
ABSTRACT
Macroautophagy/autophagy plays an important role during the development of human cancer. BECN1 (beclin 1), a core player in autophagy regulation, is downregulated in many kinds of malignancy. The underlying mechanism, however, has not been fully illuminated. Here, we found that CUL3 (cullin 3), an E3 ubiquitin ligase, could interact with BECN1 and promote the K48-linked ubiquitination and degradation of this protein; In addition, CUL3 led to a decrease in autophagic activity through downregulating BECN1. We also found that KLHL38 was a substrate adaptor of the CUL3 E3 ligase complex-mediated ubiquitination and degradation of BECN1. In breast and ovarian cancer, CUL3 could promote the proliferation of tumor cells, and the expression of CUL3 was related to poor prognosis in patients. Our study reveals the underlying mechanism of BECN1 ubiquitination and degradation that affects autophagic activity and subsequently leads to tumor progression, providing a novel therapeutic strategy that regulates autophagy to combat cancer.
Abbreviations: ATG: autophagy-related BECN1: beclin 1 CHX: cycloheximide CoIP: co-immunoprecipitation CUL3: cullin 3 IP: immunoprecipitation MS: mass spectrometry PtdIns3K: phosphatidylinositol 3-kinase UPS: ubiquitin-proteasome system
Introduction
Autophagy is an evolutionarily conserved degradation pathway through which damaged organelles or misfolded proteins are engulfed into a double-membrane structure and transported to the lysosome for degradation. The contents of autophagosomes are degraded into small molecules (such as nucleotides, amino acids and fatty acids) and pumped out of lysosomes to supply the cell [Citation1,Citation2]. Therefore, autophagy is an important inherent response by which cells cope with stress conditions, such as starvation, hypoxia and DNA damage. Autophagy is closely related with many other physiological and pathological processes, including cancer. The process of autophagy is strictly controlled by a series of genes called autophagy-related (ATG) genes to maintain cellular homeostasis [Citation3,Citation4].
BECN1 (beclin 1), the first tumor-associated ATG protein identified in mammals, is named for the homology domain of the BH3 only protein family. BECN1 forms one of the most important protein complexes in the autophagy pathway, which acts from the formation of autophagosomes to their extension and maturation [Citation5,Citation6]. The core factor of the BECN1 complex is PIK3C3/Vps34, a class III phosphatidylinositol 3-kinase (PtdIns3K) that facilitates the production of phosphatidylinositol-3–phosphate to recruit other factors to the autophagy membrane [Citation6]. Different factors of the BECN1 complex can modify the kinase activity of PIK3C3/Vps34 through interacting with BECN1 [Citation7]. ATG14 can promote production of autophagosomes through positive regulation of the BECN1 complex; however, the interaction between BCL2 and BECN1 can disassociate PIK3C3/Vps34 from the complex and block autophagy. Another factor, RUBCN (rubicon autophagy regulator), can suppress the maturation of autolysosomes by binding BECN1 [Citation5,Citation8]. Many cellular signals mediate autophagic activity due to direct regulation of BECN1 and its complex [Citation9–11]. It has been widely reported that BECN1 is heterozygously deleted at an elevated rate in prostate, ovarian and breast cancer samples. The BECN1 protein is expressed at lower level in tumor samples than in normal samples, while BECN1 deficiency could significantly promote proliferation and tumorigenesis in vitro and in vivo [Citation12-14]. These results indicate that BECN1, a tumor-suppressor gene that plays a central role in the autophagy pathway, is a prospective target for tumor therapy via/through autophagy regulation. Although heterozygously deletion can lead to low expression of BECN1 in many tumor tissues, low expression of BECN1 protein is not correlated with low BECN1 mRNA levels, based on our analyses of TCGA data. Other mechanisms should be involved in regulation of BECN1 protein expression.
In addition to the autophagy-lysosome pathway, the ubiquitin-proteasome system (UPS) is a degradative system in cells. Ubiquitin was identified as the common degron adopted by both degradative systems. Ubiquitin is a small protein containing seven lysine sites, so targeted protein can undergo at least seven kinds of polyubiquitination. K11- and K48-linked polyubiquitination mediates the degradation of proteins through the proteasome, while K63-linked polyubiquitination targets substrates for autophagy degradation and participates in signal transduction and protein translocation [Citation15].
The process of ubiquitination involves a cascade of three enzymes, the third of which is an E3 ligase that transfers the ubiquitin from an E2 ubiquitin conjugating enzyme to specific substrates. Thus, it is the E3 ubiquitin ligases that’s the most important one of these three enzymes as it determines substrate specificity and variety [Citation16]. Due to the specificity of E3 ligases, UPS inhibition could be a good strategy for targeted cancer therapy [Citation17,Citation18].
Comprising more than 200 multi-subunit E3 ubiquitin ligase complexes assembled from the eight different scaffold proteins in the cullin proteins family, the cullin-RING family is the largest family of E3 ligases in mammals. As a member of the cullin-RING family, CUL3 (cullin 3) E3 ligase complex participates in the ubiquitination of many other proteins. Among the three components of the CUL3 complex, RBX1 is responsible for the interaction of CUL3 and E2, and the core member of the complex, CUL3, connects with E2 and the substrate to facilitate the transfer of ubiquitin from E2 to the target protein. Since CUL3 cannot interact with substrate proteins directly, a series of proteins containing kelch and BTB domains are required for the recognition and recruitment of substrate proteins [Citation19,Citation20]. More than 50 kinds of kelch domain- or BTB domain-containing proteins have been discovered to target hundreds of protein substrates for ubiquitination by the CUL3 complex in mammalian cells [Citation21].
In our study, we found that the E3 ligase CUL3-KLHL38 interacted with BECN1 and mediated its K48-linked ubiquitination, which resulted in degradation of BECN1 via the proteasome. CUL3 was highly expressed in both breast cancer and ovarian cancer samples, and its expression was related to poor prognosis in patients. High expression of CUL3-KLHL38 inhibited autophagic flux and promoted malignant behavior in breast cancer due to the degradation of BECN1. MLN4924, a neddylation inhibitor of Cullin-Ring E3 ligases, dramatically reduced the proliferation of tumor cells, especially CUL3-overexpressing cells, which showed that BECN1 degradation mediated by CUL3 could be an effective therapeutic target for breast cancer.
Results
BECN1 protein level is regulated through the ubiquitin-proteasome pathway
Based on sequencing data from The Cancer Genome Atlas, we found little overlap between samples from both ovarian cancer and breast cancer patients with low BECN1 protein level and those with low BECN1 mRNA level (). The protein level of BECN1 was not correlated with its mRNA expression or gene copy number (). Therefore, we hypothesized that posttranslational modifications play a role in BECN1 protein degradation. In mammalian cells, proteins can be degraded via either the ubiquitin-proteosome system (UPS) or the autophagy pathway. Using inhibitors of these different protein degradation pathways, we found that the proteasome inhibitor MG132 could increase the BECN1 protein level in HeLa cells, while the lysosome inhibitor bafilomycin A1 (BafA1) could not (). The protein level of BECN1 was also elevated in MDA-MB-231 cells following MG132 treatment in a dose-dependent manner (), indicating that BECN1 is degraded through the UPS.
Figure 1. CUL3 interacts with BECN1. (A) BECN1 protein or mRNA downregulation overlap in different tumor samples. (B) The low expression of BECN1 was not due to the transfectional repression or gene copy number deletion. (C) HeLa cells were treated with the indicated inhibitors for 24 h and immunoblotting was performed. (D) 231 cells were treated with indicated concentration of MG132 for 24 h and the protein extracts was subjected to immunoblotting. (E) 293 t cells transfected with plasmids encoding Flag-BECN1, treated with 10 μM MG132 for 6 h, and the BECN1 immunoprecipitation were subjected to SDS-PAGE and then analyzed by mass spectrum. The peptide fragment of CUL3 was shown. (F) 293 t cells were transfected with indicated plasmids, the immunoprecipitation was performed by using Flag affinity gels or anti-MYC-tag antibody, and then bloted for the indicated proteins. (G) The illustration of BECN1 deletion mutants. (H) The binding site of BECN1 and CUL3 is near BH3 domain. 293 t cells were transfected with indicated plasmids, the immunoprecipitation of Flag affinity gels was subjected to immunoblotting assay
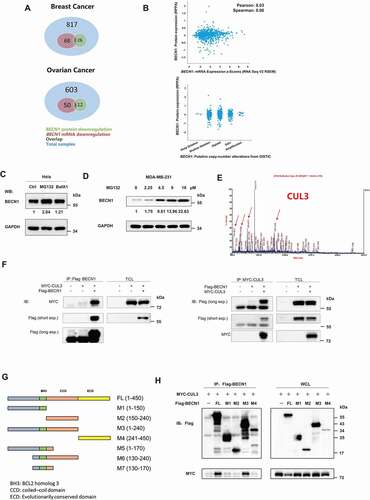
Mass spectrometry (MS) analyses was used to identify proteins that might participate in degradation of the BECN1 protein through the UPS. HEK293T cells transfected with Flag-tagged BECN1 were treated with MG-132. Then, BECN1 protein was isolated through immunoprecipitation (IP) and analyzed by MS. CUL3, an E3 ligase in the Cullin-RING family, could bind BECN1 (). Coimmunoprecipitation (CoIP) of Flag-tagged BECN1 and MYC-tagged CUL3 validated their interaction (). A complex of endogenous BECN1-CUL3 (Fig. S1A and S1B) was also detected in the MDA-MB-231 cell line, which indicated that the binding between CUL3 and BECN1 is not an artifact of overexpression.
To map the CUL3-binding site on BECN1, cells expressing one of four BECN1 deletion mutants and ectopic MYC–CUL3 expression were used for the CoIP assays. The M1 fragment (AAs 1–150) is not evolutionarily conserved. The M2 fragment (AAs 150-240) included a coiled-coil domain that has been reported to interact with ATG14 and UVRAG. In addition to the first 150 nonconserved amino acids and the coiled–coil domain, the BH3 domain between the M1 fragment and M2 fragment was included in the M3 fragment (AAs 1–240). The M4 fragment (AAs 241-450) contained an evolutionarily conserved domain that has been reported to bind PIK3C3/VPS34 (). CoIP assays showed that only the M3 fragment had the same affinity to CUL3 as the full-length BECN1 protein, suggesting that the BH3 domain may mediate the interaction of BECN1 with CUL3 (). To further confirm that the interaction between BECN1 and CUL3 is via BH3 domain, the GFP-conjugated BH3 domain of BECN1 protein (GFP-BH3) and the BECN1 mutant with the BH3 domain deleted (dBH3) was cloned into the pcDNA3.1B plasmid and transfected into the HEK293T cells. Subsequent CoIP assays detected that the BH3 domain alone had the same affinity to CUL3 as the full-length BECN1 protein (Fig. S1C), while the BECN1 mutant with the BH3 domain deleted showed extremely weak affinity to CUL3 compared with the full-length BECN1 protein (Fig. S1D). Thus, we speculated that the BH3 domain alone was sufficient to mediate the interaction between BECN1 and CUL3. Taken together, these results suggested that CUL3 interacts with BECN1 through the BH3 domain and very likely participates in the proteasomal degradation of BECN1.
CUL3 mediates the degradation of BECN1 through the ubiquitin-proteasome pathway
To further explore the regulatory effect of CUL3 on the BECN1 protein, several different breast cancer cell lines were collected, and the expression of CUL3 and BECN1 was detected. A negative correlation between the protein expression of BECN1 and CUL3 was found based on the extracts of different breast cancer cell lines (). However, there was no correlation between their mRNA expression levels (). Moreover, siRNA to knock down CUL3 and plasmid to overexpress CUL3 were designed. Knockdown of CUL3 using siRNA in MDA-MB-231 cells increased level of the BECN1 protein, while CUL3 overexpression decreased BECN1 protein level (). However, the mRNA expression of BECN1 was not significantly altered when CUL3 was knocked down (). Therefore, we speculated that CUL3 mediates the degradation of BECN1 rather than altering its transcription. Cycloheximide (CHX) has been widely used to measure the half-lives of specific proteins within cells because it can block the biosynthesis of proteins without selectivity. After treating HeLa cells with CHX, we found that the knockdown of CUL3 with siRNA prolonged the half-life of the BECN1 protein, further validating our hypothesis (). The proteasome inhibitor MG132 rescued the degradation of BECN1 mediated by CUL3 overexpression, but the lysosome inhibitor BafA1 did not, further confirming that the degradation of BECN1 mediated by CUL3 occurs via the UPS ().
Figure 2. CUL3 attenuates proteasomal degradation of BECN1. (A) The protein lysates of the indicated cell lines were analyzed by western blot by using anti-BECN1 or CUL3. (B) The RNA of the indicated cell lines was extracted and the expression of BECN1 and CUL3 were analyzed by qRT-PCR. (C) CUL3 affects the protein expression of BECN1. MDA-MB-231 cells were transfected with siRNA targeting CUL3, or plasmids encoding CUL3, for 48 h. The protein extracts were subjected to the immunoblotting with indicated antibodies. (D) Knockdown of CUL3 does not affect the transcription of BECN1. MDA-MB-231 cells were transfected with siRNA targeting CUL3, and the expression of BECN1 or CUL3 was detected by qRT-PCR. (E) CUL3 regulates the degradation of BECN1 protein. Hela cells transfected with CUL3 siRNA were treated with 20 μM CHX for the indicated time, and the protein extracts were subjected to the immunoblotting assay. (F) CUL3 promotes the proteasomal degradation of BECN1. MDA-MB-231 cells transient overexpressing with MYC-CUL3 were treated with 100 nM BafA1 or 10 μM MG132 for 6 h, and then assayed for immunoblotting. CUL3 ubiquitin ligase inhibits autophagy. (G) MDA-MB-231 cells were transfected with siRNA targeting CUL3 for 48 h. Cells were treated with BafA1 for 6 h and the cell lysates were detected by immunoblot. (H) Hela cells overexpressing EYFP-LC3 were transfected with the indicated siRNAs for 48 h, and then be fixed and subjected to immunofluorescence assay. Scale bars: 10 μm. (I) MDA-MB-231 cells were transfected with the indicated siRNA for 48 h. Cell lysates were subjected to immunoblotting assay. (J) CUL3 does not affect the formation of BECN1 complex. 293 t cells transfected with siRNA targeting CUL3 and the immunoprecipitation was performed by using anti-BECN1 antibody. The formation of BECN1 complex was analyzed by western blot using the indicated antibodies
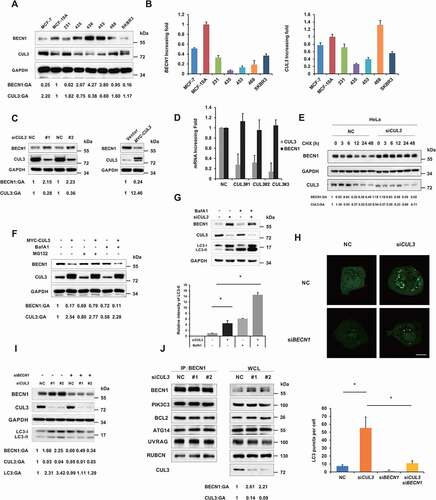
BECN1, one of the most important ATG, and its complex function from the formation of autophagosomes to their maturation. Since CUL3 could mediate the proteasomal degradation of BECN1, we wondered whether it would have an effect on the activity of the autophagy process. LC3 is a characteristic molecular marker of autophagosomes. During the formation of autophagosomes, LC3 is cleaved at its C terminus to form LC3-I in the cytoplasm, and LC3-I then binds phosphatidylethanolamine, which attaches LC3-I to the phagophore membrane as the LC3-II form. After CUL3 was knocked down with siRNA in HeLa cells, the accumulation of LC3-II, which can be explained by either increased or blocked autophagic flux within cells, was detected. After treatment with the inhibitor BafA1, which inhibits the H+ pump on the lysosomal membrane, the accumulation of LC3-II was further increased, indicating that autophagic flux was enhanced rather than blocked when CUL3 was knocked down within the cells (). To further verify this result, an immunofluorescence assay was adopted to observe the formation of LC3 punctate foci. When autophagy occurs, LC3 accumulates on the autophagosome membrane, forming LC3 punctate foci that can indicate the autophagy level more intuitively. Consistent with our above results, knock down of CUL3 with siRNA greatly increased the formation of LC3-II puncta labeled with green fluorescence (). These results demonstrated that CUL3 could inhibit the formation of autophagosomes and thereby block autophagic flux.
To prove that the increased autophagic flux induced by CUL3 knockdown was due to elevated BECN1 protein level, we knocked down BECN1 at the same time. As expected, the increase in the accumulation of LC3-II was reversed, and the formation of LC3-II punctate foci was also decreased when BECN1 was knocked down ). During the early stage of autophagy, BECN1 binds PIK3C3/Vps34, ATG14, and UVRAG, and the resultant complex promotes the autophagy process. In contrast, BCL2 and RUBCN can bind BECN1 as negative factors that block autophagic flux. Therefore, we wondered whether CUL3 could also alter the function of the BECN1 complex in addition to promoting degradation of the BECN1 protein. HEK293T cells were co-transfected with Flag-tagged BECN1 and siRNA against CUL3. CoIP assays of cells expressing Flag-tagged BECN1 demonstrated that CUL3 knockdown had no effect on the binding between BECN1 and PIK3C3/Vps34, ATG14, UVRAG, BCL2, or RUBCN (). Therefore, CUL3 mediated the degradation of BECN1 and thereby blocked autophagic flux without affecting formation of the BECN1 complex.
CUL3 promoted the K48-linked ubiquitination of BECN1
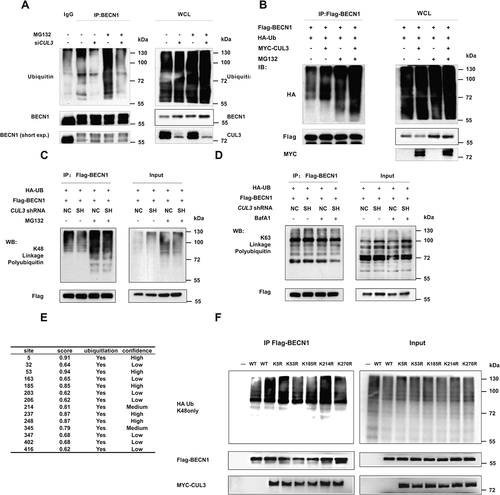
Then, we wanted to further elucidate the mechanism by which CUL3 mediates BECN1 degradation. Ubiquitination is a signal for protein degradation via the proteasome pathway in mammalian cells, and CUL3 is an E3 ligase of the Cullin-RING family. Therefore, we speculated that CUL3 can mediate the ubiquitination of BECN1. IP assays were performed to examine ubiquitination of the BECN1 protein. The ubiquitination of endogenous BECN1 decreased and the BECN1 protein accumulated when CUL3 was knocked down (), while the ubiquitination of exogenous BECN1 increased and the BECN1 protein decreased when CUL3 was overexpressed (). Many different kinds of ubiquitin chains with different biological functions have been observed in mammalian cells. Among them, K29- and K48-linked polyubiquitination lead to proteasome-mediated degradation of substrate proteins, while substrate proteins modified via K63-linked polyubiquitination are recognized by specific regulatory factors, triggering the corresponding signaling pathways. Specific anti-K63-linkage or anti-K48-linkage polyubiquitin antibodies were used to identify the kind of ubiquitin chain that leads to BECN1 degradation. BECN1 showed basal level of K48-linked polyubiquitination instead of K63-linked polyubiquitination. Meanwhile, K48-linked polyubiquitination was attenuated in cells with stable silencing of CUL3 and accumulated after treatment with MG132, while K63-linked polyubiquitination was not (). These findings indicated that CUL3 mediates the degradation of BECN1 through K48-linked ubiquitination. Then, we sought to determine which lysine residues of BECN1 are the major ubiquitination sites that result in BECN1 degradation. Among the 14 lysine residues in the human BECN1 protein, 7 might be ubiquitinated, as predicted by Ubipred (http://www.ubpred.org/) with high or medium confidence (). Thus, 7 Flag-tagged BECN1 mutants in which the corresponding lysine residues were individually mutated to arginine were overexpressed in HEK293T cells with MYC-tagged CUL3 and an HA-tagged only K48-linked ubiquitin mutant (only able to form K48-linked chains). Subsequent IP assays demonstrated that the ubiquitination of BECN1 K53R, K185R and K270R was decreased, suggesting that these three lysine residues are essential for the K48-linked ubiquitination of BECN1 mediated by CUL3 (). To further confirm these 3 ubiquitination sites of BECN1, BECN1 mutants with all the 3 lysines above mutated was constructed. Subsequent IP and WB assays demonstrated that the K48-linked ubiquitination level of BECN1 mutant with all the 3 lysines above mutated was significantly lower than the wild type and the degradation of BECN1 mediated by CUL3 was significantly attenuated with the 3KR mutant (Fig. S2A and S2B). Collectively, these results revealed that CUL3 promotes the K48-linked ubiquitination of BECN1 at residues K53, K185, and K270, which targets BECN1 for degradation via the UPS.
Figure 3. CUL3 promotes K48-linked ubiquitination of BECN1. (A) The ubiquitination of BECN1 decreased with CUL3 deletion. 293 t cells were transient transfected with siRNA targeting CUL3, incubated with 10 μM MG132 for 6 h, and then anti-BECN1 immunoprecipitation and immunoblotting was performed. (B) CUL3 increases BECN1 ubiquitination. 293 t cells were transfected with HA-ubiquitin, MYC-CUL3 and Flag-BECN1, and treated with 10 μM MG132 for 6 h. The immunoprecipitation and immunoblotting was performed by using the indicated antibodies. (C,D) The ubiquitination of BECN1 is K48-linked. 293 t cells were transfected with indicated plasmids, and immunoprecipitation with Flag affinity gels and immunoblotting were performed. (E) The prediction of BECN1 ubiquitination site by using Ubpred. (F) 293 t cells were transfected with HA-ubiquitin and different mutant BECN1 plasmids, and the immunoprecipitation with Flag affinity gels and immunoblotting were performed
KLHL38 is an adaptor of CUL3-mediated ubiquitination and degradation of BECN1
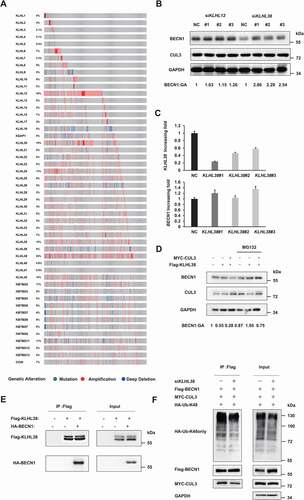
Similar to other E3 ligases of the Cullin-Ring family, CUL3 cannot interact with its substrate directly. Kelch-like (KLHL) proteins, a group of proteins with a BTB domain, is required for the recognition between CUL3 and its substrate. At least 42 different KLHL proteins with similar structures have been discovered in mammals. The arrangement of the Kelch domain in the N terminus differs among KLHL proteins, corresponding to their different substrates. Based on a transcriptome sequencing project from TCGA, KLHL12 and KLHL38 were overexpressed in triple-negative breast cancer samples (). We found that the BECN1 protein significantly accumulated only when KLHL38 was knocked down (). Real-time PCR assays confirmed KLHL38 silencing, and the mRNA level of BECN1 was not affected by KLHL38 knockdown in the cells (). Consistently, the BECN1 protein was significantly decreased after either KLHL38 or CUL3 was overexpressed, which could be enhanced if both KLHL38 and CUL3 was overexpressed at the same time (Fig. S3A). The proteasome inhibitor MG132 reversed the decrease in BECN1 protein mediated by KLHL38 and CUL3, suggesting that KLHL38 and CUL3 act synergistically to promote the degradation of BECN1 through the proteasome (). Moreover, the endogenous and exogenous CoIP assays confirmed that KLHL38 could interact with BECN1 and CUL3 (, S1a and S1B). In addition, silencing KLHL38 impaired the K48-linked polyubiquitination of BECN1 mediated by CUL3 in vivo (). In vitro ubiquitination assays were also performed to further prove that KLHL38 regulate BECN1 ubiquitination directly. Flag-tagged KLHL38 was overexpressed in HEK293T cells altogether with MYC-tagged CUL3 and RBX1. The CUL3-KLHL38-RBX1 complex was affinity-purified using Flag affinity gel and then incubated with purified, baculoviral expressed GST-tagged BECN1 protein in the in vitro ubiquitination reaction. And WB assays were adopted to detect the ubiquitination of the BECN1 protein subsequently. The ubiquitination level of the BECN1 protein significantly increased after incubated with the affinity-purified CUL3-KLHL38-RBX1 complex in vitro (Fig. S3B), suggesting that KLHL38 participates in the recognition between CUL3 and BECN1 to regulate the ubiquitination of BECN1 directly.
Figure 4. KLHL38 is the adaptor that targets BECN1 for ubiquitination and degradation. (A) The expression of CUL3 adaptors in breast cancer tissue compared to normal tissue. (B) KLHL38 affects the expression of BECN1. MDA-MB-231 cells were transfected with indicated siRNAs, and the expression of BECN1 is determined by immunoblotting. (C) The effect of KLHL38 siRNAs on the transcription of BECN1 was determined by qRT-PCR. Data are represented as the mean ± SD. (D) CUL3 complex promotes the proteasomal degradation of BECN1. MDA-MB-231 cells were transfected with indicated plasmids, treated with MG132 for 6 h, and the expression of BECN1 is determined by immunoblotting. (E) KLHL38 interacts with BECN1. 293 t cells overexpressing KLHL38 and BECN1 were treated with 10 μM MG132 for 6 h, and the immunoprecipitation was performed. (F) Knockdown of KLHL38 affects BECN1 ubiquitination. 293 t cells were transfected with the indicated plasmids or siRNAs and the immunoprecipitation was performed
CUL3-KLHL38 promotes cell proliferation in breast cancer both in vitro and in vivo
Given that BECN1 is widely recognized as a tumor-suppressor gene with high deletion and mutation rates in many kinds of carcinomas, we next examined the effect of BECN1 degradation mediated by CUL3 on cell proliferation and tumor growth. Compared with the negative control, MDA-MB-231 cells and HeLa cells in which CUL3 was silenced showed a significantly decreased proliferation rate, while MDA-MB-231 cells overexpressing CUL3 showed increased cell proliferation (). Consistent with the results above, compared with the negative control, the cells in which CUL3 was knocked down showed significantly decreased colony-forming ability, while the cells overexpressing CUL3 showed enhanced colony formation (). To explore whether CUL3 promotes the proliferation of MDA-MB-231 cells through altering the cell cycle or affecting apoptosis, after CUL3 was knocked down with siRNA, the distribution of cells in three major phases of the cell cycle (G1, S and G2/M) and the apoptosis of the MDA-MB-231 cells were analyzed with flow cytometry. After CUL3 was knocked down, the apoptosis of the cells was not affected, but the proportion of cells in G1 phase of the cell cycle decreased, while the proportion of cells in S phase increased (Fig. S4A and S4B). Given that cells in which CUL3 was silenced showed significantly decreased proliferation rate and significantly decreased colony-forming ability, we speculated that the cell division may be blocked in S phase of the cell cycle after CUL3 was silenced. Furthermore, the proliferation of MDA-MB-231 cells was inhibited after treatment with MLN4924, a Cullin-Ring inhibitor, especially when CUL3 was overexpressed (), suggesting that CUL3 is a potential drug target in breast cancer, especially for patients with high CUL3 expression. Notably, the proliferation and colony formation of MDA-MB-231 cells were also impaired when KLHL38 was silenced, indicating the coordinated effect of CUL3 and KLHL38 (). All these results indicated that CUL3-KLHL38 deficiency decreased breast cancer malignant behavior in vitro.
Figure 5. CUL3 mediated tumorigenesis through BECN1 degradation. (A), (B) Hela or 231 cells were transfected with siRNAs targeting CUL3 for the indicated days. Cell viability was measured by MTT assay. (C) 231 cells are transfected with siRNA targeting KLHL38 for the indicated days. Cell viability is measured by MTT assay. (D-F,H), Cells were transfected with indicated siRNAs or plasmids and the clone formation assay was performed. Cells were fixed and stained by crystal violet. (G) 231 cells were transfected with plasmids encoding MYC-CUL3 for the indicated days. Cell viability was measured by MTT assay. (I) left, 231 cells stable transfected with shRNA targeting CUL3 were treated with the indicated amount of CQ. Cell viability was measured by MTT assay. right, 231 cells overexpressing CUL3 were treated with indicated amount of MLN4924 for 24 h. Cell viability was measured by MTT assay
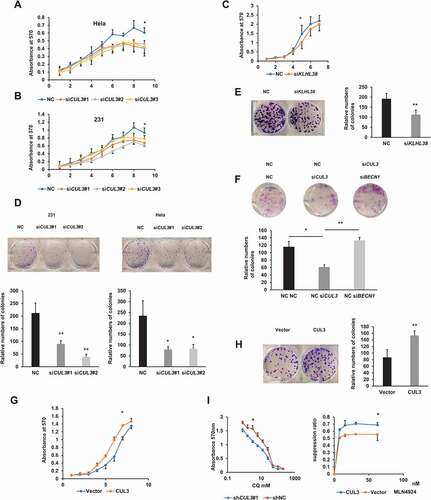
Next, we investigated whether the effect of CUL3 in promoting tumor proliferation was due to the degradation of BECN1 and blockade of autophagic flux. First, BECN1 and CUL3 were knocked down at the same time. As expected, the impairment of colon-formation ability caused by CUL3 silencing was rescued (). Then, MDA-MB-231 cells in which CUL3 was stably silenced by shRNA and negative control cells were treated with CQ, an autophagy inhibitor. Cell proliferation was inhibited when CUL3 was silenced and rescued after treatment with CQ (). To block the autophagy process more specifically, ATG5, a key protein involved in the extension of the phagophore membrane in autophagic vesicles, was knocked out using CRISPR-Cas9. Based on the ATG5 deficient cells, CUL3 was knocked down to further explore if the biological function of CUL3 was through the suppression of autophagy flux. In vitro colony-formation assays demonstrated that the impaired colony-formation by silencing CUL3 was rescued in ATG5 deficient cells (Fig. S5A). Altogether, we concluded that CUL3 and KLHL38 acted synergistically to promote cell proliferation in breast cancer due to the degradation of BECN1 and blockade of autophagic flux.
To further explore the involvement of autophagy pathway in CUL3 mediated tumor progression in vivo, the tumorigenicity model of nude mice was constructed. Consistent with the results of in vitro assays, the tumorigenic ability of MDA-MB-231 cells in vivo was significantly attenuated after CUL3 was stably knocked down, which was rescued if BECN1 was knocked down or ATG5 was knocked out at the same time. Thus, xenograft animal model of nude mice further confirmed that CUL3 promotes the tumor proliferation through degradation of BECN1 and subsequent blockade of autophagic flux (, I, J S5B and S5C).
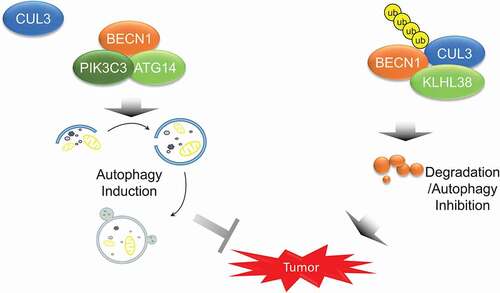
Figure 6. The model that CUL3-KLHL38-mediated BECN1 degradation inhibits autophagy and promotes tumor progression
CUL3 is aberrantly expressed in cancer and predicts poor patient prognosis
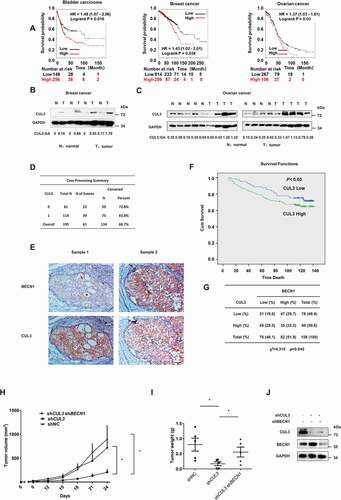
Since CUL3-KLHL38 has been observed to promote proliferation through the degradation of BECN1 in vitro, we next sought clinical evidence in support of this finding. Immunoblotting demonstrated higher CUL3 protein expression in tumor samples than in paired normal samples from breast cancer patients (). A similar phenomenon was also observed in nonpaired samples from ovarian cancer patients (). In addition, Kaplan-Meier plotter was used to assess the relationship between CUL3 protein expression and the clinical outcomes of patients with breast cancer, ovarian cancer and bladder cancer. High CUL3 expression was identified as a negative prognostic factor in patients with these cancers (). These data were corroborated by our own immunohistochemical analyses from 195 human breast cancer specimens, which showed that high CUL3 expression may affect neoplastic malignancies and clinical outcomes (). We also sought to determine the clinical relevance of CUL3-mediated regulation of the BECN1 protein. Immunohistochemical analyses of 158 human breast cancer specimens was performed to further consolidate the association between CUL3 and BECN1 expression, which demonstrated a statistically significant inverse correlation between BECN1 and CUL3 protein level (). In summary, high CUL3 expression appeared to be a predictor of poor patient prognosis in several cancers, which is consistent with the proliferation-promoting effect of CUL3-KLHL38 in vitro.
Discussion
Our research revealed that the E3 ligase CUL3 can interact with BECN1, a widely recognized tumor-suppressor gene, mediating its K48-linked ubiquitination at the sites K53, K185, and K270 and subsequent proteasomal degradation. The Kelch-BTB domain protein KLHL38 is required for the recognition and interaction of CUL3 and BECN1 in breast cancer cells. Through the degradation of BECN1, CUL3-KLHL38 blocks autophagic flux, promotes cell proliferation and predicts poor prognosis in breast cancer. The proliferation of breast cancer cells was impaired by a CUL3 inhibitor and rescued by an autophagy inhibitor ().
Figure 7. CUL3 mediated BECN1 degradation is relevant both in patients and in tumorigenicity model of nude mice. (A) Online analyses of survival in Bladder carcinoma, Breast cancer or Ovarian cancer patients with high or low CUL3 expression. The number of surviving patients at different time points is indicated below the graphs. HR, hazard ratio. (B,C) Immunoblotting analyses of CUL3 protein expression in tumor samples and normal samples of breast cancer or ovarian cancer patients. (D,F) Expression of CUL3 of breast cancer patients correlates with poor overall survival. Scale bars: 50 μm. (E) Immunohistochemical analyses of 158 specimens from breast cancer patients using anti-BECN1 and anti-CUL3 antibodies were performed. Representative images of IHC staining of tumors form two human breast cancer patients are presented. (G) Correlation study of BECN1 and CUL3 expression in the breast cancer consisting of 158 samples. (H,I,J) The tumorigenicity model of nude mice was constructed with the indicated cells, and then the presence or absence of a visible or palpable tumor was evaluated and tumor growth was monitored every 3 d (H). After the tumor volume exceeded 800 mm3, the mice were sacrificed, and the tumors were excised to determine the weight of them (I). The expression of BECN1 or CUL3 in the MDA-MB-231 cells subcutaneously inoculated into nude mice was detected by western blot analyses (J)
BECN1 associates with PIK3C3/VPS34 and ATG14 to form a complex during the formation, extension and maturation of autophagosomes. Posttranslational modifications of BECN1 and its complex are crucial for autophagy control and regulation. Phosphorylation is the most intensively studied modification of BECN1 in the autophagy process. Autophagy triggered by nutrient starvation relies on the phosphorylation of BECN1 by ULK1 and AMPK and subsequent activation of the PIK3C3/VPS34 kinase complex. Upon starvation, especially under conditions of amino acid depletion, MTORC1 is inactivated, relieving its inhibitory effect on ULK1 [Citation22]. Then, ULK1 phosphorylates BECN1 at S15 to activate PIK3C3/VPS34 lipid kinase and trigger autophagy [Citation23]. Meanwhile, AMPK, an energy sensor kinase activated by glucose depletion and low-energy states, directly phosphorylates BECN1 at S90 and S93, also contributing to the activation of the PtdIns3K complex [Citation24]. In addition to activating the PtdIns3K complex, phosphorylation alters the interactome of BECN1 to inhibit autophagy. Phosphorylation by MST1, EGFR and AKT promotes formation of the autophagy-inhibitory BECN1 complex [Citation9,Citation25,Citation26]. In addition to phosphorylation, other posttranslational modifications of BECN1, such as its ubiquitination and acetylation, have been reported. Proautophagic K63-linked polyubiquitination of the BECN1 protein is mediated by TRAF6, TRIM16 and TRIM50 and eliminated by the deubiquitinating enzyme A20 [Citation27,Citation28]. The acetyltransferase EP300/p300 mediates the acetylation of BECN1 at K430 and K437 to block autophagosome maturation, while SIRT1 deacetylates the BECN1 protein to eliminate it [Citation29]. Furthermore, dynamic interplay between the phosphorylation and other posttranslational modifications of the BECN1 protein has been discovered. Phosphorylation of BECN1 at S90 mediated by CAMK2/CaMKII promotes the K63-linked ubiquitination of BECN1 and activation of autophagy [Citation30]. In addition, it was reported that the acetylation of BECN1 mediated by EP300 is dependent on its phosphorylation at S409 mediated by CK1 [Citation29].
Here, we identified the K48-linked polyubiquitination of BECN1 protein mediated by CUL3, which was also mentioned by Liu et al. in their research [Citation31]. Instead of affecting the formation and function of the BECN1 complex, it facilitated the proteasomal degradation of BECN1 and thereby blocked autophagic flux, contrary to the proautophagic K63-linked polyubiquitination of BECN1. While Liu et al. only detected the ubiquitination and protein level of BECN1 with KLHL20 manipulated, our study provided more direct evidence of the CUL3 mediated ubiquitination and degradation of BECN1 protein through manipulating CUL3 directly. Besides, its autophagy regulatory function and proliferation-promoting effect in breast cancer cells was also explored in detail. These findings provide novel insights into the modification of BECN1 and its role in autophagy control and regulation.
The UPS and autophagy-lysosome pathway are two major degradative systems adopted by eukaryotic cells to eliminate misfolded proteins and damaged organelles. Although these two distinct systems have been viewed as independent for a long time due to the great differences in their nature, mode of action and core machinery, emerging research has revealed their crosstalk and interplay for the maintenance of cellular homeostasis [Citation32].
Ubiquitin was identified as the common degron that targets substrates for degradation via both systems, but the exact types of ubiquitination recognized by these systems appeared to be different in early work [Citation33]. While K48-linked polyubiquitination was thought to be recognized by the UPS [Citation34,Citation35], and substrates destined for autophagic degradation were either modified by K63–linked chains or just monoubiquitinated [Citation36,Citation37]. However, some overlap was detected because of the incomplete specificity of the different adaptor molecules linking ubiquitylated cargoes and the machinery of these two degradative systems. Thus, the UPS and autophagy seem to cooperate closely with each other to eliminate unwanted waste modified by various types of ubiquitination.
Moreover, perturbations in the activity of the UPS have been reported to affect autophagic flux and vice versa. With impairment of the UPS, autophagy was observed to be compensatorily activated to relieve the accumulation of ubiquitinated substrates [Citation38,Citation39]. Although the catalytic activity of UPS was not affected while autophagy was inhibited, accumulated SQSTM1/p62 could form protein aggregates with ubiquitinated substrates to delay their delivery for proteasomal degradation [Citation40-42]. In addition, aberrant SQSTM1-containing aggregates might also recruit and functionally inactivate regulators of the UPS, such as VCP/p97, to potentiate the widespread disruption of proteasomal flux [Citation32,Citation43].
In addition, the major components of both systems have been reported to be degraded through the other system. Proteasomes can be polyubiquitinated and selectively degraded through autophagy, which is called proteaphagy [Citation44,Citation45]. In turn, some ATG and other proteins related to autophagy, such as LC3B, MTORC1, etc, are destroyed by the UPS to block autophagic flux [Citation46].
As the largest family of E3 ligases in mammalian, Cullin-RING family and its role in the ubiquitination and degradation of proteins related to autophagy and subsequent alteration in the autophagy activity is attracting extensive attention [Citation47,Citation48]. AMBRA1, a protein functions in the early stage of autophagy by favoring formation of the autophagosome core complex, was reported to be ubiquitinated by CUL4 and released from CUL4 in a ULK1-dependent manner during the induction of autophagy. The accumulated AMBRA1 not only promotes the autophagy directly, but also binds and inhibits CUL5 to promote the accumulation of the MTOR inhibitor DEPTOR and establishes a positive feedback loop that ensures the rapid onset of autophagy [Citation49]. Besides CUL5, DEPTOR was also reported to be ubiquitinated by CUL1-FBXW11/β-TRCP2 after phosphorylation by the activated MTOR [Citation50]. Additionally, DEPDC5 subunit of GATOR1, another MTOR inhibitor, is ubiquitinated by CUL3-KLHL22 to downregulate autophagy upon amino acid stimulation [Citation51]. CUL3 was also found to mediate the ubiquitination and degradation of ULK1, and components of PIK3C3/VPS34 complex including BECN1 with KLHL20 as the adaptor during prolonged starvation to avoid overactivation of autophagy [Citation31]. And BECN1 is also ubiquitinated by CUL1-SKP2 to attenuate autophagy. While the ubiquitination mentioned above mainly regulates the abundance of the proteins related to autophagy, the ubiquitination of SQSTM1 mediated by CUL3-KEAP1 enhances its sequestering activity and autophagic degradation [Citation52].
In our study, CUL3-KLHL38 was observed to mediate the K48-linked polyubiquitination and subsequent proteasomal degradation of BECN1, which blocked autophagic flux and promoted cell proliferation in breast cancer. Clinical data also showed that CUL3 was aberrantly expressed in cancer and predicted poor patient prognosis. Silencing CUL3 with siRNA rescued the impaired autophagy and abolished the proliferation-promoting effect of CUL3 both in vitro and in vivo. Although the CUL3-KLHL20-mediated ubiquitination of BECN1 has been reported previously, neither the expression of KLHL20 nor the interaction between endogenous KLHL20 and endogenous BECN1 was detected in breast cancer (Fig. S6A, S6C and S1A). Moreover, the proliferation of the breast cancer cells was not affected after KLHL20 was overexpressed (Fig. S6A, S6B). Thus, KLHL38 might act alternatively to KLHL20 as the substrate adaptor for the CUL3 mediated ubiquitination of BECN1 in breast cancer. While the ubiquitination of BECN1 mediated by CUL3-KLHL20 is mainly responsible for autophagy termination during prolonged starvation and impairment of this autophagy termination mechanism potentiates starvation-induced cell death and aggravates diabetes-associated muscle atrophy, CUL3-KLHL38 mediated the ubiquitination of BECN1 to block the autophagy in normal conditions and enhanced the cell proliferation subsequently in breast cancer.
In conclusion, our research provided more direct evidence of the K48-linked polyubiquitination of the BECN1 protein mediated by CUL3 with KLHL38 as the substrate adaptor. Instead of affecting the formation and function of the BECN1 complex, this modification blocks autophagic flux through facilitating the proteasomal degradation of BECN1. CUL3-KLHL38 mediated the ubiquitination of BECN1 independently of the autophagy induction to block the autophagy process and enhanced the cell proliferation subsequently in breast cancer. These findings not only provide novel insights into the modification of BECN1 and its role in autophagy control and regulation but also expand our knowledge about crosstalk between the UPS and autophagy. Aberrantly expressed in cancer, CUL3 promoted cell proliferation in vitro and predicted poor patient prognosis. Silencing CUL3 rescued the impairment of autophagy and abolished the proliferation-promoting effect both in vitro and in vivo, indicating that CUL3 and KLHL38 are potential therapeutic targets in breast cancer.
Materials and methods
Cell culture
The HEK293T, HeLa, MDA-MB-231/435/436/453/468, MCF-7 and SK-BR3 cell lines were grown in DMEM (GIBCO, C11995500BT) supplemented with 10% FBS (Gibco, 10,099,141) and an appropriate amount of penicillin-streptomycin (100 IU/ml). An immortalized human mammary epithelial cell line (MCF-10A) was kindly gifted by Prof. Qiang Liu (Sun Yat-sen University Cancer Center, Guangzhou, China). The MCF–10A cell line was grown in DMEM/F12 medium (Wisent, 319-085-CL) supplemented with 20% horse serum (GIBCO, 16,050,122) and an appropriate amount of penicillin-streptomycin (100 IU/ml). All the cell lines were cultured in a 37°C incubator with a humidified 5% CO2 atmosphere. All experiments were performed in cells at 70% confluence.
Antibodies and chemicals
The following antibodies were used in our research: anti-PIK3C3/Vps34 (4263), anti-BECN1/beclin1 (3738), anti-HA tag (rabbit; 3724), anti-HA tag (mouse; 2367), anti-CUL3/cullin 3 (2759s), anti-Flag (rabbit; 14793S), anti-MYC tag (rabbit; 2278S), anti-MYC tag (mouse, 2276S), anti-ubiquitin (3936s), anti-K48-linked ubiquitin (8081s) and anti-K63-linked ubiquitin (5621s) antibodies purchased from Cell Signaling Technology; anti-UVRAG (NBP1-18,885) and anti-LC3 (NB100-2220) antibodies purchased from NOVUS; anti-RUBCN/rubicon (ab92388) antibody purchased from Abcam; anti-SQSTM1/p62 (sc-28,359), anti-BCL2 (sc-7382), anti-KLHL20 (sc-515,381), goat anti-mouse IgG-HRP (sc-2005; secondary antibody) and goat anti-rabbit IgG-HRP (sc-2004; secondary antibody) antibodies purchased from Santa Cruz Biotechnology; anti-Flag (mouse; F3165) and anti-ATG14 (A6358) antibodies purchased from Sigma-Aldrich; anti–GAPDH (60,004-1) antibody purchased from Proteintech; and Alexa Fluor 488-labeled goat anti-rabbit IgG antibody (A11008; fluorescence secondary antibody) purchased from Invitrogen. MG132 (S2619), BafA1 (S1413), CHX (S7418) and CQ (S6999) were purchased from Selleck. A CUL3 inhibitor (MLN4924, B1036) were purchased from APExBio. Penicillin-Streptomycin (0916704), ampicillin (02190146) and puromycin (0219453925) were purchased from MP biomedicals. Two conformation-specific secondary antibodies, Rabbit TrueBlot* Ultra Anti–Rabbit IgG HRP (18-8816-33) and Mouse TrueBlot* Ultra Anti–Mouse IgG HRP antibodies (18-8817-33) were purchased from Rockland.
Western blotting and IP
Cells and tissues were collected and lysed in RIPA lysis buffer (Cell Signaling Technology, 9806) containing complete protease inhibitor cocktail (Bimake, B14002-1). Then, the lysates were centrifuged at 15,000 × g for 15 min at 4°C, and the supernatant was collected. The protein concentration was determined using a Pierce BCA protein assay kit (Thermo, 23,227).
For western blotting, equal amounts of proteins (20 to 40 μg) were electrophoresed on SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, IPVH00010). The membranes were then blocked in a 5% nonfat milk solution in Tris-buffered saline (150 mM NaCl, 10 mM Tris, pH 7.6) containing 0.1% Tween (MP, 02194724; TBST) for 2 h and incubated with a primary antibody at 4°C overnight. Next, the membranes were incubated with the corresponding HRP-conjugated secondary antibodies for 2 h at room temperature. After 3 washes with TBST for 5 min, immunoreactivity was detected using Clarity Western ECL substrate (Bio–Rad, 1,705,061) according to the manufacturer’s instructions.
For IP, equal amounts of protein samples were incubated with a commercial antibody (1-4 µg/ml) at 4°C overnight. After washing with RIPA lysis buffer 3 times, protein A/G agarose beads (Thermo, 20,421) were mixed and incubated with the antibody-protein complex at 4°C for 4 h. For proteins tagged with Flag, Flag affinity gel (Bimake, B23102) was directly mixed with the protein lysates and incubated at 4°C overnight after 3 washes with RIPA lysis buffer. After centrifugation, the pellet fraction was washed 4 times with RIPA lysis buffer and resuspended in 50 µl of 2× SDS-PAGE buffer. The suspension was boiled for 10 min at 100°C and centrifuged, and the supernatant was subjected to western blotting or MS to analyze the coprecipitated proteins or modification of the target protein. Conformation specific secondary antibodies were adopted to attenuate the interference of the heavy chain and light chain of IgG during the detection of immunoreactivity if necessary.
For ubiquitination IP assays, a specific kind of lysis buffer containing SDS (1%) was adopted in ubiquitination IP assays to break the interaction between BECN1 and its interacting partners. To prevent the impact of SDS on the binding between Flag-tagged BECN1 and Flag affinity gel, the lysates were 10 times diluted using a specific kind of dilution buffer before the Flag affinity gel was added. The formulas of the lysis buffer and the dilution buffer were listed as follows:
Lysis buffer
Tris Buffer, pH 8.0 50 mM
SDS 1%
EDTA 10 mM
Dilution buffer
Tris Buffer, pH 8.0 16.7 Mm
SDS 0.01%
NaCl 167.2 mM
EDTA 1.2 mM
Triton X-100 (MP, 219,485,480) 0.2%
In vitro ubiquitination assays
All reagents related to the in vitro ubiquitination assays were purchased from R&D Systems. For in vitro ubiquitination assays, CUL3-KLHL38-RBX1 complex was affinity-isolated from lysates of HEK293T cells using anti-Flag affinity gel (Bimake, B23102) 48 h after transfected with the Flag-tagged KLHL38, MYC-tagged CUL3 and RBX1 plasmids. And baculoviral expressed GST-tagged BECN1 protein was purified. Then the affinity-purified CUL3-KLHL38-RBX1 complex was incubated with purified GST-tagged BECN1 protein in a 20-μl in vitro ubiquitination reaction mixture at 37°C for 4 h. And WB assays were adopted to detect the ubiquitination of the BECN1 protein subsequently. The in vitro ubiquitination reaction mixture contains 50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 1 μM ubiquitin, 20 μM MG132, 2 mM ATP, 2 mM NaF, 1 mM DTT (MP, 0210059780), 10 μg ubiquitin, 40 ng E1, and 200 ng E2. The ATP, ubiquitin, E1 enzyme, E2 enzyme and the Reaction Buffer were obtained from Human MDM2/HDM2 Ubiquitin Ligase Kit– p53 Substrate (R&D, K-200B) [Citation53,Citation54].
MS
To analyze proteins that interact with BECN1, affinity-isolated BECN1 protein was electrophoresed on SDS-polyacrylamide stacking gels, and the bromophenol blue band and a nearby region were excised from the gels. To analyze the posttranslational modification of BECN1, affinity-isolated BECN1 protein was electrophoresed on SDS-polyacrylamide separating gels, and the BECN1 band and a nearby region were excised from the gels. The excised gel bands and regions were subjected to trypsin digestion and liquid chromatography-coupled tandem MS. Identification of proteins and their modification was performed with a database search, and identified peptides were validated with Peptide Prophet.
RNA extraction and real-time RT-PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, 15,596,018). One microgram of total RNA from each sample was subjected to reverse transcription using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara, RR047B) to synthesize cDNA. Real-time PCR was performed with SYBR® Premix Ex Taq™ II (Takara, RR820B) on a CFX96 real-time PCR system (Bio-Rad, USA) following the manufacturer’s instructions. All genes were assayed in triplicate to ensure reproducibility.
The primers 5′-AAGCCTGCCGGTGACTAAC-3′ (forward) and 5′-GTTAAAAGCAGCCCTGGTGAC-3′ (reverse) were used to generate a 174-bp GAPDH fragment as an internal control. The primers used to detect CUL3 mRNA were 5′- TGTCGAATCTGAGCAAAGGC-3′ (forward) and 5′-CATCCATGGTCATCGGAAAGG-3′ (reverse). The primers used to detect BECN1 mRNA were 5′-GAGGGATGGAAGGGTCTA-3′ (forward) and 5′-GCCTGGGCTGTGGTAAGT-3′ (reverse). The primers used to detect KLHL38 mRNA were 5′-TGAAGGAGCTCTGTGCCTTG-3′ (forward) and 5′-ATCGTTGGCGATGAAGTGGT-3′ (reverse).
Plasmids
The pcDNA4-Flag-BECN1 (24,388; Qing Zhong), four plasmids encoding Flag-tagged deletion mutants of BECN1 (24,389, 24,390, 24,391, 24,392; deposited by Qing Zhong), pcDNA3.1-MYC-CUL3 (19,893; deposited by Yue Xiong), pRK5-HA-Ubiquitin-WT (17,608; deposited by Ted Dawson) and pRK5-HA-Ubiquitin-K48 (17,605; deposited by Ted Dawson) plasmids were obtained from Addgene. Flag-KLHL38 (CH812939) and Flag-KLHL20 (CH825033) were obtained from Vigene. and was cloned into the pcDNA3.1 vector (Invitrogen, V80020). Site-directed mutations in the Flag-BECN1 plasmid were generated by using specific primers, PrimeSTAR Max DNA Polymerase (Takara, R045B) and a ClonExpress II One Step Cloning Kit (Vazyme, C112). The construct coding for BECN1 with the HA tag was cloned into the pBABE vector (Addgene, 1764; deposited by Hartmut Land, Jay Morgenstern, Bob Weinberg) between the BamHI and EcoRI restriction sites. All the newly constructed plasmids were verified by DNA sequencing. All plasmids were transfected into the cell lines with Lipofectamine 2000 (Invitrogen, 11,668,019) and Opti-MEM medium (Gibco, 31,985,070) according to the manufacturer’s instructions.
siRNA
The targeted siRNA sequences were as follows:
si NC: 5′-UUCUCCGAACGUGUCACGUTT-3′
si CUL3 #1: CAUGAAGACUAUAGUAGAATT
si CUL3 #2: GUCGUAGACAGAGGCGCAATT
si CUL3 #3: GAGAUCAAGUUGUACGGUATT
si KLHL12 #1: GUACCUGGAAGUUGCAGAATT
si KLHL12 #2: GGGAAACCUUAUGUUGACATT
si KLHL12 #3: CCAAGCAUCACUCGUAAGATT
si KLHL38 #1: GGGAGAAGAGUGAAGCCAATT
si KLHL38 #2: GGAGUCUGGUCAGUCACAATT
si KLHL38 #3: CCCUCAAUCCAACAAAUUUTT
si BECN1: UGGAAUGGAAUGAGAUUAATT
All siRNAs were synthesized by RiboBio and transfected into the cells using Lipofectamine 2000 and Opti-MEM medium according to the manufacturer’s instructions.
shRNA
The sequence for short hairpin RNA (shRNA) targeted against CUL3 and a nontargeting shRNA control were 5′-TTCTCCGAACGTGTCACGT-3′ and 5′- GCTTGGAATGATCATCAAACA −3′, respectively. The shRNA expression plasmids were ordered from GeneChem and transfected into HEK293T cells at 40% confluence with packaging plasmids (psPAX2 and pMD2.G; from addgene, 12,260, 12,259; deposited by Didier Trono) using Lipofectamine 2000 and Opti-MEM medium according to the manufacturer’s instructions to generate retroviruses. The medium was changed at 6 h and 24 h after transfection, and the cell supernatant was collected at 48 h and 72 h after transfection. The cleared supernatant was filtered through a 0.45-μm filter, aliquoted and stored at −80°C. Target cells were infected with retrovirus supernatant in the presence of 8 μg/ml Polybrene every 12 h for three rounds. After infection, the cells were selected using puromycin (1 μg/ml) for 3 days.
Immunofluorescence
Cells at 30% confluence were seeded on glass coverslips and transfected with the indicated siRNA. Forty-eight hours after transfection, the coverslips were washed briefly with PBS (136.89 mM NaCl, 2.68 mM KCl, 10.14 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4), fixed with 4% paraformaldehyde at room temperature for 30 min, blocked in a 4% BSA (MP, 2,180,549,910) solution in PBS for 1 h and incubated with anti-LC3 antibody at 4°C overnight. The coverslips were then incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG antibody at room temperature for 1 h, stained with 1 μg/ml DAPI (Sigma-Aldrich, D9542) for 5 min, washed with PBS and dried. The cells were observed at 100× magnification using an Olympus FV-1000 confocal microscope.
Cell proliferation assay
Twenty-four hours after transfection, cells were seeded in triplicate in 96–well plates (500 cells/well). The cells were counted every day by MTT (5 mg/mL; MP, 0210222710) assay over 8-9 days.
Colony-formation assay
Twenty-four hours after transfection, cells were seeded in 6-well plates (500 cells/well). After incubation in a 37°C incubator for 2 weeks, the cells were briefly incubated with PBS, fixed with 4% paraformaldehyde at room temperature for 30 min and dyed with 0.1% crystal violet at room temperature for 30 min. Then, the 6-well plates were scanned to observe and count the colonies.
Cell viability assays
Cells were seeded in triplicate in 96-well plates and incubated in a 37°C incubator overnight. Then, the cells were treated with the indicated drugs for another 24 h. Following treatment, MTT (5 mg/mL) was added to each well and incubated for another 4 h at 37°C. After that, the MTT solution was removed and replaced with 100 µl of DMSO. The absorbance at 570 nm was measured by a microplate reader (Bio-Rad, USA) to estimate the cell viability.
Cell cycle analyses
Flow cytometry was adopted to analyze the distribution of cells in three major phases of the cell cycle (G1, S and G2/M) using the Cell Cycle and Apoptosis Analyses Kit (C1052) purchased from Beyotime. The cells were digested with trypsin and collected into microcentrifuge tubes, washed once with PBS, and then fixed in 70% ice-cold ethanol at 4°C overnight. After fixation, the cells were washed once with PBS again, and the cell pellets were resuspended in 0.5 ml of pre-prepared PI/RNase staining buffer and incubated at 37°C for 30 min. The staining buffer was a mixture of the following three components: 500 μl staining buffer (Beyotime, C1052-1), 25 μl 20× Propidium (Beyotime, C1052-2), and 10 μl 50× RNase A (Beyotime, C1052-3). The staining cells were analyzed with a flow cytometer immediately, and the data were analyzed with the ModFit LT software.
Cell apoptosis analyses
The apoptosis of the cells was analyzed with the Annexin V-FITC Apoptosis Detection Kit purchased from Beyotime (C1062). The adherent cells were digested with trypsin and collected into microcentrifuge tubes together with the cells suspended in the culture medium. After centrifugation at 1000 g for 5 min, the supernatant was discarded, and the cell pellets were resuspended in PBS and counted with a hemocytometer. 1 × 105 cells were centrifuged to discard the PBS, and the cell pellets were resuspended in 195 μl ANXA5/annexin V-FITC binding buffer, with 5 μl ANXA5/annexin V-FITC and 10 µl PI staining buffer added. After incubated in room temperature for 20 min, the cells were analyzed with flow cytometry immediately.
Animal studies/tumorigenicity model of nude mice
The tumorigenicity model of nude mice was constructed to further explore the function of CUL3 protein and its association with autophagy in breast cancer in vivo. The nude mice (nu/nu, 6-week-old females) were randomly allocated into the groups as required with 10 mice in each group. 3 × 106 MDA-MB-231 cells were inoculated subcutaneously into the right flank region of each nude mouse. The presence or absence of a visible or palpable tumor was evaluated and tumor growth was monitored every 3 d. Tumor volume was calculated by the formula 0.5 × length × width. The mice were then sacrificed until the tumor volume exceeded 800 mm3 and the tumors were excised to determine the weight of them. All animal experiments were conducted in accordance with the institutional guidelines and approved by the Animal Care and Use Committee of Sun Yat-sen University Cancer Center (L102012020070A).
Tissue microarray and immunohistochemistry
Paraffin-embedded breast cancer tissue microarrays were obtained from Sun Yat-Sen University Cancer Center, Guangzhou, China. Immunohistochemical staining was performed to analyze expression of the CUL3 and BECN1 protein. Consecutive sections of the breast cancer tissue microarrays were obtained and incubated with anti-BECN1 and anti-Cul3 antibodies, respectively. Then the sections were incubated with goat anti-rabbit IgG-HRP antibody, developed in DAB solution and stained with hematoxylin. Each sample was scored by a pathologist with the H-score method, which combines values indicating the immunoreaction intensity and percentage of stained tumor cells.
Analyses of TCGA data
Cbioportal, an online platform for analyses and integration of TCGA data (Available at: http://cbioportal.org/), was adopted. The differential expression of BECN1 protein or RNA was assessed by z-score. The threshold of z-score for “high” and “low” expression was +2.0 and −2.0, respectively, which corresponded to a p-value smaller than 0.05.
Statistical analyses
All assays were performed in triplicate. The immunoblotting data was quantified with the imageJ software and the number of the punctate foci in the immunofluorescence data was counted manually using the Photoshop software. The relative expression of the proteins and the number of the punctate foci in the immunofluorescence data was displayed in bar charts in corresponding figures. Data are expressed as the mean ± standard deviation (SD). Statistical analyses were performed with SPSS 20.0 software. Two-tailed Student’s t-test was adopted to assess differences between two groups. Differences for which *p < 0.05 were considered statistically significant; those for which **p < 0.01 were considered highly significant.
Supplemental Material
Download MS Word (1.4 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131.
- Kliosnky D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy. 2016;12(2):443.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–995.
- Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580.
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362.
- Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–396.
- Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476.
- Wang RC, Wei YJ, An ZY, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959.
- Zalckvar E, Berissi H, Mizrachy L, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-X-L and induction of autophagy. EMBO Rep. 2009;10(3):285–292.
- Wei YJ, Zou ZJ, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284.
- Yue ZY, Jin SK, Yang CW, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082.
- Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676.
- Qu XP, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820.
- Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science (New York, NY). 1989;243(4898):1576–1583.
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67(1)::425–479.
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438–444.
- Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617.
- Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44.
- Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. Embo J. 2013;32(17):2307–2320.
- Dhanoa BS, Cogliati T, Satish AG, et al. Update on the Kelch-like (KLHL) gene family. Hum Genomics. 2013;7(1):13.
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750.
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303.
- Maejima Y, Kyoi S, Zhai PY, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478-+.
- Wei Y, Zou Z, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3(123):ra42.
- Fusco C, Mandriani B, Di Rienzo M, et al. TRIM50 regulates Beclin 1 proautophagic activity. Biochim Biophys Acta Mol Cell Res. 2018;1865(6):908–919.
- Sun T, Li X, Zhang P, et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015;6(1):7215.
- Li X, Wu XQ, Deng R, et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun. 2017;8(1):1159.
- Liu CC, Lin YC, Chen YH, et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61(1):84–97.
- Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. 2017;40(7):441–449.
- Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143(5):682–685.
- Collins GA, Goldberg AL. The logic of the 26S proteasome. Cell. 2017;169(5):792–806.
- Ciechanover A, Stanhill A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta. 2014;1843(1):86–96.
- Tan JMM, Wong ESP, Kirkpatrick DS, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17(3):431–439.
- Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–516.
- Pandey UB, Nie ZP, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447(7146):859–863.
- Cha-Molstad H, Sung KS, Hwang J, et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol. 2015;17(7):917–929.
- Korolchuk VI, Menzies FM, Rubinsztein DC. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009;5(6):862–863.
- Korolchuk VI, Mansilla A, Menzies FM, et al. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517–527.
- Qiao LY, Zhang JH. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett. 2009;456(1):15–19.
- Bayraktar O, Oral O, Kocaturk NM, et al. IBMPFD disease-causing mutant VCP/p97 proteins are targets of autophagic-lysosomal degradation. PloS One. 2016;11(10): e0164864.
- Marshall RS, McLoughlin F, Vierstra RD. Autophagic turnover of inactive 26S proteasomes in yeast is directed by the ubiquitin receptor Cue5 and the Hsp42 chaperone. Cell Rep. 2016;16(6):1717–1732.
- Marshall RS, Li FQ, Gemperline DC, et al. Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/Ubiquitin receptor RPN10 in arabidopsis. Mol Cell. 2015;58(6):1053–1066.
- Jia R, Bonifacino JS. Regulation of LC3B levels by ubiquitination and proteasomal degradation. Autophagy. 2020;16(2):382–384.
- Li Y, Zhou X, Zhang Y, et al. CUL4B regulates autophagy via JNK signaling in diffuse large B-cell lymphoma. Cell Cycle. 2019;18(4):379–394.
- Shin HJ, Kim H, Oh S, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534(7608):553–557.
- Antonioli M, Albiero F, Nazio F, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. 2014;31(6):734–746.
- Gao D, Inuzuka H, Tan MK, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44(2):290–303.
- Chen J, Ou Y, Yang Y, et al. KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature. 2018;557(7706):585–589.
- Lee Y, Chou TF, Pittman SK, et al. Keap1/cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017;20(8):1994.
- Cao Z, Xue J, Cheng Y, et al. MDM2 promotes genome instability by ubiquitinating the transcription factor HBP1. Oncogene. 2019;38(24):4835–4855.
- Lee YR, Yuan WC, Ho HC, et al. The cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. Embo J. 2010;29(10):1748–1761.