
ABSTRACT
Macroautophagy/autophagy is an evolutionarily conserved pathway responsible for clearing cytosolic aggregated proteins, damaged organelles or invading microorganisms. Dysfunctional autophagy leads to pathological accumulation of the cargo, which has been linked to a range of human diseases, including neurodegenerative diseases, infectious and autoimmune diseases and various forms of cancer. Cumulative work in animal models, application of genetic tools and pharmacologically active compounds, has suggested the potential therapeutic value of autophagy modulation in disease, as diverse as Huntington, Salmonella infection, or pancreatic cancer. Autophagy activation versus inhibition strategies are being explored, while the role of autophagy in pathophysiology is being studied in parallel. However, the progress of preclinical and clinical development of autophagy modulators has been greatly hampered by the paucity of selective pharmacological agents and biomarkers to dissect their precise impact on various forms of autophagy and cellular responses. Here, we summarize established and new strategies in autophagy-related drug discovery and indicate a path toward establishing a more efficient discovery of autophagy-selective pharmacological agents. With this knowledge at hand, modern concepts for therapeutic exploitation of autophagy might become more plausible.
Abbreviations: ALS: amyotrophic lateral sclerosis; AMPK: AMP-activated protein kinase; ATG: autophagy-related gene; AUTAC: autophagy-targeting chimera; CNS: central nervous system; CQ: chloroquine; GABARAP: gamma-aminobutyric acid type A receptor-associated protein; HCQ: hydroxychloroquine; LYTAC: lysosome targeting chimera; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; NDD: neurodegenerative disease; PDAC: pancreatic ductal adenocarcinoma; PE: phosphatidylethanolamine; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol 3-phosphate; PROTAC: proteolysis-targeting chimera; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; SQSTM1/p62: sequestosome 1; ULK1: unc-51 like autophagy activating kinase 1.
Introduction
Autophagy is the collective term covering a number of catabolic pathways that regulate cellular homeostasis via lysosomal degradation and recycling of cytoplasmic components, such as protein aggregates, damaged or unwanted organelles as well as invading pathogens. Three main subtypes of autophagy are microautophagy (direct acquisition of cytosolic cargo via invagination of the limiting membrane of the endolysosomal compartment), chaperone-mediated autophagy (chaperone-mediated recruitment of unfolded proteins to the lysosome via KFERQ-like polypeptide sequences), and macroautophagy (characterized by the double-membrane vesicle, the autophagosome, which fully engulfs the cargo prior to fusion with the lysosome to access the lytic compartment). Macroautophagy is the best-characterized form of mammalian autophagy and is often referred to as simply autophagy (throughout this manuscript, we are using the term “autophagy” when discussing the macroautophagy process).
More than 30 autophagy-related (ATG) gene products govern the formation of the autophagosome, which lies at the heart of autophagy. Disruption of ATG gene function has been linked to profound changes in cellular and tissue homeostasis, cell metabolism, immunity, and development [Citation1]. Biogenesis of autophagosomes can be triggered by diverse stressors, e.g., amino acid starvation, hyperthermia, and hypoxia, as well as under some physiological conditions, e.g. via hormones [Citation2]. At a molecular level, activation of AMP-activated protein kinase (AMPK) and/or inhibition of MTOR (mechanistic target of rapamycin kinase) kinases unleash a cascade of events culminating in the production of a primordial membrane, known as the phagophore, that extends by acquiring additional lipids to form the mature double-membrane autophagosome [Citation3]. Activation of the ULK1 (unc-51 like autophagy activating kinase 1)-ULK2 kinase complex downstream of AMPK and MTOR signaling is considered as the first step (). The ULK1-ULK2 complex activates the class III phosphatidylinositol 3-kinase (PtdIns3K) lipid kinase complex, resulting in the production and accumulation of phosphatidylinositol-3-phosphate (PtdIns3P) on the phagophore membrane and recruitment of phospholipid-binding ATG proteins, such as WIPI1 (WD repeat domain, phosphoinositide interacting 1)-WIPI2 and WDR45B/WIPI3-WDR45/WIPI4 [Citation4,Citation5] ().
Figure 1. Druggable nodes in autophagy. The process of autophagy can be separated into four phases: (a) Initiation, (b) nucleation, (c) elongation and (d) degradation. Each step is amenable to modulation at certain critical nodes of the process by activators (green) and inhibitors of autophagy (red), some of which have reached clinical testing.
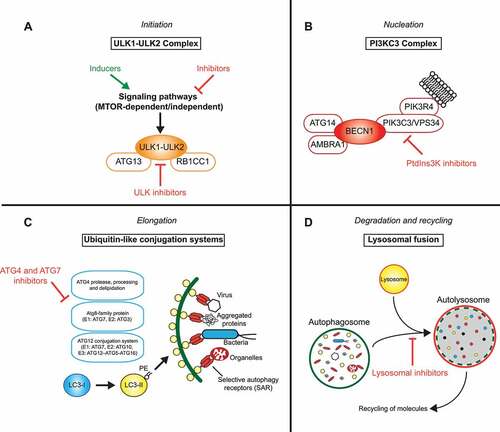
Two ubiquitin-like conjugation systems, ATG12–ATG5 and LC3/GABARAP–phosphatidylethanolamine (PE), are traditionally viewed downstream of the ULK1-ULK2 and PtdIns3K signaling complexes and are involved in the expansion of the phagophore membrane [Citation6] (). The ubiquitin-like proteins, ATG5, ATG12, and members of the LC3/GABARAP protein family share the characteristic globular 3D structure with ubiquitin as well as the ability to be conjugated to protein/lipid substrates. Whereas ATG12 is conjugated to ATG5 co-translationally via the action of E1 (ATG7) and E2 (ATG10) enzymes, LC3/GABARAP proteins are synthesized as precursor proteins that first need to be proteolytically activated by members of the ATG4 cysteine protease family [Citation7]. Through the catalytic activity of E1 (ATG7), E2 (ATG3), and E3 (ATG12–ATG5-ATG16L1 complex) enzymes, soluble LC3 (members of the MAP1LC3 [microtubule associated protein 1 light chain 3] protein family) and GABARAP (members of the GABARAP [GABA type A receptor-associated protein] protein family) are covalently conjugated to the amine group of the lipid PE enriched in the phagophore membrane (). This process mediates expansion and closure of the phagophore membrane. More recently, LC3/GABARAP proteins have been shown to play additional roles in ULK1-ULK2 complex activation [Citation8,Citation9] as well as cargo recognition [Citation10]. They interact with receptor proteins for selective autophagy, such as SQSTM1/p62 (sequestosome 1), NBR1, OPTN, CALCOCO2/NDP52, TAX1BP1 and many others, which simultaneously bind cargo molecules (e.g., protein aggregates, damaged mitochondria or cytosolic bacteria) and mediate targeted autophagosome formation and eventually lysosomal degradation [Citation10] ().
Upon sealing, autophagosomes fuse with lysosomes or late endosomes to form autolysosomes and amphisomes, respectively. Membrane fusion is under the control of soluble NSF attachment protein receptor (SNARE) proteins (e.g., STX17, VAMP8), the homotypic fusion and protein sorting (HOPS) complex, lysosomal membrane proteins (e.g., LAMP2), and RAB GTPases (e.g., RAB5, RAB7) [Citation11]. Transport proteins, such as dyneins, are involved in the movement of autophagosomes along microtubules toward lysosomes or late endosomes [Citation12]. Following the fusion, the entrapped substrates are sequestered by lysosomal acid hydrolases capable of degrading a wide range of biological polymers (proteins, lipids, polysaccharides, and nucleic acids). Metabolic products (i.e. amino acids, fatty acids, simple carbohydrates, and nucleosides) are recycled back to the cytosol to maintain a pool of metabolites required for cell metabolism [Citation13].
Activation of autophagy is an evolutionarily conserved adaptation to extrinsic (environmental) and intrinsic (metabolic) stresses, whose deregulation can lead to various disease states connected either to the failure to adapt cellular metabolism, e.g., during starvation, or to the accumulation of potentially pathogenic substrates, such as misfolded proteins, damaged mitochondria, or cytosolic bacteria. Consequently, defects in autophagy have been established in neurodegenerative diseases (NDDs) [Citation14], cardiovascular diseases [Citation15], infectious and autoimmune diseases [Citation16], and cancer [Citation17]. While details of autophagy involvement in each of the diseases are being explored using a range of genetic tools (such as ATG gene disruption in model organisms), parallel attempts have been made to discover and characterize small molecule compounds capable of activating or inhibiting autophagy. Practical use of autophagy-modulating compounds has been proposed in NDDs, where repurposed drugs capable of autophagy induction have shown some promise in preclinical models [Citation18,Citation19]. On the other hand, autophagy inhibition in various forms of cancer has been proposed with the curative intent both in animal models and in patients [Citation17]. Yet, major progress in this area of drug discovery has been hampered by the paucity of specific assays and target-engagement biomarkers that would guide preclinical and clinical development of novel autophagy-modulating drugs. Furthermore, there is a lack of more selective pharmacological agents with good bioavailability to demonstrate the power of autophagy modulation in various preclinical models. Lack of reliable pharmacodynamic (PD) biomarkers may in particular be linked to the relative lack of progress in autophagy drug discovery compared to other fields.
Here, we review the current status of autophagy drug discovery and development with the focus on preclinical models, assays, tool compounds, and drugs already available. We highlight aspects that can be improved in order to provide potent and selective small molecule compounds. This group advocates that identification and use of more robust autophagy assays and better biomarkers will enable translation and help deliver on the hopes of patients that were sparked by the innumerous discoveries and vindicate the fundamental role autophagy plays in human health and disease.
Relevance of autophagy to human diseases
Cancer
Early studies pointed to the association between the lowered state of autophagy and tumorigenesis, suggesting autophagy as a potent tumor suppressor mechanism. The text book example is BECN1/VPS30/ATG6 (part of the PtdIns3K complex), whose mutations are a frequent event in breast [Citation20] and ovarian cancers [Citation21]. Genetic changes in some other ATG genes, such as allelic loss [Citation22], frameshift mutations [Citation23], and somatic mutations [Citation24] have also been reported. It has been hypothesized, and was indeed shown experimentally, that a significantly reduced level of autophagy can lead to accumulation of dysfunctional mitochondria, increased reactive oxygen species production and DNA mutagenesis, and when coupled with rewired cellular signaling (e.g., increase in NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) signaling [Citation25]), is conducive to neoplastic transformation [Citation26–28]).
Conversely, a great body of research using systemic or organ/tumor-specific loss of Atg genes, such as Atg7 or Rb1cc1/Fip200, in genetically engineered mouse models has demonstrated that functional autophagy is a prerequisite for tumor progression [Citation29]. Here, autophagy has been instrumental in maintaining sufficient levels of critical metabolites, such as tricarboxylic acid cycle (TCA) intermediates [Citation30] and nucleotides [Citation31–33], sustaining mitochondrial respiration [Citation34], and maintaining an inflammatory state [Citation16], thus supporting tumor cell metabolism and survival. More recent work also revealed that non-canonical functions of autophagy, e.g. in unconventional secretion, can be essential for tumor-stromal interactions [Citation35]. Lack of evidence that ATG genes (with the notable exception of BECN1 and some other genes) would be common target of mutagenesis [Citation36] lends additional support to the notion that autophagy is an essential pathway in tumors once they have been established.
The context-dependent roles of autophagy in cancer have been confusing and partially responsible for the lack of a major success in progressing autophagy-modulating compounds toward clinical trials. A recent study using inducible and reversible inhibition of autophagy in mice (shAtg5 mice) attempted to reconcile the various facets of autophagy in tumorigenesis and tumor progression. Thus, in line with its tumor-suppressive role, transient inhibition of autophagy in mice accelerated spontaneous tumor formation; however, reflecting its tumor-promoting role, restoration of its function was critical for the higher incidence of tumors as compared to mice in which autophagy was not reactivated [Citation37].
Current research efforts focus on further understanding the non-canonical roles of autophagy in tumor-host interactions, where additional support for pro-tumorigenic properties of the pathway has been mustered. Autophagic flow has been shown to impinge on a number of pathways, such as endocytosis and secretory pathways, affecting tumor antigen presentation and function of a number of immune cells [Citation38,Citation39]. This renewed interest in autophagy in oncology [Citation40] fuels efforts in drug discovery, and a number of biotech companies have been active in this field in the past few years (e.g., Sprint Bioscience and Vescor Therapeutics, see ).
Table 2. Biotech companies developing autophagy modulators to treat a range of diseases
NDDs
Autophagy fulfills an essential quality-control function inside the cells, which has been clearly linked to aging [Citation41]. Over time, gradual failure of cellular repair mechanisms leads to deterioration of the cell´s functions due to the accumulation of molecular and cellular damage. The cell’s capacity for autophagic degradation has also been found to decrease with age, leading to the loss of the homeostatic function, which may contribute to the aging process [Citation41] and lead to age-related diseases of the central nervous system (CNS) and the cardiovascular system [Citation42,Citation43].
NDDs like Parkinson, Huntington, and amyotrophic lateral sclerosis (ALS) present with mutations that affect the autophagy machinery, e.g., during autophagosome formation or at the cargo receptor level, leading to accumulation of undesired proteins and defective organelles in the cytoplasm of neurons [Citation14,Citation43–46]. In addition, some impaired autophagy markers, such as MTOR activation, autophagosome accumulation and limited degradation of SQSTM1 have been detected in samples from patients with multiple forms of neurodegeneration [Citation47,Citation48]. Examples of genes involved in autophagy and repeatedly found in NDD include PRKN/PARK2 [Citation49], PINK1 [Citation50], LRRK2 [Citation51], PSEN1 [Citation52], SQSTM1 [Citation53], and OPTN [Citation54].
Downregulation of autophagy in several transgenic models was used to reveal the link between autophagy and neurodegeneration. Studies in Drosophila showed that genetic ablation of genes, such as Atg7, Atg5, Atg1/Ulk1, or Atg17/Rb1cc1, results in neurodegeneration [Citation55]. In mice, knockout of Atg7 [Citation56] or Atg5 [Citation57] in CNS, similarly led to neurodegeneration, as manifested by behavioral phenotypes, neuronal cell death and accumulation of ubiquitinated proteins. Evidence exists that stimulation of autophagy can alleviate symptoms related to neurodegeneration in transgenic animal models. Upregulation of autophagy proteins, such as AMPK or Atg8a in Drosophila, or ATG5, ATG7, BECN1 or TFEB (transcription factor EB; a transcriptional regulator of autophagy and lysosome genes) in mice, not only induces autophagy, but also reduces age-associated phenotype/pathologies and extended the lifespan [Citation18,Citation58,Citation59]. Along the same line, pharmacological or genetic upregulation of autophagy is protective in several in vitro and in vivo transgenic models of NDD, as evidenced by reduced accumulation of protein aggregates and improved behavioral tests [Citation18].
A number of startup companies are targeting autophagy in neurodegeneration, e.g., Caraway Therapeutics, Autophagy Neurotherapeutics, PhoreMost, and Casma Therapeutics. ().
Infection, inflammation, and autoimmunity
Autophagy exerts immunomodulatory functions by influencing development and differentiation of cells involved in innate immunity and adaptive immunity as well as by promoting production of pro-inflammatory cytokines (e.g., TNF and IL6) [Citation60,Citation61]. In addition, autophagy-mediated elimination of dysfunctional mitochondria, which limits the release of mitochondrial reactive oxygen species and DNA into the cytosol, can regulate innate immune responses by suppressing inflammasome activation [Citation62].
More directly, autophagy mediates degradation of cytosolic pathogens, including bacteria, viruses, and intracellular protozoa [Citation63]. Mechanisms by which intracellular microbes are detected and directed to autophagic degradation (so-called xenophagy) have been elucidated [Citation64]. For instance, Salmonella, which escapes Salmonella-containing vacuoles into the cytosol, are initially ubiquitinated, followed by the recruitment of selective autophagy receptors SQSTM1, CALCOCO2 (calcium binding and coiled-coil domain 2), and OPTN (optineurin), and are finally engulfed by the phagophore for degradation [Citation65].
Interestingly, the core autophagy machinery can interact with components of the intracellular pathogens and, paradoxically, even be hijacked to support the pathogen´s replication cycle. Thus, RNA viruses including the equine arteritis virus, the hepatitis C virus (HCV), and coronaviruses tend to hijack intracellular membranes to support viral replication [Citation66–68]. Soluble, non-lipidated LC3 is observed on the cytosolic surface of double-membranes reported to support coronavirus replication independently of autophagy [Citation68,Citation69]. Moreover, the ongoing pandemic has led to intensified research on SARS-CoV-2 and other coronaviruses which contributed to our understanding of the interplay between autophagy and COVID-19 pathogenesis [Citation70–72]. Accordingly, some autophagy-essential membrane proteins, e.g. TMEM41B [Citation73] and VMP1 [Citation74] were shown to serve as host factors for SARS-CoV-2 and other coronaviruses. In addition, viral proteins such as ORF3a [Citation75] were shown to interfere with autolysosome formation. Therapeutic strategies targeting non-canonical roles of autophagy proteins may help to improve the efficacy of anti-viral drugs and potentially compromise viral replication. On a side note, understanding the exact mechanism behind how viruses use autophagic pathways for replication could also contribute to the development of therapeutic strategies against pathogenic infections promoting carcinogenesis, such as hepatitis B virus (causing hepatocellular carcinoma) [Citation76] and human papillomavirus (causing cervical carcinoma) [Citation77].
Mutations in autophagy genes have been linked to autoimmune diseases with complex etiology, such as Crohn (associated with ATG16L1 and IRGM gene polymorphism [Citation78],) and systemic lupus erythematosus (associated with ATG5 gene polymorphism [Citation79],). Similar to cancer, the role of autophagy in autoimmune diseases is highly complex and context-specific. For instance, in Crohn, a mild autophagy defect caused by the ATG16L1T300A allele translates into reduced secretory granule size and decreased lysozyme staining in Paneth cells responsible for bacterial killing in the small intestine. Furthermore, dendritic cells isolated from lamina propria of sick mice displayed increased IL1B/IL-1β secretion, associated with increased susceptibility to bacteria-induced inflammation [Citation80]. How known autophagy functions relate to the above findings is presently not clear, but the defect can be linked to an overall elevated pro-inflammatory response normally counterbalanced by active autophagy. Treating autoimmune diseases with autophagy modulators seems to be an active area of research by several early biotech companies, such as Biophagy and Casma Therapeutics ().
Other diseases
Emerging evidence suggests that autophagy can play a central role in the maintenance of homeostasis in wide variety of tissues [Citation81]. Metabolites produced through autophagic activity, such as carbohydrates, amino acids, nucleosides, and fatty acids, are often essential for maintenance of tissue homeostasis. Accordingly, dysregulation of autophagy has been associated with metabolic diseases, such as obesity, diabetes, and myopathies [Citation82]. For instance, previous studies in this area of research have revealed a role for autophagy in the regulation of lipid metabolism and insulin sensitivity [Citation83]. Type II diabetes is characterized by insulin resistance and anomalies in pancreatic islet β-cells. Disruption of Atg7 in mouse β cells resulted in reduced β-cell mass and decreased insulin secretion [Citation84]. In contrast to these findings, the liver-targeted deletion of Atg7 in mice prevented diet-induced obesity and insulin resistance [Citation85]. These results indicate that autophagy impacts differently on the progression of different metabolic symptoms. Therefore, further research is currently being conducted prior to defining precise drug development strategies.
Malfunction of autophagy has been linked to a variety of other important human diseases, including metabolic dysfunction [Citation86], vascular instability [Citation87], cardiomyopathies and myopathies [Citation88,Citation89] and nonalcoholic fatty liver disease [Citation90]. Determining whether the autophagic alterations in these diseases are causative or secondary to pathological changes will potentially shape out new therapeutic approaches in the future.
Small-molecule autophagy modulators
Therapeutic exploitation of autophagy has been considered in parallel to the characterization of its diverse roles in human diseases. Early small molecules that possess autophagy-modulating properties were discovered by serendipity as they interfere with the upstream signaling pathways, such as rapamycin (which inhibits MTOR and unleashes a noticeable autophagy response [Citation91]), wortmannin and 3-methyladenine (inhibit PIK3C3/VPS34 activity and the autophagic flux [Citation92]), or block degradation of the autophagic cargo by interfering with the lysosomal function, such as chloroquine (prevents lysosomal acidification [Citation93]). The use of these compounds to study and target autophagy, even though rapamycin and chloroquine analogs are actively exploited in clinical practice, is somewhat complicated by the fact that they show pleiotropic pharmacologic effects and are considered to be not that specific to autophagy [Citation94,Citation95].
Recently, rational drug design and screening campaigns have delivered more selective agents targeting the core autophagy machinery. These are now used in research and can serve as starting points for drug development. However, their preclinical and clinical development may be hampered by the relative paucity of reliable autophagy assays and target engagement biomarkers. Below, we will discuss several of the small molecule compounds targeting autophagy and summarize important limitations. We will further introduce principles that should govern development of more selective and potent drugs to tackle autophagy in disease. All compound-related information of this paragraph is summarized in , containing alternative names, information on respective primary target(s), solubility, polar surface area and experimentally derived blood-brain barrier penetrating characteristics, most potent autophagy-related efficacy reported in vitro and in vivo and furthest phases of clinical testing.
Table 1. Compound-related information for autophagy modulators
Inhibitors of PI3K-AKT-MTOR signaling axis
The PI3K-AKT-MTOR signaling pathway is downstream of many growth factor receptors and regulates cell proliferation, growth, differentiation, and survival [Citation96]. It continuously inhibits autophagy, and compounds that interfere with kinases constituting this pathway are potent inducers of autophagy.
The well-known MTOR inhibitor rapamycin shows potent activation of autophagy, though its use in the clinic is limited due to poor aqueous solubility and strong immunosuppressive properties [Citation97]. However, rapamycin analogs (so-called rapalogs), tacrolimus (FK-506), temsirolimus (CCI779), everolimus (RAD001), and deforolimus (AP23573), are used in clinical and preclinical settings due to their higher solubility and potency [Citation98]. In therapy, rapamycin and rapalogs have been introduced as immune-suppressive drugs to avoid transplant rejection, and for the therapy of cardiovascular diseases, vasculopathies, angiomyolipomas, gastrointestinal tumors and pancreatic neuroendocrine tumors [Citation99–101]. In particular, temsirolimus was the first FDA-approved MTOR inhibitor for cancer treatment, used as the first-line treatment for advanced renal cell carcinoma patients with poor prognostic features [Citation102]. Studies have encouragingly demonstrated that rapalogs are able to cross the blood-brain barrier, which may endow them with a critical therapeutic advantage when targeting diseases of the CNS [Citation97]. Thus, rapamycin and rapalogs are currently studied in a clinical setting for their ability to alleviate the severity of neuronal loss related to proteinopathies [Citation103].
Pan-MTOR ATP-competitive inhibitors, targeting both MTORC1 and MTORC2 complexes (Torin1 [Citation104], Torin2 [Citation104], PP242 [Citation105,Citation106], PP30 [Citation105], Ku-0063794 [Citation107,Citation108] AZD8055 [Citation109], AZD2014 [Citation110], WYE-354 [Citation111]) have been discovered. Torin1 is a potent, and widely used tool compound inducing autophagy. However, rapalogs were described as cytostatic with less toxicity compared to pan-MTOR inhibitors, thereby more promising in the treatment of neurodegenerative or metabolic diseases [Citation112].
In addition, dual inhibitors of MTOR and PI3K (PI-103 [Citation113], NVP-BGT226 [Citation114,Citation115], NVP-BEZ235 [Citation116], PF-04691502 [Citation117], PKI-587 [Citation118], GDC-0980 [Citation119]) have been identified and characterized. They offer promising alternatives to rapalogs, but their activity on autophagy will still be deprived of necessary specificity, because MTOR-PI3K inhibition will affect other key cellular pathways, such as protein synthesis, apoptosis regulation, immune cell activation and differentiation [Citation120]. Therefore, the toxicity profile of such agents might not be compatible with chronic administration in patients with NDDs, which require autophagy activation.
AKT inhibition provides another key regulatory node for autophagy activation. Screening studies have revealed many synthetic and natural AKT-targeting compounds. Many of these compounds, including ATP-competitive AZD5363 [Citation121], GSK690693 [Citation122], GDC0032 [Citation123], GDC0068 [Citation124] and allosteric antagonist MK-2206 [Citation125], caused robust autophagy stimulation, possibly by inhibiting MTORC1 [Citation126] or by regulating BECN1 [Citation127], and also induced apoptosis. AKT inhibitors showed synergistic anti-tumor activity with lysosomal inhibitors [Citation121,Citation128]. Moreover, perifosine, another autophagy-activating AKT inhibitor targeting the pleckstrin homology domain of AKT, is currently under a phase III clinical trials for the treatment of colorectal cancer (CRC) [Citation129]. However, involvement of autophagy stimulation in the anti-tumor response of AKT inhibitors observed in the clinic still needs to be elucidated.
Unlike MTOR, MTOR-PI3K and AKT inhibitors, compounds that target the PIK3C3 lipid kinase have the potential to inhibit autophagy. A group of pan-PI3K-PtdIns3K inhibitors (i.e. those that target class I and II PI3K, and class III PtdIns3K, including PIK3C3/VPS34, which is the class III PtdIns3K), such as 3-methyladenine [Citation130,Citation131], wortmannin [Citation132], and Ly294002 [Citation133]) have been reported to block autophagy. However, these compounds suffer from poor solubility, and, more importantly, they also activate the apical events in the autophagic flux due to their inhibitory activity against class I PI3K [Citation92]. Of note, new 3-methyladenine derivatives with higher solubility and without inhibiting class I PI3K, and thus improved autophagy inhibition, are now available [Citation130].
Although the aforementioned compounds have helped to understand the importance and therapeutic potential of the PI3K-AKT-MTOR pathway, they are still considered insufficient in terms of chronic autophagy inducers, as they have been linked to important adverse effects in patients, such as infections of the respiratory and urinary tracts, gastrointestinal pain, thrombocytopenia and dyslipidemia [Citation134]. For therapeutic exploitation of autophagy, it is essential to find alternative autophagy inducers and MTOR-independent autophagy modulators, which would be generally favorable and considered safer. For instance, rilmenidine promotes autophagy and mitophagy (selective degradation of mitochondria) in an MTOR-independent manner and in the mutant SOD1 (superoxide dismutase 1) mouse model of ALS via an unknown mechanism [Citation135]. Elucidation of its mode of action might reveal its real potential as an autophagy activator.
AMPK activators
AMPK is a serine/threonine protein kinase that serves as a pleiotropic regulator of intracellular energy homeostasis in response to alterations of AMP/ATP ratio [Citation136]. Activated AMPK regulates autophagy, dependent on both cell type and metabolic conditions. Under glucose starvation, AMPK promotes autophagy by phosphorylating autophagy-related proteins in the MTORC1, ULK1, and PIK3C3 complexes [Citation136]. So far, some direct and indirect modulators of AMPK have been described and associated with autophagy induction. Also, activation of CALM (calmodulin) by intracellular Ca2+ stimulates AMPK signaling [Citation137]. Therefore, any small molecule modulator triggering AMP and Ca2+ accumulation in the cell should have the potential to activate AMPK signaling [Citation138] and autophagy.
Metformin, an FDA-approved anti-diabetic drug, is such an AMPK and autophagy activator. Although metformin is reported to be involved in autophagy induction through regulating AMPK activity [Citation139], some reports also described AMPK-independent activation of autophagy by this drug [Citation140]. Moreover, metformin has been reported to modulate the mitochondrial bioenergetics through inhibiting complex I of the respiratory electron transport chain, which increases the AMP:ATP ratio [Citation141]. Currently, several clinical trials are conducted to explore metformin as a potential drug in cancer and neurodegeneration [Citation142,Citation143].
Newer potent AMPK activators, such as A-769,662 [Citation144], GSK621 [Citation145], PT1 [Citation146], Compound C/dorsomorphin [Citation136], and AICAR [Citation147,Citation148] have been shown to activate autophagy. Also, resveratrol, a natural polyphenol and caloric restriction mimetic [Citation149], is thought to promote autophagy through AMPK-dependent inhibition of MTOR [Citation150]. These compounds help understand the importance and the therapeutic potential of the AMPK-regulated autophagy, but additional studies are required to elucidate whether and how autophagy per se is involved in pathophysiological mechanisms underlying the diseases treated by AMPK activators.
Lysosomal inhibitors
Lysosomal lumen alkalizers act by neutralizing the acidic pH in the lumen of lysosomes, thereby inhibiting various hydrolases that require low pH for their activity [Citation92]. As autophagosomes must fuse with lysosomes or late endosomes to deliver their contents for degradation, small molecules that interfere with the lysosomal function effectively block autophagy at its late stage. This effect can be visualized by accumulation of the autophagic substrate, e.g., misfolded and aggregated proteins, damaged mitochondria, but also by amassing LC3-positive autophagosomes that fail to fuse and be cleared by lysosomes [Citation151,Citation152].
Two main examples of lysosomal lumen alkalizers are chloroquine (CQ) and its less toxic derivative hydroxychloroquine (HCQ), which are both drugs used for treatment of infectious diseases, such as malaria and, more recently, cancer [Citation153]. They are the first and only purported inhibitors of the autophagy pathway approved for clinical use so far. Although short-term CQ/HCQ treatment has been considered safe, some prevalence of toxicity, such as retinopathy and cardiotoxicity, have been reported depending on dosage and duration of exposure [Citation93]. It is thought that CQ and HCQ act as weak bases and deacidify the lysosome [Citation93]. However, some autophagy-independent activities of these agents have been reported, such as disorganization of the Golgi and endo-lysosomal systems [Citation152] and release of cathepsins from lysosomes [Citation154]. Additionally, even though CQ and HCQ often produce a similar level of cytotoxicity compared to genetic inhibition of autophagy, and may increase the efficacy of anti-cancer drugs (e.g., temozolomide, bortezomib, temsirolimus, vorinostat, doxorubicin, etc.) [Citation93], recent reports raised a major concern in that they exhibit autophagy-independent toxicity in the cells. Thorburn et al. showed that CQ sensitized cancer cells against anti-cancer drugs independently from autophagy [Citation94]. Similar findings were made by research teams from AstraZeneca, Novartis, and Pfizer, who showed an autophagy-independent tumor-suppressor activity of CQ in preclinical models [Citation155,Citation156].
Toxicity-associated issues along with the autophagy-independent activities of CQ and HCQ led to the effort of identifying new and more potent CQ derivates to be used as autophagy inhibitors. Lys01, for example, is a dimeric form of CQ exhibiting a 10-fold higher potency in cellular autophagy assays compared to HCQ [Citation157]. Moreover, a water-soluble analog of Lys01, Lys05, was developed as a new lysosomal alkalizer [Citation158]. It increases lysosomal pH more potently compared to HCQ resulting in an impairment of autophagy in vitro and in vivo [Citation157]. Of note, comparative studies with HCQ showed that Lys05 also had higher anti-tumor efficacy both in melanoma and colon cancer xenograft models, with Paneth cell dysfunction and intestinal toxicity observed at high doses in vivo [Citation157]. These results might allow progression of Lys05 to a clinical candidate in the near future.
Other lysosome-targeting autophagy inhibitors include the HCQ/lucanthone derivative ROC-325 [Citation159], the polyether ionophore monensin [Citation160], the antibiotic azithromycin [Citation161], the vacuolar-type H+-ATPase inhibitor bafilomycin A1 [Citation162], as well as lysosomal protease inhibitors E64d, pepstatin A, and leupeptin [Citation163]. However, none of these compounds has been used to target autophagy in a clinical setting due to issues including solubility, toxicity and lack of knowledge about their precise mechanism-of-action.
Compounds targeting autophagy selectively
The next generation of more selective autophagy inhibitors includes compounds that target components of the core autophagy machinery proteins, such as the ULK1 and PIK3C3 kinases, as well as the enzymes involved in LC3/GABARAP–PE conjugation pathway, the ATG4 proteases, and the E1-like enzyme ATG7.
ULK1-ULK2 complex inhibitors: SBI-0206965 was discovered as a small molecule pyrimidine analog ULK1 kinase inhibitor, with biochemical IC50 of 108 nM for ULK1 kinase activity and 711 nM for ULK2 [Citation164]. Notably, rigorous cellular assays confirmed that SBI-0206965 shows good selectivity for ULK1, and inhibits only 10 out of 456 kinases without impairing endogenous PTK2/FAK, AMPK, MTOR, AKT, or MAPK/ERK signaling [Citation164]. Experiments also demonstrated in vivo activity of SBI-0206965, blocking autophagy and providing protection against acute axonal degeneration [Citation165]. Two closely related derivatives with different substitution patterns on the pyrimidine ring, MRT67307 and MRT68921, were identified as selective inhibitors of ULK1-ULK2 kinases [Citation166]. MRT67307 and MRT68921 exert higher potency against ULK1 with IC50 of 45 nM and 2.9 nM, respectively, when compared to SBI-0206965. However, these compounds have poorer selectivity and show activity against a number of kinases [Citation166]. Yet, they are able to inhibit ATG13 phosphorylation and reduce the LC3-II/LC3-I ratio in vitro. Recently, a ULK1 agonist LYN-1604 (EC50 of 18.94 nM) was identified, which increased phosphorylation of ATG13 in an in vitro kinase assay [Citation167]. While further improvement is likely required, these compounds might be useful for attaining a proof-of-principle in developing small molecule inhibitors/activators for the treatment of autophagy-related diseases. One important caveat in exploiting ULK1-ULK2 inhibition is that autophagy may be activated also in the absence of the ULK1-ULK2 complex, e.g., during glucose starvation due to ammonia accumulation [Citation168]. Further validation of the ULK1-ULK2 as a therapeutic target may therefore be necessary to demonstrate under which conditions its inhibition would lead to significant inhibition of the autophagy pathway and a specific therapeutic potential [Citation169].
PIK3C3/VPS34 complex inhibitors: PIK3C3 phosphorylates phosphatidylinositol (PtdIns) at endosomal and autophagosomal membranes to generate PtdIns3P that regulates membrane trafficking processes, such as endocytosis and autophagy [Citation170,Citation171]. In recent years, several co-crystal structures of the PIK3C3 kinase domain with different types of specific inhibitors have been reported and contributed to the rational design of selective PIK3C3 inhibitors [Citation172]. The bis-aminopyrimidine compound VPS34-IN1 is a highly potent cell-permeable PIK3C3 inhibitor with IC50 of 25 nM for the phosphorylation of PtdIns [Citation173]. This compound did not significantly inhibit other kinases, including all isoforms of class I and II PI3Ks, and showed strong and rapid inhibition of PtdIns phosphorylation in cells. VPS34-IN1 has not been used in in vivo studies indicating possible issues with solubility or metabolic stability. Another potent and selective PIK3C3 inhibitor PIK-III (IC50 of 18 nM) showed at least a 100-fold selectivity against related lipid kinases and some additional protein kinases [Citation174]. Co-structure analysis of human PIK3C3 in complex with PIK-III suggested a binding mode over a unique hydrophobic pocket on the ATP-binding site of PIK3C3, which is not present in related kinases such as PIK3CA/PI3Kα. PIK-III robustly inhibits de novo lipidation of LC3 which leads to the inhibition of autophagy and stabilization of autophagy substrates in vitro. Sprint Bioscience has previously shown that their SB02024, ATP-competitive PIK3C3 inhibitor, has a similar PIK3C3 selectivity profile to PIK-III [Citation175]. It inhibits the catalytic function of PIK3C3 by binding in its active site with an IC50 of 14 nM and also blocks PtdIns3P production as shown by the use of a GFP-2xFYVE puncta formation assay. SB02024 completely inhibits turnover of LC3-II in vitro. It significantly reduces tumor growth in vivo with no observed side effects and also sensitizes breast cancer cells to tyrosine kinase inhibitors sunitinib or erlotinib [Citation175,Citation176]. Sanofi reported preclinical work with their PIK3C3 inhibitor SAR405, which is a potent (IC50 1 nM for the phosphorylation of PtdIns) and selective inhibitor with regard to PtdIns3Ks [Citation177]. Recent studies with melanoma and CRC tumor models showed that genetic or pharmacological (SB02024 or SAR405) inhibition of PIK3C3 decreased the tumor growth by enhancing infiltration of immune cells in the tumor bed [Citation176,Citation178]. Combination of PIK3C3 inhibitors with anti-CD274/PD-L1/-PD-1 immune therapy improved the therapeutic benefit and prolonged survival by turning cold tumors hot for immune cells [Citation176,Citation178]. These findings open possibilities for future treatments with PIK3C3 inhibitors as a clinical strategy to enhance the efficacy of immune-oncological drugs. Interestingly, Spautin-1-mediated specific and potent inhibition of deubiquitinating activity of USP10 and USP13 was shown to promote proteasomal degradation of PIK3C3 and thereby inhibit autophagy [Citation179]. The combination of PIK3C3 degrader Spautin-1 with PIK3C3 inhibitors might therefore facilitate quicker and more acute autophagy inhibition and exert synergistic activity in preclinical models.
ATG4 protease inhibitors: ATG4 is the family of four enzymes (ATG4A, ATG4B, ATG4C, and ATG4D) with overlapping specificities for LC3/GABARAP proteins [Citation7,Citation180]. Their inhibition either by genetic ablation, via expression of the dominant-negative ATG4BC74A mutant [Citation181], or pharmacologically leads to a block in LC3/GABARAP processing and lipidation, thereby interfering with autophagosome maturation and cargo engulfment. Representing a tractable class of cellular enzymes (proteases have been successfully drugged in a number of human diseases [Citation182],), targeting ATG4 may be one of the most attractive approaches in modulating autophagy. As of now, the development of specific antagonists of ATG4B (considered to be the major isoform) has been pursued by several groups. Thus, Akin et al. used in silico drug design to identify an ATG4B antagonist capable of inhibiting LC3B-GST cleavage (NSC185058, IC50 of 51 μM) and autophagy both in vitro and in vivo without affecting MTOR or class III PtdIns3K pathways [Citation183]. NSC185058 exhibited antitumor activity both in vitro and in vivo against osteosarcoma cells. Although the LC3B-GST cleavage assay was useful for mechanistic studies, it may not be compatible with a high throughput screening format. Therefore, a time-resolved Förster resonance energy transfer (TR-FRET) assay was developed as a robust platform in which ATG4B inhibitors showed an enhanced FRET signal [Citation184]. On this basis, Z-FA-FMK was identified in a high throughput screen with an IC50 of 14.8 μM. Further chemistry modifications resulted in more potent compounds Z-FG-FMK (IC50 of 1.13 μM) and FMK-9a (IC50 of 0.26 μM) [Citation185,Citation186]. Cell-based luciferase reporter assays confirmed ATG4B inhibitory activity of these compounds, in line with results obtained in the TR-FRET assay. However, Z-FA-FMK and Z-FG-FMK have some selectivity issues, inhibiting several other cysteine proteases, such as CTSB and CAPN (calpain) [Citation184]. Additional ATG4 inhibitors include UAMC-2526 [Citation187], Tioconazole [Citation188], LV-320 [Citation189] and S130 [Citation190] and have been described to show both in vivo and in vitro efficacy on autophagy and tumor cell killing. Some of these compounds will hopefully enter clinical development in the near future, provided they display suitable drug-like profile, including good solubility, metabolic stability, and bioavailability.
ATG7 inhibitors: ATG7 is the E1 enzyme for both ATG12 and LC3/GABARAPs, and its targeting has been tackled by scientists at Takeda for several years. Recently, the results of this campaign have been reported [Citation191]. Pyrazolopyrimidine sulfamates act as potent and selective ATG7 inhibitors and exhibit cellular and in vivo modulation of some autophagic markers, such as inhibition of LC3B puncta formation and accumulation of SQSTM1 aggregates. Some of the compounds possess remarkable cellular potency (e.g., compounds 18, 19 and 37 inhibited LC3B puncta formation in a neuroglioma cell line with IC50 around 50 nM). However, they also reported several issues, such as the lack of an effect on ATG12–ATG5 conjugate formation and some inhibitory effect toward other E1 enzymes. Takeda compounds can serve as tool compounds for further validation of ATG7 as a target, e.g. in cancer. However, the hurdle of rendering the early lead compounds to clinical candidates still seems to be high due to the challenges in translating biochemical inhibition into the full-blown and selective block on the autophagy pathway in cancer cells [Citation191].
Other autophagy modulators
Multiple classes of drug compounds exert an effect on the autophagy pathway. Thus, several Ca2+ channel antagonists, e.g., fluspirilene [Citation192], verapamil [Citation193], and nicardipine [Citation194], and ATP2A/SERCA (ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting) inhibitor thapsigargin [Citation195] were reported to induce autophagic flux in cells. The ubiquitous intracellular messenger Ca2+ has rather a complex impact on autophagy. For instance, although the Ca2+-CALM-AMPK cascade activates autophagy, recent studies have implicated the Ca2+-PtdIns3P receptor-BCL2 axis in autophagy repression [Citation196]. Therefore, careful evaluation of pharmacological properties of Ca2+ signaling modulating compounds is warranted before their use as autophagy drugs can be expanded in the clinic.
Another way to affect autophagy is via various lipid species (e.g., sphingolipids, sterols, and phospholipids), which play important roles in autophagosome formation [Citation197]. In particular, accumulating evidence suggests that activation of PtdIns signaling pathways increases the levels of free inositol and myo-inositol-1,4,5-triphosphate levels, which in turn negatively regulates autophagy [Citation198]. Lithium, which interferes with the PtdIns cycle, leads to depletion of free inositol, and thereby enhances autophagosome formation in cells [Citation199]. A combination approach in enhancing autophagy with MTOR-dependent (rapamycin) and MTOR-independent (lithium) autophagy inducers, may thus represent a promising strategy to reduce the toxicities connected to severe MTOR inhibition [Citation200,Citation201].
Small molecules interfering with the assembly of microtubules (e.g., vinblastine, nocodazole, and cytochalasin B/D) block the maturation of autophagosomes [Citation202]. Conversely, taxol-mediated stabilization of microtubules seems to enable effective autophagosome trafficking, thereby increasing the fusion of autophagosomes with lysosomes [Citation202]. However, these drugs produce many autophagy-independent activities and show severe toxicity. Clearly, less toxic approaches, such as the use of a caloric restriction mimetic, natural polyamine spermidine [Citation203] may provide a greater therapeutic benefit by enhancing autophagy in neurodegenerative models [Citation204].
Use of autophagy modulators in the clinic
Oncology
While potent and selective compounds targeting autophagy are being discovered and developed preclinically, clinical investigators have attempted to take advantage of the only approved autophagy inhibitors, CQ and HCQ, to study the safety and early efficacy of autophagy inhibition across several oncology indications. In preclinical studies, CQ/HCQ alone or in combination with other anti-cancer therapies impaired tumor growth, which has encouraged its use in clinical trials [Citation93].
The initial phase I/II studies were conducted to evaluate tolerability, safety, and early efficacy of CQ/HCQ at suppressing autophagy in human tissues either as mono or combination therapy as well as to provide their pharmacokinetic and pharmacodynamic assessment. These studies demonstrated that approved doses of CQ/HCQ could inhibit autophagy (i.e. increase in numbers of autophagosomes and LC3-II were observed in peripheral blood mononuclear cells) and were well tolerated in most patients [Citation134]. However, some studies also reported that clinical benefit by CQ/HCQ treatment might be blunted by inconsistent and inadequate autophagy inhibition in patients [Citation205,Citation206].
In pancreatic tumors, elevated levels of basal autophagy are observed, and targeting the RAS pathway results in a massive activation of autophagic flux in pancreatic ductal adenocarcinoma cells (PDAC), theoretically making them more sensitive to autophagy inhibitors [Citation207,Citation208]. In vivo combination treatments with HCQ and RAS pathway inhibitors (e.g., trametinib, vemurafenib) resulted in more effective elimination of established tumors in mice bearing patient-derived xenografts of KRAS-mutant pancreatic cancer [Citation209,Citation210]. A recent study also demonstrated that a patient with KRAS-mutated PDAC who had exhausted all approved treatments showed a striking response to HCQ plus a targeted inhibitor of MAP2K/MEK (trametinib) [Citation209]. However, another phase II clinical study in patients with metastatic pancreatic cancer treated with HCQ monotherapy showed inconsistent autophagy inhibition (i.e. various levels of LC3-II accumulation in patient lymphocytes) and resulted in a negligible therapeutic activity [Citation206]. The inefficient activity of HCQ in the study may have occurred due to the advanced metastatic cancer stage of patients enrolled into the study. These results suggested that concurrent inhibition of autophagy along with enzymes of the RAS pathway, instead of either RAS pathway or autophagy targeting monotherapies, might be an appropriate way forward. This approach may also target cancer stem cells, as silencing of autophagy genes (ATG5, ATG7, BECN1) or the administration of CQ markedly reduced the PDAC stem cell population and resistance against the standard-of-care drug gemcitabine in vitro and in vivo [Citation211].
Similar combination studies have been carried out in glioma patients, where BRAF (V600) mutant pediatric glioma cells showed high dependency on autophagy, while BRAF WT gliomas are autophagy-independent [Citation212,Citation213]. Levy et al. have confirmed the context-dependent role of autophagy in glioma tumors, where CQ showed synergistic activity with the BRAF inhibitor vemurafenib in ex vivo tumor samples of glioma patients harboring BRAFV600E mutations but not in BRAF WT cells [Citation213]. Also, the same group showed that administration of CQ after acquisition of vemurafenib-resistance by glioma patients resulted in clinical improvement and decreased growth of metastatic tumor sites [Citation213,Citation214].
Recent studies indicated that histone deacetylase inhibitors can induce autophagy [Citation215]. In a phase I study, the combination of HCQ with the histone deacetylase inhibitor vorinostat showed promising results in patients with advanced solid tumors [Citation216] and colorectal carcinoma (NCT01023737). The combination of HCQ with rapamycin or temsirolimus also exerted limited but encouraging results in clinic [Citation217,Citation218].
More than 70 trials pairing HCQ and CQ with a variety of anti-cancer agents (with different modes of action) and radiotherapy in cancer patients have been completed or are ongoing. However, full translation of preclinical results into clinical practice has yet to be achieved. Although HCQ/CQ addition to the chemotherapy regimen is generally well tolerated, only modest improvement of the progression of cancer was achieved in most cases. The fact that CQ and HCQ lack sufficient specificity to the autophagy pathway [Citation22] may explain this limited clinical success.
Arguably, more selective and potent small molecules targeting core autophagy machinery or parts of the selective autophagy system may allow for a better therapeutic window and demonstration of the clinical proof of concept. Several pharmaceutical companies, such as Vescor Therapeutics, Deciphera Pharmaceuticals, and Sprint Bioscience, are primarily seeking to find more targeted autophagy inhibitors that can be exploited in clinics safely to obtain durable anti-tumor response (). On the other hand, consistent with the context-dependent role of autophagy in cancer, autophagy activation rather than inhibition may sometimes be favorable in certain settings [Citation219]. Some drugs (e.g., gemcitabine) are reported to require autophagy to exert their anti-tumor activity provoking autophagy-associated cell death, which may be of interest in apoptosis-resistant tumors [Citation219–221].
NDDs
The neuroprotective activity of small molecules stimulating autophagic activity in neurons is being heavily investigated in preclinical models. Initial data using MTOR inhibitors, lithium, and repurposed drugs deliver encouraging results [Citation222].
Huntington is caused by a mutation in HTT (huntingtin), the protein product of which is degraded by autophagy [Citation223]. Rapamycin and temsirolimus have been found to attenuate HTT accumulation and cell death in neuronal cell culture and the fruit fly model of Huntington by facilitating autophagosome formation and enhancing lysosomal biogenesis [Citation224]. Moreover, subsequent enhancement of MTOR-independent (lithium) and MTOR-dependent (rapamycin) autophagy pathways exerted greater protection against neurodegeneration in Drosophila compared with the stimulation of either pathway alone [Citation200].
Inspired by the promising preclinical work and strong advocates of this approach [Citation225], several phase 2 clinical trials have now been planned to study the biological and clinical effects of rapamycin on ALS (NCT03359538) and Alzheimer (NCT04629495) patients. Moreover, 95% of sporadic cases of ALS present accumulation of fragmented TARDBP (TAR DNA binding protein) [Citation226], and the activation of autophagy to remove misfolded proteins has been raised as a potential therapeutic strategy. In this regard, therapeutic efficacy of three autophagy inducers on ALS are currently under clinical investigations: rapamycin (NCT03359538), colchicine (NCT03693781) and tamoxifen (NCT02166944).
Upregulation of autophagy by rilmenidine, an MTOR-independent autophagy inducer, improves the severity of the disease course in animal models of Huntington [Citation227]. Therefore, as a new therapeutic approach for Huntington, the safety, tolerability and feasibility of rilmenidine were measured in a small cohort study, where no drug-related adverse events were reported [Citation228] (EudraCT number 2009–018119-14). The efficacy of the treatment is to be tested in larger, randomized, placebo-controlled trials.
Subjective cognitive decline (SCD), which is the individual experience of a decline in cognitive function [Citation229], could evolve into objective cognitive impairments or Alzheimer [Citation230]. In order to assist in the maintenance of brain function, memory protection was studied by the use of spermidine in a phase II pilot trial [Citation231]. This polyamine was reported to maintain memory in aged fruit flies in an autophagy-dependent manner [Citation232]. Likewise, spermidine intake in older adults suffering from SCD slightly enhanced their memory performance and mnemonic discrimination compared to a placebo group [Citation233]. It has also been suggested that dietary intake of spermidine lowers all-cause mortality in a prospective study [Citation234]. Clearly, more work is needed to strengthen the link between spermidine, autophagy induction, and positive effects on well-being and cognition.
Spinocerebellar ataxia type 2 is caused by an CAG expansion in the ATXN2 gene, which is responsible for the oligomerization and cytoplasmic accumulation of ATXN2 (ataxin 2) proteins [Citation235]. The autophagy inducer lithium is used in a clinical trial (NCT00998634) including patients suffering from spinocerebellar ataxia type 2. However, no differences were found in the progression of the disease, although lithium intervention significantly improved the depressive symptoms of the patients.
One concern with the autophagy activators is connected to the association of some forms of autophagy with cell death [Citation236]. It appears that overstimulation of autophagy in some instances can sensitize neurons to apoptosis [Citation237]. Also, the intactness of the lysosomal compartment seems to be crucial to the ability of neurons to degrade cargo supplied by the autophagosomes [Citation238]. Although, some reports suggest that upregulation of the master transcription factor for lysosomal biogenesis TFEB may overcome the lysosomal deficiency and confer neuronal protection in NDDs [Citation239–241].
Infectious diseases
Studies suggest the activation of autophagy via rapamycin and a BECN1-derived peptide can limit the virulence of some viruses, including the human immunodeficiency virus (HIV), West Nile virus and chikungunya virus [Citation242–244]. More recently, we have witnessed a significant interest of using CQ/HCQ for the treatment of SARS-CoV-2. In March 2020, FDA issued an emergency authorization for the use of CQ and its derivative HCQ as experimental treatments for SARS-CoV-2 but revoked this authorization in July 2020 due to the lack of benefits and serious side effects experienced by the patients [Citation245]. Nevertheless, this approach has attracted the attention of many researchers as well as general public to the autophagy field, and there is a substantial newly-generated scientific evidence connecting autophagy to the coronavirus, which allows one to speculate that hijacking autophagy might treat SARS-CoV-2, or ameliorate the infection-related symptoms.
Several concepts emerged in the context of targeting autophagy pathways to treat COVID-19 [Citation246]. Whether this coronavirus actually benefits from autophagy inhibition/activation, and if its modulation will be effective in controlling the viral replication and regulation of the inflammatory response, remains to be further investigated [Citation71].
New drug approaches exploiting selective autophagy
A recent innovative approach in the context of autophagy drug discovery is the use of targeted protein degradation strategies. This milestone had been inspired by the earlier development of a class of small pharmacological agents called proteolysis-targeting chimeras (PROTACs), a rapidly emerging modality in targeted therapeutics [Citation247]. PROTACs comprise two covalently linked protein-binding modules: (1) a ubiquitin E3 ubiquitin ligase-binding ligand and (2) a covalently linked moiety binding the substrate protein. This binary molecule then binds to the target protein and induces its ubiquitination via the E3 ligase (e.g., CRBN or VHL), therefore enabling degradation of the substrate protein by the proteasome [Citation248].
There are some important limitations to the PROTAC technology: these are relatively high molecular weight (>800 Da) compounds with potentially poor cell permeability, lower bioavailability, and blood-brain barrier diffusion coefficient [Citation249]. In the field of NDDs, protein aggregates are often sizable structures (~ 100 nm [Citation250]), which would make them incompatible with the current PROTAC approaches, which rely on protein unfolding in order to enter the narrow barrel-shaped proteasome with a small central pore of only a few nanometers in diameter [Citation251]. Targeting aggregated proteins to the autophagy-lysosome pathway represents an attractive alternative ().
Figure 2. Targeted protein degradation strategies. Visual summary of current alternative strategies (PROTACs, LYTACs, AUTACs and ATTECs) to degrade specific proteins or organelles by exploiting cellular degradation pathways.
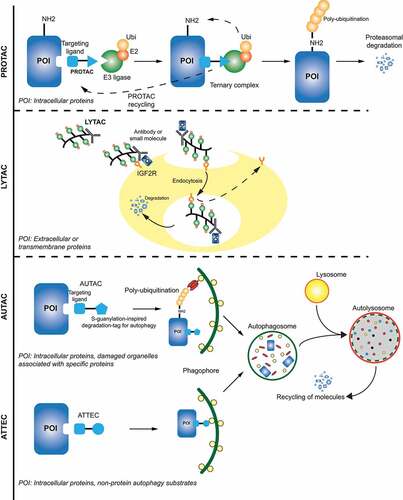
Lysosome targeting chimeras (LYTACs) are early prototypes of a lysosomal targeting molecule that would bring proteins directly to the lysosome. They consist of a small molecule or antibody fused to chemically synthesized glycopeptide ligands recognized by the lysosomal trafficking receptor IGF2R/CI-M6PR (insulin like growth factor 2 receptor) expressed on trans-Golgi network, endosomes and the cell surface. Using this strategy, extracellular (IgG and APOE/APOE4) and membrane-bound (EGFR and TFRC/CD71) proteins could be forced into the lysosomal degradation pathway [Citation252] (). Due to the large size (kDa range) of LYTACs and their dependence on the endosomal pathway to enter the cell, it is so far unclear how this approach could work with intracellular targets, such as protein aggregates characteristic of ALS and other NDDs.
Engagement of the autophagy machinery could offer a way to target autophagosomes to bulky cytosolic cargo. A recent project coordinated by the Arimoto group led to the discovery of autophagy-targeting chimeras (AUTACs), which facilitate the clearance of intracellular disease-related debris through selective autophagy [Citation253] (). AUTACs are made of a guanine degradation tag, which binds to autophagosomal membranes, and a warhead that can target specific intracellular components and accelerate their removal from specific organs. The group successfully targeted fragmented mitochondria in Down syndrome using AUTACs and showed that this improved overall mitochondrial function, which could potentially prevent DS-associated symptoms, such as heart disease, hearing loss and Alzheimer-like dementia. Mechanistically, to induce selective autophagy of their cargo, AUTACs trigger and require the activity of S-guanylation-mediated K63 ubiquitination, which occurs via a KEAP1-dependent mechanism [Citation254,Citation255]. An advantage of AUTACs over PROTACs could be that it can be also applied to large structures, such as invading bacteria, mitochondria, and protein aggregates.
An early proof-of-concept for hijacking selective autophagy mechanisms by small molecules was further provided by the authors which proposed autophagosome-tethering compounds (ATTECs) [Citation256] (). In this approach, a screening and optimization campaign yielded compounds that interacted with both LC3 and mutant HTT proteins [Citation257]. These compounds directed mutant HTT, but not WT HTT proteins, to autophagosomal degradation and rescued disease-relevant phenotypes in cellular and animal models of Huntington. In contrast to PROTAC and AUTAC, ATTEC molecules are independent of ubiquitination. ATTEC drive the protein of interest directly to phagophores by simultaneous binding to the target protein and LC3, which is associated with the phagophore membrane and assists in incorporating the cargo into the nascent autophagosome. Presently, ATTECs have proven to be efficacious in clearance of mutant HTT and other polyQ expansion proteins [Citation256], but future studies are needed to develop a general degradation tool to target other proteins and bulky cargos.
In yet another new approach, Jose et al. attempted to stimulate mitophagy using a rapamycin-mediated FRB-FKBP dimerization system attached to the selective autophagy receptor CALCOCO2. Like other selective autophagy receptors, CALCOCO2 binds both LC3/GABARAP and the cargo (e.g., ubiquitinated mitochondria), thereby mediating targeted autophagosome formation [Citation10]. These investigators showed that FKBP-GFP-CALCOCO2 (mitophagy receptor), dimerized with FRB-FIS1 (mitochondrial protein) in the presence of rapamycin, were capable of inducing mitophagy in the cell [Citation258]. Targeting selective autophagy receptors, rather than LC3/GABARAP, may have the advantage of a greater specificity to a particular target and increase the safety of the approach due to the redundancy of the selective autophagy receptor system [Citation10]. The above studies have improved our understanding of selective autophagy degradation systems. These new concepts might spark clinical interest in the future if selective disease-specific protein degradation is robustly achieved.
Current challenges in design of drugs targeting autophagy
To date, despite the ongoing efforts, there are no autophagy-selective drugs found in clinical development. Clinically useful autophagy-modulating drugs, such as rapamycin and CQ discussed above, are targeting a broad range of molecules and/or pathways and cannot be considered selective to the autophagy pathway. On the other hand, more specific autophagy inhibitors targeting ULK1, PIK3C3, and ATG4 may require further optimization before they can enter clinical assessment. In this respect, it is exciting to see that current efforts yield additional starting points for developing new ULK1 and PIK3C3 inhibitors [Citation259], as much better autophagy-targeting tool compounds are needed not only to develop better drugs (more potent and selective drugs with optimal metabolic stability and cellular permeability), but also to obtain additional preclinical evidence for the significance of autophagy modulation in various disease models. Indeed, results obtained from clinical trials with the current autophagy modulators (mostly CQ and HCQ) are encouraging enough to warrant an investment in the development of potent and safe autophagy modulators to produce robust anti-tumor effects or promote clearance of pathological aggregates in NDDs.
An unsolved issue in current drug discovery approaches in the field of autophagy is the lack of robust autophagy assays and translational biomarkers. The present overreliance on LC3 puncta formation and LC3-based flux assays has the caveat that LC3 has been increasingly associated with a number of novel autophagy-related pathways. Some of those, such as LC3-associated phagocytosis (LAP [Citation260]) and LC3-associated endocytosis (LANDO [Citation261]), also lead to degradation of their substrates in the lysosome, while others, such as non-canonical secretion [Citation262], lead to changes in the proteome but would have no effect on metabolites produced by cargo degradation. In addition, as reviewed before [Citation263], LC3 accumulation cannot discriminate between the induction of an autophagic flux and the inhibition of the lysosomal activity. There is consequently an acute need for specific reporters and markers for canonical autophagy. Selective autophagy receptors, such as SQSTM1 and NBR1, being constantly targeted to the canonical autophagy pathway via their oligomerization and LC3/GABARAP binding, may provide a more reliable system to read out autophagy activity and must be combined with LC3 assays as per current guidelines [Citation264]. However, even SQSTM1 and NBR1 were reported to be degraded via microautophagy-like endosomal pathway [Citation265], so that even more specific markers may be required. Use of powerful comparative proteomics may yield pathway-selective markers in the near future [Citation266].
Translation of a drug effect from in vitro cellular assays to efficacy in preclinical in vivo experiments and, ultimately, to a clinical benefit is the backbone of contemporary evidence-based drug discovery and development. Translational biomarkers should include those for the stratification and selection of the right patient cohort (i.e. predictive biomarker) and for demonstrating target engagement (i.e. pharmacodynamic biomarker) of a given drug. In autophagy-centric drug discovery there is currently a profound lack of both. LC3-positive puncta cannot be reliably used either as predictive or pharmacodynamic biomarkers due to the aforementioned reasons. Utility of SQSTM1 abundance is also limited due to the transcriptional regulation of the protein during certain stress conditions [Citation267]. Therefore, any progress of drug discovery and development in the field of autophagy is contingent upon the advent of new and robust assays and translational biomarkers, also amenable to clinical use.
Concluding remarks
Despite the great progress in our understanding of the autophagy pathway, there is an apparent lack in translating mechanistic studies into clinically active drugs compared to other fields, e.g. growth factor or cytokine signaling. One reason for that is the recent realization that autophagy is a highly complex and branched pathway with a plethora of canonical (e.g., catabolic) and non-canonical (e.g., secretory and trafficking) functions. In addition, seemingly autophagy-specific core ATG proteins have been shown to exert roles outside autophagy pathways (e.g., LC3/GABARAP proteins have scaffolding functions [Citation268]). This growing complexity is not matched by the discovery and validation of differentiating biomarkers that are needed to build the right assays and profile lead compounds to nominate clinical candidates. Modern assay technologies will have to be harnessed to improve the way autophagy-selective drugs are being discovered and validated in the future.
Roles of autophagy in human diseases are manifold, with some validated approaches available based on extensive preclinical work (activation of autophagy in NDD to clear aggregates is one such example [Citation269]). With more private companies () venturing in rigorous drug screening campaigns, we will see an improved pharmacological toolbox being available in the near future to provide additional validation of the therapeutic concept of targeting autophagy in cancer. Here, encouraging preclinical data using autophagy inhibition in combination with agents stimulating the anti-tumor immune response provide an inspiration for modern oncology [Citation176]. It is key for the field that academia and industry join their efforts in providing proof-of-concept for targeting autophagy across many other diseases where the causative role of autophagy dysregulation has been surmised based on early genetic studies.
Chronic pan-autophagy inhibition may come with serious limitations, such as toxicity on multiple organ systems (CNS, muscles, liver, and bone marrow), hypoglycemia, increased infection rates, and potential tumorigenesis as described in mouse models in which key autophagy genes are disabled [Citation270,Citation271]. Therefore, in parallel to the design of better autophagy inhibitors, targeting of selective autophagy pathways should be pursued, which may bring a broader therapeutic window especially in the chronic disease setting. Conversely, overactivation of the autophagy pathway may lead to pathology (overstimulation of autophagy has sensitized some cell types to cell death [Citation237]). Utility of selective autophagy activators, such as AUTACs and ATTECs, may be a safer alternative to pan-autophagy inducers currently poised to enter the clinic [Citation249].
One of the lessons of the ongoing SARS-CoV-2 pandemic has been the previously underestimated value of collaboration between academia, industry, clinicians and regulators, which ensured an unprecedented response to the global public health challenge. The field of autophagy would equally benefit from a coordinated effort of multiple academic, clinical and industrial laboratories. Driving Next Generation of Autophagy Scientists (DRIVE [Citation272]) is one example of such a consortium, which aims at improving the cross-discipline and cross-laboratory collaboration to assess modulation of autophagy as a disease-modifying strategy.
Overcoming the above hurdles in parallel to the ongoing unraveling of autophagy biology will undoubtedly unlock the massive therapeutic potential of targeting autophagy pathways in human disease.
Disclosure of interest
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):349–364.
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Ann Rev Genet. 2009;43(1):67–93.
- Ryter SW, Koo JK, Choi AM. Molecular regulation of autophagy and its implications for metabolic diseases. Curr Opin Clin Nutr Metab Care. 2014 Jul;17(4):329–337.
- Sridharan S, Jain K, Basu A. Regulation of autophagy by kinases. Cancers (Basel). 2011 Jun 9; 3(2):2630–2654.
- Proikas-Cezanne T, Takacs Z, Donnes P, et al. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015 Jan 15 128;(2)207–217.
- Kaufmann A, Wollert T. Scaffolding the expansion of autophagosomes. Autophagy. 2014 Jul;10(7):1343–1345.
- Maruyama T, Noda NN. Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot (Tokyo). 2017 Sep 13.
- Grunwald DS, Otto NM, Park JM, et al. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy. 2020 Apr 16;(4)600–614.
- Birgisdottir AB, Mouilleron S, Bhujabal Z, et al. Members of the autophagy class III phosphatidylinositol 3-kinase complex I interact with GABARAP and GABARAPL1 via LIR motifs. Autophagy. 2019 Aug 15;(8)1333–1355.
- Kirkin V, Rogov VVA. Diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019 Sep; 76(2):21.
- Lorincz P, Juhasz G. Autophagosome-Lysosome Fusion. J Mol Biol. 2020 Apr 3;432(8):2462–2482.
- Kast DJ, Dominguez R. The cytoskeleton-autophagy connection. Curr Biol. 2017 Apr 24; 27(8):R318–R326.
- Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020; 6(1):6.
- Conway O, Akpinar HA, Rogov VV, et al. Selective autophagy receptors in neuronal health and disease. J Mol Biol. 2020 Apr 3 432;(8)2483–2509.
- Gatica D, Chiong M, Lavandero S, et al. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015 Jan 30 116;(3)456–467.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737.
- Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discovery. 2019 Sep;9(9):1167–1181.
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017 Mar 8;93(5):1015–1034.
- Rubinsztein DC, Bento CF, Deretic V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med. 2015 Jun 29; 212(7):979–990.
- Tang H, Sebti S, Titone R, et al. Decreased BECN1 mRNA expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis. EBioMedicine. 2015 Mar 2;(3)255–263.
- Delaney JR, Patel CB, Bapat J, et al. Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet. 2020 Jan;16(1):e1008558.
- Cuomo F, Altucci L, Cobellis G. Autophagy function and dysfunction: potential drugs as anti-cancer therapy. Cancers (Basel). 2019 Sep 29; 11(10):1465.
- Kang MR, Kim MS, Oh JE, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009 Apr 217;(5)702–706.
- Wible DJ, Chao HP, Tang DG, et al. ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 2019; 5(1):42.
- Saito T, Ichimura Y, Taguchi K, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016 Jun 27 7;(1)12030.
- Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017 May 2; 25(5):1037–1043.
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010 May 1; 70(9):3431–3434.
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016 Sep 1; 30(17):1913–1930.
- Mainz L, Rosenfeldt MT. Autophagy and cancer - insights from mouse models. FEBS J. 2018 Mar;285(5):792–808.
- Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014 Jul;4(7):766–772.
- Vanzo R, Bartkova J, Merchut-Maya JM, et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2020 Mar;27(3):1134–1153.
- Elliott IA, Dann AM, Xu S, et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci U S A. 2019 Apr 2 116;(14)6842–6847.
- Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016 Aug 1 30;(15)1704–1717.
- Roca-Agujetas V, De Dios C, Leston L, et al. Recent Insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid Med Cell Longev. 2019 2019;2019: 3809308.
- Goruppi S, Clocchiatti A, Dotto GP. A role for stromal autophagy in cancer-associated fibroblast activation. Autophagy. 2019 Apr;15(4):738–739.
- Lebovitz CB, Robertson AG, Goya R, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11(9):1668–1687.
- Cassidy LD, Young ARJ, Young CNJ, et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat Commun. 2020 Jan 16 11;(1)307.
- Jiang GM, Tan Y, Wang H, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019 Jan 24 18;(1)17.
- Kocaturk NM, Akkoc Y, Kig C, et al. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. 2019 Jun;15(134):116–137.
- Dolgin E. Anticancer autophagy inhibitors attract ‘resurgent’ interest. Nat Rev Drug Discov. 2019 Jun;18(6):408–410.
- Barbosa MC, Grosso RA, Fader CM. Hallmarks of aging: an autophagic perspective. Front Endocrinol (Lausanne). 2019 Jan 9;(9):790.
- Mei Y, Thompson MD, Cohen RA, et al. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. 2015 Feb;1852(2):243–251.
- Corti O, Blomgren K, Poletti A, et al. Autophagy in neurodegeneration: new insights underpinning therapy for neurological diseases. J Neurochem. 2020 Aug;154(4):354–371.
- Stamatakou E, Wrobel L, Hill SM, et al. Mendelian neurodegenerative disease genes involved in autophagy. Cell Discov. 2020;6(1):24.
- Scrivo A, Bourdenx M, Pampliega O, et al. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018 Sep;17(9):802–815.
- Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017 Jul;27(7):491–504.
- Crews L, Spencer B, Desplats P, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010 Feb 19 5;(2)e9313.
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun 118;(6)2190–2199.
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012 Feb 15; 125(4):795–799.
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015 Jan 21; 85(2):257–273.
- Bang Y, Kim KS, Seol W, et al. LRRK2 interferes with aggresome formation for autophagic clearance. Mol Cell Neurosci. 2016 Sep;75:71–80.
- Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25 141;(7)1146–1158.
- Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011 Nov 68;(11)1440–1446.
- Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010 May 13 465;(7295)223–226.
- Kim M, Ho A, Lee JH. Autophagy and human neurodegenerative diseases-A fly’s perspective. Int J Mol Sci. 2017 Jul 23 18;(7)1596.
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006 Jun 15 441;(7095)880–884.
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006 Jun 15 441;(7095)885–889.
- Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009 Oct 28 29;(43)13578–13588.
- Decressac M, Mattsson B, Weikop P, et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013 May 7 110;(19)E1817–26.
- Moscat J, Karin M, Diaz-Meco MT. p62 in Cancer: signaling adaptor beyond autophagy. Cell. 2016 Oct 20; 167(3):606–609.
- Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy. 2018;14(2):190–198.
- Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011 Mar 12;(3)222–230.
- Wileman T. Autophagy as a defence against intracellular pathogens. Essays Biochem. 2013;55:153–163.
- Mao K, Klionsky DJ. Xenophagy: a battlefield between host and microbe, and a possible avenue for cancer treatment. Autophagy 2017 Feb;13(2):223–224.
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014 Feb;12(2):101–114.
- Monastyrska I, Ulasli M, Rottier PJ, et al. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy. 2013 Feb 1 9;(2)164–174.
- Shrivastava S, Devhare P, Sujijantarat N, et al. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J Virol. 2016 Feb 1 90;(3)1387–1396.
- Reggiori F, Monastyrska I, Verheije MH, et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010 Jun 25 7;(6)500–508.
- Zhao Z, Thackray LB, Miller BC, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007 Nov-Dec;3(6):581–585.
- Carmona-Gutierrez D, Bauer MA, Zimmermann A, et al. Digesting the crisis: autophagy and coronaviruses. Microb Cell. 2020 May 4 7;(5)119–128.
- Delorme-Axford E, Klionsky DJ. Highlights in the fight against COVID-19: does autophagy play a role in SARS-CoV-2 infection? Autophagy. 2020 Dec; 16(12):2123–2127.
- Miller K, McGrath ME, Hu Z, et al. Coronavirus interactions with the cellular autophagy machinery. Autophagy. 2020 Dec;16(12):2131–2139.
- Hoffmann HH, Schneider WM, Rozen-Gagnon K, et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell. 2021 Jan 7 184;(1)133–148 e20.
- Schneider WM, Luna JM, Hoffmann HH, et al. Genome-Scale Identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell. 2021 Jan 7 184;(1)120–132 e14.
- Miao G, Zhao H, Li Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. 2021 Feb 22; 56(4):427–442. e5.
- Poh Z, Goh B-BG, Chang P-EJ, et al. Rates of cirrhosis and hepatocellular carcinoma in chronic hepatitis B and the role of surveillance: a 10-year follow-up of 673 patients. Eur J Gastroenterol Hepatol. 2015 Jun 27;(6)638–643.
- Rijkaart DC, Berkhof J, Rozendaal L, et al. Human papillomavirus testing for the detection of high-grade cervical intraepithelial neoplasia and cancer: final results of the POBASCAM randomised controlled trial. Lancet Oncol. 2012 Jan;13(1):78–88.
- Palomino-Morales RJ, Oliver J, Gomez-Garcia M, et al. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes Immun. 2009 Jun 10;(4)356–364.
- Ciccacci C, Perricone C, Alessandri C, et al. Evaluation of ATG5 polymorphisms in Italian patients with systemic lupus erythematosus: contribution to disease susceptibility and clinical phenotypes. Lupus. 2018 Aug 27;(9)1464–1469.
- Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014 May 27 111;(21)7741–7746.
- Migneault F, Hebert MJ. Autophagy, tissue repair, and fibrosis: a delicate balance. Matrix Biol. 2021 Jan 14.
- Moulis M, Vindis C. Autophagy in metabolic age-related human diseases. Cells 2018 Sep 24; 7(10):149.
- Xie Y, Li J, Kang R, et al. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431.
- Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008 Oct;8(4):325–332.
- Kim KH, Jeong YT, Oh H, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013 Jan 19;(1)83–92.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009 Apr 30 458;(7242)1131–1135.
- Ryter SW, Lee SJ, Smith A, et al. Autophagy in vascular disease. Proc Am Thorac Soc. 2010 Feb;7(1):40–47.
- Xie M, Morales CR, Lavandero S, et al. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011 May;26(3):216–222.
- Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009 Dec;10(6):507–515.
- Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol. 2014 Mar;11(3):187–200.
- Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015 Jan;125(1):25–32.
- Vakifahmetoglu-Norberg H, Xia HG, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015 Jan;125(1):5–13.
- Verbaanderd C, Maes H, Schaaf MB, et al. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017;11:781.
- Maycotte P, Aryal S, Cummings CT, et al. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012 Feb 1 8;(2)200–212.
- Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014 Mar 4; 19(3):373–379.
- Xu F, Na L, Li Y, et al. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020;10(1):54.
- Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017 Jul;16(7):487–511.
- Limpert AS, Lambert LJ, Bakas NA, et al. Autophagy in cancer: regulation by small molecules. Trends Pharmacol Sci. 2018 Dec;39(12):1021–1032.
- MacDonald AS, Group RGS. A worldwide, phase III, randomized, controlled, safety and efficacy study of a sirolimus/cyclosporine regimen for prevention of acute rejection in recipients of primary mismatched renal allografts. Transplantation. 2001 Jan 27; 71(2):271–280.
- Waksman R, Ajani AE, Pichard AD, et al. Oral rapamycin to inhibit restenosis after stenting of de novo coronary lesions: the Oral Rapamune to Inhibit Restenosis (ORBIT) study. J Am Coll Cardiol. 2004 Oct 6 44;(7)1386–1392.
- Barbieri F, Albertelli M, Grillo F, et al. Neuroendocrine tumors: insights into innovative therapeutic options and rational development of targeted therapies. Drug Discov Today. 2014 Apr;19(4):458–468.
- Kwitkowski VE, Prowell TM, Ibrahim A, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15(4):428–435.
- Sarkar S, Ravikumar B, Floto RA, et al. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009 Jan;16(1):46–56.
- Liu Q, Xu C, Kirubakaran S, et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013 Apr 15 73;(8)2574–2586.
- Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009 Feb 10 7;(2)e38.
- Janes MR, Limon JJ, So L, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010 Feb 16;(2)205–213.
- Cordaro M, Paterniti I, Siracusa R, et al. KU0063794, a Dual mTORC1 and mTORC2 inhibitor, reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. Mol Neurobiol. 2017 May 54;(4)2415–2427.
- Zhang H, Berel D, Wang Y, et al. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS One. 2013; 8(1):e54918.
- Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010 Jan 1 70;(1)288–298.
- Liao H, Huang Y, Guo B, et al. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res. 2015;5(1):125–139.
- Wang L, Zhu YR, Wang S, et al. Autophagy inhibition sensitizes WYE-354-induced anti-colon cancer activity in vitro and in vivo. Tumour Biol. 2016 Sep 37;(9)11743–11752.
- He S, Li Q, Jiang X, et al. Design of small molecule autophagy modulators: a promising druggable strategy. J Med Chem. 2018 Jun 14 61;(11)4656–4687.
- Park S, Chapuis N, Bardet V, et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia. 2008 Sep 22;(9)1698–1706.
- Simioni C, Cani A, Martelli AM, et al. The novel dual PI3K/mTOR inhibitor NVP-BGT226 displays cytotoxic activity in both normoxic and hypoxic hepatocarcinoma cells. Oncotarget. 2015 Jul 10 6;(19)17147–17160.
- Chang KY, Tsai SY, Wu CM, et al. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res. 2011 Nov 15 17;(22)7116–7126.
- Fan QW, Cheng C, Hackett C, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010 Nov 9 3;(147)ra81.
- Fei HR, Tian H, Zhou XL, et al. Inhibition of autophagy enhances effects of PF-04691502 on apoptosis and DNA damage of lung cancer cells. Int J Biochem Cell Biol. 2016 Sep;78:52–62.
- Mallon R, Feldberg LR, Lucas J, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011 May 15 17;(10)3193–3203.
- Powles T, Lackner MR, Oudard S, et al. Randomized open-label phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016 May 10 34;(14)1660–1668.
- Fruman DA, Chiu H, Hopkins BD, et al. The PI3K pathway in human disease. Cell. 2017;170(4):605–635.
- Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013 Jul;9(7):1119–1120.
- Jeong EH, Choi HS, Lee TG, et al. Dual Inhibition of PI3K/Akt/mTOR pathway and role of autophagy in non-small cell lung cancer cells. Tuberc Respir Dis (Seoul). 2012 Apr 72;(4)343–351.
- Zorea J, Prasad M, Cohen L, et al. IGF1R upregulation confers resistance to isoform-specific inhibitors of PI3K in PIK3CA-driven ovarian cancer. Cell Death Dis. 2018 Sep 20 9;(10)944.
- Lin J, Sampath D, Nannini MA, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013 Apr 1 19;(7)1760–1772.
- Cheng Y, Ren X, Zhang Y, et al. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011 Apr 1 71;(7)2654–2663.
- Dan HC, Ebbs A, Pasparakis M, et al. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J Biol Chem. 2014 Sep 5 289;(36)25227–25240.
- Wang RC, Wei Y, An Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012 Nov 16 338;(6109)956–959.
- Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008 Oct 6 183;(1)101–116.
- Richardson PG, Eng C, Kolesar J, et al. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin Drug Metab Toxicol. 2012 May;8(5):623–633.
- Wu Y, Wang X, Guo H, et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy. 2013 Apr 9;(4)595–603.
- Jing CH, Wang L, Liu PP, et al. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012 Jun;213(213):144–153.
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, et al. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996 Apr 16;(4)1722–1733.
- Gharbi SI, Zvelebil MJ, Shuttleworth SJ, et al. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007 May 15 404;(1)15–21.
- Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017 Jun 16; 18(6):6.
- Perera ND, Sheean RK, Lau CL, et al. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy. 2018;14(3):534–551.
- Jang M, Park R, Kim H, et al. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci Rep. 2018 Aug 23 8;(1)12637.
- Cardenas C, Foskett JK. Mitochondrial Ca(2+) signals in autophagy. Cell Calcium. 2012 Jul; 52(1):44–51.
- Guigas B, Viollet B. Targeting AMPK: from ancient drugs to new small-molecule activators. Exp Suppl. 2016;107:327–350.
- Wang Y, Xu W, Yan Z, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res. 2018 Mar 20 37;(1)63.
- Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011 Apr;9(1):33.
- Fontaine E. Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol (Lausanne). 2018;9:753.
- Rotermund C, Machetanz G, Fitzgerald JC. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol (Lausanne). 2018;9:400.
- Saraei P, Asadi I, Kakar MA, et al. The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manag Res. 2019;11:3295–3313.
- Walter C, Clemens LE, Muller AJ, et al. Activation of AMPK-induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology. 2016 Sep;108:24–38.
- Liu W, Mao L, Ji F, et al. Targeted activation of AMPK by GSK621 ameliorates H2O2-induced damages in osteoblasts. Oncotarget. 2017 Feb 7 8;(6)10543–10552.
- Huang L, Dai K, Chen M, et al. The AMPK Agonist PT1 and mTOR Inhibitor 3HOI-BA-01 Protect Cardiomyocytes After Ischemia Through Induction of Autophagy. J Cardiovasc Pharmacol Ther. 2016 Jan 21;(1)70–81.
- Lee JW, Park S, Takahashi Y, et al. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010 Nov 3 5;(11)e15394.
- Robert G, Ben Sahra I, Puissant A, et al. Acadesine kills chronic myelogenous leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PLoS One. 2009 Nov 18 4;(11)e7889.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006 Jun;5(6):493–506.
- Pineda-Ramirez N, Alquisiras-Burgos I, Ortiz-Plata A, et al. Resveratrol activates neuronal autophagy through AMPK in the Ischemic Brain. Mol Neurobiol. 2020 Feb;57(2):1055–1069.
- Redmann M, Benavides GA, Berryhill TF, et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017 Apr;11:73–81.
- Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455.
- Ding ZB, Hui B, Shi YH, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res. 2011 Oct 1 17;(19)6229–6238.
- Boya P, Gonzalez-Polo RA, Poncet D, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003 Jun 19 22;(25)3927–3936.
- Eng CH, Wang Z, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016 Jan 5 113;(1)182–187.
- King MA, Ganley IG, Flemington V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene. 2016 Aug 25; 35(34):4518–4528.
- McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012 May 22 109;(21)8253–8258.
- Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 2012 Sep; 8(9):1383–1384.
- Carew JS, Espitia CM, Zhao W, et al. Disruption of Autophagic Degradation with ROC-325 Antagonizes Renal Cell Carcinoma Pathogenesis. Clin Cancer Res. 2017 Jun 1 23;(11)2869–2879.
- Choi HS, Jeong EH, Lee TG, et al. Autophagy Inhibition with monensin enhances cell cycle arrest and apoptosis induced by mTOR or Epidermal Growth Factor Receptor Inhibitors in Lung Cancer Cells. Tuberc Respir Dis (Seoul). 2013 Jul;75(1):9–17.
- Renna M, Schaffner C, Brown K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011 Sep;121(9):3554–3563.
- Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998 Feb 23;(1)33–42.
- Yang YP, Hu LF, Zheng HF, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013 May;34(5):625–635.
- Egan DF, Chun MG, Vamos M, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015Jul 1659;(2)285–297.
- Vahsen BF, Ribas VT, Sundermeyer J, et al. Inhibition of the autophagic protein ULK1 attenuates axonal degeneration in vitro and in vivo, enhances translation, and modulates splicing. Cell Death Differ. 2020 Oct 27;(10)2810–2827.
- Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015 May 1 290;(18)11376–11383.
- Zhang L, Fu L, Zhang S, et al. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem Sci. 2017 Apr 1 8;(4)2687–2701.
- Cheong H, Lindsten T, Wu J, et al. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011 Jul 5 108;(27)11121–11126.
- Liu L, Yan L, Liao N, et al. A review of ULK1-mediated autophagy in drug resistance of cancer. Cancers (Basel). 2020 Feb 4 12;(2)352.
- Ohashi Y, Tremel S, Williams RL. VPS34 complexes from a structural perspective. J Lipid Res. 2019 Feb; 60(2):229–241.
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol.2010 Jun20;(6):355–62.
- Miller S, Tavshanjian B, Oleksy A, et al. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010 Mar 26 327;(5973)1638–1642.
- Bago R, Malik N, Munson MJ, et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J. 2014 Nov 1 463;(3)413–427.
- Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014 Nov 16;(11)1069–1079.
- Dyczynski M, Yu Y, Otrocka M, et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018 Oct 28;435:32–43.
- Noman MZ, Parpal S, Van Moer K, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv. 2020 May;6(18):eaax7881.
- Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014 Dec 10;(12)1013–1019.
- Janji B, Hasmim M, Parpal S, et al. Firing up the cold tumors by targeting Vps34. Oncoimmunology. 2020 Aug 31 9;(1)1809936.
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011 Sep 30 147;(1)223–234.
- Agrotis A, On KR. ATG4B as drug target for treatment of solid tumours-the knowns and the unknowns. Cells. 2019 Dec 24;91.
- Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008 Nov 19;(11)4651–4659.
- Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010 Sep;9(9):690–701.
- Akin D, Wang SK, Habibzadegah-Tari P, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10(11):2021–2035.
- Qiu Z, Kuhn B, Aebi J, et al. Discovery of Fluoromethylketone-Based Peptidomimetics as Covalent ATG4B (Autophagin-1) Inhibitors. ACS Med Chem Lett. 2016 Aug 11 7;(8)802–806.
- Xu D, Xu Z, Han L, et al. Identification of New ATG4B inhibitors based on a novel high-throughput screening platform. SLAS Discov. 2017 Apr 22;(4)338–347.
- Chu J, Fu Y, Xu J, et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch Biochem Biophys. 2018 Apr 15;644:29–36.
- Kurdi A, Cleenewerck M, Vangestel C, et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem Pharmacol. 2017 Aug 15;138:150–162.
- Liu PF, Tsai KL, Hsu CJ, et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 inhibitor that suppresses autophagy and sensitizes cancer cells to chemotherapy. Theranostics. 2018;8(3):830–845.
- Bosc D, Vezenkov L, Bortnik S, et al. A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease ATG4B. Sci Rep. 2018 Aug 3 8;(1)11653.
- Fu Y, Hong L, Xu J, et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019 Feb 15;(2)295–311.
- Huang SC, Adhikari S, Brownell JE, et al. Discovery and optimization of pyrazolopyrimidine sulfamates as ATG7 inhibitors. Bioorg Med Chem. 2020 Oct 1 28;(19)115681.
- Xia HG, Zhang L, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010 Jan 6;(1)61–66.
- Kania E, Pajak B, O’Prey J, et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. FEBS J. 2017 May;284(9):1370–1387.
- Ochi M, Kawai Y, Tanaka Y, et al. Characterization of nicardipine hydrochloride-induced cell injury in human vascular endothelial cells. J Toxicol Sci. 2015 Feb;40(1):71–76.
- Ganley IG, Wong PM, Gammoh N, et al. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011 Jun 24 42;(6)731–743.
- Sun F, Xu X, Wang X, et al. Regulation of autophagy by Ca(2). Tumour Biol. 2016 Nov 1837;(12)15467–15476.
- Martens S, Nakamura S, Yoshimori T. Phospholipids in Autophagosome Formation and Fusion. J Mol Biol. 2016 Oct 27428;(24 Pt A)4819–4827.
- Motoi Y, Shimada K, Ishiguro K, et al. Lithium and autophagy. ACS Chem Neurosci. 2014 Jun 185;(6)434–442.
- Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005 Sep 26 170;(7)1101–1111.
- Sarkar S, Krishna G, Imarisio S, et al. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet. 2008 Jan 15 17;(2)170–178.
- Pupyshev AB, Tikhonova MA, Akopyan AA, et al. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol Biochem Behav. 2019 Feb;177:1–11.
- Mackeh R, Perdiz D, Lorin S, et al. Autophagy and microtubules - new story, old players. J Cell Sci.2013 Mar 1 126;(5)1071–1080.
- Madeo F, Bauer MA, Carmona-Gutierrez D, et al. Spermidine: a physiological autophagy inducer acting as an anti-aging vitamin in humans? Autophagy. 2019 Jan;15(1):165–168.
- Singh S, Kumar R, Garg G, et al. Spermidine, a caloric restriction mimetic, provides neuroprotection against normal and D-galactose-induced oxidative stress and apoptosis through activation of autophagy in male rats during aging. Biogerontology. 2021 Feb;22(1):35–47.
- Rosenfeld MR, Ye X, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014 Aug;10(8):1359–1368.
- Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. The Oncologist. 2014 Jun;19(6):637–638.
- Yang S, Kimmelman AC. A critical role for autophagy in pancreatic cancer. Autophagy. 2011 Aug;7(8):912–913.
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011 Apr 1 25;(7)717–729.
- Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019 Apr 25;(4)620–627.
- Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019 Apr;25(4):628–640.
- Yang MC, Wang HC, Hou YC, et al. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer. 2015 Oct;14(1):179.
- Levy JM, Thorburn A. Modulation of pediatric brain tumor autophagy and chemosensitivity. J Neurooncol. 2012 Jan;106(2):281–290.
- Levy JM, Thompson JC, Griesinger AM, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014 Jul 4;(7)773–780.
- Mulcahy Levy JM, Zahedi S, Griesinger AM, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017 Jan;17:6.
- Mrakovcic M, Kleinheinz J, Frohlich LF. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: implications for p53. Int J Mol Sci. 2017 Aug 31; 18(9):9.
- Mahalingam D, Mita M, Sarantopoulos J, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014 Aug;10(8):1403–1414.
- Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014 Aug;10(8):1391–1402.
- Chi MS, Lee CY, Huang SC, et al. Double autophagy modulators reduce 2-deoxyglucose uptake in sarcoma patients. Oncotarget. 2015 Oct 6 6;(30)29808–29817.
- Masuelli L, Granato M, Benvenuto M, et al. Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overexpressing breast cancer cells in vitro and in vivo in nude mice while counteracts it in immune competent mice. Oncoimmunology. 2017;6(11):e1356151.
- Mukubou H, Tsujimura T, Sasaki R, et al. The role of autophagy in the treatment of pancreatic cancer with gemcitabine and ionizing radiation. Int J Oncol. 2010 Oct;37(4):821–828.
- Zhu Y, Xian X, Wang Z, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. 2018 Aug 238;(3)3.
- Suresh SN, Chakravorty A, Giridharan M, et al. Pharmacological Tools to Modulate Autophagy in Neurodegenerative Diseases. J Mol Biol. 2020 Apr 3 432;(8)2822–2842.
- Ravikumar B, Rubinsztein DC. Role of autophagy in the clearance of mutant huntingtin: a step towards therapy? Mol Aspects Med. 2006 Oct-Dec;27(5–6):520–527.
- Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004 Jun;36(6):585–595.
- Kaeberlein M, Rapamycin GV. Alzheimer’s disease: time for a clinical trial? Sci Transl Med. 2019 Jan 23; 11(476):476.
- Prasad A, Bharathi V, Sivalingam V, et al. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019;12:25.
- Rose C, Menzies FM, Renna M, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010 Jun 1 19;(11)2144–2153.
- Underwood BR, Green-Thompson ZW, Pugh PJ, et al. An open-label study to assess the feasibility and tolerability of rilmenidine for the treatment of Huntington’s disease. J Neurol. 2017 Dec;264(12):2457–2463.
- Jessen F, Amariglio RE, Buckley RF, et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020 Mar;19(3):271–278.
- Bessi V, Mazzeo S, Padiglioni S, et al. From subjective cognitive decline to alzheimer’s disease: the predictive role of neuropsychological assessment, personality traits, and cognitive reserve. A 7-Year Follow-Up Study. J Alzheimers Dis. 2018;63(4):1523–1535.
- Wirth M, Benson G, Schwarz C, et al. The effect of spermidine on memory performance in older adults at risk for dementia: a randomized controlled trial. Cortex 2018 Dec;109:181–188.
- Gupta VK, Scheunemann L, Eisenberg T, et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci. 2013 Oct;16(10):1453–1460.
- Wirth M, Schwarz C, Benson G, et al. Effects of spermidine supplementation on cognition and biomarkers in older adults with subjective cognitive decline (SmartAge)-study protocol for a randomized controlled trial. Alzheimers Res Ther. 2019 May 1 11;(1)36.
- Kiechl S, Pechlaner R, Willeit P, et al. Higher spermidine intake is linked to lower mortality: a prospective population-based study. Am J Clin Nutr. 2018 Aug 1108;(2)371–380.
- Alves-Cruzeiro JM, Mendonca L, Pereira De Almeida L, et al. Motor dysfunctions and neuropathology in mouse models of spinocerebellar Ataxia Type 2: a comprehensive review. Front Neurosci. 2016;10:572.
- Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J Cell Sci. 2018 Sep 20; 131(18):18.
- Button RW, Luo S, Rubinsztein DC. Autophagic activity in neuronal cell death. Neurosci Bull. 2015 Aug;31(4):382–394.
- Malik BR, Maddison DC, Smith GA, et al. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019 Nov 29;12(1):100.
- Xiao Q, Yan P, Ma X, et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci. 2015 Sep 2 35;(35)12137–12151.
- Arotcarena ML, Bourdenx M, Dutheil N, et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight. 2019 Aug 22 4;(16)16.
- Torra A, Parent A, Cuadros T, et al. Overexpression of TFEB drives a pleiotropic neurotrophic effect and prevents parkinson’s disease-related neurodegeneration. Mol Ther. 2018 Jun 6 26;(6)1552–1567.
- Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013 Feb 14 494;(7436)201–206.
- Roy J, Paquette JS, Fortin JF, et al. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother. 2002 Nov 46;(11)3447–3455.
- Nicoletti F, Lapenta C, Donati S, et al. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin Exp Immunol. 2009 Jan 155;(1)28–34.
- Boulware DR, Pullen MF, Bangdiwala AS, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. 2020 Aug 6 383;(6)517–525.
- Shojaei S, Suresh M, Klionsky DJ, et al. Autophagy and SARS-CoV-2 infection: apossible smart targeting of the autophagy pathway. Virulence. 2020 Dec 11;(1)805–810.
- Nalawansha DA, Crews CM. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem Biol. 2020 Aug 20; 27(8):998–1014.
- Sakamoto KM, Kim KB, Kumagai A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001 Jul 17 98;(15)8554–8559.
- Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020 Jul;41(7):464–474.
- Bauerlein FJB, Fernandez-Busnadiego R, Baumeister W. Investigating the structure of neurotoxic protein aggregates inside cells. Trends Cell Biol. 2020 Dec;30(12):951–966.
- Wolf DH, Hilt W. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004 Nov 29; 1695(1–3):19–31.
- Banik SM, Pedram K, Wisnovsky S, et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020 Aug;584(7820):291–297.
- Takahashi D, Moriyama J, Nakamura T, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019 Dec 576;(5)797–810 e10.
- Sawa T, Zaki MH, Okamoto T, et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3ʹ,5ʹ-cyclic monophosphate. Nat Chem Biol. 2007 Nov 3;(11)727–735.
- Ito C, Saito Y, Nozawa T, et al. Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Mol Cell. 2013 Dec 26 52;(6)794–804.
- Li Z, Zhu C, Ding Y, et al. ATTEC: a potential new approach to target proteinopathies. Autophagy. 2020 Jan;16(1):185–187.
- Li Z, Wang C, Wang Z, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019 Nov 575;(7781)203–209.
- Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell. 2019 Apr 18 74;(2)347–362 e6.
- Zachari M, Rainard JM, Pandarakalam GC, et al. The identification and characterisation of autophagy inhibitors from the published kinase inhibitor sets. Biochem J. 2020 Feb 28 477;(4)801–814.
- Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci. 2019 Feb 20;132;(5)536:–jcs222984.
- Heckmann BL, Teubner BJW, Tummers B, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in Murine Alzheimer’s Disease. Cell. 2019 Jul 25 178;(3)536–551 e14.
- Leidal AM, Huang HH, Marsh T, et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol. 2020 Feb 22;(2)187–199.
- Yoshii SR, Mizushima N. Monitoring and Measuring Autophagy. Int J Mol Sci. 2017 Aug 28; 18(9):1865.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021 Feb;8:1–382.
- Mejlvang J, Olsvik H, Svenning S, et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol. 2018 Oct 1 217;(10)3640–3655.
- Zellner S, Schifferer M, Behrends C. Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Mol Cell. 2021 Mar; 18 81(6):1337–1354.e8.
- Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014 Mar; 10(3):431–441.
- Habisov S, Huber J, Ichimura Y, et al. Structural and functional analysis of a novel interaction motif within UFM1-activating Enzyme 5 (UBA5) Required for Binding to Ubiquitin-like Proteins and Ufmylation. J Biol Chem. 2016 Apr 22 291;(17)9025–9041.
- Zatyka M, Sarkar S, Barrett T. Autophagy in rare (nonlysosomal) neurodegenerative diseases. J Mol Biol. 2020 Apr 3; 432(8):2735–2753.
- Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017 Oct 3; 13(10):1619–1628.
- Kim KW, Hwang M, Moretti L, et al. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy. 2008 Jul 4;(5)659–668.
- Kraft C, Boya P, Codogno P, et al. Driving next-generation autophagy researchers towards translation (DRIVE), an international PhD training program on autophagy. Autophagy. 2019 Feb;15(2):347–351.
- Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem. 2006,49; 49(26):7559–7583.