACTA2 in PDGF-BB-treated neovessels showed that neovessel structures stained for ACTA2 but not PECAM1 (), indicating that migrating cells were mainly VSMCs. Then we cultured WT and ulk1 KO aortic rings in Matrigel and measured the sprout distance and the area under the sprout on day 7. Compared with control WT rings, Ulk1 deletion induced significant (P < 0.01) reductions in the sprout distance and the area under sprouts (). These results suggest that Ulk1 deletion inhibits ex vivo VSMC migration.
Figure 4. Deletion of ULK1 inhibits VSMC migration ex vivo. (A) Aortic rings without adventitia and endothelial cells from wild-type (WT) mice were treated with PDGF (20 ng/ml) for 7 d, then VSMC migration was analyzed by microscopy. Red arrow indicates the sprout VSMC. Scale bar: 100 µm. (B) The identities of migration cells were determined by immunostaining with antibodies against ACTA2 or PECAM1. Scale bar: 50 µm. (C-D) The aortic rings from WT and ulk1 KO mice were embedded in Matrigel and incubated in DMEM/F-12 medium. Images were taken with a microscope equipped with a digital camera and cell migration was determined by measuring the distance between the rings and the leading front of migrating cells. (C) Representative images of aortic ring assay. Red arrow indicates the sprout VSMC. Scale bar: 100 µm. (D) Quantification of sprout distance. n = 4, **P< 0.01. (E) Quantification of the area under sprout. n = 4, ***P< 0.001
Ulk1 deletion increases TUBA acetylation and microtubule stability, inhibiting the directional migration of VSMCs.
To gain insight into the mechanisms by which Ulk1 deletion inhibited VSMC migration and neointima formation, we examined whether Ulk1 deletion increased TUBA acetylation. Because starvation-induced autophagy leads to reduction of histone H4 lysine 16 acetylation (H4K26ac) [Citation33], acetylation of TUBA is associated with microtubule stability and cell motility [Citation23]; and increased TUBA acetylation inhibits endothelial cell migration [Citation34], we examined the acetylation of TUBA in WT and ulk1 KO VSMCs. As expected, TUBA acetylation significantly (P< 0.05) increased in ulk1 KO VSMCs, but Ulk1 deletion did not affect TUBA expression ( and ). We further verified the effect of ULK1 deficiency on TUBA acetylation using immunofluorescence staining in HASMCs transfected with ULK1 siRNA. Similar to the finding with ulk1 KO VSMCs, silencing of ULK1 significantly increased the abundance of acetylated TUBA (Fig. S3A and S3B) but had no effect on the expression of total TUBA.
Figure 5. Ulk1 deletion increases acetylated TUBA protein levels. (A-B) Western blot analysis of Ac-TUBA in WT and ulk1 KO mouse VSMCs. n = 6, *P < 0.05 vs. WT. (C) HASMCs transfected with siCtrl (control siRNA) or siULK1 (ULK1 siRNA) were incubated at 0°C for 0, 5, 10, or 15 min. The morphology of microtubules was examined by immunostaining of TUBA. (D) Quantitative analysis of microtubule intensity (n= 25/group, *P< 0.05, ***P< 0.001). (E) HASMCs were transfected with siCtrl or siULK1 and subjected to scratch wound assay. HASMCs stained with TUBA (green) and Ac-TUBA (red) antibodies were analyzed using a confocal microscope. Nuclei were stained with DAPI (blue). Scale bar: 50 µm. (F) WT or ulk1 KO mice were subjected to carotid artery ligation (CAL), and carotid artery sections were stained with Ac-TUBA antibody. Representative images of immunofluorescence staining of Ac-TUBA. Scale bar: 100 µm. (G) Quantification of immunostaining for Ac-TUBA. n = 5, *P< 0.05, **P< 0.01 vs. WT sham. #P< 0.05 vs. ulk1 KO sham
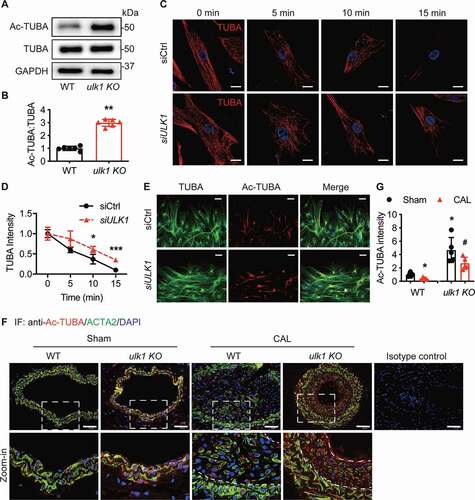
Since acetylation of TUBA is a well-established marker of more stable microtubules [Citation18], we determined the effect of ULK1 deficiency on microtubule stability by examining the morphology of microtubules using immunofluorescence microscopy. We transfected HAMSCs with control or ULK1 siRNA, and then incubated the cells at 0°C for 5, 10, or 15 min. We found that cold treatment led to the depolymerization of microtubules in a time-dependent manner. Microtubules were completely depolymerized at 15 min after cold treatment in cells transfected with control siRNA. Conversely, microtubules were still present in cells transfected with ULK1 siRNA ( and ), suggesting that deletion of ULK1 increased microtubule stability, which is required for directional cell migration [Citation35]. We then examined the effect of ULK1 deficiency on cell polarity using the scratch wound healing assay and immunofluorescence staining. In control siRNA-treated HASMCs, the first two layers of leading cells were clearly polarized toward the leading edge. ULK1 silencing, however, dramatically increased the staining of acetylated TUBA in the first two layers of leading cells and resulted in a loss of orientation toward the leading edge in these cells (). These data suggest that acetylation of TUBA increases microtubule stability, inhibits directional migration. It was reported that posttranslational modifications of TUBA, including acetylation, detyrosination, and polyglutamylation, regulates the function and stability of microtubules [Citation16,Citation17], affecting cell motility. We also studied if deletion of ULK1 affected detyrosination and polyglutamylation of TUBA. As shown in Fig. S3C and S3D, silencing of ULK1 did not influence detyrosination and polyglutamylation of TUBA, supporting that the increase in microtubule stability in ULK1-deficient conditions is due to the acetylation of TUBA.
We had shown that deletion of Ulk1 inhibited VSMC migration in cultured VSMCs () and aortic rings (). To establish the relationship between acetylated TUBA and neoinitima formation, we evaluated how Ulk1 deletion affected TUBA acetylation in mouse carotid arteries after ligation surgery. Immunostaining for acetylated TUBA was significantly (P< 0.05) lower in carotid arteries from WT mice subjected to carotid artery ligation than in the sham group ( and ). Consistent with results in cultured cells, ulk1 KO mouse carotid arteries had stronger staining for acetylated TUBA than WT mice, and Ulk1 deletion prevented the decreased acetylated TUBA in ulk1 KO mice subjected to carotid artery ligation ( and ). Taken together with the finding that Ulk1 deletion impeded neointima formation ( and ), our results indicate an inverse relationship between TUBA acetylation and neointima formation and suggest that TUBA acetylation inhibits VSMC migration and consequently reduces neointima formation.
Ulk1 deletion results in KAT2A accumulation that mediates TUBA acetylation
To further characterize how Ulk1 deletion increased the levels of acetylated TUBA protein, we measured mRNA levels of 17 deacetylases and 20 acetyltransferases genes in WT and ulk1 KO VSMCs by RT-PCR, we found that mRNA levels of Hdac2, Hdac4, Hdac6, and Hadc11 increased by approximately 2-fold, Tap1 increased by 1.55-fold, and Rac3 reduced by 0.25-fold in ulk1 KO VSMCs (Fig. S4). We further examined the protein levels of acetyltransferases and deacetylases that are reported to be related to TUBA acetylation, including KAT2A, KAT8/hMOF, (lysine acetyltransferase 8), EP300 (E1A binding protein p300), ATAT1 (TUBA N-acetyltransferase 1), HDAC6 (histone deacetylase), SIRT1, and SIRT2. In mouse VSMCs, the absence of ULK1 did not affect protein expression of HDAC6, SIRT1, SIRT2, EP300, ATAT1, and KAT8 (Fig. S5A), but increased KAT2A protein levels ( and S5B) without changing its mRNA levels (Fig. S5C), suggesting that Ulk1 deletion regulates KAT2A at the posttranslational level by reducing KAT2A autophagic degradation.
Figure 6. Ulk1 deletion leads to KAT2A accumulation that mediates the acetylation of TUBA. (A) Western blot analysis of the expression of KAT2A, TUBA, and Ac-TUBA in WT and ulk1 KO VSMCs, n= 6. (B) HASMCs were transfected with an adenovirus encoding GFP-LC3 for 24 h and then treated with chloroquine (CQ, 5 µM) for 16 h. KAT2A was stained by immunocytochemistry, and the co-localization (yellow) of KAT2A (red) and GFP-LC3 puncta was evaluated by immunofluorescence microscopy. Scale bar: 10 µm. (C) VSMCs isolated from WT or ulk1 KO mice were treated with CQ (5 µM) for 24 h and the interaction of KAT2A and LC3 were analyzed by immunoprecipitation (IP) and western blot (WB). (D) HASMCs were transfected with siCtrl (control siRNA) or siULK1 (ULK1 siRNA), and levels of ULK1, KAT2A, TUBA, and Ac-TUBA were measured by western blot. n = 6. (E-G) HASMCs were starved with Hank’s balanced salt solution (HBSS) for 3 h, and protein levels of SQSTM1 and LC3 were analyzed by western blot. n = 3, *P< 0.01 vs. Ctrl. (H-J) Levels of KAT2A, TUBA, and Ac-TUBA were measured using western blot. *P < 0.01, n = 3. (K) Purified porcine brain TUBA protein was incubated with recombinant KAT2A-catalytic domain protein in acetylation buffer at 37°C for 1 h. The acetylation of TUBA was assessed by western blot (n= 3, *P< 0.05, **P< 0.01). (L-M) WT and ulk1 KO VSMCs were transfected with siCtrl (control siRNA) or siKat2a (Kat2a siRNA) for 72 h, and protein levels of ULK1, KAT2A, TUBA, and Ac-TUBA were detected by western blot. * P< 0.05 vs. WT siCtrl, #P< 0.05 vs. ulk1 KO siCtrl, n = 4
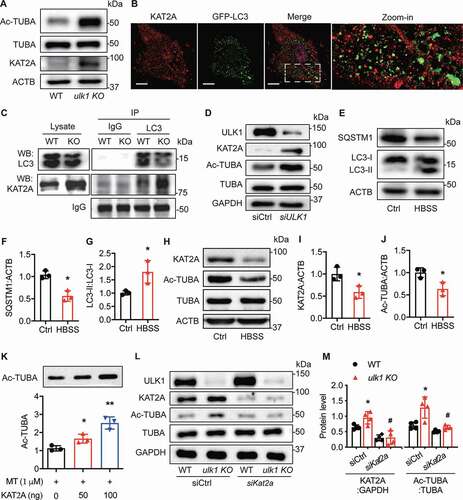
Our previous study showed that KAT2A has a LIR domain [Citation25], we therefore examined if KAT2A localizes in autophagosomes by immunostaining of KAT2A in HASMCs transfected with GFP-LC3 adenovirus as a probe for detection of autophagosome formation. We found that KAT2A (red) was localized in both nucleus and cytoplasm. Notably, some of the KAT2A protein in the cytoplasm co-localized with GFP-LC3 puncta (), suggesting that KAT2A may interact physiologically with LC3. We further examined the interaction between KAT2A and LC3 in WT and ulk1 KO VSMCs by immunoprecipitation with anti-LC3 antibody and immunoblotting with KAT2A antibody. The result revealed that KAT2A associated with LC3 in WT VSMCs and the association was increased in ulk1 KO VSMCs (), indicating that KAT2A physically associates with LC3 in VSMCs.
We then tested how manipulating autophagic activity in cultured HASMCs affected KAT2A protein levels. First, we verified whether autophagy inhibition increased KAT2A protein levels in HASMCs. Transfecting HAMSCs with ULK1 siRNA reduced ULK1 levels ( and S5D), inhibited the conversion of LC3-I to LC3-II, and increased SQSTM1 protein levels (). The suppression of autophagy increased KAT2A protein levels ( and S5E) and concomitantly increased TUBA acetylation ( and S5F). Then, we investigated if autophagy activation affected KAT2A protein regulation. We induced autophagy by starvation with Hank’s buffered salt solution (HBSS), which is typically used as an autophagy trigger [Citation36]. We found that this starvation significantly (P< 0.05) increased the conversion of LC3-I to LC3-II and reduced SQSTM1 levels (), indicating autophagy activation. The activation of autophagy by starvation significantly (P< 0.05) reduced levels of KAT2A and acetylated TUBA protein ().
To investigate whether KAT2A directly acetylated TUBA, we incubated purified porcine brain TUBA protein (>99% pure) with recombinant KAT2A-catalytic domain protein in acetylation buffer. We found that the KAT2A catalytic domain alone increased acetylated TUBA in the purified porcine brain TUBA protein in a dose-dependent manner (). To address whether KAT2A accumulation mediated TUBA acetylation in ULK1-deficient VSMCs, we transfected WT and ulk1 KO mouse VSMCs with control or Kat2a siRNA and then measured the levels of acetylated TUBA protein. As shown in and , Ulk1 deletion increased KAT2A protein levels in cells transfected with control siRNA compared to WT VSMCs. Consistent with the change in KAT2A expression, we saw that acetylated TUBA levels also increased. But in cells transfected with Kat2a siRNA, there was a dramatic inhibition in the ulk1 KO-induced increase in acetylated TUBA, supporting the notion that KAT2A mediates TUBA acetylation in ULK1-deficient VSMCs.
Local transfection of Kat2a increases neointima formation in ulk1 KO mice subjected to carotid artery ligation.
Given that KAT2A mediated TUBA acetylation in ULK1-deficient VSMCs, and TUBA acetylation is negatively associated with neointima formation, we investigated whether silencing the Kat2a gene could prevent the attenuation of neointima formation in ulk1 KO mice. To this end, we performed siRNA transfection. In mice transfected with control siRNA, Ulk1 deletion prevented the decrease in staining for KAT2A (, , S6A and S6B) and acetylated TUBA (, , S6C and S6D). However, local transfection of Kat2a siRNA blocked the effect of Ulk1 deletion.
Figure 7. Silencing Kat2a increases neointima formation in ulk1 KO mice subjected to carotid artery ligation. Left carotid arteries of WT and ulk1 KO mice were ligated and right carotid arteries were collected as sham controls. Both left and right arteries were treated with siCtrl (control siRNA) or siKat2a (Kat2a siRNA) after the surgery. Four weeks after ligation, hematoxylin and eosin staining was performed on the paraffin sections of carotid arteries to detect neointimal formation, and immunofluorescence staining was used to detect protein levels of KAT2A and acetylation of TUBA (Ac-TUBA). (A) Representative images of immunofluorescence staining of KAT2A. Scale bar: 50 µm. (B) Quantification of immunofluorescence staining for KAT2A. *P< 0.05, **P< 0.01, n = 5. (C) Representative images of immunofluorescence staining of Ac-TUBA. Scale bar: 50 µm. (D) Quantification of immunofluorescence staining for Ac-TUBA, *P< 0.05, **P< 0.01, n = 5. (E) Representative H&E staining to show neointima formation. Scale bar: 100 µm. (F) Quantitative analysis of lumen:media ratio in WT and ulk1 KO mice. n = 5. **P< 0.01 vs. WT sham, ##P< 0.01 vs. WT CAL
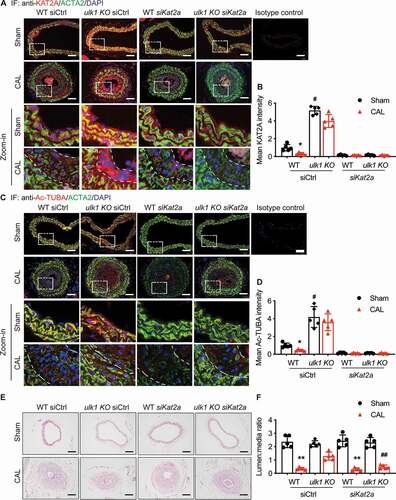
H&E staining of carotid artery cross-sections indicated that carotid artery ligation significantly (P< 0.01) increased the intima area in the control siRNA group compared to sham-operated WT mice, and the increase was significantly (P< 0.01) attenuated in ulk1 KO mice ( and ). After Kat2a silencing in WT mice, we found that carotid artery ligation still induced neointima formation to an extent similar to that observed in WT mice without Kat2a silencing. However, Kat2a silencing in ulk1 KO mice dramatically increased the intima area compared to observations of ulk1 KO mice without Kat2a silencing. Further, in WT and ulk1 KO mice subjected to carotid artery ligation, Kat2a siRNA treatment did not affect reedothelialization rate, an important factor determining neointima size in response to vascular injury [Citation32] (Fig. S6E and S6F). Taken together, these data suggest that Ulk1 deletion inhibits neointima formation via a KAT2A-dependent pathway.
ULK1 deficiency does not influence VSMC phonotypic switching and foam cell formation in cultured VSMCs
We also studied if deletion of ULK1 affected foam cell formation and VSMC phenotypic switching, two important processes in the development of atherosclerosis in culture VSMCs. Smooth muscle cells are the most abundant cells in human atherosclerotic lesions and are suggested to contribute at least 50% of atheroma foam cells. Smooth muscle cells within the arterial wall develop into foam cells by taking up oxidized low-density lipoprotein (ox-LDL) via scavenger receptors, which is one of the hallmarks of atherosclerotic plaques [Citation37]. We checked the morphological change of cells after incubation of WT and ulk1 KO VSMCs with 80 mg/L ox-LDL for 72 h and found that lipid staining became positive compared with the control group. However, the lipid staining was comparable between WT and ulk1 KO VSMCs treated with ox-LDL (Fig. S7A). Phenotypic switching of VSMCs plays a key role in atherosclerosis [Citation38]. To study if Ulk1 deletion affected VSMC phenotypic switching, we detected markers of contractile and synthetic VSMC phenotypes by western blot. As shown in Fig. S7A, ACTA2, MYH11 (myosin heavy chain 11), and CNN (calponin), which are three canonical contractile markers of VSMC, were not significantly changed in ulk1 KO VSMC compared to those from WT. Similarly, the protein levels of SPP1/OPN (secreted phosphoprotein 1), and VIM (vimentin), two well-characterized markers for synthetic VSMCs, were comparable between WT and ulk1 KO VSMCs (Fig. S7B and S7C). These results suggest that lack of ULK1 does not influence VSMC phonotypic switching and foam cell formation in cultured VSMCs.
Discussion
In this study, we demonstrated that loss of ULK1, a protein required for both ATG5-ATG7-dependent and -independent autophagy, inhibited VSMC migration and vascular injury-induced neointima formation. Mechanistically, autophagy suppression by Ulk1 deletion led to KAT2A accumulation, which increased TUBA acetylation and subsequently inhibited VSMC migration and neointima formation. Our findings suggest that accumulation of TUBA acetylation mediated by KAT2A is an important mechanism to explain how Ulk1 deletion inhibits VSMC motility.
The proposed model implies that enhanced autophagic clearance can be initially protective, but that overactive autophagy over time may promote cardiovascular disease. However, the role of autophagy in the development of vascular diseases remains controversial. Reportedly, autophagy activation by growth factors and oxidized lipids inhibits the expression of contractile proteins and upregulates the expression of synthetic smooth muscle cell markers, which is concurrent with enhanced migration and proliferation of VSMCs [Citation39–41]. In addition, autophagy activated by SHH (sonic hedgehog) protein secretion increases VSMC proliferation and critically contributes to the pathogenesis of proliferative vascular disease, such as restenosis [Citation42]. However, VSMC-specific deletion of Atg7 leads to VSMC senescence, enhances cell migration, and promotes ligation-induced neointima formation and diet-induced atherogenesis [Citation7]. The explanation for this discrepancy may be that ATG7 can directly regulate TRP53/p53-dependent cell cycle and death pathways [Citation43], thus affecting cell survival and proliferation. In addition, autophagy can also occur in the absence of ATG7 through unconventional biogenesis of canonical autophagosomes. In this study, we found that VSMC-specific deletion of Ulk1 inhibited cell migration in cultured HASMCs, reduced sprout distance in an aortic ring assay, and prevented vascular injury-induced neointima hyperplasia. Taken together, our findings indicate that autophagy suppression inhibits VSMC migration and neointima hyperplasia.
Autophagy defects induced by BECN1 deficiency promote neointima formation after vascular injury [Citation31], which contradicts our finding that neointima formation was inhibited by autophagy suppression induced by VSMC-specific ulk1 KO. However, the BECN1 deficiency study using a carotid artery injury model of endothelial denudation suggests that the increase in neointima formation is mainly due to inhibited reendothelialization [Citation31]. In contrast, our study induced vascular injury by carotid artery ligation. Moreover, we saw that the lack of ULK1 in VSMCs did not inhibit reendothelialization, but inhibited VSMC migration and consequently reduced neointima formation. One possible reason for this discrepancy is that BECN1 not only mediates autophagy, but also links autophagy and apoptosis because BECN1 deficiency in vessels induces endothelial apoptosis [Citation31].
VSMCs and ECs are important for both initiation and development of neointima hyperplasia, and both cells express autophagy markers. In most cases, autophagy is an important process by which VSMCs remove damaged protein and organelles to prevent cell death and promote VSMC development into a highly proliferative and migratory phenotype [Citation40,Citation42,Citation44]. In ECs, however, impaired autophagy is associated with endothelial dysfunction and apoptotic cell death that may lead to neointima hyperplasia. Although autophagy dysregulation could cause neointima hyperplasia, it is important to consider that autophagic players may have different roles in different cell types and at different stages of vascular disease.
Autophagy has been considered a nonselective degradation mechanism; however, recent findings indicate that autophagy can selectively degrade mitochondria and other specific organelles, bacteria, and protein aggregates [Citation2]. We have shown that KAT2A contains an LIR motif [Citation25]. In this study, we found that KAT2A associated with LC3 and localized in autophagosomes. Suppressing autophagy by deleting Ulk1 increased KAT2A protein levels but did not affect Kat2a mRNA expression. In contrast, starvation-activated autophagy reduced KAT2A protein expression, suggesting that KAT2A was selectively degraded by an autophagy-lysosomal pathway in VSMCs.
Increased TUBA acetylation reportedly inhibits endothelial cell migration, but little is known about the enzyme responsible for this reaction [Citation34]. Although ATAT1 was identified as a critical player in TUBA acetylation in neurons [Citation20], ATAT1 may not be involved in TUBA acetylation in ULK1-deficient conditions because Ulk1 deletion did not alter ATAT1 expression. Our results suggest that KAT2A is the enzyme mediating TUBA acetylation in VSMCs because 1) in a cell-free system, KAT2A catalytic domain alone increased acetylated TUBA in a dose-dependent manner; 2) Increased KAT2A protein levels were associated with higher acetylated TUBA levels, whereas starvation-activated autophagy reduced protein levels of KAT2A and acetylated TUBA; and 3) Silencing Kat2a in ulk1 KO cells decreased TUBA acetylation. TUBA can be deacetylated by both HDAC6 and SIRT2 [Citation22,Citation23]. However, in our study, Ulk1 deletion did not alter HDAC6 or SIRT2 expression, suggesting that the increased acetylation of TUBA in ULK1-deficient conditions is not mediated by these deacetylases.
Because acetylation is essential for stable microtubule populations, altering TUBA acetylation levels may regulate cell migration. However, TUBA acetylation has different effects on cell behavior in different cell types. For example, decreased TUBA acetylation impairs migration of neuronal [Citation21,Citation45] and breast cancer cells [Citation46]. By contrast, increased TUBA acetylation by HDAC6 inhibitors reduces cell migration in estrogen receptor-positive breast cancer [Citation47]. Moreover, hyperacetylation of TUBA suppresses endothelial cell migration [Citation34]. In our study of cultured VSMCs, we found that high levels of acetylated TUBA were associated with increased microtubule stability, inhibited VSMC directional migration, and reduced neointima formation. Importantly, Kat2a silencing decreased TUBA acetylation and inhibited the attenuation of neointima formation in ulk1 KO mice.
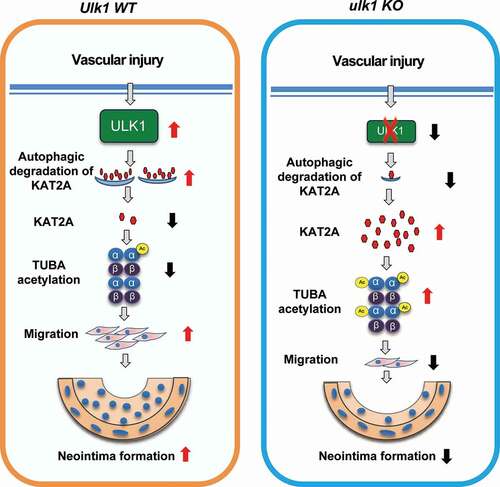
In summary, ULK1 upregulation was associated with neointima hyperplasia in response to vascular injury. Ulk1 deletion reduced autophagic flux, inhibited VSMC migration, and attenuated vascular injury-induced neointima hyperplasia. Gene silencing of Kat2a reduced TUBA acetylation and prevented the attenuation of neointima formation in ulk1 KO mice. Therefore, we propose that increased KAT2A-mediated acetylation of TUBA is an essential mechanism by which autophagy suppression inhibits VSMC migration and neointima formation (). Our work suggests that suppressing autophagy by inhibiting ULK1 is a valid strategy to develop treatments for neointima hyperplasia.
Figure 8. Proposed scheme for the role of ULK1 in neointima formation following vascular injury. Deletion of Ulk1 in VSMCs results in KAT2A accumulation and thus increases TUBA acetylation, which inhibits VSMC migration and neointima formation through modulating microtubule stability and cell motility
Materials and Methods
Reagents
Antibodies and reagents were from the following companies: SQSTM1 (Abcam, ab56416), acetyl TUBA (Cell Signaling Technology, 5335), TUBA (Cell Signaling Technology, 3873), LC3B (Cell Signaling Technology, 4108; Santa Cruz Biotechnology, sc-398822), ULK1 (Cell Signaling Technology, 8054; GeneTex, GTX80551), KAT2A/GCN5 (Cell Signaling Technology, 3305; Abcam, ab137515), HDAC6 (Cell Signaling Technology, 7612), SIRT1 (Cell Signaling Technology, 9475), SIRT2 (Santa Cruz Biotechnology, sc-28298), PECAM1/CD31 (Cell Signaling Technology, 77699), ACTB (Santa Cruz Biotechnology, sc-1616 HRP), EP300/p300 (Santa Cruz Biotechnology, sc-48343), GAPDH (glyceraldehyde-3-phosphate dehydrogenase, Santa Cruz Biotechnology, sc-32233), ULK1 siRNA (Santa Cruz Biotechnology, 44849), siRNA delivery reagent Lipofectamine RNAiMAX (Life Technologies, 13778150), chloroquine (Sigma-Aldrich, 6628), EnVision® + Dual Link System-512 HRP (DAB+) (Dako Cytomation, 3468), and elastic Stain Kit (Sigma-Aldrich, HT25A-1KT).
Mouse model of carotid artery ligation injury
Ulk1flox/flox and Tagln/Sm22-cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME). We generated ulk1-SMC-specific knockout mice (ulk1 KO, ulk1flox/flox/Tagln-Cre+) by mating female Ulk1flox/flox mice with male Tagln-Cre mice to generate hemizygous mice (ulk1/f/wt:Cre/wt mice). Next, we assigned hemizygous breeding pairs to obtain homozygous mice. We PCR genotyped offspring using DNA from tail-snip biopsies. We selected two groups of mice for this study: (a) VSMC ulk1 KO (ulk1 KO) mice with genotype ulk1flox/flox/Tagln-Cre+ and (b) littermate control mice (WT) with genotype Ulk1flox/flox/Tagln-Cre−/-. We used 8 to 10-week-old male mice for the experiment. Mice were housed in temperature-controlled cages under a 12-h light-dark cycle and given free access to water and normal chow.
The carotid artery ligation (CAL) was performed as in our previous study [Citation48]. Briefly, male WT and ulk1 KO mice were anesthetized with an intraperitoneal injection of 100 mg/kg ketamine-HCl and 10 mg/kg xylazine-HCl. Both left and right carotid arteries were isolated. Ligation was performed in the left common carotid artery proximal to the bifurcation, and the right artery was treated as a sham control. For in vivo siRNA transfection, 15 μg scrambled Stealth RNAi siRNA (Invitrogen, 12,935–112) or Kat2a siRNA (Santa Cruz Biotechnology, sc-37,947) dissolved in 30% pluronic gel solution (Sigma-Aldrich, P2443) was peri-vascularly delivered to the common carotid arteries immediately after ligation. Four weeks after surgery, we harvested carotid arteries for histological and molecular biological analyses. For morphometric measurement, we fixed the carotid arteries in 10% formaldehyde and embedded them in paraffin. Sections were obtained at 3–4 mm proximal to the ligation site from each animal and stained with hematoxylin and eosin. In addition, the sections were subjected to Verhoeff Van Gieson staining (Sigma, HT25A) to delineate elastin layers. We measured the cross-sectional areas of the media and neointima using a Bioquant analysis system (Bioquant, Nashville, TN) and calculated the ratio between lumen and media area [Citation49]. All animal protocols were performed in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of Georgia State University.
Vascular functional assay
Arterial blood pressure (BP) was determined via carotid artery catheterization under the anesthesia of ketamine (50 mg/kg) and xylazine (10 mg/kg). After BP was recorded, the mesenteric arteries were collected and cut into 5-mm rings. The mesenteric arterial rings were mounted onto a wire myography chambers (DMT 620 M, Denmark) and equilibrated in oxygenated Krebs-buffer (118.3 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, pH 7.0, 0.026 mM EDTA, 11 mM glucose (Sigma-Aldrich, G7021)) for 1 h under a resting tension of 3 mN. The mesenteric arterial rings were pre-contracted with 40 nM of U46619, and the contraction force was recorded. At the plateau of contraction, endothelium-dependent and – independent vasodilation responses were determined in the presence of acetylcholine (Ach, 10−9 to 10−5 M) and sodium nitroprusside (SNP, 10−10 to 10−6 M), respectively.
In vivo vascular smooth muscle cell migration assay
We performed the in vivo VSMC migration assay as described previously [Citation26,Citation27]. Briefly, 5 d after ligation, we fixed carotid arteries with 10% formalin, and cut 0.5 cm of the common carotid artery near its bifurcation, and fixed in cold acetone for 10 min. Arteries were then opened longitudinally and spread onto an agar plate with the luminal surface facing upward. Arteries were washed in phosphate-buffered saline (PBS; Sigma-Aldrich, P3813) and incubated in 0.3% hydrogen peroxide (VWR, BDH7690-1) for 1 h to block endogenous peroxidase activity. After blocking with 3% normal goat serum (BioGenex, HK112-9 K), we incubated arteries with smooth muscle ACTA2 antibody (abcam, 7817) overnight at 4°C, followed by incubation with secondary antibody at room temperature for 1 h. Antibody signal was visualized using the Liquid DAB+ Substrate Chromogen System (Dako, K3468). We detected the intimal surface of vessels using a microscope and measured smooth muscle ACTA2 staining areas.
Ex vivo aortic explant migration
Ex vivo smooth muscle cell migration was determined using an aortic ring assay as previously described [Citation28]. We anesthetized C57BL/6 mice (8 weeks) with ketamine and xylazine, then isolated thoracic aortas. After removing the connective tissues surrounding the aorta, we rinsed aorta fragments in PBS, stripped the adventitia by collagenase type II (Worthington, LS004176) digestion (2 mg/mL), and removed the endothelium mechanically. The vessels were cut into 1 mm rings and then embedded in 48-well plates coated with Matrigel (Life Sciences, 354,248). We incubated embedded rings in DMEM/F-12 (Corning, 10–092-CV) medium supplemented with 20 ng/mL PDGF-BB (RD System, 220-BB) for 7 d. To determine whether Ulk1 deletion inhibited VSMC migration ex vivo, we cultured WT and ulk1 KO aortic rings. Images were taken using a microscope (IX71, Olympus) equipped with a digital camera. We determined cell migration by measuring the sprout distance and the area under sprout and determined the identity of migrating cells in neovessel structure by whole mount immunostaining. Briefly, aortic ring explants were formalin-fixed, blocked with 1% BSA (RPI, A30075-100.0) and 5% normal goat serum, and permeabilized with 0.5% Triton X-100 (Sigma, T8787). We detected endothelial cells using a PECAM1-specific antibody and identified VSMCs by reactivity with an antibody against ACTA2. Tissues were mounted and imaged using an Olympus BX-51 fluorescence microscope (Olympus, Genter Valley, PA).
Cell culture and transfection
We purchased human aortic smooth muscle cells (HASMCs) from Invitrogen (Life Technologies, C0075 C) and maintained them in Medium 231 (Gibco, M-231-500) supplemented with 5% smooth muscle cell growth supplements (Invitrogen, M231500). Mouse aortic smooth muscle cells (MASMCs) were isolated from thoracic aortas of WT and ulk1 KO mice as described previously, and were grown in DMEM/F12 medium supplemented with 5% fetal bovine serum (FBS, Gibco, 16–000-044) and smooth muscle growth supplements [Citation50]. During the migration experiment, HASMCs were cultured in complete DMEM medium. All culture media were supplemented with penicillin (100 units/mL) and streptomycin (100 µg/mL) (Thermo Fisher, 15,140,148). Cultured cells were incubated in a humidified atmosphere of 5% CO2 at 37°C.
We obtained ULK1 and KAT2A siRNA from Cell Signaling Technology. For siRNA knockdown, we plated HASMCs or mouse VSMCs in 6-well plates. After 70% confluence, cells were treated with the indicated siRNA at a final concentration of 50 nM using Lipofectamine RNAiMAX reagent in serum-free media. After 6 h, we removed the media and replaced with fresh media containing 10% FBS, and cells were incubated in fresh media for 48 h. We measured protein expression by immunoblotting with specific antibodies.
Autophagy analysis
Cultured cells were treated with chloroquine, transfected with adenovirus or siRNA, or subjected to starvation by incubation in Hanks’ balanced salt solution (HBSS; Invitrogen, 14,025). We assessed autophagy activity by measuring GFP-LC3 puncta formation, LC3 cleavage, and SQSTM1 degradation. To determine autophagosome formation, we cultured the cells on glass coverslips and allowed them to adhere to the coverslip overnight. The cells were transfected with GFP-LC3 adenovirus for 24 h followed by incubation with 5 μM chloroquine diphosphate (CQ) for another 24 h. After treatment, we fixed cells and obtained fluorescence images using a fluorescent microscope (Olympus, Genter Valley, PA). Autophagy was measured by quantifying the average number of autophagosomes per cell for each sample. We counted a minimum of 100 cells per sample.
Real-time PCR analysis
Total RNA was extracted from cultured cells using an RNeasy Mini Kit (Qiagen, 74,104). For reverse transcription, 1 µg total mRNA was converted to first strand complementary DNA in 20 µL reactions using iScript cDNA synthesis kit (Bio-Rad Laboratories, 1,708,890). Primers used for gene amplification were as follows: Kat2a/Gcn5 (mouse) 5ʹ-CGAGTTGTGCCGTAGCTGTGA-3ʹ (forward) and 5ʹ-ACCATTCCCAAGAGCCGGTTA-3ʹ (reverse); Gapdh (mouse) 5ʹ-CCACTCCTCCACCTTTGAC-3ʹ (forward) and 5ʹ-ACCCTGTTGCTGTAGCCA-3ʹ (reverse). Quantitative RT-PCR reactions were performed using aiQ™ SYBRGreen Supermix Kit (Bio-Rad Laboratories, 1,708,880) and a CFX96 real-time PCR detection system (Applied Biosystems), as described previously [Citation51,Citation52]. We quantified results using the 2-∆∆Ct method with Gapdh as an internal control [Citation53].
Western blot
We lysed cells and carotid arteries in radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology, sc-24,948) and measured protein concentration with a bicinchoninic acid protein assay (Pierce, 23,225). Cell lysates were analyzed by western blotting. Briefly, proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes (Bio-rad, 1,620,112), and probed with specific antibodies. We detected antibody signal using enhanced chemiluminescence (Thermo Fisher Scientific, 34,578), then stripped the membranes and probed with total protein and/or ACTB or GAPDH to verify equal loading [Citation54].
Scratch wound assay
We performed monolayer smooth muscle cell migration according to a previously described method [Citation55]. Briefly, HASMCs or mouse VSMCs were plated onto glass coverslips or 6-well plates at 2.5 × 105 cells/well and transfected with either siRNA or adenovirus. After 48 h, we gently made a linear wound in the center of the confluent cell monolayer with a 200-μL pipette tip, then washed with fresh media to remove cellular debris. Repeated observations of the edge of the same scratched lesion were performed. After 10 h, we captured images using a microscope (IX71, Olympus) equipped with a digital camera. We calculated the distance of gap closure as the total distance of the gap closed over the migration time.
Transwell migration assay
We measured cell migration using transwells fitted with a polycarbonate membrane (8-μm pore size; Corning Inc., CLS3464). Briefly, growth-arrested HASMCs were trypsinized and resuspended at 1 × 106 cells/mL in DMEM supplemented with 0.5% FBS. We placed the HASMC suspension (100 μL) in the upper chamber and 500 μL DMEM containing 10% FBS in the lower chamber. Cells were allowed to migrate through membrane pores for 6 h at 37°C and 5% CO2. We then removed the filters and fixed filter membranes in 4% paraformaldehyde. The cells on the upper side of the filter were scraped off with cotton swabs, and the cells that migrated to the lower surface of the filter were stained with 0.2% crystal violet in 10% methanol for 30 min. We washed the chambers thoroughly with water and counted cells on the lower surface of filters using a light microscope. Experiments were performed at least three times for each group, and cell motility is presented as the number of migrated cells/field.
Immunohistochemistry and immunofluorescence staining
For immunohistochemical staining, carotid arteries were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into sections. We deparaffinized sections and stained with indicated antibodies as previously described [Citation56,Citation57]. The signal was visualized using the Liquid DAB+ Substrate Chromogen System and slides were analyzed with an Olympus microscope or LSM 510 Zeiss confocal microscope. Immunohistochemically staining density of ULK1 and SQSTM1 were assessed with a Histoscore system (H-score) [Citation58]. Briefly, we estimated the extent of positively stained cells and classified staining on a four-point scale (0%, no staining; 1, 1–10%; 2, 11–50%; 3, 51–80%; and 4, 81–100%). The intensity of the immuno-expression was categorized into three groups: weak (+1), moderate (+2), and strong (+3). A final IHScore was obtained by multiplying the score for extent and the score for intensity. For immunofluorescence staining, we permeabilized sections by incubating in 1% Triton X-100 in phosphate-buffered solution at 25°C for 15 min. After blocking with goat serum for 1 h at room temperature, we incubated slides with primary antibodies overnight in a humidity chamber. We diluted the ACTA2 antibody 1:400, PECAM1 antibody 1:200, and all other antibodies 1:50 in blocking buffer (BioGenex, HK112-9 K). The isotype controls were used as negative controls. To evaluate reendothelialization, we collected carotid arteries 7 d after ligation and performed IHC staining with a PECAM1 antibody. The intensity of immunofluorescence staining was assessed using ImageJ software (NIH). Values represent mean protein intensity, which was calculated by the ratio of total protein intensity to total area in each sample.
Statistical analysis
Data are presented as mean ± SEM. The differences between two groups were analyzed by Student’s t test. The comparisons between multiple groups were performed with ANOVA (one-way analysis of variance) followed by a Bonferroni post hoc analysis. A value of P< 0.05 was considered statistically significant.
Supplemental Material
Download Zip (81.5 MB)Acknowledgments
This study was supported by grants from the following agencies: NHLBI (HL079584, HL080499, HL089920, HL110488, HL128014, HL132500, HL137371, and HL142287), NCI (CA213022), NIA (AG047776), and AHA (16GRANT29590003). Dr. Zou is the Eminent Scholar in Molecular and Translational Medicine of the Georgia Research Alliance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132.
- Kim PK, Hailey DW, Mullen RT, et al. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008 Dec;105(52):20567–20574.
- Lu K, Psakhye I, Jentsch S. A new class of ubiquitin-Atg8 receptors involved in selective autophagy and polyQ protein clearance. Autophagy. 2014;10(12):2381–2382.
- Steele S, Brunton J, Kawula T. The role of autophagy in intracellular pathogen nutrient acquisition. Front Cell Infect Microbiol. 2015;5:51.
- Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009 Feb;104(3):304–317.
- He C, Zhu H, Zhang W, et al. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am J Pathol. 2013 Aug;183(2):626–637. .
- Grootaert MO, Da Costa Martins PA, Bitsch N, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. 2015 Nov; 11(11):2014–2032. .
- LaRocca TJ, Gioscia-Ryan RA, Hearon CM Jr., et al. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. 2013 Jul;134(7–8):314–320.
- Sprott D, Poitz DM, Korovina I, et al. Endothelial-specific deficiency of ATG5 (Autophagy Protein 5) attenuates ischemia-related angiogenesis. Arterioscler Thromb Vasc Biol. 2019 Jun;39(6):1137–1148. .
- Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009 Oct;461(7264):654–658. .
- Wong PM, Puente C, Ganley IG, et al. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013 Feb;9(2):124–137.
- Lane JD, Korolchuk VI, Murray JT. Signalling mechanisms in autophagy: an introduction to the issue. Essays Biochem. 2017 Dec;61(6):561–563.
- Liu L, Yan L, Liao N, et al. A Review of ULK1-Mediated Autophagy in Drug Resistance of Cancer. Cancers (Basel). 2020 Feb;12(2):352.
- Lacolley P, Regnault V, Nicoletti A, et al. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012 Jul;95(2):194–204.
- Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007 Mar;100(5):607–621.
- Song Y, Brady ST. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 2015 Mar;25(3):125–136.
- Fukushima N, Furuta D, Hidaka Y, et al. Post-translational modifications of tubulin in the nervous system. J Neurochem. 2009 May;109(3):683–693.
- Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987 Feb;104(2):289–302.
- Janke C, Montagnac G. Causes and consequences of microtubule acetylation. Curr Biol. 2017 Dec;27(23):R1287–R1292.
- Akella JS, Wloga D, Kim J, et al. MEC-17 is an alpha-tubulin acetyltransferase. Nature. 2010 Sep;467(7312):218–222. .
- Creppe C, Malinouskaya L, Volvert ML, et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009 Feb;136(3):551–564. .
- North BJ, Marshall BL, Borra MT, et al. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003 Feb;11(2):437–444.
- Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002 May;417(6887):455–458. .
- Burgess RJ, Zhou H, Han J, et al. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell. 2010 Feb;37(4):469–480.
- Ouyang C, Mu J, Lu Q, et al. Autophagic degradation of KAT2A/GCN5 promotes directional migration of vascular smooth muscle cells by reducing TUBA/alpha-tubulin acetylation. Autophagy. 2020 Oct;16(10):1753–1770. .
- Singh NK, Kundumani-Sridharan V, Kumar S, et al. Protein kinase N1 is a novel substrate of NFATc1-mediated cyclin D1-CDK6 activity and modulates vascular smooth muscle cell division and migration leading to inward blood vessel wall remodeling. J Biol Chem. 2012 Oct;287(43):36291–36304. .
- Bendeck MP, Zempo N, Clowes AW, et al. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994 Sep;75(3):539–545.
- Baker M, Robinson SD, Lechertier T, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012 Jan;7(1):89–104. .
- Jawien A, Bowen-Pope DF, Lindner V, et al. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. 1992 Feb;89(2):507–511.
- Iwai-Kanai E, Yuan H, Huang C, et al. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008 4; Apr(3): 322–329. .
- Ye LX, Yu J, Liang YX, et al. Beclin 1 knockdown retards re-endothelialization and exacerbates neointimal formation via a crosstalk between autophagy and apoptosis. Atherosclerosis. 2014 Nov;237(1):146–154.
- Hutter R, Sauter BV, Reis ED, et al. Decreased reendothelialization and increased neointima formation with endostatin overexpression in a mouse model of arterial injury. Circulation. 2003 Apr;107(12):1658–1663. .
- Fullgrabe J, Lynch-Day MA, Heldring N, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013 Aug;500(7463):468–471. .
- Wang YH, Yan ZQ, Qi YX, et al. Normal shear stress and vascular smooth muscle cells modulate migration of endothelial cells through histone deacetylase 6 activation and tubulin acetylation. Ann Biomed Eng. 2010 Mar;38(3):729–737. .
- Kaverina I, Straube A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 2011 Dec;22(9):968–974.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007 Nov;21(22):2861–2873.
- Wang Y, Dubland JA, Allahverdian S, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2019 May;39(5):876–887. .
- Chen Y, Su X, Qin Q, et al. New insights into phenotypic switching of VSMCs induced by hyperhomocysteinemia: role of endothelin-1 signaling. Biomed Pharmacother. 2020 Mar;123:109758.
- Salabei JK, Cummins TD, Singh M, et al. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013 May;451(3):375–388.
- Salabei JK, Hill BG. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radic Biol Med. 2013 Dec;65:693–703.
- Cherepanova OA, Pidkovka NA, Sarmento OF, et al. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ Res. 2009 Mar;104(5):609–618. .
- Li H, Li J, Li Y, et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2012 Dec;303(11):H1319–H1331. .
- Lee IH, Kawai Y, Fergusson MM, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012 Apr;336(6078):225–228. .
- Hill BG, Haberzettl P, Ahmed Y, et al. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J. 2008 Mar;410(3):525–534.
- Li L, Wei D, Wang Q, et al. MEC-17 deficiency leads to reduced alpha-tubulin acetylation and impaired migration of cortical neurons. J Neurosci. 2012 Sep;32(37):12673–12683. .
- Boggs AE, Vitolo MI, Whipple RA, et al. alpha-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015 Jan;75(1):203–215. .
- Saji S, Kawakami M, Hayashi S, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005 Jun;24(28):4531–4539. .
- Song P, Zhou Y, Coughlan KA, et al. Adenosine monophosphate-activated protein kinase-alpha2 deficiency promotes vascular smooth muscle cell migration via S-phase kinase-associated protein 2 upregulation and E-cadherin downregulation. Arterioscler Thromb Vasc Biol. 2013 Oct;33(12):2800–2809. .
- Indolfi C, Avvedimento EV, Rapacciuolo A, et al. Inhibition of cellular ras prevents smooth muscle cell proliferation after vascular injury in vivo. Nat Med. 1995 Jun;1(6):541–545.
- Ray JL, Leach R, Herbert JM, et al. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23(4):185–188.
- Tedesco L, Valerio A, Cervino C, et al. Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesis through endothelial nitric oxide synthase expression in white adipocytes. Diabetes. 2008 Aug;57(8):2028–2036. .
- Li H, Lee J, He C, et al. Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids. Am J Physiol Endocrinol Metab. 2014 Jan;306(2):E197–E209.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001 May;29(9):e45.
- Xie Z, Singh M, Siwik DA, et al. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: role of protein kinase C-zeta. J Biol Chem. 2003 Dec;278(49):48546–48552.
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333.
- He C, Choi HC, Xie Z. Enhanced tyrosine nitration of prostacyclin synthase is associated with increased inflammation in atherosclerotic carotid arteries from type 2 diabetic patients. Am J Pathol. 2010 May;176(5):2542–2549.
- He C, Li H, Viollet B, et al. AMPK suppresses vascular inflammation in vivo by inhibiting signal transducer and activator of transcription-1. Diabetes. 2015 Dec;64(12):4285–4297.
- Soslow RA, Dannenberg AJ, Rush D, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000 Dec;89(12):2637–2645. .