
ABSTRACT
Defects in macroautophagy/autophagy are implicated in the pathogenesis of neuromuscular and heart diseases. To precisely define the roles of autophagy-related genes in skeletal and cardiac muscles, we generated muscle-specific rb1cc1- and atg14-conditional knockout (cKO) mice by using Ckm/Ckmm2-Cre and compared their phenotypes to those of ulk1 ulk2-conditional double-knockout (cDKO) mice. atg14-cKO mice developed hypertrophic cardiomyopathy, which was associated with abnormal accumulation of autophagic cargoes in the heart and early mortality. Skeletal muscles of both atg14-cKO and rb1cc1-cKO mice showed features of autophagic vacuolar myopathy with ubiquitin+ SQSTM1+ deposits, but only those of rb1cc1-cKO mice showed TARDBP/TDP-43+ pathology and other features of the inclusion body myopathy–like disease we previously described in ulk1 ulk2-cDKO mice. Herein, we highlight tissue-specific differences between skeletal and cardiac muscles in their reliance on core autophagy proteins and unique roles for ULK1-ULK2 and RB1CC1 among these proteins in the development of TARDBP+ pathology.
ABBREVIATIONS:AVM: autophagic vacuolar myopathy; cDKO: conditional double knockout; cKO: conditional knockout; H&E: hematoxylin and eosin; IBM: inclusion body myopathy; mtDNA: mitochondrial DNA; PFA: paraformaldehyde; RNP: ribonucleoprotein; TBST: Tris-buffered saline with 0.2% Triton X-100
Introduction
Macroautophagy (hereafter referred to as autophagy) is a highly conserved catabolic process wherein cytoplasmic materials such as ubiquitinated proteins and organelles are sequestered within double-membrane bound vesicles and then delivered to lysosomes for degradation [Citation1]. Although the process occurs under basal physiological conditions, flux through the autophagy pathway is modulated in response to changes in intracellular and extracellular environments. Dysregulation of autophagy has been linked to the pathogenesis of numerous diseases, such as cardiovascular disease and neuromuscular degeneration [Citation2]. Skeletal and cardiac muscles are constantly challenged by fluctuations in nutrients, metabolites, and oxygen; therefore, anomalies in the autophagy pathway are observed in many muscle-related diseases, such as autophagic vacuolar myopathy (AVM), lysosomal storage disease, and inclusion body myopathy (IBM) [Citation3]. Activating autophagy by either a low amino acid diet or resveratrol supplementation ameliorates cardiac and skeletal muscle phenotypes in some mouse models of muscular dystrophy [Citation4,Citation5]. However, the precise contribution of specific autophagy-related genes and pathways in the pathophysiological manifestations of these disorders remains poorly characterized.
The most direct experimental evidence supporting a protective role for autophagy in mammalian muscle physiology primarily comes from analyses of mice lacking the expression of nonredundant essential autophagy genes. For example, the muscle-specific conditional knockout (cKO) of Atg5 or Atg7, which encodes key components of the ubiquitin-like conjugation machinery that are responsible for the lipidation of LC3B and other Atg8-related proteins, results in age-dependent muscle atrophy and weakness associated with the accumulation of abnormal mitochondria and ubiquitinated protein aggregates typically observed in autophagy-defective models [Citation6,Citation7]. Similarly, inhibition of autophagy in the heart leads to accumulation of SQSTM1/p62 and ubiquitin, cardiac dysfunction, and shortened lifespan in mice [Citation8,Citation9].
Other muscle-specific autophagy gene knockout mice, such as those lacking Pik3r4/Vps15 or Pi3k3c/Vps34, show additional histopathological features not found in atg5- or atg7-cKO mice. PI3K3C/VPS34 is a class III phosphatidylinositol 3-kinase (PtdIns3K), and its activity is regulated by the serine-threonine kinase PIK3R4/VPS15. The PIK3C3/VPS34 kinase complex regulates various processes, such as autophagosome nucleation, membrane fusion events, and endolysosomal trafficking of proteins [Citation10]. Consistent with the role of the PIK3C3/VPS34 complex in regulating flux through the autophagy pathway, the SQSTM1+, ubiquitin+ pathology observed in atg5- and atg7-cKO models is also present in muscle-specific pik3r4/vps15- and pi3k3c/vsp34-cKO mice [Citation11,Citation12]. However, the latter 2 models also show accumulation of glycogen and sarcoplasmic vacuolation typically found in the muscle of patients with AVMs and lysosomal storage diseases [Citation11,Citation12]. Vacuoles in these mice also have sarcolemmal features (i.e., positive for DMD [dystrophin, muscular dystrophy] and other proteins typically localized to the sarcolemma).
Muscle-specific depletion of ULK1 (unc-51 like kinase 1) and ULK2 (unc-51 like kinase 2), two functionally redundant serine-threonine protein kinases involved in stress-induced autophagy [Citation13] and autophagosome maturation [Citation14,Citation15], leads to the development of a vacuolar myopathy with SQSTM1+, ubiquitin+ pathology in mice [Citation16]. Vacuoles in ulk1 ulk2-cDKO mice do not have sarcolemmal features. However, ulk1 ulk2-cDKO mice show aberrant redistribution of the RNA-binding protein TARDBP from the nucleus to the cytoplasm and in pathological inclusions [Citation16]. The TARDBP+ pathology in ulk1 ulk2-cDKO mice is similar to that observed in subsets of patients with IBM, amyotrophic lateral sclerosis, or frontotemporal dementia; it most likely results from the aberrant persistence of RNA–protein assemblies [Citation16]. In patients, TARDBP+ pathology is associated with mutations in RNA-binding proteins (e.g., TIA1, FUS, TARDBP) or autophagy-related proteins (e.g., SQSTM1, UBQL2, OPTN1, VCP) [Citation17,Citation18]. Although tight regulation of ribonucleoprotein (RNP) granule dynamics is clearly essential for muscle homeostasis [Citation19], the precise role of autophagy in resolving RNP granules and TARDBP+ pathology remains poorly understood.
Given the importance of autophagy in muscle homeostasis and evidence that defects in ULK1-ULK2 function affect IBM pathogenesis, we investigated the consequences of muscle-specific deletion of core autophagy-related genes Atg14 and Rb1cc1. Both genes encode proteins that are functionally related to ULK1-ULK2: RB1CC1 is a component of the canonical autophagy-inducing ULK1-ULK2 complex [Citation20], and ATG14 is a component of the autophagy-initiating PI3K3C/VPS34 PtdIns3K complex and is directly phosphorylated by ULK1-ULK2 [Citation13,Citation21]. The contributions of ATG14 and RB1CC1 in muscle physiology have not been previously studied.
Results
Atg14 deficiency causes shortened life span and cardiac hypertrophy
We first examined the expression levels of core autophagy genes Ulk1, Ulk2, Atg14, Rb1cc1, and Atg7 in the skeletal and cardiac muscles of adult mice. Interestingly, the abundance of all autophagy genes in the heart was approximately 4- to 5-fold higher than that in the skeletal muscle, perhaps indicative of a higher demand for autophagy in the heart (Figure S1A). Next, mice harboring floxed alleles of Atg14 or Rb1cc1 were crossed with mice carrying the Ckm-Cre transgene, wherein the expression of Cre recombinase is driven by Ckm (creatine kinase, muscle) promoter starting at embryonic day 13.5 [Citation22]. Quantitative reverse-transcriptase real-time PCR (qRT-PCR) analyses confirmed efficient knockout of Atg14 and Rb1cc1 in both the cardiac and skeletal muscles (Figure S1B and C). Similar to ulk1 ulk2-cDKO (ulk1flox/flox;ulk2−/-;Ckm-Cre), both atg14-cKO (atg14flox/flox;Ckm-Cre) and rb1cc1-cKO (rb1cc1flox/flox;Ckm-Cre) mice were indistinguishable from age-matched littermate controls (atg14+/flox;Ckm-Cre and rb1cc1+/flox;Ckm-Cre) at birth and weaning. Body weight (Table S1) and metabolic parameters (i.e., serum M-type levels of albumin, alkaline phosphatase, glucose) were not significantly different in cKO mice compared to littermate controls at 2 months (Table S2). The viability of atg14-cKO mice decreased sharply beginning at 38 weeks, with all animals reaching humane endpoints by 55 weeks. In contrast, ulk1 ulk2-cDKO and rb1cc1-cKO mice exhibited normal life spans up to 2 years () [Citation16].
Figure 1. Atg14 deficiency in the heart causes cardiomyopathy and shortened life span. (A) Kaplan–Meier curve of control (n = 54), ulk1 ulk2-cDKO (n = 21), atg14-cKO (n = 15), and rb1cc1-cKO (n = 18) mice within 100 weeks. P < 0.0001 for control vs. atg14-cKO; P > 0.05 for control vs. ulk1 ulk2-cDKO and control vs. rb1cc1-cKO. (B) Cross sections of hearts from control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice at 5 months stained with H&E and antibodies against ubiquitin and UQCRC2. (C) Mitochondrial DNA (mtDNA) copy number in the hearts of ulk1 ulk2-cDKO, and atg14-cKO, rb1cc1-cKO mice and their corresponding controls at 5 months assessed by quantitative PCR analyses of mt-Nd2 (mtDNA). Data were normalized to Rn18s and are presented as mean ± SEM; n = 3 for each genotype. *P < 0.05 for control vs. atg14-cKO; ns: not significant. (D) The mRNA levels of Ppargc1 in the hearts of control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice at 5 months were determined by qRT-PCR. Data were normalized to Gapdh and presented as mean ± SEM; n = 3 for each genotype; ns: not significant
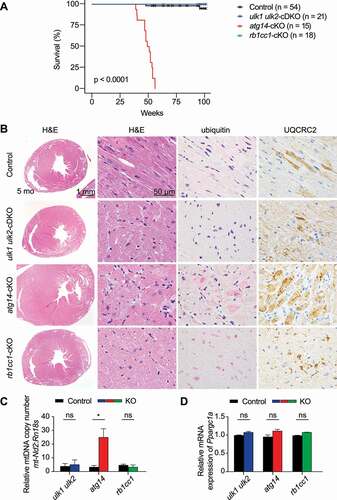
To determine the cause of early mortality in atg14-cKO mice, we performed necropsies on a cohort of 5-month-old mice. Consistent with the expression of the Ckm-Cre transgene in cardiac and skeletal muscles, gross examination revealed enlarged hearts in atg14-cKO mice compared to littermate controls (Figure S1D). H&E staining showed that the ventricular walls of atg14-cKO hearts were thickened approximately 2 fold, with marked hypertrophy of cardiomyocytes. The majority of affected cardiomyocytes had enlarged rounded nuclei with multiple prominent nucleoli and vacuolated sarcoplasm (). The normal linear arrangement of myofibrils in myocytes of atg14-cKO mice was disrupted by scattered ubiquitin+ inclusions and aggregates, and numerous large, irregular MTCO1+ and UQCRC2+ mitochondria ( and data not shown), similar to that in atg7-cKO mice (Figure S1E). Consistent with the increase in mitochondrial content suggested by the immunohistochemical stains, quantitative PCR analyses revealed an increase in mitochondrial DNA (mtDNA) copy number in atg14-cKO hearts (). In contrast, mtDNA copy number analyses showed no significant increase in atg7-cKO hearts compared to controls (Figure S1F). Unlike in atg14-cKO hearts, where the UQCRC2 staining and mtDNA copy both increased, atg7-cKO hearts showed a discrepancy between the UQCRC2 staining and mtDNA copy number. This finding suggests that the mitochondria accumulating in atg7-cKO (but not atg14-cKO) hearts are depleted of mtDNA. Although steady-state mitochondrial content may be elevated due to impaired degradation, it can also result from increased biogenesis. Therefore, we also examined the expression of Ppargc1a, which encodes a well-characterized activator of the mitochondrial biogenesis program in cardiomyocytes (and other cell types) and causes cardiomyopathy when overexpressed [Citation23]. We observed no significant differences in Ppargc1a mRNA levels by in situ hybridization or qRT-PCR between atg14-cKO hearts and those of their littermate controls ( and S1G). Thus, the accumulation of ubiquitin+ deposits and UQCRC2+ mitochondria in cardiomyocytes, similar to that observed in atg5-cKO (atg5flox/flox; α-MyHC-Cre) [Citation8] and atg7-cKO (atg7flox/flox;Ckm-Cre) mice (Figure S1E), was consistent with the expected defect in autophagy.
Unlike the ventricular walls in the hearts of atg14-cKO mice, those of rb1cc1-cKO and ulk1 ulk2-cDKO mice had normal thickness, and myocardiocytes in H&E-stained sections were mostly unremarkable at 5 months (). Rare scattered cardiomyocytes with clusters of enlarged UQCRC2+ mitochondria were observed. Ubiquitin+ inclusions were extremely small and rare (). Examination of the hearts of 24-month-old mice revealed mild interstitial fibrosis in myocardial fibers of aged rb1cc1-cKO and ulk1 ulk2-cDKO mice (Figure S1H). Myocytes also showed vacuolization, with numerous large, irregular mitochondria and scattered ubiquitin+/SQSTM1+ aggregates (Figure S1H and data not shown).
Together, these findings suggest that ULK1-ULK2 and RB1CC1 play a relatively minor role, compared to those of ATG14 and ATG7, in regulating autophagy activity in cardiomyocytes of unstressed animals.
Skeletal muscle deficient in Atg14 and Rb1cc1 displays features of AVM and IBM
Next, we compared skeletal muscle phenotypes of mice lacking Atg14, Rb1cc1, or Ulk1 Ulk2 expression. The ulk1 ulk2-cDKO mice exhibited muscle weakness; myopathy with vacuolar degeneration; ubiquitin+, SQSTM1+, and TARDBP+ inclusions; and tubulofilamentous aggregates in electron microscope (EM) analysis, which together indicated IBM-like disease [Citation3,Citation16]. Similar to ulk1 ulk2-cDKO mice, atg14-and rb1cc1-cKO mice had significantly higher levels of serum M-type CKM (creatine kinase, muscle) compared to controls at 2 months (). Hanging wire tests showed significant decreases in grip strength by 8 months (but not at 2 or 5 months) in atg14-cKO and rb1cc1-cKO mice compared to controls, indicative of the progressive nature of muscle weakness () [Citation16,Citation24]. H&E and modified Gomori trichrome staining revealed vacuoles in muscles of 5-month-old atg14- and rb1cc1-cKO mice, similar to those in ulk1 ulk2-cDKO mice (). Evident variations in fiber diameters and scattered myocytes with centralized nuclei suggested myogenic myopathy (Figure S2A). Membrane localization of DMD and CAV3 was normal in muscle fibers of atg14-cKO, rb1cc1-cKO, or ulk1 ulk2-cDKO mice (Figure S2B). There was no obvious glycogen accumulation (as indicated by PAS staining) or inflammation in these muscles, and degeneration/necrosis of myocytes was rare (data not shown). Immunostaining performed on different skeletal muscles using antibodies against the muscle type–specific myosin heavy chain revealed that vacuoles were predominantly present in fast-switch (type II) fibers (Figure S2C). Hence, we focused on the quadricep muscles as they have a predominance of type II fibers.
Figure 2. Loss of Atg14 and Rb1cc1 causes skeletal vacuolar myopathy. (A) Serum M-type (CKM/creatine kinase) levels of control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice at 2 months. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t-test. All quantitative data are presented as mean ± SEM. (B) Muscle strength of control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice at the age of 8 months. *P < 0.05, **P < 0.01, ns: not significant by Student’s t-test. All quantitative data are presented as mean ± SEM arbitrary units (a.u.). (C) Muscle cross sections of control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice were stained with H&E, modified Gomori trichrome, and an anti-ubiquitin antibody. (D) Immunostaining of quadriceps in 5-month-old mice showing accumulation of SQSTM1, which colocalized with ubiquitin in ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice. (E) Percentages of myofibers from quadriceps with SQSTM1+ deposits were quantified from 5- and 24-month-old control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t-test. All quantitative data are presented as mean ± SEM, n = 3 for each genotype. (F) Size distribution of SQSTM1+ deposits observed in the 5-month-old control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice. n = 3 for each genotype. (G) Muscle cross sections of 24-month-old control, ulk1 ulk2-cDKO, and rb1cc1-cKO mice stained with H&E and anti-SQSTM1 antibodies. (H) Ultrastructural studies of quadriceps from 5-month-old control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice. (I) Protein extracts prepared from quadriceps of 5-month-old control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice and analyzed by immunoblot against SQSTM1, LC3B, TARDBP, and GAPDH. (J) Fold-change in protein levels of SQSTM1, LC3-II, LC3-II, LC3-I and TARDBP in 5-month-old ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice compared to the corresponding control, as determined by densitometry. n = 3 for each genotype. ns, not significant; *P < 0.05 and **P< 0.01 by Student’s t-test. Note: In (A), (E), and (F), data for ulk1 ulk2-cDKO mice and littermate controls were previously reported17 and replotted to facilitate direct comparison
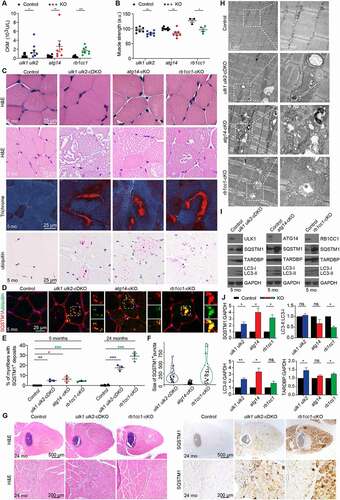
Immunohistochemical studies revealed ubiquitin+ and SQSTM1+ inclusions in the quadriceps of 5-month-old atg14- and rb1cc1-cKO mice (). These inclusions were also positive for LC3B and LAMP1 (Figure S2D). Although SQSTM1+ deposits were identified in approximately 5% of myofibers in ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice (), those in the atg14-cKO muscles were smaller and more dispersed than the deposits in rb1cc1-cKO and ulk1 ulk2-cDKO muscles () but similar in size and distribution to those in atg7-cKO mice [Citation16]. The variations in size and distribution of the deposits may reflect differences in their trafficking. There was no reproducible increase in the number of MTCO1+ and UQCRC2+ mitochondria or mtDNA copy number in the muscles of any of the knockout models (Figure S2E and data not shown). Myopathic features became more prominent in aged (24 months old) rb1cc1-cKO and ulk1 ulk2-cDKO mice and affected almost all myocytes in type II fibers (), with 20%–30% of these myofibers containing SQSTM1+ inclusions, thereby confirming the progressive nature of the disease ().
The EM studies revealed abnormal sarcomeric architecture in atg14- and rb1cc1-cKO mice, including extensive accumulation of membranous structures, debris, and mitochondrial elements (degenerate and intact) (). These features are frequently detected in autophagy-defective muscles [Citation6,Citation7]. Tubulofilamentous structures similar to those in muscles of ulk1 ulk2-cDKO mice [Citation16] were present in Rb1cc1-cKO mice but not in muscles of atg14-cKO mice ().
Immunoblot analyses of extracts prepared from quadriceps of these mice confirmed significantly higher steady-state levels of SQSTM1 in all 3 sets of cKO mice compared to their corresponding controls (). Immunoblot analyses of LC3B showed significant increases in LC3-II levels in atg14-cKO and ulk1 ulk2-cDKO mice compared to littermate controls, which together with the increase in SQSTM1 levels was consistent with the roles of Atg14 and Ulk1 Ulk2 in autophagosome maturation [Citation14,Citation25]. The increase in LC3-II levels also most likely masked the expected defects in LC3 lipidation associated with Ulk1 Ulk2 or Atg14 deficiency [Citation14,Citation25,Citation26], resulting in minimal changes in the LC3-II:LC3-I ratio. The LC3-II levels were unchanged in muscles of rb1cc1-cKO mice compared to controls, and there was an expected decrease in the LC3-II:LC3-I ratio (). Interestingly, both Sqstm1 and Map1lc3 mRNA levels were elevated in all 3 sets of cKO mice (Figure S2F). These compensatory transcriptional responses may contribute to pathological accumulation of ubiquitin+ and SQSTM1+ deposits in the muscles of autophagy-defective mice.
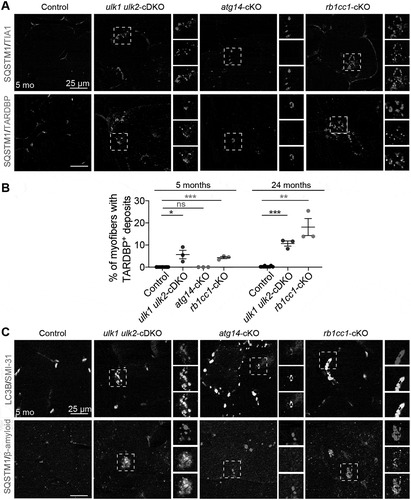
A subset of IBM cases are characterized by abnormal redistribution of RNA-binding proteins, such as TARDBP, from the nucleus to the cytoplasm and the presence of TARDBP+ inclusions [Citation16,Citation27]. Although Tardbp mRNA expression levels were slightly increased in all 3 sets of cKO animals (Figure S3A), immunoblotting analyses showed increases in TARDBP protein abundance in rb1cc1-cKO and ulk1 ulk2-cDKO but not in atg14-cKO mice (). Immunostaining for TARDBP and TIA1 revealed the accumulation of both RNA-binding proteins within sarcoplasmic SQSTM1+ inclusions in rb1cc1-cKO and ulk1 ulk2-cDKO mice but not in atg14-cKO mice (). Examination of aged (24 months old) rb1cc1-cKO and ulk1 ulk2-cDKO mice showed that the TARDBP pathology in these animals worsened over time (). In contrast to the abnormal distribution pattern of TARDBP and TIA, other RNA-binding proteins such as HNRNPA2B1 and FUS had normal nuclear localization in muscles of all 3 sets of cKO mice (Figure S3B). Finally, inclusion bodies in rb1cc1-cKO and ulk1 ulk2-cDKO mice (but not in atg14-cKO mice) were also positively labeled by antibodies reactive to phosphorylated MAPT/Tau (SMI-31) [Citation16,Citation28] and β-amyloid (Peptide 1–42) ().
Figure 3. Deficiency of Rb1cc1 and Ulk1 Ulk2 but not Atg14 in the skeletal muscle causes TARDBP+ pathology. (A) Frozen sections of quadriceps from control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice at 5 months were co-stained with antibodies against SQSTM1 and TIA1 or SQSTM1 and TARDBP. (B) Percentages of myofibers in quadriceps with TARDBP+ deposits quantified from 5-month-old and 24-month-old controls, ulk1 ulk2-cDKO, atg14-KO, and rb1cc1-cKO mice. n = 3 for each genotype; ns: not significant. *P < 0.05, **P < 0.01, ***P < 0.001 by student’s t-test. (C) Frozen sections of quadriceps from 5-month-old control, ulk1 ulk2-cDKO, atg14-cKO, and rb1cc1-cKO mice co-stained for LC3B and phosphorylated MAPT/Tau (SMI-31) or SQSTM1 and β-amyloid (peptide 1–42)
Discussion
The comparison of cardiac and skeletal muscle phenotypes in atg14-cKO, rb1cc1-cKO, and ulk1 ulk2-cDKO mice presented in this study provides several new insights into the functions of these autophagy-related genes. For example, skeletal muscles of all the genetic models (atg14-cKO, rb1cc1-cKO, ulk1 ulk2-cDKO, atg5-cKO and atg7-cKO mice) [Citation6,Citation7] showed the typical SQSTM+ ubiquitin+ deposits associated with impaired autophagy; however, targeting Atg14 expression in the heart resulted in more rapid and prominent accumulation of mitochondria and SQSTM1+ ubiquitin+ deposits than did targeting Ulk1 Ulk2 or Rb1cc1 expression. The accumulation of autophagic cargo in the hearts of atg14-cKO mice was similar to that in atg7-cKO, atg5-, and pi3k3c/vps34-cKO mice [Citation8,Citation12,Citation29]. Although the autophagy defect arising from the Atg14 deficiency in hearts contributed to the accumulation of mitochondria, it was not severe enough to cause the mtDNA depletion observed in atg7-cKO mice and autophagy-defective starved yeast [Citation30]. Also, although there is a precedent for ULK1 ULK2 not being essential for autophagy in physiological settings (e.g., in the central nervous system) [Citation31], RB1CC1 is essential for the autophagy-mediated turnover of SQSTM1 and ubiquitinated proteins in most tissues, including the central nervous system and liver [Citation32,Citation33]. To the best of our knowledge, these observations provide the first example of constitutive autophagy occurring in the absence of RB1CC1. Together, these results highlight tissue-specific differences in the configuration of the autophagy pathway and its context-dependent reliance on the function of specific autophagy-related genes.
We observed that atg14-cKO mice developed hypertrophic cardiomyopathy, which was most likely the cause of their premature death. The cardiac hypertrophy was similar to that seen in pi3k3c/vps34-cKO mice [Citation34] but was not observed in rb1cc1-cKO, ulk1 ulk2-cDKO, atg5-cKO [Citation8], or atg7-cKO mice. It is possible that there are differences in the pathological consequences associated with impaired autophagy in autophagy-defective cells that retain the ability for lipidation of LC3 (e.g., atg14- or pi3k3c/vps34-deficient cardiomyocytes) versus those that do not (e.g., atg5- or atg7-deficient cardiomyocytes). In addition to its role in autophagy nucleation (mediated by the ATG14-containing PIK3C3/VPS34 complex), PI3K3C/VPS34 regulates ESCRT-mediated proteolysis [Citation34] and endolysosomal trafficking [Citation35], but ATG14 has not been implicated in these processes. Therefore, an alternative (although not mutually exclusive) possibility is that there are as-yet undescribed functions of the ATG14-containing PIK3C3/VPS34 complex that are unrelated to autophagy, the disruption of which contributes to cardiac hypertrophy.
The sarcoplasmic accumulation of CAV3, DMD, and glycogen is attributed to impaired endocytosis and lysosomal function [Citation36] and observed in muscle-specific pik3r4/vps15- and pi3k3c/vps34-cKO mice [Citation11,Citation12], which is consistent with the role of core class III PI3K complex constituents (i.e., PIK3C/VPS34 and PIK3R4/VPS15 but not ATG14) in endolysosomal trafficking [Citation35]. Therefore, it is not surprising that these features were not observed in atg14-cKO or other autophagy-defective mouse models (i.e., rb1cc1-cKO, ulk1 ulk2-cDKO, atg7-cKO) [Citation11].
Deletion of Ulk1-Ulk2, Atg14, or Rb1cc1 in the skeletal muscle resulted in the accumulation of vacuoles, which is typically not observed in the muscles of atg7-cKO mice [Citation11,Citation16]. That the loss of Ulk1-Ulk2, Atg14, or Rb1cc1 impairs autophagy flux but does not completely eliminate the lipidation of ATG8 family members and the generation of LC3B+ autophagosomes (as does deletion of Atg5 or Atg7) suggests that the presence of LC3B+ autophagosomes (albeit at reduced levels) is required for the development of vacuolar pathology in the muscle.
In addition to their roles in autophagy, ULK1-ULK2 and RB1CC1 have been implicated in several autophagy-independent processes [Citation31,Citation37]. For example, ULK1-ULK2 are required for the efficient disassembly of TARDBP+ and TIA1+ stress granules in cells in culture [Citation16]. Persistent TARDBP+ RNP granules (including stress granules), such as those observed in ulk1 ulk2-deficient cells, are thought to serve as crucibles for the seeding of poorly dynamic TARDBP+ amyloid-like structures characteristically seen in patients with IBM or related neurodegenerative disorders [Citation27] and in ulk1 ulk2-cDKO mice [Citation16]. The disassembly of stress granules in acutely stressed cells does not involve autophagy (or other components of the autophagy-inducing ULK1-ULK2 kinase complex such as ATG13 and RB1CC1); however, emerging evidence supports a role for autophagy in the degradation of persistent stress granules and other RNA–protein assemblies under certain conditions [Citation38,Citation39]. Although we could not detect TARDBP+ pathology in atg14-cKO mice, its presence in rb1cc1-cKO mice (similar to that in ulk1 ulk2-cDKO mice) raises the possibility that a noncanonical autophagy-related pathway [Citation40–42] is involved in the clearance of RNP granules in muscle. The molecular machinery involved in targeting RNP granules to lysosomes for degradation remains to be elucidated. Nevertheless, it is intriguing that the autophagy-related gene TBK1, which is mutated in patients with TARDBP+ ALS [Citation43], has also been implicated in an alternative lysosomal-targeting pathway [Citation42].
Materials and methods
Animals
Animal experiments were performed per the guidelines of the Institutional Animal Care and Use Committee, St. Jude Children’s Research Hospital. The ulk1flox/flox [Citation16], ulk2–/ – [Citation44], atg14flox/flox [Citation45], rb1cc1flox/flox [Citation32], and atg7flox/flox [Citation16] mice have been described previously. The Ckm-Cre line was purchased from The Jackson Laboratory (stock 006475). Both male and female mice were used in the analyses. Mice were bred in a C57BL/6 background.
Immunostaining and histology
Mice were transcardially perfused with 4% paraformaldehyde (PFA). Quadriceps were dissected and postfixed in 4% PFA for 1 h at 4°C. Next, 30% sucrose was used as cryoprotectant in PBS overnight at 4°C and embedded in optimum cutting temperature for cryosectioning. Frozen sections were washed with 0.2% Triton X-100 (Sigma Aldrich, T8787) in Tris-buffered saline (Thermo Fisher Scientific, 28358) and incubated in blocking solution 5% normal goat (Sigma Aldrich, G9023) or donkey (Sigma Aldrich, G9663) serum M-type in TBST for 1 h at room temperature. Sections were incubated with primary antibodies diluted in the blocking solution overnight at 4°C. Then, they were washed with TBST and incubated with Alexa Fluor-conjugated secondary antibodies (Invitrogen, A-11008, A21422, A21426, A21434, A21244) diluted at 1:1000 in the blocking solution for 2 h at room temperature. This was followed by mounting sections in ProLong Gold Antifade Reagent with DAPI (Invitrogen, P-36931). Primary antibodies used were mouse anti-SQSTM1 (Abnova, H00008878-M01), rabbit anti-ubiquitin (DAKO, Z0458), rabbit anti-LC3B (MBL, PM036), rat anti-LAMP1 (Developmental Studies Hybridoma Bank, 1D4B), mouse anti–phosphorylated MAPT/Tau (SMI-31) (Covance, SMI-31 R), rabbit anti–β-amyloid (peptide 1–42) (Millipore, AB5078P), rabbit anti-FUS (Bethyl Laboratories, IHC-00074), rabbit anti-TARDBP/TDP-43 (Proteintech, 10782-2-AP), goat anti-TIA1 (Santa Cruz Biotechnology, sc-1751), and mouse anti–HNRNPA2B1 (Santa Cruz Biotechnology, sc-53531). Next, 12-μm cross sections were prepared from fresh-frozen muscle and stained with modified Gomori trichrome stain [Citation46].
For histological studies, paraffin-embedded tissues were sectioned at 4 μm and mounted on positively charged glass slides (Thermo Fisher Scientific, Superfrost Plus). Primary antibodies against SQSTM1/p62 (Abcam, ab194720), ubiquitin (Enzo Biochem, BML-PW8810-0500), LC3B (Millipore, L7543), MHCf (Leica Microsystems, NCL-MHCf), MHCs (Leica Microsystems, NCL-MHCs), UQCRC (Abcam, ab14745) mt-CO1(Abcam, ab14705) and TARDBP/TDP-43 (Proteintech, 10782-2-AP) were used for immunohistochemical analyses. Antigen retrieval for all targets (except SQSTM1) required pretreatment with Cell Conditioning Solution 1 (Ventana Medical Systems, 950–500) for 32 min on the automated Discovery Ultra platform (Ventana-Roche). Antigen retrieval for SQSTM1 required pretreatment with the Cell Conditioning Solution 2 (Ventana Medical Systems, 760–107). Tissues were incubated with primary antibodies, and the OmniMap anti-rabbit HRP kit (Ventana Medical Systems, 760–4311) was used as the secondary antibody. Primary antibody binding was detected by the DISCOVERY ChromoMap DAB Kit (Ventana Medical Systems, 760–159). For ubiquitin, DISCOVERY Purple (Ventana Medical Systems, 760–229) was used as the chromogen, followed by hematoxylin counter-staining. A pathologist blinded to experimental groups analyzed the histology images. To quantify myofibers with centralized nuclei, SQSTM1+, and TARDBP+ aggregates, muscle sections were stained with CAV3, SQSTM1, TARDBP, and DAPI and imaged at 20 × . For each section, 4 × 4 fields were captured. For each animal, the mean value was obtained from 2 separate sections, and for each genotype, 3 animals were used. To quantify the area having SQSTM1+ deposits, images were masked with SlideBook 6.0 based on SQSTM1 staining. The average area was calculated from ≥50 SQSTM1+ objects for each genotype.
Immunoblot analyses
Quadriceps were lysed in RIPA buffer supplemented with protease inhibitor (Roche, 1836170001) and phosphatase inhibitor (Sigma Aldrich, P5726). Protein in cleared lysates was separated by electrophoresis on 4%–12% Bis-Tris gels (Life Technologies, NP0335BOX) and transferred to PVDF membranes. This was followed by blocking the membrane with 5% skim milk. Blots were probed with antibodies directed against ULK1 (Sigma Aldrich; A7481), SQSTM1 (Sigma Aldrich; P0067), LC3B (Novus Biologicals; NB100–2220), ATG13 (Sigma Aldrich; SAB4200100), RB1CC1 (Cell Signaling Technology; 12436), ATG14 (MBL; PD026), TARDBP/TDP-43 (Proteintech; 10782-2-AP), and GAPDH (Sigma Aldrich; G9545). Membranes were incubated with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, RPN4301, NA931), and bands were detected by chemiluminescence-detection kits (Amersham, RPN2232).
Electron microscopy
Mice were anesthetized with CO2 and transcardially perfused with phosphate buffer and then 2.5% glutaraldehyde, 2% PFA in 0.1 M CaCO4. Quadriceps were removed, fixed in the same fixative, and postfixed in 2% osmium tetroxide in 0.1 M sodium cacodylate buffer with 0.3% potassium ferrocyanide overnight. Samples were dehydrated using a series of graded ethanol-to-propylene oxide solutions, infiltrated and embedded in epoxy resin, and polymerized at 70°C overnight. Semithin (0.5-μm) sections were stained with toluidine blue for light microscopy studies. Ultrathin (80-nm) sections were cut and imaged using a FEI Tecnai F 20 TEM FEG electron microscope with an AT XR41 camera.
Muscle strength measurement
The force of forepaws and hindpaws was measured as previously described [Citation16]. Grip strength was measured as grams of force in 6 repeated measurements for forepaws and hindpaws of each animal by using a grip-strength meter (Bioseb). Mean muscle strength for each animal was calculated from 4 independent measurements, after excluding the maximal and minimal reads.
Blood chemistry
Blood samples were obtained by periorbital bleeding in anaesthetized mice. Serum M-type levels of chemicals (e.g., CKM/creatine kinase, glucose) were determined and analyzed by the Veterinary Pathology Laboratory Core at St. Jude Children’s Research Hospital.
Quantitative real-time PCR
Total RNA was isolated from the skeletal or cardiac muscles. The reverse transcription reaction was carried out using SuperScript IV VILO with ezDNase Enzyme (Invitrogen; 11766050). Taqman Gene expression assay containing FAM-labeled probes for Ppargc1a (Mm01208835_m1), Sqstm1 (Mm00448091_m1), Map1lc3 (Mm00458724_m1), Tardbp (Mm00523866_m1), Ulk1 (Mm00437236_m1), Ulk2 (Mm03048846_m1), Atg14 (Mm01322951_m1), Atg7 (Mm00512204_m1), and Gapdh (Mm99999915_g1) were purchased from Thermo Fisher. The probes for Rb1cc1 (APMF29D) were custom designed. Relative expression was normalized to Gapdh RNA and calibrated to the respective controls.
Analyses of mtDNA copy number
Total genomic DNA was prepared from the cardiac muscle tissues by using QIAamp DNA FFPA Tissue kit (QIAGEN; 56404). Samples were assayed using PowerUp SYBR green master mix (Applied Biosystem; A25741) and primers complementary to mt-Nd2 and Rn18s. Mitochondrial DNA content was quantified by log2 as the ratio of mitochondrial genome to nuclear genome.
Statistical analysis
All analyses were performed blinded to genotype. All quantitative data are shown as mean ± SEM from n ≥ 3 biological replicates, unless otherwise specified. Statistical significance was determined by two-tailed Student’s t-tests or ANOVA, and P < 0.05 was considered statistically significant.
Supplemental Material
Download Zip (47 MB)Acknowledgments
We are grateful to Dr. Doug Green (St. Jude) for providing the Atg14flox/flox mouse line, to Dr. Jun-Lin Guan (University of Cincinnati) for sharing the Rb1cc1flox/flox mouse line, and Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for the gift of Atg7flox/flox mouse line. We are also grateful to the staffs in the St. Jude Veterinary Pathology Core and Cell and Tissue Imaging Core for technical assistance and to Dr. Vani Shanker and Dr. Angela J McArthur for editing the manuscript.
Disclosure statement
The authors declare that they have no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109.
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–1416.
- Margeta M. Autophagy Defects in Skeletal Myopathies. Annu Rev Pathol. 2020;15:261–285.
- Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–1320.
- Kuno A, Hosoda R, Sebori R, et al. Resveratrol ameliorates mitophagy disturbance and improves cardiac pathophysiology of dystrophin-deficient mdx mice. Sci Rep. 2018;8:15555.
- Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515.
- Raben N, Hill V, Shea L, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908.
- Taneike M, Yamaguchi O, Nakai A, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606.
- Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624.
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362.
- Nemazanyy I, Blaauw B, Paolini C, et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med. 2013;5:870–890.
- Reifler A, Li X, Archambeau AJ, et al. Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am J Pathol. 2014;184:1819–1830.
- Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61:585–596.
- Wang C, Wang H, Zhang D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018;9:3492.
- Kraft C, Kijanska M, Kalie E, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. Embo J. 2012;31:3691–3703.
- Wang B, Maxwell BA, Joo JH, et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol Cell. 2019;74:742–57 e8.
- Majcher V, Goode A, James V, et al. Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci. 2015;66:43–52.
- Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27:491–504.
- Vogler TO, Wheeler JR, Nguyen ED, et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature. 2018;563:508–513.
- Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510.
- Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12:547–564.
- Bruning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569.
- Russell LK, Mansfield CM, Lehman JJ, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533.
- Hilton-Jones D, Brady S. Diagnostic criteria for inclusion body myositis. J Intern Med. 2016;280:52–62.
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–566.
- Bernard A, Klionsky DJ. Toward an understanding of autophagosome-lysosome fusion: the unsuspected role of ATG14. Autophagy. 2015;11:583–584.
- Taylor JP. Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology. 2015;85:658–660.
- Mirabella M, Alvarez RB, Bilak M, et al. Difference in expression of phosphorylated tau epitopes between sporadic inclusion-body myositis and hereditary inclusion-body myopathies. J Neuropathol Exp Neurol. 1996;55:774–786.
- Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008.
- Medeiros TC, Thomas RL, Ghillebert R, et al. Autophagy balances mtDNA synthesis and degradation by DNA polymerase POLG during starvation. J Cell Biol. 2018;217:1601–1611.
- Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol. 2017;45:47–54.
- Liang CC, Wang C, Peng X, et al. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499–3509.
- Ma D, Molusky MM, Song J, et al. Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol. 2013;27:1643–1654.
- Kimura H, Eguchi S, Sasaki J, et al. Vps34 regulates myofibril proteostasis to prevent hypertrophic cardiomyopathy. JCI Insight. 2017;2:e89462.
- Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J. 2016;473:2251–2271.
- Jaber N, Zong WX. Class III PI3K Vps34: essential roles in autophagy, endocytosis, and heart and liver function. Ann N Y Acad Sci. 2013;1280:48–51.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177:1682–1699.
- Buchan JR, Kolaitis RM, Taylor JP, et al. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–1474.
- Wang Z, Zhang H. Phase separation, transition, and autophagic degradation of proteins in development and pathogenesis. Trends Cell Biol. 2019;29:417–427.
- Scarlatti F, Maffei R, Beau I, et al. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318–1329.
- Mauthe M, Jacob A, Freiberger S, et al. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 2011;7:1448–1461.
- Goodwin JM, Dowdle WE, DeJesus R, et al. Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017;20:2341–2356.
- Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10:5.
- Joo JH, Wang B, Frankel E, et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62:982.
- Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396.
- Tanji K, Bonilla E. Light microscopic methods to visualize mitochondria on tissue sections. Methods. 2008;46:274–280.