
ABSTRACT
Different types of autophagy co-exist in all mammalian cells, however, the specific contribution of each of these autophagic pathways to the maintenance of cellular proteostasis and cellular function remains unknown. In this work, we have investigated the consequences of failure of chaperone-mediated autophagy (CMA) in neurons and compared the impact, on the neuronal proteome, of CMA loss to that of macroautophagy loss. We found that these autophagic pathways are non-redundant and that CMA is the main one responsible for maintenance of the metastable proteome (the one at risk of aggregation). We demonstrate that loss of CMA, as the one that occurs in aging, has a synergistic effect with the proteotoxicity associated with neurodegenerative conditions such as Alzheimer disease (AD) and, conversely, that, pharmacological enhancement of CMA is effective in improving both behavior and pathology in two different AD mouse models.
Cellular protein quality control and continuous turnover are essential in post-mitotic cells, such as neurons. Autophagy is one of the surveillance systems that contributes to protein homeostasis (proteostasis) through their degradation in lysosomes. Loss of proteostasis occurs with age and in neurodegenerative diseases such as Alzheimer disease (AD). Defective autophagy has been proposed to contribute to accumulation of protein aggregates in the elderly brain and brains of patients with neurodegenerative conditions
Various types of autophagy, differing in the mechanism of substrate delivery to lysosomes, coexist in most mammalian cells. In chaperone-mediated autophagy (CMA), substrate targeting requires recognition of a pentapeptide motif (KFERQ-like) in the substrate protein by the cytosolic chaperone HSPA8/HSC70. The substrate/chaperone complex binds at the lysosome surface to LAMP2A (lysosomal-associated membrane protein 2A) triggering LAMP2A multimerization into a translocation complex which, after substrate unfolding, mediates its lysosomal internalization for degradation.
In our recent work [Citation1], we investigated the role of CMA in neuronal homeostasis by generating a mouse model with selective deletion of Lamp2a in excitatory neurons (those expressing CAMK2A [calcium/calmodulin-dependent protein kinase II alpha]). CAMK2A-lamp2a-/- (CKL2A-/-) mice display behavioral defects, including short-term memory loss. Histological analysis revealed severe brain proteotoxicity in the form of neuronal protein inclusions, lipofuscin deposits and elevated levels of ubiquitinated and oxidized proteins. Quantitative proteomics of the detergent-insoluble fraction of CKL2A-/- mouse brains revealed an overall switch of the proteome toward insolubility. Interestingly, proteins enriched in the insoluble fraction of CMA-deficient brains display a 20-fold higher supersaturation score indicating that they are part of the metastable proteome (the one at risk of aggregation). Furthermore, we noted that proteins containing KFERQ-like motifs are at higher risk of misfolding, indicating a tight relationship between the collapse of the metastable proteome and CMA deficiency. These findings support the idea that the protein inclusions observed in the brains of CKL2A-/- mice originate from the inability to timely degrade soluble forms of prone-to-aggregate proteins that leads to their subsequent precipitation.
Because accumulation of protein aggregates and neurodegeneration also occur upon blockage of neuronal macroautophagy – a type of autophagy in which cargo sequestered in double-membrane vesicles is delivered to lysosomes through vesicular fusion – we analyzed the status of macroautophagy in CMA-deficient brains. Contrary to the upregulation of macroautophagy previously observed upon CMA blockage in liver, macroautophagy activity was comparable in CKL2A-/-and control littermate mice, indicating the absence of compensatory activation for neuronal CMA loss. Next, to assess the level of redundancy between these two autophagic pathways, we performed comparative proteomics on the insoluble fractions of brains from mice defective in CMA (CKL2A-/-) or macroautophagy (CAMK2A-atg7-/-) in the same neuronal population. Approximately half of the proteins in the inclusions are the same in both mouse groups, while the most enriched proteins are highly selective to a given autophagic pathway. Proteins related to metabolic processes such as glycolysis, calcium metabolism and protein trafficking and assembly are enriched in inclusions from CKL2A-/- mice. Entrapment of these proteins in aggregates leads to their loss of function as we confirmed profound defects, for example in glycolysis in CKL2A-/- neurons. We previously found that blockage of CMA in liver also reduces glycolytic enzymes degradation, but the absence of protein aggregation in this organ (through compensatory activation of other proteolytic systems) preserves the undegraded enzymes in solution and leads instead to increased functional output. Here, we propose a model in which neuronal loss of function upon CMA blockage is due to entrapment of functional proteins in protein aggregates ().
Figure 1. Contribution of chaperone-mediated autophagy (CMA) to neuronal protein homeostasis. (A) Aging and/or pathogenic proteins reduce CMA activity, which in turn lead to the accumulation of undegraded CMA substrates in the form of protein aggregates. Neuronal aggregates upon CMA blockage have two direct consequences: (1) a gain of toxic function due to the presence of the aggregate itself, and (2) a loss function due to the entrapment of functional proteins inside the aggregate and their subsequent loss of function. Furthermore, failure to timely degrade proteins by CMA to terminate their function can lead to dysregulation of other neuronal functions. (B) CMA loss leads to a switch of the proteome toward insolubility, whereas CMA activation, even in the context of preexisting neuropathology, restores proteostasis and contributes to the neuronal defense against proteotoxicity. LAMP2A: lysosomal-associated membrane protein 2A
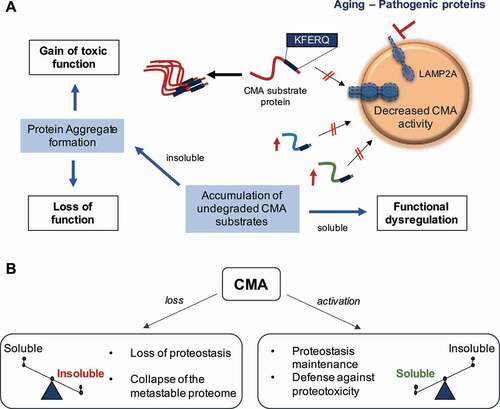
The contribution of CMA to neuronal proteostasis identified in this new study [Citation1] led us to analyze CMA activity in neurodegenerative conditions such as AD. We used transgenic mice expressing the human protein MAPT/tau, involved in AD, and a fluorescent reporter (KFERQ-Dendra) that allows monitoring CMA activity in situ and in vivo. We observed a significant decrease in neuronal CMA activity in the hippocampus of MAPT/tau-expressing mice compared to control littermates. To determine if reduced CMA also occurred in AD patients’ brains, we developed an activity index based on transcriptional expression of genes involved in CMA. Using data from single-nucleus RNA sequencing of brains of AD and healthy individuals, we found a gradual decrease in the CMA transcriptional index of AD patients with disease progression. Interestingly, as in the AD mouse model, CMA changes were more evident in neurons, especially in excitatory ones.
We next proposed that declined CMA with age could contribute to neurodegenerative disease progression. To test this possibility, we imposed CMA loss in a mouse model expressing pathogenic forms of MAPT/tau and APP (amyloid beta (A4) precursor protein) and found that CMA deficiency accelerates the accumulation of these AD-related proteins. Quantitative proteomics revealed that changes induced by CMA loss are synergistic with AD proteotoxicity and that CMA deficiency increases the similarity between the brain proteome of AD mice and AD patients. Thus, CMA loss mimics part of the disease that is not reproduced by the conventional AD mouse models.
The reduced CMA activity in AD brains and the aggravating effect of CMA blockage on AD pathology motivated us to test if CMA activation could be beneficial against AD-related proteotoxicity. We used a newly designed small molecule CMA activator (CA) optimized for in vivo studies. CA has favorable brain permeability and is well-tolerated after months of daily treatment. CA administration to a mouse model expressing mutant MAPT/tau leads to strong beneficial behavioral, histopathological and biochemical effects. In a second AD mouse model that combines both MAPT/tau and amyloid-β pathologies, CA also proves effective in improving both behavior and pathology even when administered after amyloid-β plaque formation (). Thus, CA restores CMA function in both mouse models demonstrating beneficial effects in AD-related pathology.
Altogether, we identified the contribution of CMA to the maintenance of the metastable neuronal proteome in physiological conditions and showed that CMA loss (due to aging and/or pathogenic proteins) accelerates neurodegenerative disease progression. Last, our work highlights the potential value of pharmacological activation of CMA in age-related neurodegenerative diseases, such as AD.
Disclosure statement
AMC and EG are co-founders and scientific advisors of Selphagy Therapeutics now a program of Life Biosciences LLC (Boston, MA, USA).
Reference
- Bourdenx M, Martin-Segura A, Scrivo A, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell. 2021;184(10):2696–2714.e25.