
ABSTRACT
The eukaryotic-type protein kinase G (PknG), one of the eleven eukaryotic type serine-threonine protein kinase (STPK) in Mycobacterium tuberculosis (Mtb), is involved in mycobacterial survival within macrophages, presumably by suppressing phagosome and autophagosome maturation, which makes PknG an attractive drug target. However, the exact mechanism by which PknG inhibits pathogen clearance during mycobacterial infection remains largely unknown. Here, we show that PknG promotes macroautophagy/autophagy induction but inhibits autophagosome maturation, causing an overall effect of blocked autophagy flux and enhanced pathogen intracellular survival. PknG prevents the activation of AKT (AKT serine/threonine kinase) via competitively binding to its pleckstrin homology (PH) domain, leading to autophagy induction. Remarkably, PknG could also inhibit autophagosome maturation to block autophagy flux via targeting host small GTPase RAB14. Specifically, PknG directly interacts with RAB14 to block RAB14-GTP hydrolysis. Furthermore, PknG phosphorylates TBC1D4/AS160 (TBC1 domain family member 4) to suppress its GTPase-activating protein (GAP) activity toward RAB14. In macrophages and in vivo, PknG promotes Mtb intracellular survival through blocking autophagy flux, which is dependent on RAB14. Taken together, our data unveil a dual-functional bacterial effector that tightly regulates host autophagy flux to benefit pathogen intracellular survival.
Abbreviations: AKT: AKT serine/threonine kinase; ATG5: autophagy related 5; BMDMs: bone marrow-derived macrophages; DTT: dithiothreitol; FBS: fetal calf serum; GAP: GTPase-activating protein; MOI: multiplicity of infection; Mtb: Mycobacterium tuberculosis; MTOR: mechanistic target of rapamycin kinase; OADC: oleic acid-albumin-dextrose-catalase; PC, phosphatidylcholine; PH: pleckstrin homology; PI3K: phosphoinositide 3-kinase; PknG: protein kinase G; PtdIns(3,4,5)P3: phosphatidylinositol(3,4,5)-trisphosphate; SQSTM1: sequestosome 1; STPK: serine-threonine protein kinase; TB: tuberculosis; TBC1D4: TBC1 domain family member 4; TPR: tetratricopeptide repeat; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type
Introduction
Tuberculosis (TB) is a global health threat caused by the pathogen Mycobacterium tuberculosis (Mtb) [Citation1], which is an intracellular pathogen that secretes various effector proteins to evade host immune clearance in order to survive in macrophages [Citation2–8]. However, the detailed molecular mechanisms underlying Mtb-host interactions still remain largely unclear, while such information is crucial for developing novel anti-TB treatments to maximize therapeutic benefit and minimize unwanted side effect.
In macrophages, Mtb-containing phagosomes are processed by a series of interactions with early and late endosomes, respectively, resulting in phagosome maturation [Citation9]. Interestingly, studies have also revealed that Mtb can damage the phagosomal membrane and gain access to the cytosol [Citation10], and the escaped cytosolic Mtb can then be recaptured into double-membraned vesicles called autophagosomes through a type of selective autophagy called xenopahgy, which represents a conserved host defense response against intracellular pathogens through innate immune clearance [Citation11]. As an intracellular clearance pathway that delivers cytoplasmic contents to the lysosome for degradation, autophagy is controlled by a conserved ATG (autophagy related) gene family. In the initial steps of autophagy, a double-membraned and cup-shaped precursor called the phagophore is formed within the cytoplasm. Then the phagophore expands and engulfs substrates and eventually forms a double-membraned vesicle, the autophagosome. Afterward, the autophagosome progresses through similar maturation stages as the phagosome and also culminates in fusion with the lysosome to form a degradative autolysosome, and the complete process of autophagy is termed autophagy flux [Citation12,Citation13]. Though host autophagy plays key roles in the defense against invading bacterial infection, intracellular pathogens have developed a series of immune evasion strategies that interfere with host autophagy processes [Citation14,Citation15]. For example, the enhanced intracellular survival (Eis) protein from Mtb suppresses host innate immune defenses by modulating autophagy in a redox-dependent manner [Citation16], and a further study indicates that Eis inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3 [Citation17]. Another study showed that the ESX-1 secretion system of Mycobacterium marinum, which is a close relative to Mtb, could stimulate autophagosome formation while blocking the autophagic flux, leading to enhanced pathogen intracellular survival [Citation18]. Several proteins (such as PhoP and ESAT-6) from Mtb have been reported to inhibit autophagy flux in macrophages, but the detailed mechanisms are not fully elucidated [Citation19,Citation20]. Therefore, the regulatory roles of Mtb effectors involved in host autophagy process and the underlying molecular mechanisms need to be further explored to provide new insights for the development of novel therapeutic strategies for TB.
Mtb genome encodes for eleven eukaryotic-like serine-threonine protein kinases (STPKs) that serve as key regulators of the virulence, metabolic process, growth and host-pathogen interaction [Citation21]. Among these STPKs, only protein kinase G (PknG) is secreted into host cells during infection [Citation22–24]. Furthermore, PknG is essential for TB pathogenicity by blocking phagosome maturation of macrophages to promote pathogen intracellular survival during Mtb infection [Citation24,Citation25]. Thus, PknG could be an attractive drug target for TB treatment. However, the regulatory roles of Mtb PknG on host autophagy, another important pathway of Mtb intracellular clearance [Citation26], remain largely ignored. Thus, understanding the regulatory roles and the underlying mechanism of PknG on host autophagy and identifying PknG-interacting host factors involved in this process during mycobacterial infection is helpful to provide new targets for TB treatments based on pathogen-host interacting interfaces.
In this study, adopting multiple experimental techniques as well as in vitro and in vivo models, we demonstrate that Mtb PknG, through its C-terminal region, suppresses the activation of the serine/threonine protein kinase AKT/protein kinase B by binding to its conserved pleckstrin homology (PH) domain, thus promoting host autophagy activation [Citation27]. Furthermore, at the later stage of the autophagy process, PknG inhibits autophagosome maturation to block autophagy flux in two ways. First, PknG, through its tetratricopeptide repeat (TPR) domain, binds to host small GTPase RAB14 and blocks RAB14-GTP hydrolysis. Second, PknG phosphorylates TBC1D4/AS160 (TBC1 domain family member 4) to suppress its GTPase-activating protein (GAP) activity toward RAB14 in a kinase activity-dependent manner [Citation28]. Thus, Mtb PknG represents a dual-functional bacterial effector protein that tightly regulates host autophagy flux to benefit pathogen survival in the non-acidified autophagosomes in macrophages. Our findings reveal novel insights into the intricate interactions between Mtb effector proteins and the host autophagy process, and provide potential TB treatment strategies via specifically targeting the kinase activity and PknG-RAB14 interacting interface while avoiding PknG-AKT interacting interface of PknG.
Results
Mtb PknG promotes autophagy induction and inhibits autophagosome maturation during mycobacterial infection
To explore the underlying mechanisms by which PknG modulates autophagy flux, we took advantage of the tandem mCherry-GFP-LC3 construct, which is detected as yellow puncta in immature autophagosomes and as red puncta in autolysosomes, for confocal microscopy experiments using multiple Mtb strains, including ΔpknG (the pknG gene-deletion Mtb strain), ΔpknG:pknG (the pknG gene-deletion Mtb strain complemented with WT pknG) and ΔpknG:pknGK181M (which is a kinase-dead mutant of PknG). We confirmed that PknG could be secreted during Mtb cultivation (Fig. S1A) before conducting the confocal microscopy analysis, and found that wild-type (WT) Mtb, ΔpknG:pknG and ΔpknG:pknGK181M strains, but not the MtbΔpknG strain, markedly increased the number of the total LC3 puncta (including both red and yellow LC3 puncta) in infected U937 cells. This result suggests that PknG may promote autophagy induction, which is represented by increased autophagosome formation, in a kinase activity-independent manner. Furthermore, the yellow LC3 puncta significantly increased in cells infected with WT Mtb or ΔpknG:pknG strain, and slightly increased in cells infected with ΔpknG:pknGK181M strain, as compared with that in cells infected with ΔpknG strain. This result indicates that Mtb PknG might block autophagy flux, partially depending on its kinase-activity (). Consistenly, treatment by AX20017, a kinase inhibitor of PknG [Citation24], confirmed that Mtb PknG could promote autophagy induction independent of its kinase activity but inhibit autophagosome maturation depending on its kinase activity (Fig. S1B). To further confirm PknG-mediated regulation of autophagy flux during Mtb infection, we overexpressed PknG in U937 cells for Mtb infection, and found that all Mtb strains could increase the total and yellow LC3 puncta to comparable levels in the infected cells (Fig. S1C).
Figure 1. Mtb PknG promotes autophagy induction and inhibits autophagosome maturation during mycobacterial infection. (A) Representative confocal images of fixed U937 cells stably expressing mCherry-GFP-LC3. Cells were infected with WT Mtb, ΔpknG, ΔpknG:pknG and ΔpknG:pknGK181M at a MOI of 1 for 6 h. The uninfected cells were used as the control. Yellow LC3 puncta, autophagosomes; red LC3 puncta, autolysosomes. Scale bars: 5 μm. The numbers of yellow and red LC3 puncta were quantified from 100 cells for each biological replicate. Data represent means ± SEM of three independent experiments. ** P < 0.01 denotes significant differences between total LC3 puncta and yellow LC3 puncta, as analyzed by two-way ANOVA and Sidak multiple comparisons test; *P < 0.05 and ** P < 0.01 denote significant differences of the total LC3 puncta (or yellow LC3 puncta) among cells infected with different Mtb strains, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (B) Immunoblotting of LC3 and SQSTM1 in U937 macrophages uninfected or infected by various Mtb strains. Differentiated U937 cells were incubated with the indicated Mtb strains at a MOI of 1 for 0 − 12 h with DMSO or 50 nM bafilomycin A1, and analyzed by immunoblotting. (C) Densitometry of triplicate samples was performed for quantification, and the ratios of LC3-II to ACTB were presented. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains at the indicated infection time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (D) Immunoblotting of LC3 and SQSTM1 in WT (ATG5+/+) and ATG5-knockout (ATG5−/-) U937 cells. Differentiated U937 cells were uninfected or infected with WT Mtb, Mtb ΔpknG strain for 0 − 12 h at a MOI of 1 and analyzed by immunoblotting. (E) Densitometry of triplicate samples was performed for quantification, and the ratios of LC3-II to ACTB were presented. Data are shown as mean ± SEM of three independent experiments. ** P < 0.01 denotes significant differences among cells infected with different Mtb strains, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (F) Confocal microscopy analysis of Mtb-containing autophagosomes maturation in macrophages. U937 cells were uninfected or infected with the indicated Alexa FluorTM 405-labeled Mtb strains (blue) at a MOI of 1 for 6 h. Cells were incubated with 75 nM LysoTracker for an additional 30 min. Cells were fixed and stained with the antibody against LC3 (green), and then subjected to confocal microscopy analysis (Left). Scale bars: 5 μm. Right, Percent colocalizations of LC3-positive Mtb with LysoTracker. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
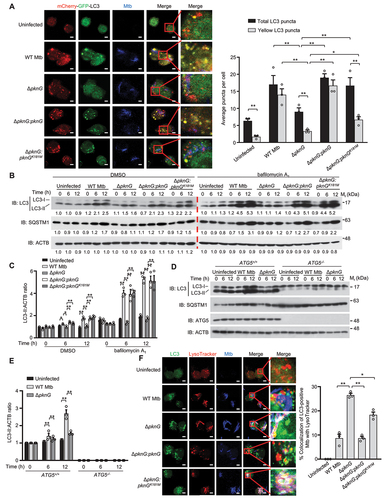
We then performed western blotting analysis to detect the protein levels of the 16-kD lipidated autophagosome-bound form of LC3 (LC3-II) and SQSTM1(sequestosome 1)/p62, because measuring LC3 lipidation by western blotting is an established method for measuring autophagy flux [Citation13], and the degradation of SQSTM1 is another widely used marker for monitoring autophagy flux because SQSTM1 binds to LC3 and is selectively degraded by autophagy [Citation29]. As shown in , increased protein levels of LC3-II and SQSTM1 were detected in cells infected with WT Mtb, ΔpknG:pknG or ΔpknG:pknGK181M strain, and the increased LC3-II can be caused either by an increase in autophagosome formation or a blockade of autophagic flux, while the accumulated SQSTM1 is usually resulted from reduced degradation due to autophagic flux blockade. To discriminate the effect of PknG on autophagic flux, including autophagosome formation and autophagosome maturation, we further conducted western blotting in the presence of bafilomycin A1, a proton ATPase inhibitor which prevents the lysosomal degradation by inhibiting lysosome acidification, and found that LC3-II levels increased further in cells infected with WT Mtb, ΔpknG:pknG or ΔpknG:pknGK181M strain, but not the Mtb ΔpknG strain, which result further demonstrates that Mtb PknG induces autophagy during mycobacterial infection (). It should be noted that SQSTM1 was accumulated concurrently with the increase of LC3-II, further indicating an impaired autophagy flux in Mtb-infected cells. To determine if PknG induced host autophagy depending on ATG5 (autophagy related 5), a critical molecule for the classical autophagy pathway [Citation30,Citation31], we infected WT (ATG5+/+) and ATG5-knockout (ATG5−/-) U937 cells with WT Mtb or Mtb ΔpknG strain, respectively, and found that WT PknG could increase the levels of LC3-II in ATG5+/+ U937 cells, but not ATG5−/- U937 cells. This result indicates that Mtb PknG induces host autophagy through ATG5-dependent autophagy pathway. Meanwhile, SQSTM1 levels were higher in ATG5+/+ U937 cells infected with WT Mtb than that in cells infected with Mtb ΔpknG strain, which difference disappeared in ATG5−/- U937 cells, indicating that PknG blocks autophagy flux in a ATG5-dependent manner (). Consistently, the confocal microscopy analysis showed that infection with WT Mtb and ΔpknG:pknG strain significantly inhibited the colocalization of LysoTracker with Mtb-containing autophagosomes, while infection with ΔpknG:pknGK181M strain partially inhibited this colocalization, as compared with ΔpknG strain infection, confirming that PknG inhibits autophagosome maturation in a kinase activity-dependent manner (). Collectively, Mtb PknG promotes autophagy induction independent of its kinase activity, and inhibits autophagosome maturation in a kinase activity-dependent manner, thus leading to autophagy flux blockade in macrophages during mycobacterial infection.
Mtb PknG promotes autophagy induction through interacting with host AKT
To further investigate the underlying mechanism by which pknG promotes host autophagy induction through identifying crucial host factor involved in this process, we conducted a yeast two-hybrid screening of a mouse cDNA library, and found that Mtb PknG interacts with the serine/threonine protein kinase AKT (). To verify this interaction, we challenged U937 cells with the indicated Mtb strains for confocal microscopy analysis and immunoprecipitation assay. The results showed that PknG could be secreted into host cells and interact with AKT in macrophages during Mtb infection (Fig. S2A and B). To determine the interaction of PknG with AKT, we also performed affinity-isolation or co-immunoprecipitation assays, and demonstrated that PknG bound to the conserved PH domain of AKT via its carboxyl (C)-terminal region (amino acids: 569–750) ( and S2C). Because the phosphoinositide 3-kinase (PI3K)-AKT-MTOR (mechanistic target of rapamycin kinase) signaling pathway is critical for the switch of the autophagy induction [Citation32,Citation33], we speculated that Mtb PknG may regulate host autophagy induction by interacting with AKT. As expected, confocal microscopy analysis and quantification assay showed that PknG, PknGK181M and PknG(C) (the C-terminal region of PknG) could induce autophagosome formation both in the infected macrophages and transfected HeLa cells, and the RFP-LC3 puncta formation was further increased by bafilomycin A1 treatment ( and S2D). We then further analyzed autophagy flux based on the turnover of LC3-II by western blotting in the presence or absence of 50 nM bafilomycin A1, and the results showed that the protein levels of LC3-II were upregulated in U937 cells infected with WT Mtb, Mtb ΔpknG:pknG (C) or Mtb ΔpknG:pknGK181M strain compared with that in U937 cells infected with Mtb ΔpknG strain, which phenomenon was further enhanced by bafilomycin A1 treatment (). Taken together, PknG promotes autophagy induction depending on the AKT-binding C-terminal region of PknG, but not its kinase activity.
Figure 2. Mtb PknG promotes autophagy induction through interacting with host AKT. (A) Yeast two-hybrid assay for the interaction of PknG with AKT. Yeast strains were transformed with the indicated plasmids in which MAP3K7/TAK1-TAB2 interaction serves as a positive control. Top, low-stringency. Bottom, high stringency. (B) Affinity-isolation assay of His-tagged AKT (10 μg each) by GST-tagged PknG (20 μg) or its truncations (6 μg each). (C) Affinity-isolation assay of His-tagged PknG (15 μg each) by GST-tagged AKT (15 μg) or its truncations (8 μg). (D) Formation of RFP-LC3 (red) puncta in RFP-LC3-overexpressing U937 cells. Cells were uninfected or infected with the indicated Alexa FluorTM 488-labeled Mtb strains (green) at a MOI of 1 for 6 h. Scale bars: 5 μm (Left). Right, Mean number of LC3 puncta per cell. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. ** P < 0.01 denotes significant differences between DMSO- and bafilomycin A1-treated cells infected with the same Mtb strain, as analyzed by two-way ANOVA and Sidak multiple comparisons test; ** P < 0.01 denotes significant differences among cells infected with different Mtb strains under DMSO or bafilomycin A1 treatment, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (E) Immunoblotting of LC3 and total ACTB (loading control) in U937 cells. Cells were uninfected or infected with WT Mtb, ΔpknG, ΔpknG:pknG(C) or ΔpknG:pknGK181M strain at a MOI of 1 and treated with or without bafilomycin A1 (50 nM) for 0 − 12 h, and then were lysed and analyzed by immunoblotting (Left). Right, Densitometry of triplicate samples was performed for quantification, and the ratios of LC3-II to ACTB in the presence or absence of bafilomycin A1 were presented. Data are shown as mean ± SEM of three independent experiments. ** P < 0.01 denotes significant differences among cells infected with different Mtb strains at the indicated infection time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test.
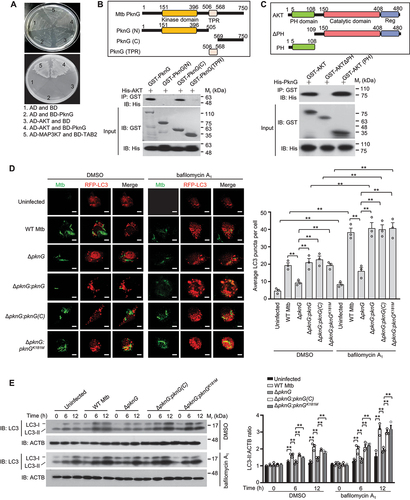
To confirm the role of AKT in the PknG-mediated host autophagy induction, we treated U937 cells with MK2206 (an AKT inhibitor), and first verified that MK2206 could inhibit AKT phosphorylation followed by autophagy activation (Fig. S3A). We then further validated that the percentage of LC3-positive Mtb was increased in cells infected with WT Mtb, Mtb ΔpknG:pknG, Mtb ΔpknG:pknGK181M and Mtb ΔpknG:pknG(C) strains, but not the Mtb ΔpknG strain, which phenomenon was abolished by MK2206 treatment (Fig. S3B). Together, these results indicate that Mtb PknG promotes autophagy induction in mycobacterial-infected U937 cells through targeting AKT, which effect is independent of the kinase activity of PknG.
It is reported that GarA is the substrate of PknG. Specifically, PknG directly phosphorylates GarA to control the activities of three enzymes that link glutamate metabolism to the tricarboxylic acid cycle, and thus contributing to metabolism regulation in Mtb [Citation34]. To exclude the possibility that host autophagy induction is regulated by disordered metabolism of Mtb caused by the inhibition of GarA phosphorylation after PknG knockout, we performed a western blotting analysis to detect whether the phenotype caused by PknG knockout could be rescued by expressing phosphomimetic GarA (GarAT21E) in ΔpknG background, and we found that GarAT21E could not alter AKT phosphorylation status and autophagy induction (Fig. S3C). Thus, PknG promotes host autophagy induction, which process is independent of its kinase activity and GarA phosphorylation.
Mtb PknG prevents AKT activation via competitively binding to its PH domain
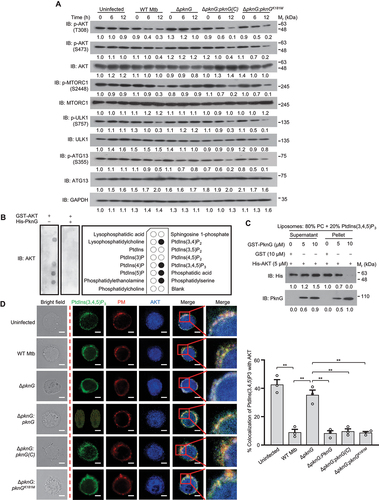
Cytosolic AKT is recruited to the plasma membrane via interacting with the membrane phosphatidylinositol(3,4,5)-trisphosphate (PtdIns[3,4,5]P3) through its PH domain, and then AKT is activated by phosphoinositide-dependent protein kinases (PDKs) [Citation27], we thus speculated that PknG might trigger autophagy by impairing AKT activation through competitively binding to its PH domain to disrupt the interaction between AKT and phosphoinositides (PIs). To test this hypothesis, we first detected AKT activation in cells infected with WT Mtb and various PknG mutant strains, and we found that the phosphorylation level of threonine 308 (T308) and serine 473 (S473) in AKT, which play an important role in AKT activation, significantly decreased in U937 cells infected with WT Mtb, Mtb ΔpknG:pknG(C) or Mtb ΔpknG:pknGK181M strain, compared with that in cells infected with Mtb ΔpknG strain, leading to the inactivation of the downstream MTOR and the activation of key initiators for mammalin autophagy, including ULK1 (unc-51 like autophagy activating kinase 1) and ATG13 (autophagy related 13) (). In vitro assays further demonstrated that PknG blocked the interaction between AKT and PIs, mainly including PtdIns(3,4)P2 and PtdIns(3,4,5)P3, in a concentration-dependent manner ( and S4). Consistently, confocal microscopy analysis results further showed that PknG, PknG(C) and PknGK181M could disrupt the colocalization of AKT with PtdIns(3,4,5)P3 in macrophage cells (). These data indicate that Mtb PknG suppresses the activation of AKT by competitively binding to its PH domain to block the AKT-PI interaction, thus promoting autophagy induction.
Figure 3. Mtb PknG prevents AKT activation via competitively binding to the PH domain of AKT to block the interaction between AKT and PIs. (A) Immunoblotting of phosphorylated AKT, MTORC1, ULK1, ATG13 and total GAPDH in U937 cells. Cells were uninfected or infected for 0 − 12 h with WT, ΔpknG, ΔpknG:pknG(C), ΔpknG:pknGK181M Mtb strains at a MOI of 1 and then analyzed by immunoblotting. (B) Immunoblotting of the interaction between purified GST-tagged AKT and immobilized phosphoinositides with or without PknG using PIP Strips membrane. (C) Binding of AKT to PtdIns(3,4,5)P3 in the presence of various concentrations of PknG. Purified His-AKT proteins were incubated with liposomes with 80% PC and 20% PtdIns(3,4,5)P3 in the presence of GST or various concentrations of GST-PknG. The mixture was ultracentrifugated and the liposome-free supernatant and the liposome pellet were analyzed by immunoblotting. (D) Representative confocal images of U937 cells. Cells were infected with the indicated Mtb strains at a MOI of 1 for 6 h. Cells were then incubated with 2 μM DiI plasma membrane probe (PM, red) for an additional 20 min and were fixed and stained with antibodies against PtdIns(3,4,5)P3 (green) and AKT (blue), and then were subjected to confocal microscopy analysis (Left). Scale bars: 5 μm. Right, Percent colocalizations of AKT with PtdIns(3,4,5)P3. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. ** P < 0.01 denotes significant differences among cells infected with different Mtb strains, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
Mtb PknG inhibits autophagosome and phagosome maturation through interacting with host RAB14
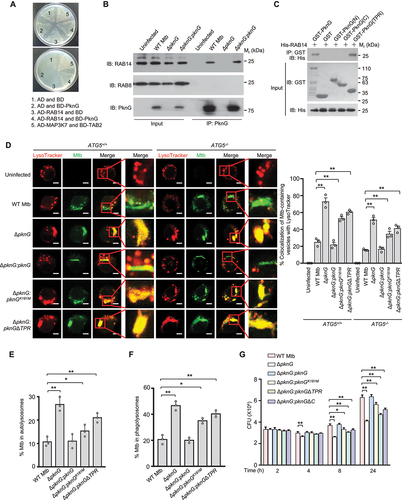
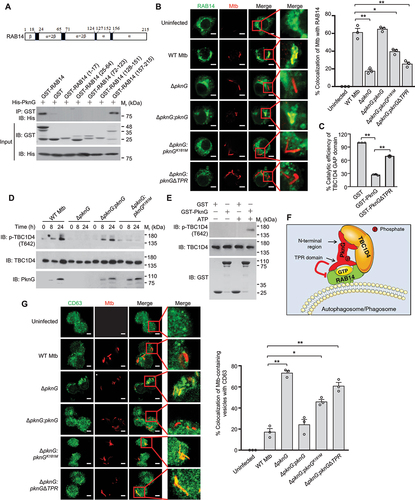
Our data from indicated that PknG could not only promote autophagy induction, but also inhibit autophagosome maturation. We thus further sought to explore the underlying molecular mechanisms. Through the yeast two-hybrid screening, we found that PknG cold also interact with RAB14 (), a small GTPase reported to play an important role in maintaining early endosomal characteristics of Mtb-containing phagosomes [Citation35]. We then conducted confocal microscopy and co-immunoprecipitation analysis to confirm that PknG could be secreted into host cells and interact with RAB14 in Mtb-infected macrophages ( and S5A). Furthermore, we found that PknG directly interacted with RAB14 through its TPR domain (). To determine whether PknG interferes with Mtb-containing vesicles maturation by interacting with RAB14, we infected ATG5+/+ and ATG5−/- U937 cells with the indicated Mtb strains for confocal microscopy analysis. The results showed that WT PknG, but not PknGΔTPR caused the inhibition of colocalization of Mtb-containing vesicles with LysoTracker both in ATG5+/+ and ATG5−/- U937 cells, suggesting that PknG might inhibit autophagosome and phagosome maturation through targeting RAB14 (). To further examine whether autophagosome maturation is inhibited by Mtb PknG through its interaction with RAB14, we quantified the colocalization of LC3-positive (LC3+, autophagosomes) Mtb with LysoTracker in U937 cells, and the results showed that PknG inhibited the colocalization of LC3+ Mtb with LysoTracker, depending on the TPR domain and partially depending on the kinase activity of PknG (). Since RAB14 has been reported to be involved in the maturation of mycobacterial phagosome, we thus also examined the effect of PknG on the maturation of mycobacterial phagosome. Consistently, PknG inhibited the colocalization of LC3-negative (LC3−, phagosomes) Mtb with LysoTracker in a TPR domain- and kinase activity-dependent manner (). Thus, RAB14 plays a critical role in both autophagosome and phagosome maturation, which could be hijacked by PknG to evade host immune clearance. Mtb survival assay results further showed that WT PknG could promote mycobacterial intracellular survival in macrophage cells, mainly depending on its TPR domain, and partially depending on its kinase-activity and the C-terminal region (). Taken together, these results indicate that Mtb PknG inhibits autophagosome maturation and thus blocks autophagy flux through interacting with host RAB14, which depends on the TPR domain and kinase activity of PknG.
Figure 4. Mtb PknG blocks autophagy flux through interacting with host RAB14 in a TPR domain and kinase activity-dependent manner. (A) Yeast two-hybrid assay for the interaction of PknG with RAB14. Yeast strains were transformed with the indicated plasmids in which MAP3K7/TAK1-TAB2 interaction serves as a positive control. Top, low-stringency. Bottom, high stringency. (B) IP of RAB14 by PknG in U937 cells. Cells were uninfected or infected with the indicated Mtb strains separately at a MOI of 1 for 6 h. Three dishes of U937 cells infected with the same Mtb strain were pooled into one sample, and were then lysed and immunoprecipitated with the antibody against PknG. IP products were immunoblotted with the antibody against RAB14. RAB8 was used as a negative control. (C) Affinity isolation of His-RAB14 (3 μg each) by GST-tagged PknG (20 μg) or its truncated forms (6 μg each). (D) Confocal microscopy analysis of Mtb-containing vesicles acidification in ATG5+/+ and ATG5−/- U937 cells. Cells were uninfected or infected with Alexa FluorTM 488-labeled WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strains (green) at a MOI of 1 for 6 h. Cells were then incubated with 75 nM LysoTracker Red for an additional 30 min and then subjected to confocal microscopy analysis (Left). Scale bars: 5 μm. Right, Percent colocalizations of Mtb with LysoTracker Red. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. ** P < 0.01 denotes significant differences among ATG5+/+ or ATG5−/- U937 cells infected with different Mtb strains, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (E and F) The percentage of Mtb that was localized in an autolysosome (LC3+ LT+ Mtb+) (E) or in a phagolysosome (LC3−LT+Mtb+) (F). U937 cells were infected and incubated with LysoTracker Red (LT) as in (D), and fixed and stained with the antibody against LC3 (green), and then subjected to count with fluorescence confocal microscope. Autophagosomes, LC3+ Mtb+; phagosomes, LC3−Mtb+; autolysosomes, LC3+ LT+ Mtb+; phagolysosomes, LC3− LT+ Mtb+ (+, positive; – negative). About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test. (G) Survival of Mtb in U937 cells infected with WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M, ΔpknG:pknGΔTPR or ΔpknG:pknGΔC Mtb strain at a MOI of 1 for 0 − 24 h. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains at the indicated infection time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test.
Mtb PknG suppresses RAB14-GTP hydrolyzation to inhibit autophagosome and phagosome maturation
In our efforts to explore more detailed molecular mechanisms underlying PknG-RAB14 interaction-mediated autophagosome maturation, we found that PknG bound to the α-helix of the C-terminus in RAB14 (amino acids 157–215) without occupying the GTP binding sites ( and S5B). Since PknG also did not disrupt the interaction between RAB14 and TBC1D4 (the GAP interacting with RAB14) (Fig. S5C), we thus hypothesized that PknG might cause the inhibition of RAB14-GTP hydrolysis, potentially through forming steric hindrance. As expected, we found that the percentage of RAB14-positive ΔpknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strains decreased, comparing with RAB14-positive WT or ΔpknG:pknG Mtb strain (). This data suggest that PknG keeps RAB14 in a RAB14-GTP state to prevent the maturation process of Mtb-containing vesicles during Mtb infection, which depends on the kinase activity and the TPR domain of PknG. Moreover, purified WT PknG protein, but not PknGΔTPR protein, could largely inhibit RAB14-GTP hydrolysis mediated by TBC1D4 (). Additionally, PknG inhibits autophagosome maturation depending on its kinase activity, we thus sought to identify the substrate phosphorylated by PknG in host cells, and found that RAB14 was not phosphorylated in vitro (Fig. S5D), but the levels of TBC1D4 phosphorylated at T642 (p-TBC1D4 (T642)) were higher in U937 cells infected with Mtb expressing WT PknG than that from cells infected with Mtb ΔPknG or Mtb ΔpknG:pknGK181M strain (). We then incubated His-tagged TBC1D4 and GST-tagged PknG recombinant proteins together in vitro, and confirmed that TBC1D4 was phosphorylated by PknG at T642 (). It was previously reported that among the four putative phosphorylated sites (S318, S588, T642 and S751) on TBC1D4, T642 is suggested to be predominantly phosphorylated by AKT and appears to be critical for mediating the vesicle transport process [Citation36,Citation37]. Consistently, when using antibodies against total phospho-Ser and phospho-Thr for the in vitro phosphorylation assay, we found that PknG phosphorylated TBC1D4 on its threonine sites instead of its serine sites (Fig. S5E). Furthermore, the in vitro PknG kinase activity of the purified WT PknG and kinase-dead mutant proteins were measured using increasing concentrations of TBC1D4 as the substrate, and the data showed that in the presence of ATP, the Vmax and Km values of WT PknG were 11 pmoles/s and 34.64 μM, respectively (Fig. S5F). Phosphorylation of TBC1D4 could inactivate TBC1D4 to cause prolonged activation of RAB14 and its retention on Mtb-containing vesicles, thereby contributing to vesicles maturation arrest [Citation38]. Thus, in addition to binding with RAB14 to block RAB14-GTP hydrolysis in a TPR domain-dependent manner, PknG could also directly phosphorylate RAB14-interacting protein TBC1D4 to suppress its GAP activity toward RAB14, leading to the blockade of autophagosome/phagosome maturation in a kinase activity-dependent manner (). As reported previously, RAB14 contributes to the arrest of mycobacterial phagosome maturation by maintaining the phagosomes in immature early endosomal-like stage to avoid their fusion with late endosomal and lysosomal degradative components. We thus further performed confocal microscopy analysis to demonstrate that there was a decrease in the colocalization between late endosomal tetraspanin protein CD63 (a robust marker of vesicle maturation) and WT Mtb or Mtb ΔpknG:pknG strain, but not Mtb ΔpknG, Mtb ΔpknG:pknGK181M or Mtb ΔpknG:pknGΔTPR strain, in U937 cells (). Taken together, these results indicate that PknG targets host RAB14 and its GAP TBC1D4 to suppress RAB14-GTP hydrolysis, thus keeping mycobacterial autophagosomes/phagosomes in immature stage without further proceeding into mature autolysosomes/phagolysosomes in Mtb-infected macrophages.
Figure 5. Mtb PknG inhibits RAB14-GTP hydrolyzation to block autophagosome and phagosome maturation. (A) Affinity isolation of His-tagged PknG (8 μg each) by GST-RAB14 (4 μg each) and its truncated forms (2 μg each). (B) Confocal microscopy analysis of U937 cells infected with pHrodoTM Red-labeled WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strain (red) at a MOI of 1 for 6 h. Cells were then fixed and stained with the antibody against RAB14 (green), and subjected to confocal microscopy analysis (Left). Scale bars: 5 μm. Right, Percent colocalizations of Mtb with RAB14. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test. (C) GAP activity of purified TBC1D4 GAP domain toward His-RAB14 in the presence of GST, GST-tagged PknG or PknGΔTPR in vitro. Data are shown as mean ± SEM of triplicate samples. ** P < 0.01 denotes significant differences among different treatments, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test. (D) Immunoblotting of p-TBC1D4 (T642) in U937 cells infected with the indicated Mtb strains separately at a MOI of 1 for 0 − 24 h. (E) Immunoblotting of PknG-mediated phosphorylation of TBC1D4 (T642) in vitro. His-tagged TBC1D4 and GST-tagged PknG recombinant proteins were incubated together at 30°C for 1 h before being processed for immunoblotting using the p-TBC1D4 (T642) antibody. (F) Proposed model depicting Mtb PknG-mediated suppression of host autophagosome/phagosome maturation by targeting RAB14 and TBC1D4. (G) Confocal microscopy analysis of autophagosome/phagosome maturation in U937 cells. Cells were infected with pHrodoTM Red-labeled WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strain (red) at a MOI of 1 for 6 h. Cells were then fixed and stained with the antibody against CD63 (green), and were subjected to confocal microscopy analysis (Left). Scale bars: 5 μm. Right, Percent colocalizations of Mtb-containing vesicles with CD63. About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among cells infected with different Mtb strains, as analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
Host RAB14 is essential for Mtb PknG-mediated arrest of autophagosome and phagosome maturation
To further confirm the physiological relevance of PknG-RAB14 interaction in regulating host autophagy flux during mycobacterial infection, we generated Rab14flox/flox (Rab14fl/fl) and rab14flox/flox-Lyz2/LyzM-Cre (rab14[Lyz2]) mice that lacked RAB14 in myeloid cells, and then collected RAB14-deficient bone marrow-derived macrophages (BMDMs) from rab14[Lyz2] mice (Fig. S6). We then infected the BMDMs from Rab14fl/fl and rab14[Lyz2] mice with Mtb strains for confocal microscopy analysis, and we found that WT PknG caused Mtb-containing vesicles being arrested in immature early endosomal-like stage in a TPR domain and kinase activity-dependent manner, which effect was abolished in the Rab14-deficient rab14[Lyz2] BMDMs (). We then further quantified the colocalization of LC3+ or LC3− Mtb with LysoTracker in the BMDMs from Rab14fl/fl and rab14[Lyz2] mice, respectively, and found that both LC3+ and LC3− WT Mtb localized less frequently to LysoTracker than the pknG-deficient strain, largely depending on the TPR domain and partially depending on the kinase activity of PknG, which effect was abolished in the rab14[Lyz2] BMDMs (). Thus, host RAB14 is essential for Mtb PknG to arrest the autophagosome and phagosome maturation during Mtb infection.
Figure 6. RAB14 is essential for Mtb PknG-mediated autophagosome/phagosome maturation arrest. (A) Confocal microscopy analysis for the acidification of Mtb-containing vesicles in BMDMs from Rab14fl/fl and rab14[Lyz2] mice. Cells were infected with Alexa FluorTM 405-labeled WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strains (blue) at a MOI of 1 for 6 h. Cells were incubated with 75 nM LysoTracker Red for an additional 30 min and fixed and stained with the antibody against Rab14 (green), and then subjected to confocal microscopy analysis. Scale bars: 10 μm. (B) Percent colocalizations of Mtb-containing vesicles with LysoTracker Red in (A). About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among BMDMs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains, as analyzed by two-way ANOVA and Sidak multiple comparisons test. (C and D) The percentage of Mtb that was localized in an autolysosome (LC3+ LT+ Mtb+) (C) or in a phagolysosome (LC3− LT+ Mtb+) (D). BMDMs from Rab14fl/fl and rab14[Lyz2] mice were infected and incubated with LT Red as in (A), and were then fixed and stained with the antibody against LC3 (green), followed by fluorescence confocal microscope analysis. Autophagosomes, LC3+ Mtb+; phagosomes, LC3− Mtb+; autolysosomes, LC3+ LT+ Mtb+; phagolysosomes, LC3− LT+ Mtb+ (+, positive; – negative). About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among BMDMs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains, as analyzed by two-way ANOVA and Sidak multiple comparisons test.
![Figure 6. RAB14 is essential for Mtb PknG-mediated autophagosome/phagosome maturation arrest. (A) Confocal microscopy analysis for the acidification of Mtb-containing vesicles in BMDMs from Rab14fl/fl and rab14[Lyz2] mice. Cells were infected with Alexa FluorTM 405-labeled WT, ΔpknG, ΔpknG:pknG, ΔpknG:pknGK181M or ΔpknG:pknGΔTPR Mtb strains (blue) at a MOI of 1 for 6 h. Cells were incubated with 75 nM LysoTracker Red for an additional 30 min and fixed and stained with the antibody against Rab14 (green), and then subjected to confocal microscopy analysis. Scale bars: 10 μm. (B) Percent colocalizations of Mtb-containing vesicles with LysoTracker Red in (A). About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among BMDMs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains, as analyzed by two-way ANOVA and Sidak multiple comparisons test. (C and D) The percentage of Mtb that was localized in an autolysosome (LC3+ LT+ Mtb+) (C) or in a phagolysosome (LC3− LT+ Mtb+) (D). BMDMs from Rab14fl/fl and rab14[Lyz2] mice were infected and incubated with LT Red as in (A), and were then fixed and stained with the antibody against LC3 (green), followed by fluorescence confocal microscope analysis. Autophagosomes, LC3+ Mtb+; phagosomes, LC3− Mtb+; autolysosomes, LC3+ LT+ Mtb+; phagolysosomes, LC3− LT+ Mtb+ (+, positive; – negative). About 100 cells were counted and analyzed for each biological replicate. Data are shown as mean ± SEM of three independent experiments. *P < 0.05 and ** P < 0.01 denote significant differences among BMDMs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains, as analyzed by two-way ANOVA and Sidak multiple comparisons test.](/cms/asset/9329b914-cfe8-4283-ad01-2e51ae114a2e/kaup_a_1938912_f0006_c.jpg)
Mtb PknG promotes mycobacteria survival in vivo by targeting host RAB14
To further determine the role of PknG-RAB14 interaction in Mtb intracellular survival in vivo, we challenged Rab14fl/fl and rab14[Lyz2] mice intratracheally with various Mtb strains to determine bacterial loads and evaluate pathologic changes in the lungs or livers of the infected mice. Our data showed that PknG promoted mycobacterial intracellular survival in Rab14fl/fl mice depending on the kinase activity and TPR domain of PknG. By comparison, PknG had no obvious effect on Mtb survival in rab14[Lyz2] mice (). Consistently, Mtb ΔpknG, Mtb ΔpknG:pknGK181M and ΔpknG:pknGΔTPR strains exhibited markedly reduced inflammation levels and cellular infiltrations in the lungs and livers of the infected Rab14fl/fl mice from 10 d after infection, as compared to the WT Mtb strain, which effect was abolished in the rab14[Lyz2] mice ( and S7A). Together, these results indicate that PknG contributes to the intracellular survival of Mtb through targeting RAB14, which depends on the TPR domain and kinase activity of PknG.
Figure 7. The TPR domain and kinase activity of PknG contribute to Mtb survival in vivo by targeting host RAB14. (A) Bacterial loads of Mtb in the lungs from Rab14fl/fl and rab14[Lyz2] mice. Mice were infected with WT or mutant Mtb strains, and lungs were harvested and analyzed at 1, 5, 10, 15 and 20 d after infection. Data are shown as mean ± SEM of n = 3 per group. *P < 0.05 and ** P < 0.01 denote significant differences of CFUs among lungs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains at the indicated time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (B) H&E staining of the lungs of Rab14fl/fl and rab14[Lyz2] mice treated as in (A). Arrows indicate the puncta of cellular infiltration. Scale bars: 200 μm. Data are representative of one experiment with two independent biological replicates. (C) The percentage of lung area occupied by inflammatory cells. *P < 0.05 and ** P < 0.01 denote significant differences among lungs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains at the indicated time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test.
![Figure 7. The TPR domain and kinase activity of PknG contribute to Mtb survival in vivo by targeting host RAB14. (A) Bacterial loads of Mtb in the lungs from Rab14fl/fl and rab14[Lyz2] mice. Mice were infected with WT or mutant Mtb strains, and lungs were harvested and analyzed at 1, 5, 10, 15 and 20 d after infection. Data are shown as mean ± SEM of n = 3 per group. *P < 0.05 and ** P < 0.01 denote significant differences of CFUs among lungs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains at the indicated time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test. (B) H&E staining of the lungs of Rab14fl/fl and rab14[Lyz2] mice treated as in (A). Arrows indicate the puncta of cellular infiltration. Scale bars: 200 μm. Data are representative of one experiment with two independent biological replicates. (C) The percentage of lung area occupied by inflammatory cells. *P < 0.05 and ** P < 0.01 denote significant differences among lungs from Rab14fl/fl or rab14[Lyz2] mice infected with different Mtb strains at the indicated time points, as analyzed by two-way ANOVA and Tukey’s multiple comparisons test.](/cms/asset/fd27ff81-58cd-49db-8c54-7ed1cd6762fb/kaup_a_1938912_f0007_c.jpg)
In summary, our study demonstrates that Mtb PknG promotes autophagy induction in a C-terminal region-dependent manner while blocking autophagy flux at the step of autophagosome/phagosome maturation in a TPR domain and kinase activity-dependent manner, leading to the overall effect of blocked autophagy flux and enhanced pathogen intracellular survival in macrophage cells (Fig. S7B). Thus, targeting the TPR domain and kinase activity of PknG to release autophagosome/phagosome maturation arrest while avoiding the C-terminal region of PknG could be an effective strategy in treating TB.
Disscussion
During infection, Mtb is engulfed by a phagocyte, which results in the formation of an intracellular vacuole termed phagosome. The phagosome containing the microbe eventually fuses with lysosomes, resulting in the formation of an acidic and oxidative phagolysosome to degrade microbes [Citation8,Citation25,Citation39,Citation40]. But under certain circumstances, Mtb could damage the phagosomal membrane and enter into the cytosol [Citation41]. The escaped pathogens could then be surrounded by various types of ubiquitin chains and subsequently recognized by host autophagy receptors including SQSTM1, NBR1 and CALCOCO2/NDP52 [Citation42–45], or the pathogens could directly interact with ubiquitin via its surface protein to recruit autophagy receptors [Citation46], both cytosolic pathogen-initiated autophagy induction pathways could lead to the formation of autophagosomes through the selective autophagy called xenophagy, which is an essential component of host innate immunity that involved in host intracellular pathogen clearance and inflammatory responses [Citation47–50]. In turn, intracellular pathogens have developed various strategies to exploit autophagy to ensure their survival [Citation51]. Actually, the autophagy flux, that is the complete process of the autophagy, is the key factor that determines the ultimate outcomes of the pathogen intracellular survival, because when the autophagy flux is blocked, the vacuolar compartments of the increased immature autophagosomes, but not the mature autolysosomes, might support the pathogen growth and establish a sheltered niche for pathogen immune evasion of cellular antibacterial mechanisms, instead of helping clear the invading pathogen [Citation51–53]. Consistently, clinical isolates of Mtb are shown to manipulate autophagy for their intracellular survival [Citation54]. Also, it was reported that Mtb is able to induce autophagosome formation and replicate in the LC3-positive compartment in resting human lymphatic epithelial cells [Citation55]. However, previous studies tend to focus only on the autophagy induction step when investigating the autophagy-regulating roles of pathogen effector proteins, while the whole picture of the autophagy flux regulation by the pathogen is obscure. A study reported that the ESX-1 secretion system of Mycobacterium marinum, which is a close relative to Mtb, could induce autophagosome formation and block the autophagic flux, leading to enhanced intracellular survival of pathogens [Citation18]. However, the specific Mtb effector proteins that regulate the host autophagy flux and the underlying molecular mechanisms need to be further explored. Here, we reveal a dual-functional bacterial effector protein PknG that tightly regulates host autophagy flux to benefit pathogen intracellular survival. Basically, PknG promotes autophagy induction but inhibits autophagy maturation during Mtb infection, leading to an overall effect of blocked autophagy flux and enhanced pathogen intracellular survival. It is worth mentioning that our study reveals that Mtb PknG inhibits both basal autophagic flux and xenophagic flux during Mtb infection, which finding is different from a preveious study reporting that Mtb PhoP and ESAT-6 specifically inhibit xenophagic flux, while leaving basal autophagic flux intact [Citation19]. We speculate that the PknG-mediated inhibition of xenophagic flux might prevent pathogen clearance in the lysosome, while the suppression of the basal autophagic flux by PknG might interfere with host immune defense against Mtb in some other ways. One possibility is that PknG might prevent the degradation of certain secreted Mtb effector proteins, such as Ag85B, to inhibit antigen presentation. Those secreted Mtb effector proteins can be captured and degraded by the autophagy-lysosomal pathway, and then the resulting peptide is presented to stimulate the adaptive immune response [Citation56].
Mtb PknG represents a potent manipulator of host autophagy flux from pathogens that hijack host autophagy process to benefit pathogen intracellular survival. In an era of increasing antibiotic resistance, the in-depth understanding of how pathogens manipulate host autophagy process will hopefully provide novel host-based strategies for combating infection. Considering the various mechanisms and multiple steps involved in the regulation of host autophagy process by pathogens, we need to correctly and precisely adopt autophagy modulators to enhance the autophagy flux to ultimately eliminate invading bacterial pathogens [Citation49].
As reported, there is a direct link between the PI3K-AKT-MTOR signaling pathway and autophagy. Specifically, activation of the PI3K-AKT-MTOR pathway inhibits autophagy induction, whereas inhibition of this signaling cascade relieves the negative repression of MTOR on autophagy induction [Citation12]. Given the pivotal role of MTOR in controlling macrophage function, pathogens have evolved elaborate strategies to target this pathway to manipulate host cells for their own benefit [Citation57–59], and it becomes evident that the consequences of stimulating or inhibiting MTOR signaling by pathogens in terms of immune evasion and pathogen intracellular replication and persistence are highly context-dependent. Whether and how Mtb regulates PI3K-AKT-MTOR pathway to modulate autophagy activation in macrophages for facilitating its intracellular survival remains unknown. During PI3K-AKT-MTOR signaling pathway activation, one of the key steps is the translocation of AKT to the plasma membrane, which process is dependent on the PH domain at the amino terminal of AKT. Once AKT is translocated to the plasma membrane, a protein kinase named PDK1 could phosphorylate its threonine residue (T308), leading to increased enzymatic activity of AKT. The activated AKT then detaches from the plasma membrane and translocate into the cytosol, resulting in the activation of MTOR, a downstream target of kinase AKT and a negative modulator of host autophagy [Citation60]. In this study, we demonstrate that Mtb PknG competitively binds to the PH domain of AKT to interrupt the interaction between AKT and PtdIns(3,4,5)P3 in macrophage cells, thus blocking the membrane translocation and activation of AKT, leading to increased formation of autophagosomes to recapture cytosolic Mtb. Phosphoinositides are an essential class of membrane lipids that can be reversibly phosphorylated at the 3,4 and/or 5 positions of the inositol ring to generate distinct PIs such as PtdIns(3,4,5)P3, and those lipids have unique distributions within the cell and play important roles in cellular processes, including membrane trafficking, signal transduction and cell migration [Citation61], thus it is not surprising that PIs are a target of intracellular bacterial pathogens [Citation62,Citation63]. Thus, our study contributes to a better understanding of the roles of Mtb effector proteins in modulating host PI3K-AKT-MTOR pathway and PIs to manipulate host autophagy process in macrophage cells.
RAB GTPases, with more than 60 members encoded within human genome, are the largest group of the RAS superfamily small GTPases, and they are the master regulators of intracellular membrane trafficking [Citation64]. In recent years, RAB GTPases have also been recognized to modulate immune responses by regulating the immune receptor transport, cytokine secretion and phagocytosis, etc [Citation65,Citation66]. Thus, some intracellular bacterial pathogens have evolved various strategies to target RAB GTPase proteins to modulate their intracellular trafficking events, thus promoting pathogen intracellular survival. For instance, Legionella pneumophila subverts the functions of RAB1 to generate an organelle favorable for bacterial replication [Citation67]. RAB11 has been shown to be necessary for the replication and cell-to-cell spreading of Shigella [Citation68]. In addition, a previous study reported that RAB14 is critical for maintaining phagosome maturation arrest during Mtb infection [Citation35]. Here, we further revealed that Mtb PknG could hijack RAB14 to cause prolonged activation of RAB14 and its retention on both autophagosomes as well as phagosomes, thereby contributing to the maturation arrest of autophagosomes/phagosomes. It was suggested that RAB14 coordinates the autophagosome maturation in Drosophila [Citation69], and here for the first time, we demonstrate the physiological role of RAB14 in regulating autophagosome maturation in mammalians. Moreover, our in vivo data from Rab14-deficient mice further indicated that RAB14 might be a host susceptible factor hijacked by Mtb PknG to benefit the intracellular survival of mycobacteria.
It was shown that Mtb PknG inhibits phagosome maturation in a kinase activity-dependent manner [Citation24]. Moreover, previous structural analysis of Mtb PknG suggested it to be a unique eukaryotic-type kinase that adopts unusual quaternary structure [Citation70]. Consistently, it was demonstrated that, unlike most other Mtb STPKs, PknG shows limited in vitro kinase activity toward a wide range of peptide and protein substrates [Citation71], indicating that it might have selective preferences toward previously undiscovered substrates, perhaps including those from host cells. Here, in this study, we reveal that PknG could phosphorylate host TBC1D4 to suppress its GAP activity toward RAB14, thus blocking autophagosome/phagosome maturation. It should be mentioned that a recent study reported that PknG interacts with cyclophilin A using the human protein microarray and phosphorylates cyclophilin A in the in vitro assay [Citation72], and another study reported that PknG targets GDP-bound RAB29/RAB7L1 and blocks transition of inactive RAB29-GDP to active RAB29-GTP in a kinase activity-dependent manner, instead of directly phosphorylating RAB29 to inhibit phagosome-lysosome fusion [Citation73]. Thus, TBC1D4 represents a newly identified physiological kinase substrate of PknG from host cells. Both RAB GTPases and their GAPs play crucial roles in the regulation of autophagy [Citation74], our study reveal that PknG could target both RAB14 and its GAP TBC1D4 to potently block RAB14-GTP hydrolysis, leading to the arrest of autophagosome/phagosome maturation. Thus, based on studies from both ours and others, PknG represents a multitasking bacterial effector protein that targets multiple steps of host autophagy process including autophagosome formation and autophagosome/phagosome maturation.
In summary, our data reveal the detailed molecular mechanisms of Mtb PknG in regulating host autophagy flux during mycobacterial infection. In addition to inhibiting phagosome maturation as reported previously, we further demonstrate that PknG, through its various regions or kinase activity, targets different stages of the autophagy process including autophagy induction stage and autophagosome maturation stage, leading to an overall effect of blocked host autophagy flux and enhanced pathogen intracellular survival. Taken together, our study highlights the intricate interactions between pathogen effector proteins and the host autophagy process, and provides potential TB treatment strategies via specifically targeting the kinase activity and PknG-RAB14 interacting interface to release the autophagy flux blockade effect while avoiding PknG-AKT interacting C-terminal region of PknG to maintain the autophagy induction effect.
Materials and methods
Bacterial strains, mammalian cell lines and plasmids
E. coli DH5α and BL21 (DE3) were used for genetic manipulation and protein overexpression. Mycobacterium tuberculosis (Mtb) H37Rv was used for infection. PknG deletion and mutant strains in Mtb were created as described previously [Citation6]. Mtb strains included wild-type (WT) Mtb, pknG-deleted Mtb (ΔpknG), Mtb ΔpknG complemented with WT pknG (ΔpknG:pknG), pknGK181M (ΔpknG:pknGK181M, which is a kinase-dead mutant), the N-terminal region of pknG (ΔpknG:pknG N), the C-terminal region of pknG (ΔpknG:pknG C), pknGΔC (ΔpknG:pknGΔC, from which the C-terminal region of pknG is deleted), and pknGΔTPR (ΔpknG:pknGΔTPR, from which the TPR domain is deleted). Gene sequencing was used to confirm the mutation, deletion or truncation of PknG in Mtb strains. Expression of PknG in infected host cells was examined by immunoblotting analysis. HEK293T (ATCC CRL-3216, RRID: CVCL_0063), HeLa (ATCC CCL-2, RRID: CVCL_0030) and the human monocytic cell line U937 cells (ATCC CRL-1593.2, RRID: CVCL_0007) were obtained from the American Type Culture Collection (ATCC). For expression in mammalian cells, pknG or its mutants were amplified by PCR and cloned into pEGFP-N1 (with GFP-tag) or p3xFlag-CMV14 (with 3xFlag-tag). Bacterial expression plasmids were constructed by cloning the cDNA into pET30a (with His6-tag) or pGEX-6P-1 (with GST-tag). All the plasmids were sequenced at the Beijing Genomics Institute (BGI) for verification. The plasmids used in this study are listed on the Table S1.
Antibodies
All of the antibodies were used according to manufacturer’s instructions and based on previous experience in the laboratory. Rabbit anti-PknG and anti-PtpA antibodies were produced and purified by GenScript Biotechnology with the recombinant GST-tagged PknG or GST-tagged PtpA protein as the immunogen, 1:10000 for immunoblotting, 1:200 for immunofluorescence. The following commercial antibodies have been used: anti-pan-AKT (Abcam, Ab8805), 1:1000 for immunoblotting, 1:200 for immunofluorescence; anti-phospho-AKT T308 (Cell Signaling Technology, 2965), 1:1000 for immunoblotting; anti-phospho-AKT S473 (Abcam, Ab66138), 1:1000 for immunoblotting; anti-MTOR (Abcam, Ab2732), 1:1000 for immunoblotting; anti-phospho-MTOR S2448 (Abcam, Ab109268), 1:1000 for immunoblotting; anti-ULK1 (Cell Signaling Technology, 8054), 1:1000 for immunoblotting; anti-phospho-ULK1 S757 (Abcam, Ab229909), 1:1000 for immunoblotting; anti-ATG13 (Abclonal Technology, A0690), 1:1000 for immunoblotting; anti-phospho-ATG13 S355 (Abclonal Technology, Ap1089), 1:1000 for immunoblotting; anti-ATG5 (Immunoway Biotechnology, YM3752), 1:1000 for immunoblotting; anti-ACTB/actin beta (Sigma-Aldrich, A2228), 1:6000 for immunoblotting; anti-LC3B (Sigma-Aldrich, L7543), 1:3000 for immunoblotting; anti-TUBA (Sigma-Aldrich, T6199); anti-SQSTM1/p62 (Cell Signaling Technology, 5114), 1:1000 for immunoblotting; anti-MYC (Santa Cruz Biotechnology, sc-40), 1:2000 for immunoblotting; anti-Flag (Sigma-Aldrich, F1804), 1:5000 for immunoblotting; anti-GST (ZSGB-BIO, TA-03), 1:5000 for immunoblotting; anti-His (ZSGB-BIO, TA-02), 1:2000 for immunoblotting; anti-GFP (Abcam, ab1218), 1:2000 for immunoblotting; anti-GAPDH (Santa Cruz Biotechnology, sc-25,778), 1:5000 for immunoblotting; anti-PtdIns(3,4,5)P3 IgG (Echelon Biosciences, Z-P345), 1:200 for immunofluorescence; anti-TBC1D4/AS160 (Cell Signaling Technology, 2670), 1:1000 for immunoblotting; anti-phospho-TBC1D4 T642 (Cell Signaling Technology, 8881), 1:1000 for immunoblotting; anti-RAB14 (Abcam, ab28639), 1:2000 for immunoblotting, 1:200 for immunofluorescence; anti-CD63 (Abcam, ab217345), 1:200 for immunofluorescence; anti-RAB8 (BD Biosciences, 610,844), 1:1000 for immunoblotting; anti-phospho-Thr (Cell Signaling Technology, 9386s), 1:1000 for immunoblotting; anti-phospho-Ser (Abcam, ab9332), 1:1000 for immunoblotting. The following chemicals and assay kits were used: AKT inhibitor MK2206 (Selleck, S1078), PknG inhibitor AX20017 (Macklin, A879559), bafilomycin A1 (Sigma-Aldrich, B1793), PIP Strips membrane (Molecular Probes, P23750) and PIP Array membrane (Molecular Probes, P23748).
Mice
Lyz2/LyzM-Cre mice were from the Jackson Laboratory. Rab14fl/fl mice were generated by Biocytogen (Beijing, China) using the CRISPR-Cas9 system. Both Lyz2-Cre mice and Rab14fl/fl mice were in C57BL/6 genetic background. Rab14fl/fl mice were first crossed with Lyz2-Cre mice to generate F1 rab14fl/+-Lyz2-Cre mice. The rab14fl/+-Lyz2-Cre mice were then crossed with rab14fl/fl mice to generate rab14fl/fl-Lyz2-Cre (rab14[Lyz2]) mice as macrophage-specific rab14-knockout mice and Rab14fl/fl mice as controls. All mice were bred and maintained in specific pathogen-free-grade cages using standard humane animal husbandry protocols. Mice were 6–8-weeks old during the course of the experiments and were age- and sex-matched in each experiment. Sample size was based on empirical data from pilot experiments. No additional randomization or blinding was used to allocate experimental groups. All experimental protocols were performed in accordance with the instructional guidelines of the China Council on Animal Care, and were approved by the Biomedical Research Ethics Committee of the Institute of Microbiology, Chinese Academy of Sciences, and the Beijing Chest Hospital, Capital Medical University.
Establishment of U937 cell line stablely expressing mCherry-GFP-LC3
HEK293T cells were co-transfected with PMSCV puro-mCherry-GFP-LC3, pVSV-G and pCMV-Gag-Pol vectors to obtain virus, the medium was then collected at 48 h post-transfection and filtered with a 0.45-μm filter. The medium containing virus was used to infect U937 cells. The medium of U937 cells was replaced with fresh RPMI 1640 medium (Gibco, 11875093) containing 2 μg/mL puromycin (Selleck, s7417) at 3, 6, 9 and 12 d after infection. Two weeks later, the cells stablely expressing mCherry-GFP-LC3 were selected.
ATG5−/- U937 cells construction using CRISPR-Cas9 system
The ATG5-targeting sequence (sgRNA-F: CACCGTCCGA TTGATGGCCCAAAAC; sgRNA-R: AAACGTTTTGG GCCATCAATCGGAC) were cloned into lentiCRISPR v2 vector. The recombinant lentiCRISPR v2 vector together with pLP1, pLP2 and pLP/VSVG plasmids were co-transfected to HEK293T cells to product large amount of lentivirus. Lentivirus was collected at 48 h and 72 h post-transfection and filtered with a 0.45 μm filter, followed by concentrating at 4°C to increase the titer of virus. Then the concentrated lentivirus was added to U937 cells, and the medium was replaced with fresh RPMI 1640 medium containing 2 μg/mL puromycin at 48 h after infection for selection. Two weeks later, the surviving cells were seeded into 96-well plates at a density of 1 cell/well to grow single-cell clones. And the knockout cells were identified with the antibody against ATG5.
Preparation of bone marrow-derived macrophages
BMDMs were obtained by flushing the tibia and femurs of mice and cultured in DMEM medium supplemented with 10% fetal calf serum (FBS), 100 U/mL penicillin, 100 µg/mL streptomycin and 30% L929 cell supernatant (containing CSF1/M-CSF) for 7 d. Mature BMDMs were plated in 24-well plates for 24 h and infected with the indicated Mtb strains separately at a multiplicity of infection (MOI) of 1. Twenty-four h later, cells were harvested for immunoblotting and colony-forming unit (CFU) counting.
Cell culture and bacteria cultivation
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (Gibco, 11995065) supplemented with 10% FBS. U937 cells were cultured in RMPI 1640 medium (Gibco, 11875093) with 10% FBS. Before infection, U937 cells were differentiated into adherent macrophage-like cells using 10 ng/mL PMA (Sigma-Aldrich, P1585) for 24 h. E. coli DH5α and BL21 (DE3) were grown in LB medium. Mtb strains were cultivated in middlebrook 7H9 broth supplemented with 10% oleic acid (Sigma-Aldrich, O1008)-albumin (5%; Roche, 738,328)-dextrose (0.2%; VWR Life Science, VWRV0188)-catalase (0.0004%; Sigma-Aldrich, C9322) (OADC) and 0.05% Tween-80 (Sigma-Aldrich, P1754), or on Middlebrook 7H10 agar (BD Biosciences, 262,710) supplemented with 10% OADC.
Affinity-isolation assay
GST-tagged protein bound to 20 μL of glutathione Sepharose 4B beads (GE Healthcare, 17–0756-05) in 500 μL of binding buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol [DTT], 0.1% NP-40 [Beyotime Biotechnology, ST366]) supplemented with 1% protease inhibitors (Bimake, B14001) for 1 h at 4°C. The beads were washed five times with binding buffer and further incubated with prey protein supplemented with 0.1 mg/mL BSA (the molar ratio of GST-tagged protein and prey protein is 1:1). After 2 h of incubation at 4°C, beads were washed and subjected to immunoblotting.
Western blotting analysis
Cells were lysed in the Cell Lysis Buffer (Beyotime Biotechnology, P0013) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF; Beyotime Biotechnology, ST505). Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, PIVH00010). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (20 mM Tris-HCl, pH 7.4, 150 mM NaCl) with 0.1% Tween-20 (Sigma-Aldrich, P1379) (TBST) for 1 h at room temperature (RT) and subsequently incubated overnight at 4°C with primary antibodies. Following three washes of 10 min each with TBST, the membranes were incubated with goat anti-mouse IgG/HRP (ZSGB-BIO, ZB-2305) or goat anti-rabbit IgG/HRP (ZSGB-BIO, ZB-2301) for 1 h at RT. After three washes with TBST, the membranes were developed by Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0500) and exposed to X-ray film. For quantifications, densitometry was performed using ImageJ to evaluate the intensity of the western blotting band signals.
Mtb infection
Cells were infected with Mtb at a MOI of 1. Before infection, Mtb in Middle brook 7H9 medium with 0.05% Tween-80 (Sigma-Aldrich, P1754) and 10% OADC enrichment were grown to an OD600 of approximately 0.6, which value corresponds to 1 × 108 CFUs/mL of Mtb [Citation75]. For infection, U937 cells were seeded at 1 × 106 cells per well and cultured overnight in fresh RPMI1640, followed by infection with 1 × 106 CFUs of Mtb (MOI = 1).
Immunoprecipitation
U937 cells were infected with Mtb strains at a MOI of 1. After 1 h, cells were washed with 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) for three times to exclude non-internalized bacteria, and were then incubated in the fresh RPMI 1640 medium for an additional 5 h. Cells were lysed in Western and Immunoprecipitation (IP) buffer (Beyotime Biotechnology, P0013) for 30 min at 4°C. Cell lysates were then incubated with the antibody against PknG (Rabbit anti-PknG antibody was produced and purified by GenScript Biotechnology with the recombinant GST-tagged PknG protein as immunogen) and protein A/G agarose (Santa Cruz Biotechnology, sc-2003) at 4°C, followed by extensively washing with the Western and IP buffer. The immunoprecipitated samples were analyzed by immunoblotting with the indicated antibodies.
CFU counting of infected cells
For macrophage infection, Mtb in Middle brook 7H9 medium with 0.05% Tween-80 and 10% OADC enrichment were grown to an OD600 of approximately 0.6. Macrophage cells were infected with Mtb strains at a MOI of 1. After 1 h, cells were washed with 1× PBS for three times to exclude non-internalized bacteria, and were then incubated in the fresh RPMI 1640 medium. For CFU counting, cells were washed with 1× PBS for three times and lysed in 7H9 broth containing 0.05% SDS for 10 min. The cell lysate was then diluted to different concentrations and plated on 7H10 agar plates.
Lipid-protein interaction assay
PIP Strips membrane (Molecular Probes, P23750) and PIP Array membrane (Molecular Probes, P23748) were used to analyze the competitive inhibition of PknG on the interaction between AKT and PtdIns(3,4,5)P3. Before the assay, the membrane was blocked with TBS-T plus 3% fatty acid-free BSA (Beyotime Biotechnology, ST025) for 1 h at RT, followed by incubation with 0.5 µg/mL of purified GST-tagged AKT protein in TBS-T plus 3% fatty acid-free BSA in the presence or absence of purified His-tagged PknG at 4°C overnight. Finally, the membrane was washed with TBS-T plus 3% fatty acid-free BSA three times with gentle agitation for 10 min each time. The bound protein AKT was detected by immoblotting using the antibody against GST-tag.
Liposome-binding assay
The liposome-binding assay was performed as described previously [Citation76]. Before the assay, phosphatidylcholine (PC; Sigma-Aldrich, 850457 C) and PtdIns(3,4,5)P3 (Sigma-Aldrich, 850156P) were dissolved in chloroform and chloroform-methanol mixture (20:9, v:v), respectively. Then 20% PC and 80% PtdIns(3,4,5)P3 were mixed in a glass vial and was evaporated under a stream of nitrogen. The dry lipid film was then resuspended in 500 μL buffer 1 (20 mM HEPES, pH 7.5, 150 mM NaCl). Liposomes were generated by extrusion of the hydrated lipids through a 100-nm polycarbonate filter (Avanti Polar Lipids Inc, 610005–1Ea) 35 times using the Mini-Extruder device (Avanti Polar Lipids Inc, 610000–1Ea). The indicated His-AKT protein (5 μM) was incubated with the liposome at RT for 30 min in a total volume of 80 μL in the presence of GST or various concentrations of GST-PknG. Samples were centrifuged in a Beckman Optima MAX-XP ultracentrifuge at 4°C for 20 min at 100,000 x g. The supernatant was collected to examine proteins not bound to the liposome. The pellets were washed twice with 100 μL buffer 1 by re-centrifugation and brought up to the same volume as the supernatant. The supernatant and pellet fractions were analyzed by immunoblotting with the indicated antibodies.
Yeast two-hybrid assay
Yeast two-hybrid assay was performed using the Matchmaker Two-Hybrid System (Clontech Laboratories, Inc., 630489) by following the manufacturer’s instructions. A mouse 7-day embryo cDNA Library (Clontech Laboratories, Inc., 630478) was used to identify host interaction proteins of Mtb PknG with yeast two-hybrid assay. Mtb PknG gene was subcloned into the plasmid pGBKT7 as the bait plasmid. Saccharomyces cerevisiae AH109 cells were cotransduced with the bait plasmids and the prey plasmids by the lithium acetate method. To test the interactions between proteins, the transformants were streaked onto low-stringency (lacking leucine and tryptophan) and high-stringency (lacking adenine, histidine, leucine and tryptophan) selection plates.
Bacteria staining and Immunofluorescence confocal microscopy
For bacteria staining, pHrodo™ Red succinimidyl (NHS) ester (Invitrogen, P3600), Alexa FluorTM 488 (green; Invitrogen, A20000) and Alexa FluorTM 405 (blue; Invitrogen, A30000) were used as described previously [Citation46]. Brifely, the bacteria that grown to an OD600 of approximately 0.6 were centrifuged to remove the supernatant, and then were washed thrice with Hanks’ Balanced Salt Solution (Beyotime Biotechnology, C0218) containing 0.05% Tween-80 by vortexing, and finlly resuspended in the buffer with adequate dye for 30 min at 37°C. Then the bacteria were extensively washed and resuspended in DMEM medium with 0.05% Tween-80 for infection. For immunofluorescence confocal microscopy assay, cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 (Solarbio, T8200), and blocked with 5% BSA for 1 h. Confocal images were taken with a Leica SP8 confocal system and processed with the Leica LAS AF Lite program. For the image quantifications, approximately 100 cells were analyzed with Imaris 9.6 (Bitplane AG) for each biological replicate, and the data was shown as the mean ± SEM of three independent experiments.
GTPase-activating protein (GAP) assay
RAB14 was loaded with GTP (Sigma-Aldrich, 371806) by incubating 2 mg of RAB14 with a 25-fold molar excess of GTP at 25°C for 1 h in buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM DTT. Free nucleotide was removed with a D-Salt column (Pierce Biotechnology, 20439) pre-equilibrated with buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl. The single-turnover kinetics of intrinsic and GAP-accelerated GTP hydrolysis was measured using the EnzChek Phosphate Assay Kit (Invitrogen, E6646). GTP-loaded RAB14 was mixed with solutions containing the assay reagents and TBC1D4-GAP domain, and dispensed into 96-well microplates (NEST, 701001). The final solution contained 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.15 mM 2-amino-6-mercapto-7-methylpurine ribonucleotide (MESG; Biotium, Inc., 10300), 0.75 U mL−1 purine nucleoside phosphorylase (PNP; Sigma-Aldrich, 40809), 10 mM MgCl2, 20 µM GTP-loaded RAB14 and TBC1D4-GAP domain with WT PknG or PknGΔTPR. The absorbance at 360 nm was monitored.
In vitro phosphorylation assay
His-tagged TBC1D4 and GST-tagged PknG recombinant proteins were expressed and batch purified from E. coli BL21 (DE3) using Ni-NTA beads (QIAGEN, 30230) and glutathione-Sepharose 4B (GE Healthcare, 17–0756-05). One μg of purified His-tagged TBC1D4 protein and 6 μg of GST or 1.5 μg of GST-PknG were incubated at 30°C for 60 min in the presence or absence of 100 μM ATP (pH 7.2; Sigma-Aldrich, 20–306) in Kinase Assay buffer containing 2 mM MnCl2, 25 mM Tris-HCl, pH 7.5, 1 mM DTT. The reactions were stopped by adding 5 × SDS sample buffer, and the mixture was boiled for 10 min and subjected to immunoblotting.
Kinase kinetics
Five μM of GST-PknG and GST-PknGK181M were pre-reacted at 30°C for 10 min in Kinase Assay buffer containing 2 mM MnCl2, 25 mM Tris-HCl, pH 7.5, 1 mM DTT, 100 μM ATP. Reactions were then initiated by the addition of purified His-tagged TBC1D4 (0 − 100 μM) and incubated for 60 min at RT. The released inorganic phosphate was measured by the EnzChek® Phosphate Assay Kit (Invitrogen, E6646). The Lineweaver-Burk plots were used to determine the Michaelis-Menten kinetic parameters (kcat and Km).
Murine infection
Mice were intratracheally infected with 1 × 105 CFU of Mtb (in 25 µL PBST) per mouse. Lungs and livers were analyzed as described previously [Citation6]. Staining with hematoxylin and eosin (H&E) stain was performed for evaluation of tissue pathologic changes in blinded fashion by a pathologist, and the percentage of lung area occupied by inflammatory cells was analyzed with QuPath software. Lung tissues were homogenized in 1 mL 6% NaOH, and then each homogenate was diluted with 7H9 medium (BD Biosciences, 271,310; supplemented with 10% OADC, 0.5% glycerol, 0.05% Tween 80) and plated onto the 7H10 agar (BD Biosciences, 262,710; supplemented with 10% OADC, 0.5% glycerol) plate for CFU counting.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 Software. All data were analyzed using an one-way or two-way ANOVA with Bonferroni posttest to correct for multiple comparisons as indicated in the corresponding figure legends. Each value of *P < 0.05 or **P < 0.01 was considered to be statistically significant.
Supplemental Material
Download Zip (21.9 KB)Acknowledgments
We thank N. Mizushima (The University of Tokyo) for Atg5flox/flox mice; X. Zhang (Institute of Microbiology, Chinese Academy of Sciences, Beijing) for helping with confocal microscopy analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- World Health Organization. Global tuberculosis report 2020. WHO Press; 2020.
- Awuh JA, Flo TH. Molecular basis of mycobacterial survival in macrophages. Cell Mol Life Sci. 2017 May;74(9):1625–1648.
- Chai QY, Wang L, Liu CH, et al. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol Immunol. 2020 Sep;17(9):901–913.
- Liu CH, Liu HY, Ge BX. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol. 2017 Dec;14(12):963–975.
- Qiang LH, Wang J, Zhang Y, et al. Mycobacterium tuberculosis Mce2E suppresses the macrophage innate immune response and promotes epithelial cell proliferation. Cell Mol Immunol. 2019 Apr;16(4):380–391.
- Wang J, Li BX, Ge PP, et al. Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system. Nat Immunol. 2015 Mar;16(3):237–U189.
- Mehra A, Zahra A, Thompson V, et al. Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog. 2013 Oct;9(10):e1003734.
- Wong D, Bach H, Sun J, et al. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011 Nov 29;108(48):19371–19376.
- Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes [Review]. Trends Immunol. 2012 Aug;33(8):397–405.
- Simeone R, Bobard A, Lippmann J, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012 Feb;8(2):e1002507.
- Wang Z, Li C. Xenophagy in innate immunity: a battle between host and pathogen [Review]. Dev Comp Immunol. 2020 Aug;109:103693.
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy [Review]. Annu Rev Genet. 2009;43(1):67–93.
- Lopez A, Fleming A, Rubinsztein DC. Seeing is believing: methods to monitor vertebrate autophagy in vivo. Open Biol. 2018 Oct;8(10):180106.
- Espert L, Beaumelle B, Vergne I. Autophagy in Mycobacterium tuberculosis and HIV infections. Front Cell Infect Mi. 2015 Jun 2;5:49.
- Hu W, Chan H, Lu L, et al. Autophagy in intracellular bacterial infection. Semin Cell Dev Biol. 2020 May;101:41–50.
- Shin DM, Jeon BY, Lee HM, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010 Dec 16;6(12):e1001230.
- Duan L, Yi M, Chen J, et al. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem Biophys Res Commun. 2016 May 13;473(4):1229–1234.
- Cardenal-Munoz E, Arafah S, Lopez-Jimenez AT, et al. Mycobacterium marinum antagonistically induces an autophagic response while repressing the autophagic flux in a TORC1- and ESX-1-dependent manner. PLoS Pathog. 2017 Apr;13(4):e1006344.
- Chandra P, Ghanwat S, Matta SK, et al. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages [Research Support, Non-U.S. Gov’t]. Sci Rep. 2015 Nov 6;5:16320.
- Romagnoli A, Etna MP, Giacomini E, et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy. 2012 Sep;8(9):1357–1370.
- Wehenkel A, Bellinzoni M, Grana M, et al. Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Bba-Proteins Proteom. 2008 Jan;1784(1):193–202.
- Av-Gay Y, Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000 May;8(5):238–244.
- van der Woude AD, Stoop EJ, Stiess M, et al. Analysis of SecA2-dependent substrates in Mycobacterium marinum identifies protein kinase G (PknG) as a virulence effector. Cell Microbiol. 2014 Feb;16(2):280–295.
- Walburger A, Koul A, Ferrari G, et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004 Jun 18;304(5678):1800–1804.
- Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PLoS Pathog. 2018 Apr;14(4):e1007011.
- Paik S, Kim JK, Chung C, et al. Autophagy: a new strategy for host-directed therapy of tuberculosis. Virulence. 2019 Jan 1;10(1):448–459.
- Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005 Feb 18;307(5712):1098–1101.
- Miinea CP, Sano H, Kane S, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005 Oct 1;391(Pt 1):87–93.
- Yoshii SR, Mizushima N. Monitoring and Measuring Autophagy. Int J Mol Sci. 2017 Sep;18(9). DOI:10.3390/ijms18091865
- Romanov J, Walczak M, Ibiricu I, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. Embo J. 2012 Nov 14;31(22):4304–4317.
- Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013 Mar;9(3):424–425.
- Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014 Dec;26(12):2694–2701.
- Singh P, Subbian S. Harnessing the mTOR Pathway for Tuberculosis Treatment. Front Microbiol. 2018 Jan;30;9. DOI:10.1111/j.1365-2133.1975.tb06468.x
- Rieck B, Degiacomi G, Zimmermann M, et al. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS Pathog. 2017 May;13(5):e1006399.
- Kyei GB, Vergne I, Chua J, et al. Rab14 is critical for maintenance of Mycobacterium tuberculosis phagosome maturation arrest. Embo J. 2006 Nov 15;25(22):5250–5259.
- Gurriaran-Rodriguez U, Al-Massadi O, Roca-Rivada A, et al. Obestatin as a regulator of adipocyte metabolism and adipogenesis. J Cell Mol Med. 2011 Sep;15(9):1927–1940.
- Peck GR, Chavez JA, Roach WG, et al. Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. J Biol Chem. 2009 Oct 30;284(44):30016–30023.
- Kuijl C, Savage NDL, Marsman M, et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature. 2007 Nov 29;450(7170):725–U10.
- Sturgillkoszycki S, Schlesinger PH, Chakraborty P, et al. Lack of acidification in mycobacterium phagosomes produced by exclusion of the vesicular proton-atpase. Science. 1994 Feb 4;263(5147):678–681.
- Vergne I, Chua J, Lee HH, et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005 Mar 15;102(11):4033–4038.
- Watson RO, Manzanillo PS, Cox JS, et al. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012 Aug 17;150(4):803–815.
- Franco LH, Nair VR, Scharn CR, et al. The ubiquitin ligase Smurf1 functions in selective autophagy of mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017 Jan 11;21(1):59–72.
- Heath RJ, Goel G, Baxt LA, et al. RNF166 determines recruitment of adaptor proteins during antibacterial autophagy. Cell Rep. 2016 Nov 22;17(9):2183–2194.
- Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013 Sep 26;501(7468):512-+.
- Ponpuak M, Davis AS, Roberts EA, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010 Mar 26;32(3):329–341.
- Chai QY, Wang XD, Qiang LH, et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun. 2019 Apr 29;10. DOI:10.1038/s41467-019-09955-8
- Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016 Nov;16(11):661–675.
- Deretic V, Kimura T, Timmins G, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015 Jan;125(1):75–84.
- Kim YS, Silwal P, Kim SY, et al. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp Mol Med. 2019 Dec 11;51. DOI:10.1038/s12276-019-0290-7
- Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015 Oct;16(10):1014–1024.
- Xiong Q, Yang M, Li P, et al. Bacteria exploit autophagy for their own benefit [Review]. Infect Drug Resist. 2019;12:3205–3215.
- Kumar Y, Valdivia RH. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe. 2009 Jun 18;5(6):593–601.
- Philips JA. Mycobacterial manipulation of vacuolar sorting. Cell Microbiol. 2008 Dec;10(12):2408–2415.
- Kumar D, Nath L, Kamal MA, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010 Mar 5;140(5):731–743.
- Lerner TR, de Souza Carvalho-Wodarz C, Repnik U, et al. Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest. 2016 Mar 1;126(3):1093–1108.
- Jagannath C, Lindsey DR, Dhandayuthapani S, et al. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009 Mar;15(3):267–276.
- Chiramel AI, Best SM. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018 Aug;2(254):34–40.
- Nouwen LV, Everts B. Pathogens MenTORing macrophages and dendritic cells: manipulation of mTOR and cellular metabolism to promote immune escape. Cells-Basel. 2020 Jan;9(1):161.
- Yang B, Xue QH, Guo JN, et al. Autophagy induction by the pathogen receptor NECTIN4 and sustained autophagy contribute to peste des petits ruminants virus infectivity. Autophagy. 2020 May 3;16(5):842–861.
- Yao H, Han XY, Han XZ. The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drug. 2014 Dec;14(6):433–442.
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics [Review]. Nature. 2006 Oct 12;443(7112):651–657.
- Hilbi H. Modulation of phosphoinositide metabolism by pathogenic bacteria. Cell Microbiol. 2006 Nov;8(11):1697–1706.
- Wasilko DJ, Mao YX. Exploiting the ubiquitin and phosphoinositide pathways by the Legionella pneumophila effector, SidC. Curr Genet. 2016 Feb;62(1):105–108.
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009 Aug;10(8):513–525.
- Prashar A, Schnettger L, Bernard EM, et al. Rab GTPases in Immunity and Inflammation. Front Cell Infect Mi. 2017 Sep 29;7:435.
- Spano S, Galan JE. Taking control: Hijacking of Rab GTPases by intracellular bacterial pathogens. Small GTPases. 2018 Mar 4;9(1–2):182–191.
- Kagan JC, Stein MP, Pypaert M, et al. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J Exp Med. 2004 May 3;199(9):1201–1211.
- Mellouk N, Weiner A, Aulner N, et al. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe. 2014 Oct 8;16(4):517–530.
- Mauvezin C, Neisch AL, Ayala CI, et al. Coordination of autophagosome-lysosome fusion and transport by a Klp98A-Rab14 complex in Drosophila. J Cell Sci. 2016 Mar 1;129(5):971–982.
- Scherr N, Honnappa S, Kunz G, et al. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):12151–12156.
- Prisic S, Husson RN. Mycobacterium tuberculosis Serine/Threonine Protein Kinases [Review]. Microbiol Spectr. 2014 Oct;2(5). DOI:10.1128/microbiolspec.MGM2-0006-2013
- Wu FL, Liu Y, Zhang HN, et al. Global profiling of PknG interactions using a human proteome microarray reveals novel connections with CypA. Proteomics. 2018 Dec;18(23):1800265.
- Pradhan G, Shrivastva R, Mukhopadhyay S. Mycobacterial PknG targets the Rab7l1 signaling pathway to inhibit phagosome-lysosome fusion. J Immunol. 2018 Sep 1;201(5):1421–1433.
- Lamber EP, Siedenburg AC, Barr FA. Rab regulation by GEFs and GAPs during membrane traffic [Research Support, Non-U.S. Gov’t Review]. Curr Opin Cell Biol. 2019 Aug;59:34–39.
- Moores RC, Brilha S, Schutgens F, et al. Epigenetic Regulation of Matrix Metalloproteinase-1 and −3 Expression in Mycobacterium tuberculosis Infection. Front Immunol. 2017;8:602.
- Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016 Jul 7;535(7610):111–116.