
ABSTRACT
PIK3C3/VPS34 is a key player in macroautophagy/autophagy and MAP1LC3/LC3-associated phagocytosis (LAP), which play critical roles in dendritic cell (DC) function. In this study, we assessed the contribution of PIK3C3 to DC function during experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS). We found that Pik3c3-deficient DCs exhibit attenuated capacity to reactivate encephalitogenic T cells in the central nervous system, leading to reduced incidence and severity of EAE in DC-specific Pik3c3-deficient mice. Additionally, animals with a DC-specific deficiency in Rb1cc1/Fip200 but not Rubcn were protected against EAE, suggesting that the EAE phenotype of DC-specific Pik3c3-deficient mice is due to defective canonical autophagy rather than LAP. Collectively, our studies have revealed a critical role of PIK3C3 in DC function and the pathogenicity of these cells during EAE, with important implications for the development of immunotherapies for autoimmune diseases such as MS.
Abbreviations: ATG: autophagy-related; CNS: central nervous system; DC: dendritic cell; DEG: differentially expressed gene; EAE: experimental autoimmune encephalomyelitis; LAP: LC3-associated phagocytosis; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MHC: major histocompatibility complex; MOG: myelin oligodendrocyte glycoprotein; MS: multiple sclerosis; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; ROS: reactive oxygen species
Introduction
Macroautophagy (called autophagy hereafter) is an evolutionarily highly conserved nutrient-sensing system that promotes the degradation of cytoplasmic proteins and damaged organelles by lysosomes [Citation1]. The resulting degradation products are then used in cellular remodeling and in regenerating molecular building blocks in normal physiology and during conditions of stress. This process is orchestrated by over 30 autophagy-related (ATG) gene products that were initially identified in yeast but are largely conserved in higher eukaryotes.
Dendritic cells (DCs) are professional antigen-presenting cells that are critical for capturing, processing, and presenting antigens to major histocompatibility complex (MHC)-restricted T cells, which requires high levels of endocytic and lysosomal activity that is tightly linked to autophagy. DCs play a critical role in the induction and progression of a variety of autoimmune diseases, including experimental autoimmune encephalomyelitis (EAE), an animal model of human multiple sclerosis (MS). The role of DC-specific autophagy in the pathogenicity of EAE has been explored previously. Bhattacharya et al. reported that Atg7-deficiency in DCs does not result in gross alterations in global DC function but leads to reduced onset and severity of MOG (myelin oligodendrocyte glycoprotein)-induced EAE by reducing in vivo priming of T cells [Citation2]. Keller et al. reported that ablation of Atg5 in DCs results in complete protection from EAE development upon adoptive transfer of primed, encephalitogenic CD4+ T cells [Citation3]. This protection appeared to be due to impaired ATG-regulated phagocytosis of injured oligodendrocytes for MHC class II antigen presentation, leading to reduced accumulation of myelin-specific T cells within the central nervous system (CNS) [Citation3]. Yet, only minor or statistically non-significant differences in disease incidence rates and clinical severity scores were observed in active EAE [Citation3]. In another report, Alissafi et al. demonstrated that DCs with a selective deletion of Atg16l1 are less efficient in priming autoreactive T cells and, as a result, co-transfer of Atg16l1-deficient DCs and 2D2 MOG35-55 antigen-specific T cells into rag−/- mice leads to less severe EAE disease [Citation4].
The PIK3C3 subunit of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex is critical for the initiation of autophagy [Citation5] and has also been implicated in other cellular processes, including endocytosis, intracellular vesicular trafficking [Citation6], and MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)-associated phagocytosis (LAP) [Citation7]. Inhibiting PtdIns3K activity has shown promising results in both preclinical and clinical trials for the treatment of immune-related disorders including MS (reviewed in [Citation8,Citation9]). We and others have previously demonstrated that Pik3c3-deficient T cells, DCs, and myeloid cells exhibit defective autophagy and have remarkably altered cellular functions [Citation10–15]. However, the role of PIK3C3 in DCs in the context of autoimmunity is unknown. In the current study, we show that mice with a DC-specific Pik3c3-deficiency exhibit significant resistance to EAE, which was associated with attenuated reactivation of primed T cells in the CNS. These effects of DC-specific PIK3C3 on EAE were independent of LAP, pointing to the autophagy defect of these cells as the cause of EAE resistance. Thus, our findings have revealed a critical role for DC PIK3C3 in CD4+ T cell-mediated autoimmune diseases.
Results
Deletion of Pik3c3 in DCs reprograms gene expression
It has been widely acknowledged that autophagy is critical for both MHC class I- and MHC class II-restricted antigen presentation [Citation16,Citation17]. To investigate the role of PIK3C3 in antigen presentation, we previously generated DC-specific Pik3c3-deficient mice by breeding mice in which exon 4 of the Pik3c3 gene was flanked by loxP sites (Pik3c3f/f) to mice expressing Cre recombinase driven by the Itgax/Cd11c promoter [Citation11]. pik3c3f/f;Itgax-Cre mice showed a DC-specific Pik3c3 ablation and DCs in these animals exhibited a partially activated phenotype, with increased surface expression of MHC class I, MHC class II, and co-stimulatory molecules, and spontaneous production of both pro- and anti-inflammatory cytokines [Citation11]. This study also revealed that mice with a DC-specific deficiency in Pik3c3 exhibit a partial impairment in the homeostatic maintenance of CD8A+ conventional DCs [Citation11], which are specialized in cross-presenting antigens on MHC class I molecules [Citation18]. Additionally, we found a modest increase in the activity of the classical MHC class I and class II antigen presentation pathways, as well as a profound defect in the cross-presentation of dead cell-associated antigens to MHC class I-restricted CD8A+ T cells owing to defective uptake of apoptotic cells [Citation11].
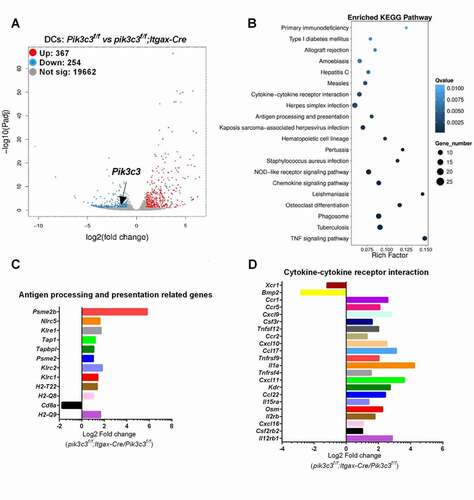
We previously showed that the pik3c3f/f;Itgax-Cre mice exhibit a selective Pik3c3 deletion in their DCs [Citation11]. DCs in these animals showed defective autophagy compared with WT DCs, as indicated by autophagosomal double-membrane structures revealed by electron microscopy [Citation11] and increased levels of LAMP1, SQSTM1/p62, and total LC3 levels revealed by western blotting (Figure S1). To obtain a better understanding of potential effects of Pik3c3 deletion on DC function, we investigated the global transcriptomic profiles of FACS-enriched DCs from both Pik3c3f/f and pik3c3f/f;Itgax-Cre mice. RNA sequencing analysis revealed significant genome-wide changes in transcript levels. Pik3c3-deficiency in DCs significantly induced (n = 367) or reduced (n = 254) gene expression, including reduced Pik3c3 mRNA expression (3.27-fold reduction, p = 5.73E-14) (). Subsequent pathway enrichment analysis identified the impacted antigen-presenting function in Pik3c3-deficient DCs (), as expected. Interestingly, impacted genes in antigen presentation were all associated with the MHC class I pathway (). Genes in the MHC class II antigen presentation pathway were unchanged or slightly downregulated. Furthermore, our RNA sequencing data confirmed the partially activated phenotype in Pik3c3-deficient DCs as indicated by enhanced expression of cytokine- and cytokine receptor-associated genes ().
Figure 1. Deletion of Pik3c3 in DCs reprograms DC gene expression. (A) Clustered heat map of RNA-seq gene counts in Pik3c3-deficient DCs and Pik3c3-sufficient DCs (n = 2 mice per group). (B) Gene set enrichment analysis showing enriched pathways from differentially expressed genes between Pik3c3-deficient DCs and Pik3c3-sufficient DCs. (C) Altered gene expression associated with antigen processing and presentation. (D) Altered gene expression associated with cytokine-cytokine receptor interactions.
Pik3c3-deficiency in DCs reduces the incidence and severity of EAE
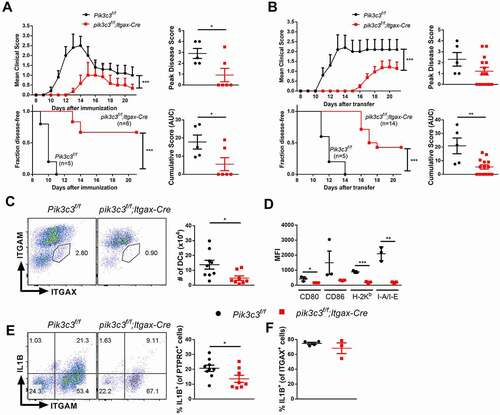
DC-specific Pik3c3-deficient mice contain a defect in the capacity of CD8A+ DCs to present dead cell-associated antigens to MHC class I-restricted T cells and, as a result, these animals show increased metastases in response to challenge with B16 melanoma cells [Citation11]. However, the role of PIK3C3 in DCs in the context of autoimmunity remains unclear. To test the role of DC-specific PIK3C3 in autoimmunity, we employed the EAE model of MS, by immunizing mice with MOG35-55 peptide to induce MOG-specific, MHC class II-restricted CD4+ T cells. We first induced active EAE in pik3c3f/f;Itgax-Cre mice with Pik3c3f/f mice as controls. We found reduced severity and delayed onset of EAE in pik3c3f/f;Itgax-Cre mice (). The incidence of EAE was also decreased in pik3c3f/f;Itgax-Cre mice (11 out of 11 in control mice versus 5 out of 12 in pik3c3f/f;Itgax-Cre mice from three independent experiments, p = 0.0024, chi-square test) (). Additionally, in pik3c3f/f;Itgax-Cre mice, the cumulative disease score and peak disease score were significantly reduced ().
Figure 2. Pik3c3-deficiency attenuates incidence and severity of EAE. (A) EAE induced by active immunization with MOG35-55 peptide. (B) EAE induced by adoptive transfer of myelin-specific T cells. Results show one experiment that is representative of three independent experiments. (C) Flow cytometric analysis of DCs in the brain of pik3c3f/f;Itgax-Cre mice at 14 days after EAE induction. Results from three independent experiments were pooled. (D) CNS leukocytes were prepared from mice, stained with anti-ITGAX, -ITGAM, -CD80, -CD86, -H-2Kb, and I-A/-E antibodies, and analyzed by flow cytometry. Results show one experiment that is representative of three independent experiments. (E) IL1B production by CNS leukocytes. Cells were isolated from the CNS and IL1B-producing ITGAM+ cells were assessed by flow cytometry. Results from two independent experiments were pooled. (F) IL1B production by ITGAX+ cells in the CNS. Results show one experiment that is representative of two independent experiments. The data shown are the average ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Previous studies have provided evidence that CNS-resident classical DCs can stimulate effector myelin-specific T cells and that selective depletion of these cells results in reduced numbers of myelin-specific T cells and reduced EAE incidence [Citation19]. Therefore, we immunized wild-type (WT) mice with MOG peptide and transferred primed myelin-specific CD4+ T cells to induce EAE in recipient Pik3c3f/f or pik3c3f/f;Itgax-Cre mice to model the effector phase of EAE, which is highly dependent on the presence of antigen-presenting cells within the CNS. Similar to our results for active EAE, pik3c3f/f;Itgax-Cre mice exhibited attenuated disease severity compared with their littermate controls upon adoptive transfer of encephalitogenic T cells (), suggesting that the reduced EAE severity in pik3c3f/f;Itgax-Cre mice is due to the lack of PIK3C3 in DCs from recipient animals.
We have previously reported that DCs from pik3c3f/f;Itgax-Cre mice exhibit an activated phenotype under steady-state conditions [Citation11], which is also reflected in the transcriptomic studies shown in . We therefore analyzed the prevalence and activation state of DCs in the CNS of pik3c3f/f;Itgax-Cre mice during EAE. We found that fewer ITGAX+ cells accumulate in the CNS of pik3c3f/f;Itgax-Cre than Pik3c3f/f mice during EAE (). Further, these cells from EAE-induced mutant animals were in a less activated state, as revealed by lower surface expression of CD80, CD86, and MHC class I and class II molecules ().
Because the cytokine IL1B plays an important role in EAE, we next analyzed IL1B production by total PTPRC+ leukocytes and ITGAX+ myeloid cells in the CNS of pik3c3f/f;Itgax-Cre mice during EAE. We found fewer IL1B+ cells among total PTPRC+ cells () but similar frequencies of IL1B+ cells among ITGAX+ cells () from pik3c3f/f;Itgax-Cre than Pik3c3f/f mice. Considering our previous finding that Pik3c3-deficient DCs produce higher levels of IL1B upon activation with heat-killed L. monocytogenes compared with Pik3c3-sufficient DCs [Citation11], we propose that reduced IL1B levels in pik3c3f/f;Itgax-Cre mice during EAE are likely a consequence rather than a cause of ameliorated EAE development in these animals.
Peripheral antigen-specific T cell recall response is unaltered in pik3c3f/f;Itgax-Cre mice
The finding that pik3c3f/f;Itgax-Cre mice showed specific defects in CD4+ T cell-mediated EAE suggested the possibility that Pik3c3-deficiency in DCs results in reduced CD4+ T cell priming. To test this possibility, we isolated splenocytes from Pik3c3f/f and pik3c3f/f;Itgax-Cre mice following EAE induction (day 10) and incubated them with MOG peptide (20 μg/ml) to measure antigen-specific cytokine production. Non-significant differences were observed for the frequency of IL17A+ CD4+ and IFNG+CD4+ T cells, although high intergroup variation was seen (). Since CD8A+ T cells can also contribute either positively or negatively to the pathogenesis of EAE in mice [Citation20–22], we further analyzed the antigen-specific cytokine production of CD8A+ T cells. However, no difference in cytokine production by CD8A+ T cells derived from MOG-immunized Pik3c3f/f and pik3c3f/f;Itgax-Cre mice was observed after in vitro MOG restimulation in our system (). To determine the extent of CNS inflammation, we examined frequencies of CNS-infiltrating T cells in pik3c3f/f;Itgax-Cre mice compared with Pik3c3f/f littermates. To this end, mice were sacrificed at day 14 following active EAE induction and T cells were analyzed by flow cytometry. pik3c3f/f;Itgax-Cre mice showed substantially lower total and CD44+ CNS-infiltrating CD4+ T cells () as well as IFNG+ CD4+ T cells, but no significant difference in the prevalence of IL17A+ CD4+ T cells ().
Figure 3. Peripheral antigen-specific T cell recall response in EAE is unaltered in pik3c3f/f;Itgax-Cre mice. (A) Splenocytes from Pik3c3f/f and pik3c3f/f;Itgax-Cre mice induced for EAE (day 10) were cultured with MOG35-55 peptide (20 μg/ml). After 18 h of culture, cells were harvested and intracellular staining was performed (IL17A and IFNG), and cytokine-producing CD4+ T and CD8+ T cells were analyzed by flow cytometry. (B) Quantification of antigen-specific cytokine production by T cells from spleen. (C) Flow cytometric analysis of CD4+ and CD4+CD44+ T cells in the brain of pik3c3f/f;Itgax-Cre mice at 14 days after EAE induction. (D) Quantification of antigen-specific cytokine production by CNS-invading CD4+ T cells. (E) Disease onset of WT mice after receiving MOG35-55-specific T cells from pik3c3f/f;Itgax-Cre or Pik3c3f/f mice. (F,G) Correlation analyses of the percentage of IFNG+ cells or IL17A+ cells within CD4+ cells with EAE peak disease score (F) and disease onset (G) after adoptive transfer EAE induction. Results from two-four independent experiments were pooled and plotted. The data shown are the average ± SEM. *p < 0.05, **p < 0.01.
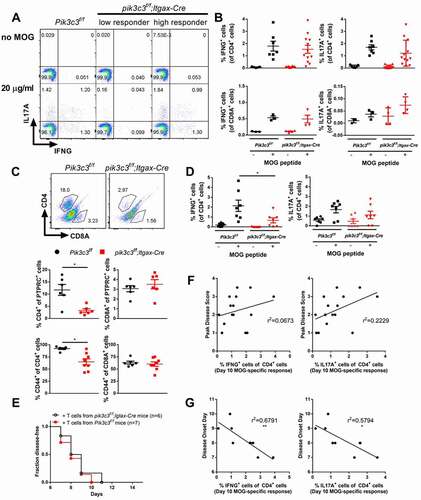
To determine whether Pik3c3-deficiency in DCs impairs CD4+ T cell pathogenicity, we immunized pik3c3f/f;Itgax-Cre mice with MOG peptide and transferred primed myelin-specific CD4+ T cells to induce EAE in WT recipient mice. We found that all mice receiving T cells from pik3c3f/f;Itgax-Cre or Pik3c3f/f donor animals develop similar clinical manifestations of EAE (). Further, antigen-specific cytokine production by T cells analyzed at day 10 after EAE induction did not correlate with peak disease score () but correlated with the kinetics of disease onset ().
Taken together, these data suggest that peripheral CD4+ T cell priming is largely unaltered but that accumulation of CD4+ T cells in the CNS is reduced in the absence of PIK3C3 in DCs, suggesting that effects of Pik3c3-deficiency on EAE might be due to DCs in the CNS rather than the periphery.
The EAE phenotype of DC-specific Pik3c3-deficient mice is associated with the lack of canonical autophagy rather than LAP
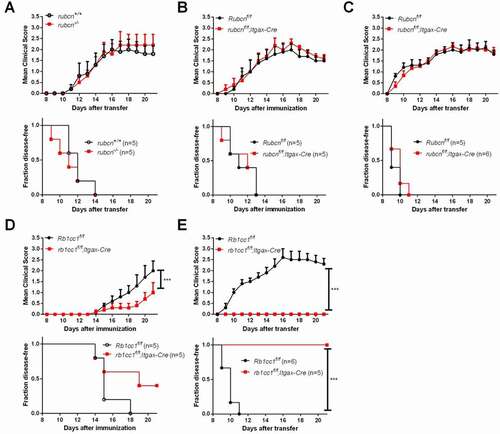
Although canonical autophagy and LAP share many molecular components including PIK3C3, LAP proceeds independently of the autophagy preinitiation complex, which is composed of ATG13, RB1CC1/FIP200, ATG101 and ULK1 [Citation23]. In addition, RUBCN, in association with the BECN1/Beclin1-PIK3C3-UVRAG-containing PtdIns3K complex mediates LAP but not autophagy [Citation7]. While basal autophagy activation was observed in rubcn-/- mice [Citation24], our previous study showed that rubcn-/- mice develop signs of EAE similar to WT control mice [Citation15], suggesting that the slight elevation in autophagy of these mice does not have a major impact on EAE disease progression. In the present study, we further transferred primed MOG-specific CD4+ T cells to induce EAE in rubcn-/- recipient mice. Again, all of the examined rubcn-/- mice developed clinical disease manifestations similar to WT control mice (). To test the role of RUBCN in DCs more directly, we induced EAE in rubcnf/f;Itgax-Cre mice. We found that rubcnf/f;Itgax-Cre mice exhibit disease symptoms similar to WT mice in both active and passive EAE models (). To confirm that the resistance of DC-specific Pik3c3-deficient mice to EAE is indeed due to defective autophagy, we induced EAE in rb1cc1f/f;Itgax-Cre mice. Similar to pik3c3f/f;Itgax-Cre mice, rb1cc1f/f;Itgax-Cre mice were resistant to EAE induction (). Collectively, these results suggest that the EAE phenotype of DC-specific Pik3c3-deficient mice is associated with the lack of canonical autophagy rather than LAP.
Figure 4. DC autophagy but not LAP is required for EAE. (A–C) EAE was induced in rubcn-/-, rubcnf/f;Itgax-Cre, and respective littermate control mice. (D,E) EAE was induced in rb1cc1f/f;Itgax-Cre and littermate control mice. Results show one experiment that is representative of two independent experiments. The data shown are the average ± SEM. ***p < 0.001.
Pik3c3-deficiency in DCs impairs their T cell stimulatory capacity
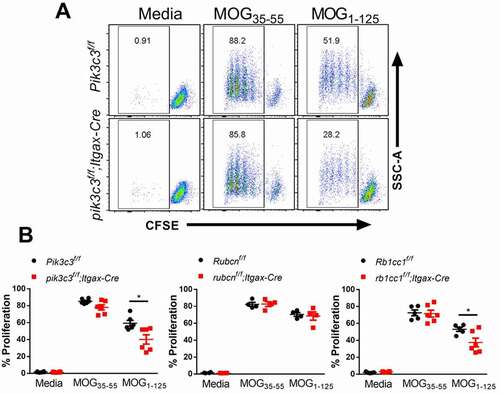
To investigate whether the absence of Pik3c3 in DCs impairs their T cell stimulatory capacity, DCs were sorted from pik3c3f/f;Itgax-Cre, rb1cc1f/f;Itgax-Cre, rubcnf/f;Itgax-Cre and their respective littermate control mice. Isolated splenic DCs were pulsed with MOG35-55 peptide or a longer fragment of MOG (MOG1-125) that requires antigen processing for its presentation on MHC class II, and then cocultured with naïve CD4+ T cells expressing a transgenic T cell receptor specific for the MOG35-55 peptide (2D2 cells). We observed similar proliferation levels of 2D2 cells cocultured with MOG35-55-primed WT, Pik3c3-, Rb1cc1-, and Rubcn-deficient DCs (). However, MOG1-125-primed WT and Rubcn-deficient DCs induced a more prominent effector T cell response than Pik3c3- and Rb1cc1-deficient DCs (). Together, these data suggest that autophagy is required for autoantigen processing and that autophagy-deficiency impairs the T cell stimulatory capacity of DCs.
Figure 5. Autophagy-deficient DCs display impaired capacity to present myelin antigens to T cells. Splenic DCs were isolated from pik3c3f/f;Itgax-Cre, rb1cc1f/f;Itgax-Cre, rubcnf/f;Itgax-Cre, and their respective littermate control mice. Sorted DCs (2 × 104) were pulsed with 20 μg/ml MOG35-55 or MOG1-125 for 4 h in complete medium and washed. CFSE-labeled 2D2 CD4+ T cells (1 × 105) were added and cocultured for 72 h to measure T cell proliferation by flow cytometry. (A) Representative FACS plots depicting proliferation of 2D2 CD4+ T cells upon coculture with MOG35-55- or MOG1-125-pulsed pik3c3-sufficient and -deficient DCs. (B) Quantification of 2D2 CD4+ T cell proliferation under different conditions. Results from two-three independent experiments were pooled and plotted. The data shown are the average ± SEM. *p < 0.05.
Discussion
The “holy grail” strategy to treat MS and other autoimmune diseases is to induce tolerance against the inciting self-antigens without affecting the capacity to respond and protect against pathogenic organisms. However, effective drugs currently available are broadly disease-modifying treatments rather than disease-specific treatments that selectively dampen autoantigen-specific immune responses, which leaves patients vulnerable to infections and possibly cancer [Citation25]. Thus, there is an urgent need to explore new modalities to treat MS and other autoimmune diseases. The most common mouse model of MS is EAE, which can be actively induced through immunization with myelin-derived antigens suspended in complete Freund’s adjuvant, followed by injection of pertussis toxin to break the blood-brain barrier. The pathogenesis of actively-induced EAE consists of an induction and an effector phase. EAE can also be induced by adoptive transfer of activated myelin-specific CD4+ T cells, which models the effector phase of EAE disease.
In this study, we found that Pik3c3-deficiency in DCs significantly reduces the onset and severity of actively induced EAE. The underlying mechanism is likely due to a disrupted effector response, as evidenced by attenuated disease severity upon adoptive transfer of encephalitogenic T cells into pik3c3f/f;Itgax-Cre mice. These data are consistent with previous studies suggesting that DCs require ATG5 to reactivate primed myelin-specific CD4+ T cells during the effector phase of EAE [Citation3]. Our results demonstrate that DC autophagy is required for the priming of autoreactive CD4+ T cells in vivo and development of EAE. Autophagy is a critical regulator of intracellular antigen loading to MHC class II molecules and antigen presentation by DCs [Citation17]. Interestingly, the autophagy machinery also contributes to MHC class II presentation of endocytosed MOG35-55 antigen. In support, a previous study showed suppressed 2D2 T cell responses after treating MOG35-55-pulsed DCs with NH4Cl (an inhibitor of autophagosome fusion with lysosomes), wortmannin (a PI3K inhibitor), or MRT67307 (a ULK1 inhibitor) [Citation4]. However, our results show similar proliferation levels of 2D2 cells cocultured with MOG35-55-primed WT, Pik3c3-, or Rb1cc1-deficient DCs. The underlying reasons for these divergent findings are currently unknown and may involve differences in experimental details and/or generic differences when protein function is disrupted by genetic deletion versus chemical inhibition. Instead, we found that autophagy is required for effective exogenous processing of recombinant MOG1-125 by DCs for presentation to MOG35-55-specific T cells. This finding is in agreement with a previous study showing that Atg16l1-deficient DCs exhibit defective processing of intact ovalbumin proteins for presentation to antigen-specific CD4+ T cells [Citation4]. The mechanism involved in MOG1-125 protein processing by DCs is currently unknown and requires further investigation. Overall, we propose that Pik3c3-deficient DCs exhibit attenuated capacity to reactivate encephalitogenic T cells in vivo, leading to reduced incidence and severity of EAE in DC-specific Pik3c3-deficient mice.
The incomplete protection afforded by conditionally knocking out Pik3c3 in DCs could be due to incomplete deletion of Pik3c3 by Itgax-Cre in plasmacytoid DCs [Citation26], which contribute significantly to priming T cells during EAE [Citation27], or the involvement of other types of antigen-presenting cells that contribute to EAE development [Citation14,Citation28]. Conversely, protection against EAE is unlikely due to defects in the homeostasis of the cDC1 compartment in these mice since batf3−/− mice lacking all peripheral ITGAM/CD11blow/− DCs show robust Th cell priming after subcutaneous immunization and are susceptible to EAE [Citation29].
It is now widely acknowledged that most, if not all, components of the autophagy apparatus can also contribute to non-autophagic functions, including LAP, endocytosis, melanogenesis, cytokinesis, and Golgi apparatus to ER transport [Citation30]. Of note, PIK3C3, BECN1, ATG7, ATG5, and ATG16L1 are shared between the autophagy pathway and LAP, whereas RB1CC1 is involved in autophagy but not LAP [Citation30]. It has recently been proposed that LAP in DCs licenses encephalitogenic CD4+ T cells to initiate and sustain EAE induction [Citation3,Citation31,Citation32]. This conclusion was based on the finding that DC-specific deletion of Cybb/Nox2 or Atg5 inhibits LC3 decoration to phagosomes, antigen presentation to encephalitogenic CD4+ T cells, CNS immune cell invasion, and clinical disease development. However, these data do not conclusively address the involvement of LAP in DCs during EAE for several reasons. First, DCs produce low levels of reactive oxygen species (ROS) and express relatively low levels of CYBB [Citation33]. Second, CYBB loss in DCs could be compensated for by other NOX family members, or possibly other forms of ROS as reported in other cell types [Citation34,Citation35]. Third, reduced CYBB-mediated ROS production in Cybb-deficient DCs may cause defects in neutrophil degranulation, resulting in reduced EAE severity [Citation36]. Moreover, CYBB may influence DC functions in various LAP-independent pathways [Citation37,Citation38]. Finally, deficiency of Cybb causes reduced cellular proliferation and leukocyte accumulation in response to tissue injury [Citation39], which could contribute for the attenuated EAE phenotype in atg5f/f;Itgax-Cre and cybbf/f;Itgax-Cre mice but was not assessed in the studies that suggested a role for LAP [Citation3,Citation31].
We demonstrated that rb1cc1f/f;Itgax-Cre mice exhibit reduced susceptibility to EAE induction. However, mice with systemic deficiency or DC-specific deficiency in RUBCN, which in association with the BECN1-PIK3C3-UVRAG-containing PtdIns3K complex mediates LAP but not autophagy [Citation7], showed WT levels of EAE susceptibility, suggesting that resistance of EAE induction in pik3c3f/f;Itgax-Cre mice is not caused by defective LAP. This conclusion is further supported by a report showing that mice deficient in the gene encoding IRGM1, an inducer of autophagy, are resistant to myelin basic protein-induced EAE [Citation40]. Nevertheless, because PIK3C3 has pleiotropic effects [Citation6], our current results cannot exclude the possibility that resistance to EAE in pik3c3f/f;Itgax-Cre mice is mediated by cellular defects besides canonical autophagy that involve PIK3C3 and other components of the autophagy machinery such as ATG5 or CYBB. We also cannot exclude the possibility that ITGAM+ ITGAX+ myeloid cells contribute to this partial protection. Indeed, we recently showed reduced EAE severity in animals with myeloid cell-specific deficiency of Pik3c3 [Citation14].
In conclusion, our data indicate that PIK3C3 in DCs has a critical role in the effector phase of EAE and potentially other autoimmune diseases. Interestingly, we recently observed a delayed EAE disease progression in mice treated with SAR405 [Citation14], a selective PIK3C3 inhibitor. These findings have important implications for the development of immunotherapies for autoimmune diseases such as MS by targeting PIK3C3 complexes.
Materials and methods
Mice
Pik3c3f/f [Citation10,Citation41], rubcn-/- [Citation7], and Rubcnf/f [Citation42] mice have been described. Rb1cc1f/f mice were obtained from Dr. Jun-Lin Guan (University of Cincinnati, Cincinnati, OH). B6.Cg-Tg(Itgax-cre)1–1Reiz/J (designated Itgax-Cre, Stock No: 008068) and C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J (designated 2D2 TCRMOG, Stock No: 006912) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). DC-specific deletion of Pik3c3, Rubcn, and Rb1cc1 was achieved by crossing the Pik3c3f/f, Rubcnf/f, and Rb1cc1f/f mice with Itgax-Cre transgenic mice. Six- to 12-week-old animals of both sexes were used in this study. All breeder and experimental mice were housed under specific pathogen-free conditions in compliance with guidelines from the Institutional Animal Care and Use Committee at Vanderbilt University.
Preparation of spleen cells
Cells from spleens were obtained by mashing the organ through a 70-μm cell strainer (Fisher Scientific International, Inc, 22–363-548) into RPMI 1640 medium (Fisher Scientific International, Inc, MT10040CM) supplemented with 5% fetal bovine serum (Sigma-Aldrich Corporation, F2442) followed by erythrocyte lysis with ACK lysing buffer (Lonza bioscience,10–548E).
Immunoblotting analyses
DCs from spleen were purified by magnetic sorting (Miltenyi Biotec, 130-100-875) and washed three times with PBS (Corning Cellgro, 21-031-CV) before preparing cellular proteins for western blotting. Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad Laboratories Ltd, 500-0116). Protein was separated by 10–15% SDS-PAGE and transferred to nitrocellulose membranes. Blot membranes were subsequently blocked with 5% milk, incubated with primary and secondary antibodies, and then visualized using ECLTM western blotting detection reagents (Amersham Bioscience) and exposure to the film. Antibodies against PIK3C3 (D9A5), LC3B (2775), SQSTM1 (5114), RUBCN (D9F7), and ACTB (13E5) were purchased from Cell Signaling Technology. Antibody against LAMP1 (1D4B) was from BioLegend.
Gene expression profiling
Fresh DCs were sorted from spleen samples using a BD FACSAria III cell sorter at Vanderbilt Technologies for Advanced Genomics. Sorted cells were pelleted and lysed with RNA lysis buffer from Quick-RNATM MiniPrep (ZYMO Research, R1054) to extract RNA. RNA quantity and purity were measured with an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Total RNA was isolated from biological replicates and provided to the Beijing Genomics Institute for library construction and sequencing following standard protocols. The library products were sequenced using a BGISEQ-500. Briefly, poly-A tailed RNA was purified using paramagnetic oligo-dT beads (Thermo Fisher Scientific, 61002) from 1 μg of total RNA. The purified mRNA was chemically fragmented and reverse transcribed to double-stranded cDNA using random primers. The adapter-ligated DNA library was amplified and sequenced. Standard bioinformatic analysis was performed by the Beijing Genomics Institute. Briefly, gene expression levels were quantified using RSEM after reads cleaning and genome mapping. Differentially expressed genes (DEGs) were detected with DEseq2, which is based on the negative binomial distribution. DEGs were classified according to official classification with the KEGG annotation result. Pathway enrichment analysis was performed using phyper, a function of R. The raw sequence data have been submitted to the Sequence Read Archive under SRA accession: PRJNA557158.
Induction and evaluation of active EAE
Six- to eight-week-old animals were used for EAE induction. Under isoflurane anesthesia, the animals were subcutaneously injected at two sites into the flank with 200 μl of 1 mg/ml MOG35-55 (MEVGWYRSPFSRVVHLYRNGK) peptide (Biomatik, 51716) emulsified in Freund’s complete adjuvant (2 mg/ml Mycobacterium tuberculosis extract H37Ra in incomplete Freund’s adjuvant [BD bioscience, 263910]). Immediately after immunization and 48 h later, all mice received 400 ng of pertussis toxin (Calbiochem, 516560) by i.p. injection. Mice were scored daily in a blind manner according to the following scale: 0, no clinical signs; 0.5, partially limp tail; 1, paralyzed tail; 1.5, paralyzed tail and hind leg inhibition; 2, loss in coordinated movement, hind limb paresis; 2.5, one hind limb paralyzed; 3; both hind limbs paralyzed; 3.5, hind limbs paralyzed, weakness in forelimbs; 4, forelimbs paralyzed; 5, moribund.
Induction of passive EAE
For induction of adoptively transferred EAE, pik3c3f/f;Itgax-Cre, rubcn-/-, rb1cc1f/f;Itgax-Cre, rubcnf/f;Itgax-Cre mice, and their respective littermate controls were used as either donors or recipients for adoptive transfer of T cells. Active EAE was induced in mice as described above. At day 10, spleens were harvested, and single-cell suspensions were prepared. The cells were stimulated with 50 μg/ml MOG35-55 at 1 × 107 cells/ml in 10 ml RPMI 1640 complete medium in a Petri dish. After 72 h, cells were harvested and resuspended in PBS (1 × 108 cells/ml) for adoptive transfer. Recipient mice (8 weeks of age) were irradiated sublethally at 400 rads to generate a lymphopenic environment 6 h prior to injection of MOG35-55-specific T cells. Cells were injected i.p. (300 μl/mouse). The clinical scores were graded similarly to active EAE.
T cell proliferation assay
Splenic DCs from pik3c3f/f;Itgax-Cre, rb1cc1f/f;Itgax-Cre, rubcnf/f;Itgax-Cre and their respective littermate control mice were isolated using a DC isolation kit according to the manufacturer’s protocols. MOG35-55-specific CD4+ T cells were isolated from 2D2 TCR-transgenic mouse splenocytes using a CD4+ T cell isolation kit (Miltenyi Biotec, 130-104-453) according to the manufacturer’s protocols. T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) according to the manufacturer’s instructions (Thermo Fisher Scientific, C34554). DCs were pulsed with 20 μg/ml MOG35-55 or MOG1-125 (AnaSpec Inc, AS-55158-100) for 4 h and cultured with T cells for 96 h at a ratio of 1:5 with media. T cell proliferation was assessed at the end of the culture.
Flow cytometry
In all experiments, dead cells were excluded from the analysis by electronic gating. Fluorescently labeled mAbs against CD3E, CD4, CD8A, CD44, PTPRC, IFNG, IL17A, ITGAM, ITGAX, IL1B, and appropriate isotype controls were obtained from BD Biosciences or eBioscience. For measuring MOG-specific IFNG and IL17A production during EAE, splenocytes or CNS leukocytes from Pik3c3f/f and pik3c3f/f;Itgax-Cre mice induced for EAE (day 10) were cultured with MOG peptide (20 μg/ml) for 18 h in the presence of GolgiStop (BD Biosciences, 554724). Flow cytometric analyses were performed using a Canto II flow cytometer (BD Biosciences). The acquired data were analyzed using FlowJo software (Version 10.0.7, Treestar, Palo Alto, CA).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad Software). Throughout the manuscript, the distribution of data points is expressed as mean ± SEM. EAE clinical scores were analyzed using ANOVA and Kaplan-Meier curves by log-rank test. EAE cumulative score, peak disease score, and cytokine production were analyzed by Student’s t-test, and non-parametric comparisons were performed by the Mann-Whitney U test. Two-tailed Pearson correlation test was applied for the correlation analysis. p < 0.05 was considered significant.
Supplemental Material
Download MS Word (74.5 KB)Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42.
- Bhattacharya A, Parillon X, Zeng S, et al. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J Biol Chem. 2014;289(38):26525–26532.
- Keller CW, Sina C, Kotur MB, et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4 + T cell pathogenicity during CNS inflammation. Proc Natl Acad Sci U S A. 2017;114(52):E11228–E11237.
- Alissafi T, Banos A, Boon L, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest. 2017;127(7):2789–2804.
- Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701.
- Backer JM. The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410(1):1–17.
- Martinez J, Malireddi RKS, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906.
- Fruman DA, Chiu H, Hopkins BD, et al. The PI3K pathway in human disease. Cell. 2017;170(4):605–635.
- Stark AK, Sriskantharajah S, Hessel EM, et al. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol. 2015;23:82–91.
- Parekh VV, Wu L, Boyd KL, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol. 2013;190(10):5086–5101.
- Parekh VV, Pabbisetty SK, Wu L, et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8alpha(+) dendritic cells. Proc Natl Acad Sci U S A. 2017;114(31):E6371–E6380.
- Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A. 2012;109(22):8670–8675.
- McLeod IX, Zhou X, Li Q-J, et al. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J Immunol. 2011;187(10):5051–5061.
- Yang G, Song W, Xu J, et al. Pik3c3 deficiency in myeloid cells imparts partial resistance to experimental autoimmune encephalomyelitis associated with reduced IL-1beta production. Cell Mol Immunol. 2020. DOI:https://doi.org/10.1038/s41423-020-00589-1
- Yang G, Song W, Postoak JL, et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function. Autophagy. 2020:1–12. DOI:https://doi.org/10.1080/15548627.2020.1752979
- Van Kaer L, Parekh VV, Postoak JL, et al. Role of autophagy in MHC class I-restricted antigen presentation. Mol Immunol. 2019;113:2–5.
- Munz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016;37(11):755–763.
- den Haan JM, Bevan MJ. Antigen presentation to CD8+ T cells: cross-priming in infectious diseases. Curr Opin Immunol. 2001;13(4):437–441.
- Giles DA, Duncker PC, Wilkinson NM, et al. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J Clin Invest. 2018;128(12):5322–5334.
- Sun D, Whitaker JN, Huang Z, et al. Myelin antigen-specific CD8 + T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166(12):7579–7587.
- Van Kaer L, Postoak JL, Wang C, et al. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol Immunol. 2019;16(6):531–539.
- Saligrama N, Zhao F, Sikora MJ, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature. 2019;572(7770):481–487.
- Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–17401.
- Nakamura S, Oba M, Suzuki M, et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun. 2019;10(1):847.
- Bonam SR, Wang F, Muller S. Autophagy: a new concept in autoimmunity regulation and a novel therapeutic option. J Autoimmun. 2018;94:16–32.
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204(7):1653–1664.
- Isaksson M, Ardesjö B, Rönnblom L, et al. Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE. Eur J Immunol. 2009;39(10):2925–2935.
- Yogev N, Frommer F, Lukas D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor+ regulatory T cells. Immunity. 2012;37(2):264–275.
- Edelson BT, Bradstreet TR, Kc W, et al. Batf3-dependent CD11b(low/-) peripheral dendritic cells are GM-CSF-independent and are not required for Th cell priming after subcutaneous immunization. PLoS One. 2011;6(10):e25660.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–1699.
- Keller CW, Kotur MB, Mundt S, et al. CYBB/NOX2 in conventional DCs controls T cell encephalitogenicity during neuroinflammation. Autophagy. 2020:1–15. DOI:https://doi.org/10.1080/15548627.2020.1756678
- Keller CW, Munz C, Lunemann JD. Autophagy pathways in CNS myeloid cell immune functions. Trends Neurosci. 2020;43(12):1024–1033.
- Elsen S, Doussière J, Villiers CL, et al. Cryptic O2- -generating NADPH oxidase in dendritic cells. J Cell Sci. 2004;117(Pt 11):2215–2226.
- Pendyala S, Gorshkova IA, Usatyuk PV, et al. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal. 2009;11(4):747–764.
- Touyz RM, Mercure C, He Y, et al. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension. 2005;45(4):530–537.
- Bhattacharya A, Wei Q, Shin J, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. 2015;12(11):1731–1739.
- Kotsias F, Hoffmann E, Amigorena S, et al. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal. 2013;18(6):714–729.
- Cachat J, Deffert C, Alessandrini M, et al. Altered humoral immune responses and IgG subtypes in NOX2-deficient mice and patients: a key role for NOX2 in antigen-presenting cells. Front Immunol. 2018;9:1555.
- Chen Z, Keaney JF, Schulz E, et al. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc Natl Acad Sci U S A. 2004;101(35):13014–13019.
- Xu H, Wu Z-Y, Fang F, et al. Genetic deficiency of Irgm1 (LRG-47) suppresses induction of experimental autoimmune encephalomyelitis by promoting apoptosis of activated CD4 + T cells. FASEB J. 2010;24(5):1583–1592.
- Jaber N, Dou Z, Chen J-S, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109(6):2003–2008.
- Sil P, Suwanpradid J, Muse G, et al. Noncanonical autophagy in dermal dendritic cells mediates immunosuppressive effects of UV exposure. J Allergy Clin Immunol. 2020;145(5):1389–1405.